
Super-Enhancers, Phase-Separated Condensates, and 3D Genome Organization in Cancer



Abstract
:Simple Summary
Abstract
1. Introduction
2. Constituents and Identification of Super-Enhancers
2.1. Super-Enhancers and Chromatin Interactions
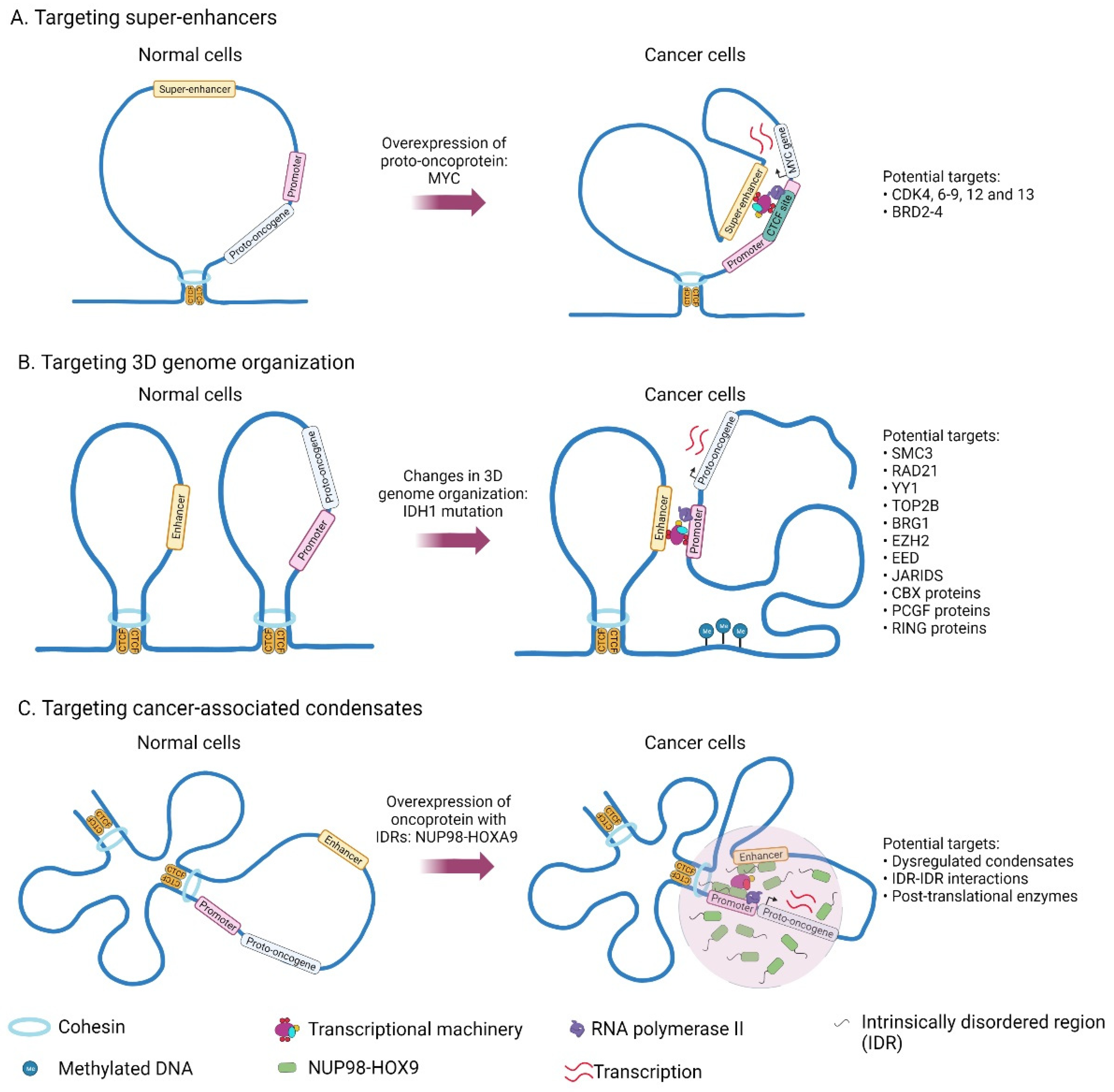
2.2. Mechanisms Related to the Acquisition of Super-Enhancers in Cancer
2.3. Targeting Transcriptional Co-Activators and Chromatin Remodelers
2.4. Drug Resistance to Super-Enhancer Drugs
3. Drugs Targeting 3D Genome Organization
4. Phase Separation Model to Explain Features of Super-Enhancers
4.1. Cooperative Interactions between Transcriptional Machinery and the Genome
4.2. Regulation of Transcriptional Condensates
5. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Furlong, E.E.M.; Levine, M. Developmental enhancers and chromosome topology. Science 2018, 361, 1341–1345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hnisz, D.; Abraham, B.J.; Lee, T.I.; Lau, A.; Saint-Andre, V.; Sigova, A.A.; Hoke, H.A.; Young, R.A. Super-enhancers in the control of cell identity and disease. Cell 2013, 155, 934–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Marco, E.; Pinello, L.; Yuan, G.C. Predicting chromatin organization using histone marks. Genome Biol. 2015, 16, 162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whyte, W.A.; Orlando, D.A.; Hnisz, D.; Abraham, B.J.; Lin, C.Y.; Kagey, M.H.; Rahl, P.B.; Lee, T.I.; Young, R.A. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 2013, 153, 307–319. [Google Scholar] [CrossRef] [Green Version]
- Hnisz, D.; Shrinivas, K.; Young, R.A.; Chakraborty, A.K.; Sharp, P.A. A Phase Separation Model for Transcriptional Control. Cell 2017, 169, 13–23. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, A.L.; Vallurupalli, M.; Chen, L.; Crompton, B.; Cowley, G.; Vazquez, F.; Weir, B.A.; Tsherniak, A.; Parasuraman, S.; Kim, S.; et al. Functional, chemical genomic, and super-enhancer screening identify sensitivity to cyclin D1/CDK4 pathway inhibition in Ewing sarcoma. Oncotarget 2015, 6, 30178–30193. [Google Scholar] [CrossRef] [Green Version]
- Riggi, N.; Knoechel, B.; Gillespie, S.M.; Rheinbay, E.; Boulay, G.; Suva, M.L.; Rossetti, N.E.; Boonseng, W.E.; Oksuz, O.; Cook, E.B.; et al. EWS-FLI1 utilizes divergent chromatin remodeling mechanisms to directly activate or repress enhancer elements in Ewing sarcoma. Cancer Cell 2014, 26, 668–681. [Google Scholar] [CrossRef] [Green Version]
- Niederriter, A.R.; Varshney, A.; Parker, S.C.; Martin, D.M. Super Enhancers in Cancers, Complex Disease, and Developmental Disorders. Genes 2015, 6, 1183–1200. [Google Scholar] [CrossRef]
- Jia, Y.; Zhou, J.; Tan, T.K.; Chung, T.H.; Chen, Y.; Chooi, J.Y.; Sanda, T.; Fullwood, M.J.; Xiong, S.; Toh, S.H.M.; et al. Super Enhancer-Mediated Upregulation of HJURP Promotes Growth and Survival of t(4;14)-Positive Multiple Myeloma. Cancer Res. 2022, 82, 406–418. [Google Scholar] [CrossRef]
- McKeown, M.R.; Corces, M.R.; Eaton, M.L.; Fiore, C.; Lee, E.; Lopez, J.T.; Chen, M.W.; Smith, D.; Chan, S.M.; Koenig, J.L.; et al. Superenhancer Analysis Defines Novel Epigenomic Subtypes of Non-APL AML, Including an RARalpha Dependency Targetable by SY-1425, a Potent and Selective RARalpha Agonist. Cancer Discov. 2017, 7, 1136–1153. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Reinberg, D. Chromatin higher-order structures and gene regulation. Curr. Opin. Genet. Dev. 2011, 21, 175–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lieberman-Aiden, E.; van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McArthur, E.; Capra, J.A. Topologically associating domain boundaries that are stable across diverse cell types are evolutionarily constrained and enriched for heritability. Am. J. Hum. Genet. 2021, 108, 269–283. [Google Scholar] [CrossRef] [PubMed]
- Banigan, E.J.; van den Berg, A.A.; Brandao, H.B.; Marko, J.F.; Mirny, L.A. Chromosome organization by one-sided and two-sided loop extrusion. Elife 2020, 9, e53558. [Google Scholar] [CrossRef] [Green Version]
- Davidson, I.F.; Peters, J.M. Genome folding through loop extrusion by SMC complexes. Nat. Rev. Mol. Cell Biol. 2021, 22, 445–464. [Google Scholar] [CrossRef]
- Dowen, J.M.; Fan, Z.P.; Hnisz, D.; Ren, G.; Abraham, B.J.; Zhang, L.N.; Weintraub, A.S.; Schujiers, J.; Lee, T.I.; Zhao, K.; et al. Control of cell identity genes occurs in insulated neighborhoods in mammalian chromosomes. Cell 2014, 159, 374–387. [Google Scholar] [CrossRef] [Green Version]
- Ji, X.; Dadon, D.B.; Powell, B.E.; Fan, Z.P.; Borges-Rivvera, D.; Shachar, S.; Weintraub, A.S.; Hnisz, D.; Pegoraro, G.; Lee, T.I.; et al. 3D Chromosome Regulatory Landscape of Human Pluripotent Cells. Cell Stem Cell 2016, 18, 262–275. [Google Scholar] [CrossRef] [Green Version]
- Hnisz, D.; Day, D.S.; Young, R.A. Insulated Neighborhoods: Structural and Functional Units of Mammalian Gene Control. Cell 2016, 167, 1188–1200. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Li, K.; Cai, W.; Liu, X.; Zhang, Y.; Orkin, S.H.; Xu, J.; Yuan, G.C. Dissecting super-enhancer hierarchy based on chromatin interactions. Nature Commun. 2018, 9, 943. [Google Scholar] [CrossRef] [Green Version]
- Shin, H.Y. The structural and functional roles of CTCF in the regulation of cell type-specific and human disease-associated super-enhancers. Genes Genom. 2019, 41, 257–265. [Google Scholar] [CrossRef]
- Mullenders, J.; Aranda-Orgilles, B.; Lhoumaud, P.; Keller, M.; Pae, J.; Wang, K.; Kayembe, C.; Rocha, P.P.; Raviram, R.; Gong, Y.; et al. Cohesin loss alters adult hematopoietic stem cell homeostasis, leading to myeloproliferative neoplasms. J. Exp. Med. 2015, 212, 1833–1850. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Mathelier, A.; Zhang, X. Super-enhancers are transcriptionally more active and cell type-specific than stretch enhancers. Epigenetics 2018, 13, 910–922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Zang, C.; Taing, L.; Arnett, K.L.; Wong, Y.J.; Pear, W.S.; Blacklow, S.C.; Liu, X.S.; Aster, J.C. NOTCH1-RBPJ complexes drive target gene expression through dynamic interactions with superenhancers. Proc. Natl. Acad. Sci. USA 2014, 111, 705–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, Y.; Lazaris, C.; Sakellaropoulos, T.; Lozano, A.; Kambadur, P.; Ntziachristos, P.; Aifantis, I.; Tsirigos, A. Stratification of TAD boundaries reveals preferential insulation of super-enhancers by strong boundaries. Nat. Commun. 2018, 9, 542. [Google Scholar] [CrossRef] [Green Version]
- Willi, M.; Yoo, K.H.; Reinisch, F.; Kuhns, T.M.; Lee, H.K.; Wang, C.; Hennighausen, L. Facultative CTCF sites moderate mammary super-enhancer activity and regulate juxtaposed gene in non-mammary cells. Nat. Commun. 2017, 8, 16069. [Google Scholar] [CrossRef] [Green Version]
- Hnisz, D.; Weintraub, A.S.; Day, D.S.; Valton, A.L.; Bak, R.O.; Li, C.H.; Goldmann, J.; Lajoie, B.R.; Fan, Z.P.; Sigova, A.A.; et al. Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science 2016, 351, 1454–1458. [Google Scholar] [CrossRef] [Green Version]
- Schuijers, J.; Manteiga, J.C.; Weintraub, A.S.; Day, D.S.; Zamudio, A.V.; Hnisz, D.; Lee, T.I.; Young, R.A. Transcriptional Dysregulation of MYC Reveals Common Enhancer-Docking Mechanism. Cell Rep. 2018, 23, 349–360. [Google Scholar] [CrossRef] [Green Version]
- Kloetgen, A.; Thandapani, P.; Ntziachristos, P.; Ghebrechristos, Y.; Nomikou, S.; Lazaris, C.; Chen, X.; Hu, H.; Bakogianni, S.; Wang, J.; et al. Three-dimensional chromatin landscapes in T cell acute lymphoblastic leukemia. Nat. Genet. 2020, 52, 388–400. [Google Scholar] [CrossRef]
- Benner, C.; Isoda, T.; Murre, C. New roles for DNA cytosine modification, eRNA, anchors, and superanchors in developing B cell progenitors. Proc. Natl. Acad. Sci. USA 2015, 112, 12776–12781. [Google Scholar] [CrossRef] [Green Version]
- Vian, L.; Pekowska, A.; Rao, S.S.P.; Kieffer-Kwon, K.R.; Jung, S.; Baranello, L.; Huang, S.C.; El Khattabi, L.; Dose, M.; Pruett, N.; et al. The Energetics and Physiological Impact of Cohesin Extrusion. Cell 2018, 173, 1165–1178.e20. [Google Scholar] [CrossRef] [Green Version]
- Tarjan, D.R.; Flavahan, W.A.; Bernstein, B.E. Epigenome editing strategies for the functional annotation of CTCF insulators. Nat. Commun. 2019, 10, 4258. [Google Scholar] [CrossRef] [PubMed]
- Pflueger, C.; Tan, D.; Swain, T.; Nguyen, T.; Pflueger, J.; Nefzger, C.; Polo, J.M.; Ford, E.; Lister, R. A modular dCas9-SunTag DNMT3A epigenome editing system overcomes pervasive off-target activity of direct fusion dCas9-DNMT3A constructs. Genome Res. 2018, 28, 1193–1206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, Y.; Zhang, X.; Su, J.; Jeong, M.; Gundry, M.C.; Huang, Y.H.; Zhou, Y.; Li, W.; Goodell, M.A. Targeted DNA methylation in vivo using an engineered dCas9-MQ1 fusion protein. Nat. Commun. 2017, 8, 16026. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.M.; Tollefsbol, T.O. Targeting cancer epigenetics with CRISPR-dCAS9: Principles and prospects. Methods 2021, 187, 77–91. [Google Scholar] [CrossRef]
- Behr, M.; Zhou, J.; Xu, B.; Zhang, H. In vivo delivery of CRISPR-Cas9 therapeutics: Progress and challenges. Acta Pharm. Sin. B 2021, 11, 2150–2171. [Google Scholar] [CrossRef]
- Jia, Q.; Chen, S.; Tan, Y.; Li, Y.; Tang, F. Oncogenic super-enhancer formation in tumorigenesis and its molecular mechanisms. Exp. Mol. Med. 2020, 52, 713–723. [Google Scholar] [CrossRef]
- Jia, Y.; Chng, W.J.; Zhou, J. Super-enhancers: Critical roles and therapeutic targets in hematologic malignancies. J. Hematol. Oncol. 2019, 12, 77. [Google Scholar] [CrossRef]
- Sengupta, S.; George, R.E. Super-Enhancer-Driven Transcriptional Dependencies in Cancer. Trends Cancer 2017, 3, 269–281. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Cairns, M.J.; Yan, J. Super-enhancers in transcriptional regulation and genome organization. Nucleic Acids Res. 2019, 47, 11481–11496. [Google Scholar] [CrossRef] [Green Version]
- Bradner, J.E.; Hnisz, D.; Young, R.A. Transcriptional Addiction in Cancer. Cell 2017, 168, 629–643. [Google Scholar] [CrossRef] [Green Version]
- Loven, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Togel, L.; Nightingale, R.; Chueh, A.C.; Jayachandran, A.; Tran, H.; Phesse, T.; Wu, R.; Sieber, O.M.; Arango, D.; Dhillon, A.S.; et al. Dual Targeting of Bromodomain and Extraterm.minal Domain Proteins, and WNT or MAPK Signaling, Inhibits c-MYC Expression and Proliferation of Colorectal Cancer Cells. Mol. Cancer Ther. 2016, 15, 1217–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Ma, P.; Jing, Y.; Yan, Y.; Cai, M.C.; Zhang, M.; Zhang, S.; Peng, H.; Ji, Z.L.; Di, W.; et al. BET Bromodomain Inhibition as a Therapeutic Strategy in Ovarian Cancer by Downregulating FoxM1. Theranostics 2016, 6, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, D.; Kannan, A.; Kern, M.; Moreno, M.A.; Vural, E.; Stack, B., Jr.; Suen, J.Y.; Tackett, A.J.; Gao, L. Disruption of BRD4 at H3K27Ac-enriched enhancer region correlates with decreased c-Myc expression in Merkel cell carcinoma. Epigenetics 2015, 10, 460–466. [Google Scholar] [CrossRef] [Green Version]
- Chapuy, B.; McKeown, M.R.; Lin, C.Y.; Monti, S.; Roemer, M.G.; Qi, J.; Rahl, P.B.; Sun, H.H.; Yeda, K.T.; Doench, J.G.; et al. Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer Cell 2013, 24, 777–790. [Google Scholar] [CrossRef] [Green Version]
- Gryder, B.E.; Yohe, M.E.; Chou, H.C.; Zhang, X.; Marques, J.; Wachtel, M.; Schaefer, B.; Sen, N.; Song, Y.; Gualtieri, A.; et al. PAX3-FOXO1 Establishes Myogenic Super Enhancers and Confers BET Bromodomain Vulnerability. Cancer Discov. 2017, 7, 884–899. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Wang, S.; Nie, D.; Lai, P.; Li, Y.; Li, Y.; Jin, Y.; Pan, J. Super-enhancer landscape reveals leukemia stem cell reliance on X-box binding protein 1 as a therapeutic vulnerability. Sci. Transl. Med. 2021, 13, eabh3462. [Google Scholar] [CrossRef]
- Jiang, Y.Y.; Lin, D.C.; Mayakonda, A.; Hazawa, M.; Ding, L.W.; Chien, W.W.; Xu, L.; Chen, Y.; Xiao, J.F.; Senapedis, W.; et al. Targeting super-enhancer-associated oncogenes in oesophageal squamous cell carcinoma. Gut 2017, 66, 1358–1368. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhang, T.; Kwiatkowski, N.; Abraham, B.J.; Lee, T.I.; Xie, S.; Yuzugullu, H.; Von, T.; Li, H.; Lin, Z.; et al. CDK7-dependent transcriptional addiction in triple-negative breast cancer. Cell 2015, 163, 174–186. [Google Scholar] [CrossRef] [Green Version]
- Chipumuro, E.; Marco, E.; Christensen, C.L.; Kwiatkowski, N.; Zhang, T.; Hatheway, C.M.; Abraham, B.J.; Sharma, B.; Yeung, C.; Altabef, A.; et al. CDK7 inhibition suppresses super-enhancer-linked oncogenic transcription in MYCN-driven cancer. Cell 2014, 159, 1126–1139. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Z.J.; Miao, D.L.; Su, Q.Y.; Tang, X.L.; Wang, X.L.; Deng, L.B.; Shi, H.D.; Xin, H.B. THZ1 suppresses human non-small-cell lung cancer cells in vitro through interference with cancer metabolism. Acta Pharmacol. Sin. 2019, 40, 814–822. [Google Scholar] [CrossRef] [PubMed]
- Alver, B.H.; Kim, K.H.; Lu, P.; Wang, X.; Manchester, H.E.; Wang, W.; Haswell, J.R.; Park, P.J.; Roberts, C.W. The SWI/SNF chromatin remodelling complex is required for maintenance of lineage specific enhancers. Nat. Commun. 2017, 8, 14648. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Parolia, A.; Qiao, Y.; Bawa, P.; Eyunni, S.; Mannan, R.; Carson, S.E.; Chang, Y.; Wang, X.; Zhang, Y.; et al. Targeting SWI/SNF ATPases in enhancer-addicted prostate cancer. Nature 2022, 601, 434–439. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Wang, L.; Zhang, S.; Bennett, B.D.; He, F.; Zhang, Y.; Xiong, C.; Han, L.; Diao, L.; Li, P.; et al. INO80 governs superenhancer-mediated oncogenic transcription and tumor growth in melanoma. Genes Dev. 2016, 30, 1440–1453. [Google Scholar] [CrossRef] [PubMed]
- Marques, J.G.; Gryder, B.E.; Pavlovic, B.; Chung, Y.; Ngo, Q.A.; Frommelt, F.; Gstaiger, M.; Song, Y.; Benischke, K.; Laubscher, D.; et al. NuRD subunit CHD4 regulates super-enhancer accessibility in rhabdomyosarcoma and represents a general tumor dependency. eLife 2020, 9, e54993. [Google Scholar] [CrossRef] [PubMed]
- Sugino, N.; Kawahara, M.; Tatsumi, G.; Kanai, A.; Matsui, H.; Yamamoto, R.; Nagai, Y.; Fujii, S.; Shimazu, Y.; Hishizawa, M.; et al. A novel LSD1 inhibitor NCD38 ameliorates MDS-related leukemia with complex karyotype by attenuating leukemia programs via activating super-enhancers. Leukemia 2017, 31, 2303–2314. [Google Scholar] [CrossRef] [PubMed]
- Tatsumi, G.; Kawahara, M.; Yamamoto, R.; Hishizawa, M.; Kito, K.; Suzuki, T.; Takaori-Kondo, A.; Andoh, A. LSD1-mediated repression of GFI1 super-enhancer plays an essential role in erythroleukemia. Leukemia 2020, 34, 746–758. [Google Scholar] [CrossRef]
- Pelish, H.E.; Liau, B.B.; Nitulescu, I.I.; Tangpeerachaikul, A.; Poss, Z.C.; Da Silva, D.H.; Caruso, B.T.; Arefolov, A.; Fadeyi, O.; Christie, A.L.; et al. Mediator kinase inhibition further activates super-enhancer-associated genes in AML. Nature 2015, 526, 273–276. [Google Scholar] [CrossRef]
- Noel, P.; Hussein, S.; Ng, S.; Antal, C.E.; Lin, W.; Rodela, E.; Delgado, P.; Naveed, S.; Downes, M.; Lin, Y.; et al. Triptolide targets super-enhancer networks in pancreatic cancer cells and cancer-associated fibroblasts. Oncogenesis 2020, 9, 100. [Google Scholar] [CrossRef]
- Ghosh, C.; Paul, S.; Dandawate, P.; Gunewardena, S.S.; Subramaniam, D.; West, C.; Anant, S.; Dhar, A. Super-enhancers: Novel target for pancreatic ductal adenocarcinoma. Oncotarget 2019, 10, 1554–1571. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Zhao, Z.; Huang, Z.; Chen, D.C.; Zhu, X.X.; Wang, Y.Z.; Yan, Y.W.; Tang, S.; Madhavan, S.; Ni, W.; et al. Super enhancer inhibitors suppress MYC driven transcriptional amplification and tumor progression in osteosarcoma. Bone Res. 2018, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Marineau, J.J.; Rajagopal, N.; Hamman, K.B.; Choi, Y.J.; Schmidt, D.R.; Ke, N.; Johannessen, L.; Bradley, M.J.; Orlando, D.A.; et al. Discovery and Characterization of SY-1365, a Selective, Covalent Inhibitor of CDK7. Cancer Res. 2019, 79, 3479–3491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwiatkowski, N.; Zhang, T.; Rahl, P.B.; Abraham, B.J.; Reddy, J.; Ficarro, S.B.; Dastur, A.; Amzallag, A.; Ramaswamy, S.; Tesar, B.; et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature 2014, 511, 616–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharifnia, T.; Wawer, M.J.; Chen, T.; Huang, Q.Y.; Weir, B.A.; Sizemore, A.; Lawlor, M.A.; Goodale, A.; Cowley, G.S.; Vazquez, F.; et al. Small-molecule targeting of brachyury transcription factor addiction in chordoma. Nat. Med. 2019, 25, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Marineau, J.J.; Hamman, K.B.; Hu, S.; Alnemy, S.; Mihalich, J.; Kabro, A.; Whitmore, K.M.; Winter, D.K.; Roy, S.; Ciblat, S.; et al. Discovery of SY-5609: A Selective, Noncovalent Inhibitor of CDK7. J. Med. Chem. 2022, 65, 1458–1480. [Google Scholar] [CrossRef]
- Tripathy, D.; Bardia, A.; Sellers, W.R. Ribociclib (LEE011): Mechanism of Action and Clinical Impact of This Selective Cyclin-Dependent Kinase 4/6 Inhibitor in Various Solid Tumors. Clin. Cancer Res. 2017, 23, 3251–3262. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Kwiatkowski, N.; Olson, C.M.; Dixon-Clarke, S.E.; Abraham, B.J.; Greifenberg, A.K.; Ficarro, S.B.; Elkins, J.M.; Liang, Y.; Hannett, N.M.; et al. Covalent targeting of remote cysteine residues to develop CDK12 and CDK13 inhibitors. Nat. Chem. Biol. 2016, 12, 876–884. [Google Scholar] [CrossRef] [Green Version]
- Rzymski, T.; Mikula, M.; Zylkiewicz, E.; Dreas, A.; Wiklik, K.; Golas, A.; Wojcik, K.; Masiejczyk, M.; Wrobel, A.; Dolata, I.; et al. SEL120-34A is a novel CDK8 inhibitor active in AML cells with high levels of serine phosphorylation of STAT1 and STAT5 transactivation domains. Oncotarget 2017, 8, 33779–33795. [Google Scholar] [CrossRef] [Green Version]
- Ceribelli, M.; Hou, Z.E.; Kelly, P.N.; Huang, D.W.; Wright, G.; Ganapathi, K.; Evbuomwan, M.O.; Pittaluga, S.; Shaffer, A.L.; Marcucci, G.; et al. A Druggable TCF4- and BRD4-Dependent Transcriptional Network Sustains Malignancy in Blastic Plasmacytoid Dendritic Cell Neoplasm. Cancer Cell 2016, 30, 764–778. [Google Scholar] [CrossRef] [Green Version]
- Amorim, S.; Stathis, A.; Gleeson, M.; Iyengar, S.; Magarotto, V.; Leleu, X.; Morschhauser, F.; Karlin, L.; Broussais, F.; Rezai, K.; et al. Bromodomain inhibitor OTX015 in patients with lymphoma or multiple myeloma: A dose-escalation, open-label, pharmacokinetic, phase 1 study. Lancet Haematol. 2016, 3, e196–e204. [Google Scholar] [CrossRef]
- Gerlach, D.; Tontsch-Grunt, U.; Baum, A.; Popow, J.; Scharn, D.; Hofmann, M.H.; Engelhardt, H.; Kaya, O.; Beck, J.; Schweifer, N.; et al. The novel BET bromodomain inhibitor BI 894999 represses super-enhancer-associated transcription and synergizes with CDK9 inhibition in AML. Oncogene 2018, 37, 2687–2701. [Google Scholar] [CrossRef]
- Albrecht, B.K.; Gehling, V.S.; Hewitt, M.C.; Vaswani, R.G.; Cote, A.; Leblanc, Y.; Nasveschuk, C.G.; Bellon, S.; Bergeron, L.; Campbell, R.; et al. Identification of a Benzoisoxazoloazepine Inhibitor (CPI-0610) of the Bromodomain and Extra-Terminal (BET) Family as a Candidate for Human Clinical Trials. J. Med. Chem. 2016, 59, 1330–1339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shu, S.; Lin, C.Y.; He, H.H.; Witwicki, R.M.; Tabassum, D.P.; Roberts, J.M.; Janiszewska, M.; Huh, S.J.; Liang, Y.; Ryan, J.; et al. Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer. Nature 2016, 529, 413–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Liu, X.; Han, L.; Chen, X.; Wu, X.; Wu, J.; Yan, D.; Wang, Y.; Liu, S.; Shan, L.; et al. BRD4-directed super-enhancer organization of transcription repression programs links to chemotherapeutic efficacy in breast cancer. Proc. Natl. Acad. Sci. USA 2022, 119, e2109133119. [Google Scholar] [CrossRef]
- Fong, C.Y.; Gilan, O.; Lam, E.Y.; Rubin, A.F.; Ftouni, S.; Tyler, D.; Stanley, K.; Sinha, D.; Yeh, P.; Morison, J.; et al. BET inhibitor resistance emerges from leukaemia stem cells. Nature 2015, 525, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Rathert, P.; Roth, M.; Neumann, T.; Muerdter, F.; Roe, J.S.; Muhar, M.; Deswal, S.; Cerny-Reiterer, S.; Peter, B.; Jude, J.; et al. Transcriptional plasticity promotes primary and acquired resistance to BET inhibition. Nature 2015, 525, 543–547. [Google Scholar] [CrossRef] [PubMed]
- Valton, A.L.; Dekker, J. TAD disruption as oncogenic driver. Curr. Opin. Genet. Dev. 2016, 36, 34–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flavahan, W.A.; Drier, Y.; Liau, B.B.; Gillespie, S.M.; Venteicher, A.S.; Stemmer-Rachamimov, A.O.; Suva, M.L.; Bernstein, B.E. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 2016, 529, 110–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akincilar, S.C.; Khattar, E.; Boon, P.L.; Unal, B.; Fullwood, M.J.; Tergaonkar, V. Long-Range Chromatin Interactions Drive Mutant TERT Promoter Activation. Cancer Discov. 2016, 6, 1276–1291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Animesh, S.; Choudhary, R.; Wong, B.J.H.; Koh, C.T.J.; Ng, X.Y.; Tay, J.K.X.; Chong, W.Q.; Jian, H.; Chen, L.; Goh, B.C.; et al. Profiling of 3D Genome Organization in Nasopharyngeal Cancer Needle Biopsy Patient Samples by a Modified Hi-C Approach. Front. Genet. 2021, 12, 673530. [Google Scholar] [CrossRef]
- Szabo, Q.; Bantignies, F.; Cavalli, G. Principles of genome folding into topologically associating domains. Sci. Adv. 2019, 5, eaaw1668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuin, J.; Dixon, J.R.; van der Reijden, M.I.; Ye, Z.; Kolovos, P.; Brouwer, R.W.; van de Corput, M.P.; van de Werken, H.J.; Knoch, T.A.; van, I.W.F.; et al. Cohesin and CTCF differentially affect chromatin architecture and gene expression in human cells. Proc. Natl. Acad. Sci. USA 2014, 111, 996–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, J.M.; Tedeschi, A.; Schmitz, J. The cohesin complex and its roles in chromosome biology. Genes Dev. 2008, 22, 3089–3114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, S.S.P.; Huang, S.C.; Glenn St Hilaire, B.; Engreitz, J.M.; Perez, E.M.; Kieffer-Kwon, K.R.; Sanborn, A.L.; Johnstone, S.E.; Bascom, G.D.; Bochkov, I.D.; et al. Cohesin Loss Eliminates All Loop Domains. Cell 2017, 171, 305–320.e24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarzer, W.; Abdennur, N.; Goloborodko, A.; Pekowska, A.; Fudenberg, G.; Loe-Mie, Y.; Fonseca, N.A.; Huber, W.; Haering, C.H.; Mirny, L.; et al. Two independent modes of chromatin organization revealed by cohesin removal. Nature 2017, 551, 51–56. [Google Scholar] [CrossRef] [Green Version]
- Antony, J.; Chin, C.V.; Horsfield, J.A. Cohesin Mutations in Cancer: Emerging Therapeutic Targets. Int. J. Mol. Sci. 2021, 22, 6788. [Google Scholar] [CrossRef]
- Kang, H.; Lieberman, P.M. Mechanism of glycyrrhizic acid inhibition of Kaposi’s sarcoma-associated herpesvirus: Disruption of CTCF-cohesin-mediated RNA polymerase II pausing and sister chromatid cohesion. J. Virol. 2011, 85, 11159–11169. [Google Scholar] [CrossRef] [Green Version]
- Dasgupta, T.; Antony, J.; Braithwaite, A.W.; Horsfield, J.A. HDAC8 Inhibition Blocks SMC3 Deacetylation and Delays Cell Cycle Progression without Affecting Cohesin-dependent Transcription in MCF7 Cancer Cells. J. Biol. Chem. 2016, 291, 12761–12770. [Google Scholar] [CrossRef] [Green Version]
- van der Lelij, P.; Lieb, S.; Jude, J.; Wutz, G.; Santos, C.P.; Falkenberg, K.; Schlattl, A.; Ban, J.; Schwentner, R.; Hoffmann, T.; et al. Synthetic lethality between the cohesin subunits STAG1 and STAG2 in diverse cancer contexts. eLife 2017, 6, e26980. [Google Scholar] [CrossRef] [Green Version]
- Weintraub, A.S.; Li, C.H.; Zamudio, A.V.; Sigova, A.A.; Hannett, N.M.; Day, D.S.; Abraham, B.J.; Cohen, M.A.; Nabet, B.; Buckley, D.L.; et al. YY1 Is a Structural Regulator of Enhancer-Promoter Loops. Cell 2017, 171, 1573–1588.e28. [Google Scholar] [CrossRef] [Green Version]
- Bonavida, B. Therapeutic YY1 Inhibitors in Cancer: ALL in ONE. Crit. Rev. Oncog. 2017, 22, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Donohoe, M.E.; Zhang, X.; McGinnis, L.; Biggers, J.; Li, E.; Shi, Y. Targeted disruption of mouse Yin Yang 1 transcription factor results in peri-implantation lethality. Mol. Cell Biol. 1999, 19, 7237–7244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nora, E.P.; Goloborodko, A.; Valton, A.L.; Gibcus, J.H.; Uebersohn, A.; Abdennur, N.; Dekker, J.; Mirny, L.A.; Bruneau, B.G. Targeted Degradation of CTCF Decouples Local Insulation of Chromosome Domains from Genomic Compartmentalization. Cell 2017, 169, 930–944.e22. [Google Scholar] [CrossRef] [Green Version]
- Hyle, J.; Zhang, Y.; Wright, S.; Xu, B.; Shao, Y.; Easton, J.; Tian, L.; Feng, R.; Xu, P.; Li, C. Acute depletion of CTCF directly affects MYC regulation through loss of enhancer-promoter looping. Nucleic Acids Res. 2019, 47, 6699–6713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasparian, A.V.; Burkhart, C.A.; Purmal, A.A.; Brodsky, L.; Pal, M.; Saranadasa, M.; Bosykh, D.A.; Commane, M.; Guryanova, O.A.; Pal, S.; et al. Curaxins: Anticancer compounds that simultaneously suppress NF-kappaB and activate p53 by targeting FACT. Sci. Transl. Med. 2011, 3, 95ra74. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Neznanov, N.; Wilfong, C.D.; Fleyshman, D.I.; Purmal, A.A.; Haderski, G.; Stanhope-Baker, P.; Burkhart, C.A.; Gurova, K.V.; Gudkov, A.V.; et al. Preclinical Validation of a Single-Treatment Infusion Modality That Can Eradicate Extremity Melanomas. Cancer Res. 2016, 76, 6620–6630. [Google Scholar] [CrossRef] [Green Version]
- Dermawan, J.K.; Hitomi, M.; Silver, D.J.; Wu, Q.; Sandlesh, P.; Sloan, A.E.; Purmal, A.A.; Gurova, K.V.; Rich, J.N.; Lathia, J.D.; et al. Pharmacological Targeting of the Histone Chaperone Complex FACT Preferentially Eliminates Glioblastoma Stem Cells and Prolongs Survival in Preclinical Models. Cancer Res. 2016, 76, 2432–2442. [Google Scholar] [CrossRef] [Green Version]
- Barone, T.A.; Burkhart, C.A.; Safina, A.; Haderski, G.; Gurova, K.V.; Purmal, A.A.; Gudkov, A.V.; Plunkett, R.J. Anticancer drug candidate CBL0137, which inhibits histone chaperone FACT, is efficacious in preclinical orthotopic models of temozolomide-responsive and -resistant glioblastoma. Neuro Oncol. 2017, 19, 186–196. [Google Scholar] [CrossRef] [Green Version]
- Carter, D.R.; Murray, J.; Cheung, B.B.; Gamble, L.; Koach, J.; Tsang, J.; Sutton, S.; Kalla, H.; Syed, S.; Gifford, A.J.; et al. Therapeutic targeting of the MYC signal by inhibition of histone chaperone FACT in neuroblastoma. Sci. Transl. Med. 2015, 7, 312ra176. [Google Scholar] [CrossRef] [Green Version]
- Burkhart, C.; Fleyshman, D.; Kohrn, R.; Commane, M.; Garrigan, J.; Kurbatov, V.; Toshkov, I.; Ramachandran, R.; Martello, L.; Gurova, K.V. Curaxin CBL0137 eradicates drug resistant cancer stem cells and potentiates efficacy of gemcitabine in preclinical models of pancreatic cancer. Oncotarget 2014, 5, 11038–11053. [Google Scholar] [CrossRef] [Green Version]
- Kantidze, O.L.; Luzhin, A.V.; Nizovtseva, E.V.; Safina, A.; Valieva, M.E.; Golov, A.K.; Velichko, A.K.; Lyubitelev, A.V.; Feofanov, A.V.; Gurova, K.V.; et al. The anti-cancer drugs curaxins target spatial genome organization. Nat. Commun. 2019, 10, 1441. [Google Scholar] [CrossRef] [Green Version]
- Xiao, T.; Li, X.; Felsenfeld, G. The Myc-associated zinc finger protein (MAZ) works together with CTCF to control cohesin positioning and genome organization. Proc. Natl. Acad. Sci. USA 2021, 118, e2023127118. [Google Scholar] [CrossRef] [PubMed]
- Uuskula-Reimand, L.; Hou, H.; Samavarchi-Tehrani, P.; Rudan, M.V.; Liang, M.; Medina-Rivera, A.; Mohammed, H.; Schmidt, D.; Schwalie, P.; Young, E.J.; et al. Topoisomerase II beta interacts with cohesin and CTCF at topological domain borders. Genome Biol. 2016, 17, 182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Q.; Yu, M.; Tirado-Magallanes, R.; Li, B.; Kong, L.; Guo, M.; Tan, Z.H.; Lee, S.; Chai, L.; Numata, A.; et al. ZNF143 mediates CTCF-bound promoter-enhancer loops required for murine hematopoietic stem and progenitor cell function. Nat. Commun. 2021, 12, 43. [Google Scholar] [CrossRef] [PubMed]
- Barisic, D.; Stadler, M.B.; Iurlaro, M.; Schubeler, D. Mammalian ISWI and SWI/SNF selectively mediate binding of distinct transcription factors. Nature 2019, 569, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Marino, M.M.; Rega, C.; Russo, R.; Valletta, M.; Gentile, M.T.; Esposito, S.; Baglivo, I.; De Feis, I.; Angelini, C.; Xiao, T.; et al. Interactome mapping defines BRG1, a component of the SWI/SNF chromatin remodeling complex, as a new partner of the transcriptional regulator CTCF. J. Biol. Chem. 2019, 294, 861–873. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Scannell, D.R.; Eisen, M.B.; Tjian, R. Control of embryonic stem cell lineage commitment by core promoter factor, TAF3. Cell 2011, 146, 720–731. [Google Scholar] [CrossRef] [Green Version]
- Pena-Hernandez, R.; Marques, M.; Hilmi, K.; Zhao, T.; Saad, A.; Alaoui-Jamali, M.A.; del Rincon, S.V.; Ashworth, T.; Roy, A.L.; Emerson, B.M.; et al. Genome-wide targeting of the epigenetic regulatory protein CTCF to gene promoters by the transcription factor TFII-I. Proc. Natl. Acad. Sci. USA 2015, 112, E677–E686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yusufzai, T.M.; Tagami, H.; Nakatani, Y.; Felsenfeld, G. CTCF tethers an insulator to subnuclear sites, suggesting shared insulator mechanisms across species. Mol. Cell 2004, 13, 291–298. [Google Scholar] [CrossRef]
- Lehman, B.J.; Lopez-Diaz, F.J.; Santisakultarm, T.P.; Fang, L.; Shokhirev, M.N.; Diffenderfer, K.E.; Manor, U.; Emerson, B.M. Dynamic regulation of CTCF stability and sub-nuclear localization in response to stress. PLoS Genet. 2021, 17, e1009277. [Google Scholar] [CrossRef]
- van de Nobelen, S.; Rosa-Garrido, M.; Leers, J.; Heath, H.; Soochit, W.; Joosen, L.; Jonkers, I.; Demmers, J.; van der Reijden, M.; Torrano, V.; et al. CTCF regulates the local epigenetic state of ribosomal DNA repeats. Epigenetics Chromatin 2010, 3, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falini, B.; Sciabolacci, S.; Falini, L.; Brunetti, L.; Martelli, M.P. Diagnostic and therapeutic pitfalls in NPM1-mutated AML: Notes from the field. Leukemia 2021, 35, 3113–3126. [Google Scholar] [CrossRef] [PubMed]
- Smits, A.H.; Jansen, P.W.; Poser, I.; Hyman, A.A.; Vermeulen, M. Stoichiometry of chromatin-associated protein complexes revealed by label-free quantitative mass spectrometry-based proteomics. Nucleic Acids Res. 2013, 41, e28. [Google Scholar] [CrossRef] [PubMed]
- Donaldson-Collier, M.C.; Sungalee, S.; Zufferey, M.; Tavernari, D.; Katanayeva, N.; Battistello, E.; Mina, M.; Douglass, K.M.; Rey, T.; Raynaud, F.; et al. EZH2 oncogenic mutations drive epigenetic, transcriptional, and structural changes within chromatin domains. Nature Genet. 2019, 51, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Zhang, Y.; Loh, Y.P.; Tng, J.Q.; Lim, M.C.; Cao, Z.; Raju, A.; Lieberman Aiden, E.; Li, S.; Manikandan, L.; et al. H3K27me3-rich genomic regions can function as silencers to repress gene expression via chromatin interactions. Nature Commun. 2021, 12, 719. [Google Scholar] [CrossRef]
- Zhang, X.; Jeong, M.; Huang, X.; Wang, X.Q.; Wang, X.; Zhou, W.; Shamim, M.S.; Gore, H.; Himadewi, P.; Liu, Y.; et al. Large DNA Methylation Nadirs Anchor Chromatin Loops Maintaining Hematopoietic Stem Cell Identity. Mol. Cell 2020, 78, 506–521.e6. [Google Scholar] [CrossRef]
- Kim, K.H.; Roberts, C.W. Targeting EZH2 in cancer. Nat. Med. 2016, 22, 128–134. [Google Scholar] [CrossRef]
- Joshi, O.; Wang, S.Y.; Kuznetsova, T.; Atlasi, Y.; Peng, T.; Fabre, P.J.; Habibi, E.; Shaik, J.; Saeed, S.; Handoko, L.; et al. Dynamic Reorganization of Extremely Long-Range Promoter-Promoter Interactions between Two States of Pluripotency. Cell Stem Cell 2015, 17, 748–757. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.L.; Zhu, K.; Zhang, H. An overview of the development of EED inhibitors to disable the PRC2 function. RSC Med. Chem. 2022, 13, 39–53. [Google Scholar] [CrossRef]
- Gil, J.; O’Loghlen, A. PRC1 complex diversity: Where is it taking us? Trends Cell Biol. 2014, 24, 632–641. [Google Scholar] [CrossRef]
- Boyle, S.; Flyamer, I.M.; Williamson, I.; Sengupta, D.; Bickmore, W.A.; Illingworth, R.S. A central role for canonical PRC1 in shaping the 3D nuclear landscape. Genes Dev. 2020, 34, 931–949. [Google Scholar] [CrossRef] [PubMed]
- Schoenfelder, S.; Sugar, R.; Dimond, A.; Javierre, B.M.; Armstrong, H.; Mifsud, B.; Dimitrova, E.; Matheson, L.; Tavares-Cadete, F.; Furlan-Magaril, M.; et al. Polycomb repressive complex PRC1 spatially constrains the mouse embryonic stem cell genome. Nat. Genet. 2015, 47, 1179–1186. [Google Scholar] [CrossRef] [PubMed]
- Kent, S.; Brown, K.; Yang, C.H.; Alsaihati, N.; Tian, C.; Wang, H.; Ren, X. Phase-Separated Transcriptional Condensates Accelerate Target-Search Process Revealed by Live-Cell Single-Molecule Imaging. Cell Rep. 2020, 33, 108248. [Google Scholar] [CrossRef] [PubMed]
- Zenk, F.; Zhan, Y.; Kos, P.; Loser, E.; Atinbayeva, N.; Schachtle, M.; Tiana, G.; Giorgetti, L.; Iovino, N. HP1 drives de novo 3D genome reorganization in early Drosophila embryos. Nature 2021, 593, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Strom, A.R.; Emelyanov, A.V.; Mir, M.; Fyodorov, D.V.; Darzacq, X.; Karpen, G.H. Phase separation drives heterochromatin domain formation. Nature 2017, 547, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Austin, C.A.; Lee, K.C.; Swan, R.L.; Khazeem, M.M.; Manville, C.M.; Cridland, P.; Treumann, A.; Porter, A.; Morris, N.J.; Cowell, I.G. TOP2B: The First Thirty Years. Int. J. Mol. Sci. 2018, 19, 2765. [Google Scholar] [CrossRef] [Green Version]
- Papillon, J.P.N.; Nakajima, K.; Adair, C.D.; Hempel, J.; Jouk, A.O.; Karki, R.G.; Mathieu, S.; Mobitz, H.; Ntaganda, R.; Smith, T.; et al. Discovery of Orally Active Inhibitors of Brahma Homolog (BRM)/SMARCA2 ATPase Activity for the Treatment of Brahma Related Gene 1 (BRG1)/SMARCA4-Mutant Cancers. J. Med. Chem. 2018, 61, 10155–10172. [Google Scholar] [CrossRef]
- Fedorov, O.; Castex, J.; Tallant, C.; Owen, D.R.; Martin, S.; Aldeghi, M.; Monteiro, O.; Filippakopoulos, P.; Picaud, S.; Trzupek, J.D.; et al. Selective targeting of the BRG/PB1 bromodomains impairs embryonic and trophoblast stem cell maintenance. Sci. Adv. 2015, 1, e1500723. [Google Scholar] [CrossRef] [Green Version]
- Paccez, J.D.; Duncan, K.; Sekar, D.; Correa, R.G.; Wang, Y.; Gu, X.; Bashin, M.; Chibale, K.; Libermann, T.A.; Zerbini, L.F. Dihydroartemisinin inhibits prostate cancer via JARID2/miR-7/miR-34a-dependent downregulation of Axl. Oncogenesis 2019, 8, 14. [Google Scholar] [CrossRef] [Green Version]
- Ren, C.; Morohashi, K.; Plotnikov, A.N.; Jakoncic, J.; Smith, S.G.; Li, J.; Zeng, L.; Rodriguez, Y.; Stojanoff, V.; Walsh, M.; et al. Small-molecule modulators of methyl-lysine binding for the CBX7 chromodomain. Chem. Biol. 2015, 22, 161–168. [Google Scholar] [CrossRef] [Green Version]
- Lamb, K.N.; Bsteh, D.; Dishman, S.N.; Moussa, H.F.; Fan, H.; Stuckey, J.I.; Norris, J.L.; Cholensky, S.H.; Li, D.; Wang, J.; et al. Discovery and Characterization of a Cellular Potent Positive Allosteric Modulator of the Polycomb Repressive Complex 1 Chromodomain, CBX7. Cell Chem. Biol. 2019, 26, 1365–1379.e22. [Google Scholar] [CrossRef]
- Wang, S.; Alpsoy, A.; Sood, S.; Ordonez-Rubiano, S.C.; Dhiman, A.; Sun, Y.; Jiao, G.; Krusemark, C.J.; Dykhuizen, E.C. A Potent, Selective CBX2 Chromodomain Ligand and Its Cellular Activity During Prostate Cancer Neuroendocrine Differentiation. ChemBioChem 2021, 22, 2335–2344. [Google Scholar] [CrossRef]
- Kreso, A.; van Galen, P.; Pedley, N.M.; Lima-Fernandes, E.; Frelin, C.; Davis, T.; Cao, L.; Baiazitov, R.; Du, W.; Sydorenko, N.; et al. Self-renewal as a therapeutic target in human colorectal cancer. Nat. Med. 2014, 20, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xing, Y.; Wang, Y.; He, Y.; Wang, L.; Peng, S.; Yang, L.; Xie, J.; Li, X.; Qiu, W.; et al. A novel BMI-1 inhibitor QW24 for the treatment of stem-like colorectal cancer. J. Exp Clin Cancer Res. 2019, 38, 422. [Google Scholar] [CrossRef] [PubMed]
- Nishida, Y.; Maeda, A.; Kim, M.J.; Cao, L.; Kubota, Y.; Ishizawa, J.; AlRawi, A.; Kato, Y.; Iwama, A.; Fujisawa, M.; et al. The novel BMI-1 inhibitor PTC596 downregulates MCL-1 and induces p53-independent mitochondrial apoptosis in acute myeloid leukemia progenitor cells. Blood Cancer J. 2017, 7, e527. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.; Xiong, X.; Crim, A.; Dwivedi, S.K.D.; Mustafi, S.B.; Mukherjee, P.; Cao, L.; Sydorenko, N.; Baiazitov, R.; Moon, Y.C.; et al. Evaluating the Mechanism and Therapeutic Potential of PTC-028, a Novel Inhibitor of BMI-1 Function in Ovarian Cancer. Mol. Cancer Ther. 2018, 17, 39–49. [Google Scholar] [CrossRef] [Green Version]
- Shukla, S.; Ying, W.; Gray, F.; Yao, Y.; Simes, M.L.; Zhao, Q.; Miao, H.; Cho, H.J.; Gonzalez-Alonso, P.; Winkler, A.; et al. Small-molecule inhibitors targeting Polycomb repressi.ive complex 1 RING domain. Nat. Chem. Biol. 2021, 17, 784–793. [Google Scholar] [CrossRef]
- Ismail, I.H.; McDonald, D.; Strickfaden, H.; Xu, Z.; Hendzel, M.J. A small molecule inhibitor of polycomb repressive complex 1 inhibits ubiquitin signaling at DNA double-strand breaks. J. Biol. Chem. 2013, 288, 26944–26954. [Google Scholar] [CrossRef] [Green Version]
- Boija, A.; Klein, I.A.; Sabari, B.R.; Dall’Agnese, A.; Coffey, E.L.; Zamudio, A.V.; Li, C.H.; Shrinivas, K.; Manteiga, J.C.; Hannett, N.M.; et al. Transcription Factors Activate Genes through the Phase-Separation Capacity of Their Activation Domains. Cell 2018, 175, 1842–1855.e6. [Google Scholar] [CrossRef] [Green Version]
- Cho, W.K.; Spille, J.H.; Hecht, M.; Lee, C.; Li, C.; Grube, V.; Cisse, I.I. Mediator and RNA polymerase II clusters associate in transcription-dependent condensates. Science 2018, 361, 412–415. [Google Scholar] [CrossRef] [Green Version]
- Sabari, B.R.; Dall’Agnese, A.; Boija, A.; Klein, I.A.; Coffey, E.L.; Shrinivas, K.; Abraham, B.J.; Hannett, N.M.; Zamudio, A.V.; Manteiga, J.C.; et al. Coactivator condensation at super-enhancers links phase separation and gene control. Science 2018, 361, aar3958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shrinivas, K.; Sabari, B.R.; Coffey, E.L.; Klein, I.A.; Boija, A.; Zamudio, A.V.; Schuijers, J.; Hannett, N.M.; Sharp, P.A.; Young, R.A.; et al. Enhancer Features that Drive Formation of Transcriptional Condensates. Mol. Cell 2019, 75, 549–561.e7. [Google Scholar] [CrossRef] [PubMed]
- Banani, S.F.; Lee, H.O.; Hyman, A.A.; Rosen, M.K. Biomolecular condensates: Organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 2017, 18, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Brangwynne, C.P.; Eckmann, C.R.; Courson, D.S.; Rybarska, A.; Hoege, C.; Gharakhani, J.; Julicher, F.; Hyman, A.A. Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science 2009, 324, 1729–1732. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.H.; Davis, E.S.; Daugird, T.A.; Zhao, S.; Quiroga, I.Y.; Uryu, H.; Li, J.; Storey, A.J.; Tsai, Y.H.; Keeley, D.P.; et al. Phase separation drives aberrant chromatin looping and cancer development. Nature 2021, 595, 591–595. [Google Scholar] [CrossRef]
- Seligson, D.; Horvath, S.; Huerta-Yepez, S.; Hanna, S.; Garban, H.; Roberts, A.; Shi, T.; Liu, X.; Chia, D.; Goodglick, L.; et al. Expression of transcription factor Yin Yang 1 in prostate cancer. Int. J. Oncol. 2005, 27, 131–141. [Google Scholar] [CrossRef]
- Chinnappan, D.; Xiao, D.; Ratnasari, A.; Andry, C.; King, T.C.; Weber, H.C. Transcription factor YY1 expression in human gastrointestinal cancer cells. Int. J. Oncol. 2009, 34, 1417–1423. [Google Scholar]
- Tsang, D.P.; Wu, W.K.; Kang, W.; Lee, Y.Y.; Wu, F.; Yu, Z.; Xiong, L.; Chan, A.W.; Tong, J.H.; Yang, W.; et al. Yin Yang 1-mediated epigenetic silencing of tumour-suppressive microRNAs activates nuclear factor-kappaB in hepatocellular carcinoma. J. Pathol. 2016, 238, 651–664. [Google Scholar] [CrossRef]
- Huang, T.; Wang, G.; Yang, L.; Peng, B.; Wen, Y.; Ding, G.; Wang, Z. Transcription Factor YY1 Modulates Lung Cancer Progression by Activating lncRNA-PVT1. DNA Cell Biol. 2017, 36, 947–958. [Google Scholar] [CrossRef]
- Wan, M.; Huang, W.; Kute, T.E.; Miller, L.D.; Zhang, Q.; Hatcher, H.; Wang, J.; Stovall, D.B.; Russell, G.B.; Cao, P.D.; et al. Yin Yang 1 plays an essential role in breast cancer and negatively regulates p27. Am. J. Pathol. 2012, 180, 2120–2133. [Google Scholar] [CrossRef] [Green Version]
- Antonio-Andres, G.; Jimenez-Hernandez, E.; Estrada-Abreo, L.A.; Garfias-Gomez, Y.; Patino-Lopez, G.; Juarez-Mendez, S.; Huerta-Yepez, S. Expression of YY1 in pro-B and T phenotypes correlation with poor survival in pediatric acute lymphoblastic leukemia. Pediatr. Hematol. Oncol. 2021, 38, 456–470. [Google Scholar] [CrossRef] [PubMed]
- de Nigris, F.; Zanella, L.; Cacciatore, F.; De Chiara, A.; Fazioli, F.; Chiappetta, G.; Apice, G.; Infante, T.; Monaco, M.; Rossiello, R.; et al. YY1 overexpression is associated with poor prognosis and metastasis-free survival in patients suffering osteosarcoma. BMC Cancer 2011, 11, 472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Qiao, S.; Li, G.; Yang, C.; Zhong, C.; Stovall, D.B.; Shi, J.; Li, D.; Sui, G. A histidine cluster determines YY1-compartmentalized coactivators and chromatin elements in phase-separated super-enhancers. bioRxiv 2021. [Google Scholar] [CrossRef]
- Sanborn, A.L.; Rao, S.S.; Huang, S.C.; Durand, N.C.; Huntley, M.H.; Jewett, A.I.; Bochkov, I.D.; Chinnappan, D.; Cutkosky, A.; Li, J.; et al. Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proc. Natl. Acad. Sci. USA 2015, 112, E6456–E6465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fudenberg, G.; Imakaev, M.; Lu, C.; Goloborodko, A.; Abdennur, N.; Mirny, L.A. Formation of Chromosomal Domains by Loop Extrusion. Cell Rep. 2016, 15, 2038–2049. [Google Scholar] [CrossRef] [Green Version]
- Lee, R.; Kang, M.K.; Kim, Y.J.; Yang, B.; Shim, H.; Kim, S.; Kim, K.; Yang, C.M.; Min, B.G.; Jung, W.J.; et al. CTCF-mediated chromatin looping provides a topological framework for the formation of phase-separated transcriptional condensates. Nucleic Acids Res. 2022, 50, 207–226. [Google Scholar] [CrossRef]
- Henninger, J.E.; Oksuz, O.; Shrinivas, K.; Sagi, I.; LeRoy, G.; Zheng, M.M.; Andrews, J.O.; Zamudio, A.V.; Lazaris, C.; Hannett, N.M.; et al. RNA-Mediated Feedback Control of Transcriptional Condensates. Cell 2021, 184, 207–225.e24. [Google Scholar] [CrossRef]
- Lee, J.H.; Wang, R.; Xiong, F.; Krakowiak, J.; Liao, Z.; Nguyen, P.T.; Moroz-Omori, E.V.; Shao, J.; Zhu, X.; Bolt, M.J.; et al. Enhancer RNA m6A methylation facilitates transcriptional condensate formation and gene activation. Mol. Cell 2021, 81, 3368–3385.e9. [Google Scholar] [CrossRef]
- Guo, Y.E.; Manteiga, J.C.; Henninger, J.E.; Sabari, B.R.; Dall’Agnese, A.; Hannett, N.M.; Spille, J.H.; Afeyan, L.K.; Zamudio, A.V.; Shrinivas, K.; et al. Pol II phosphorylation regulates a switch between transcriptional and splicing condensates. Nature 2019, 572, 543–548. [Google Scholar] [CrossRef] [Green Version]
- Larson, A.G.; Elnatan, D.; Keenen, M.M.; Trnka, M.J.; Johnston, J.B.; Burlingame, A.L.; Agard, D.A.; Redding, S.; Narlikar, G.J. Liquid droplet formation by HP1alpha suggests a role for phase separation in heterochromatin. Nature 2017, 547, 236–240. [Google Scholar] [CrossRef] [Green Version]
- Plys, A.J.; Davis, C.P.; Kim, J.; Rizki, G.; Keenen, M.M.; Marr, S.K.; Kingston, R.E. Phase separation of Polycomb-repressive complex 1 is governed by a charged disordered region of CBX2. Genes Dev. 2019, 33, 799–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatavosian, R.; Kent, S.; Brown, K.; Yao, T.; Duc, H.N.; Huynh, T.N.; Zhen, C.Y.; Ma, B.; Wang, H.; Ren, X. Nuclear condensates of the Polycomb protein chromobox 2 (CBX2) assemble through phase separation. J. Biol. Chem. 2019, 294, 1451–1463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alberti, S.; Gladfelter, A.; Mittag, T. Considerations and Challenges in Studying Liquid-Liquid Phase Separation and Biomolecular Condensates. Cell 2019, 176, 419–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulianov, S.V.; Velichko, A.K.; Magnitov, M.D.; Luzhin, A.V.; Golov, A.K.; Ovsyannikova, N.; Kireev, I.I.; Gavrikov, A.S.; Mishin, A.S.; Garaev, A.K.; et al. Suppression of liquid-liquid phase separation by 1,6-hexanediol partially compromises the 3D genome organization in living cells. Nucleic Acids Res. 2021, 49, 10524–10541. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, R.J. Therapeutics-how to treat phase separation-associated diseases. Emerg Top. Life Sci. 2020, 4, 307–318. [Google Scholar] [CrossRef]
- Fang, M.Y.; Markmiller, S.; Vu, A.Q.; Javaherian, A.; Dowdle, W.E.; Jolivet, P.; Bushway, P.J.; Castello, N.A.; Baral, A.; Chan, M.Y.; et al. Small-Molecule Modulation of TDP-43 Recruitment to Stress Granules Prevents Persistent TDP-43 Accumulation in ALS/FTD. Neuron 2019, 103, 802–819.e11. [Google Scholar] [CrossRef]
- Wheeler, R.J.; Lee, H.O.; Poser, I.; Pal, A.; Doeleman, T.; Kishigami, S.; Kour, S.; Anderson, E.N.; Marrone, L.; Murthy, A.C.; et al. Small molecules for modulating protein driven liquid-liquid phase separation in treating neurodegenerative disease. bioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Klein, I.A.; Boija, A.; Afeyan, L.K.; Hawken, S.W.; Fan, M.; Dall’Agnese, A.; Oksuz, O.; Henninger, J.E.; Shrinivas, K.; Sabari, B.R.; et al. Partitioning of cancer therapeutics in nuclear condensates. Science 2020, 368, 1386–1392. [Google Scholar] [CrossRef]
- Boija, A.; Klein, I.A.; Young, R.A. Biomolecular Condensates and Cancer. Cancer Cell 2021, 39, 174–192. [Google Scholar] [CrossRef]
- Chong, S.; Dugast-Darzacq, C.; Liu, Z.; Dong, P.; Dailey, G.M.; Cattoglio, C.; Heckert, A.; Banala, S.; Lavis, L.; Darzacq, X.; et al. Imaging dynamic and selective low-complexity domain interactions that control gene transcription. Science 2018, 361, aar2555. [Google Scholar] [CrossRef] [Green Version]
- Hung, K.L.; Yost, K.E.; Xie, L.; Shi, Q.; Helmsauer, K.; Luebeck, J.; Schopflin, R.; Lange, J.T.; Chamorro Gonzalez, R.; Weiser, N.E.; et al. ecDNA hubs drive cooperative intermolecular oncogene expression. Nature 2021, 600, 731–736. [Google Scholar] [CrossRef] [PubMed]
- Morton, A.R.; Dogan-Artun, N.; Faber, Z.J.; MacLeod, G.; Bartels, C.F.; Piazza, M.S.; Allan, K.C.; Mack, S.C.; Wang, X.; Gimple, R.C.; et al. Functional Enhancers Shape Extrachromosomal Oncogene Amplifications. Cell 2019, 179, 1330–1341.e13. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Gujar, A.D.; Wong, C.H.; Tjong, H.; Ngan, C.Y.; Gong, L.; Chen, Y.A.; Kim, H.; Liu, J.; Li, M.; et al. Oncogenic extrachromosomal DNA functions as mobile enhancers to globally amplify chromosomal transcription. Cancer Cell 2021, 39, 694–707.e7. [Google Scholar] [CrossRef] [PubMed]
- Hay, D.; Hughes, J.R.; Babbs, C.; Davies, J.O.J.; Graham, B.J.; Hanssen, L.; Kassouf, M.T.; Marieke Oudelaar, A.M.; Sharpe, J.A.; Suciu, M.C.; et al. Genetic dissection of the alpha-globin super-enhancer in vivo. Nat. Genet. 2016, 48, 895–903. [Google Scholar] [CrossRef]
- Perry, M.W.; Boettiger, A.N.; Levine, M. Multiple enhancers ensure precision of gap gene-expression patterns in the Drosophila embryo. Proc. Natl. Acad. Sci. USA 2011, 108, 13570–13575. [Google Scholar] [CrossRef] [Green Version]
- Moorthy, S.D.; Davidson, S.; Shchuka, V.M.; Singh, G.; Malek-Gilani, N.; Langroudi, L.; Martchenko, A.; So, V.; Macpherson, N.N.; Mitchell, J.A. Enhancers and super-enhancers have an equivalent regulatory role in embryonic stem cells through regulation of single or multiple genes. Genome Res. 2017, 27, 246–258. [Google Scholar] [CrossRef]
- Blobel, G.A.; Higgs, D.R.; Mitchell, J.A.; Notani, D.; Young, R.A. Testing the super-enhancer concept. Nat. Rev. Genet. 2021, 22, 749–755. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, G.; Zhang, H. Protocol for analyzing protein liquid–liquid phase separation. Biophys. Rep. 2019, 5, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Merkurjev, D.; Yang, F.; Li, W.; Oh, S.; Friedman, M.J.; Song, X.; Zhang, F.; Ma, Q.; Ohgi, K.A.; et al. Enhancer activation requires trans-recruitment of a mega transcription factor complex. Cell 2014, 159, 358–373. [Google Scholar] [CrossRef] [Green Version]
- Nair, S.J.; Yang, L.; Meluzzi, D.; Oh, S.; Yang, F.; Friedman, M.J.; Wang, S.; Suter, T.; Alshareedah, I.; Gamliel, A.; et al. Phase separation of ligand-activated enhancers licenses cooperative chromosomal enhancer assembly. Nat. Struct. Mol. Biol. 2019, 26, 193–203. [Google Scholar] [CrossRef]
- Yan, X.; Hu, Z.; Feng, Y.; Hu, X.; Yuan, J.; Zhao, S.D.; Zhang, Y.; Yang, L.; Shan, W.; He, Q.; et al. Comprehensive Genomic Characterization of Long Non-coding RNAs across Human Cancers. Cancer Cell 2015, 28, 529–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlsen, A.T.; Zahid, O.K.; Ruzicka, J.A.; Taylor, E.W.; Hall, A.R. Selective detection and quantification of modified DNA with solid-state nanopores. Nano Lett 2014, 14, 5488–5492. [Google Scholar] [CrossRef] [PubMed]
- Garalde, D.R.; Snell, E.A.; Jachimowicz, D.; Sipos, B.; Lloyd, J.H.; Bruce, M.; Pantic, N.; Admassu, T.; James, P.; Warland, A.; et al. Highly parallel direct RNA sequencing on an array of nanopores. Nat. Methods 2018, 15, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Simpson, J.T.; Workman, R.E.; Zuzarte, P.C.; David, M.; Dursi, L.J.; Timp, W. Detecting DNA cytosine methylation using nanopore sequencing. Nat. Methods 2017, 14, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Larsen, M.R.; Trelle, M.B.; Thingholm, T.E.; Jensen, O.N. Analysis of post-translational modifications of proteins by tandem mass spectrometry. Biotechniques 2006, 40, 790–798. [Google Scholar] [CrossRef] [PubMed]
- Fanning, S.W.; Mayne, C.G.; Dharmarajan, V.; Carlson, K.E.; Martin, T.A.; Novick, S.J.; Toy, W.; Green, B.; Panchamukhi, S.; Katzenellenbogen, B.S.; et al. Estrogen receptor alpha somatic mutations Y537S and D538G confer breast cancer endocrine resistance by stabilizing the activating function-2 binding conformation. eLife 2016, 5, e12792. [Google Scholar] [CrossRef] [PubMed]
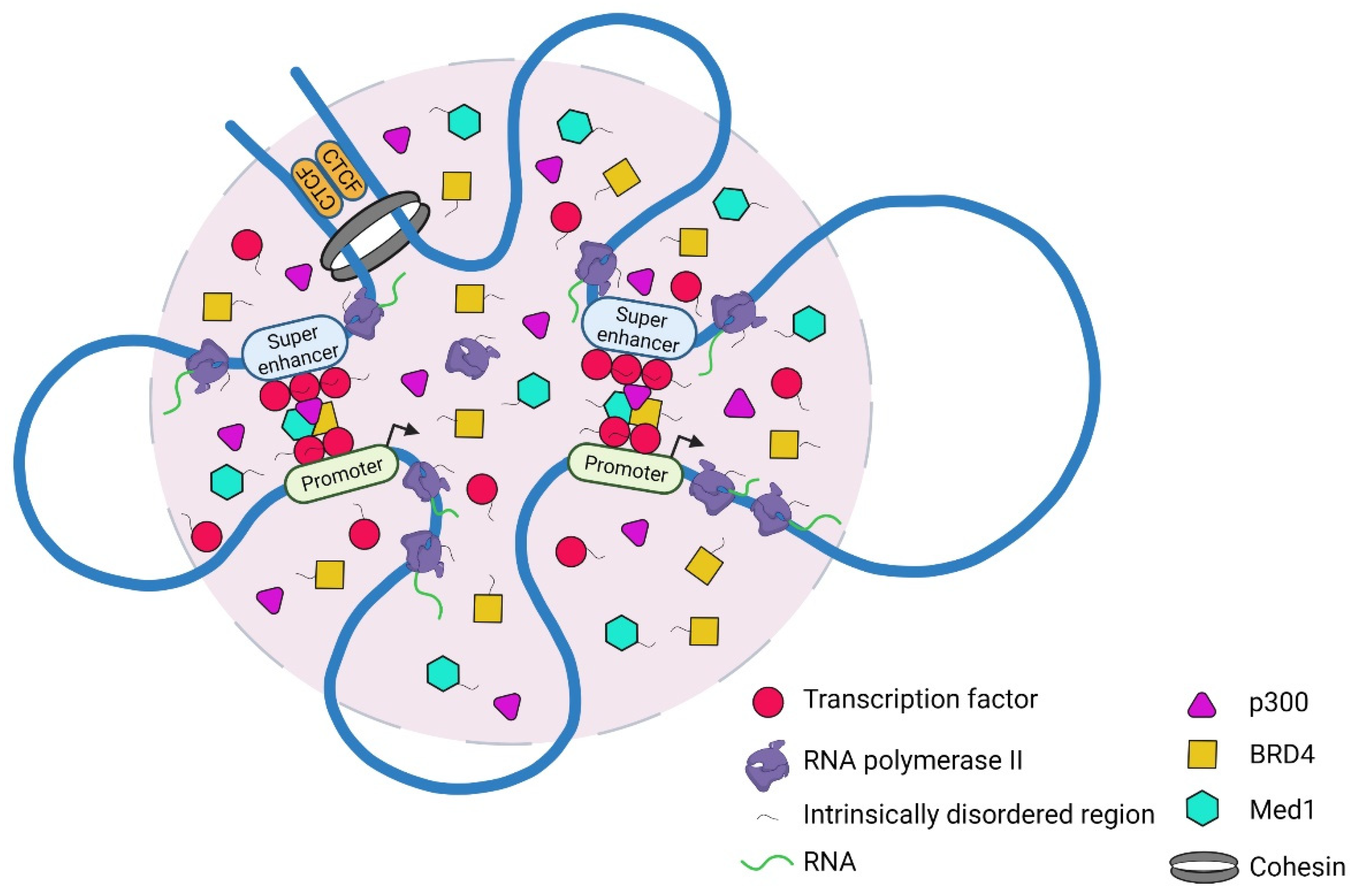

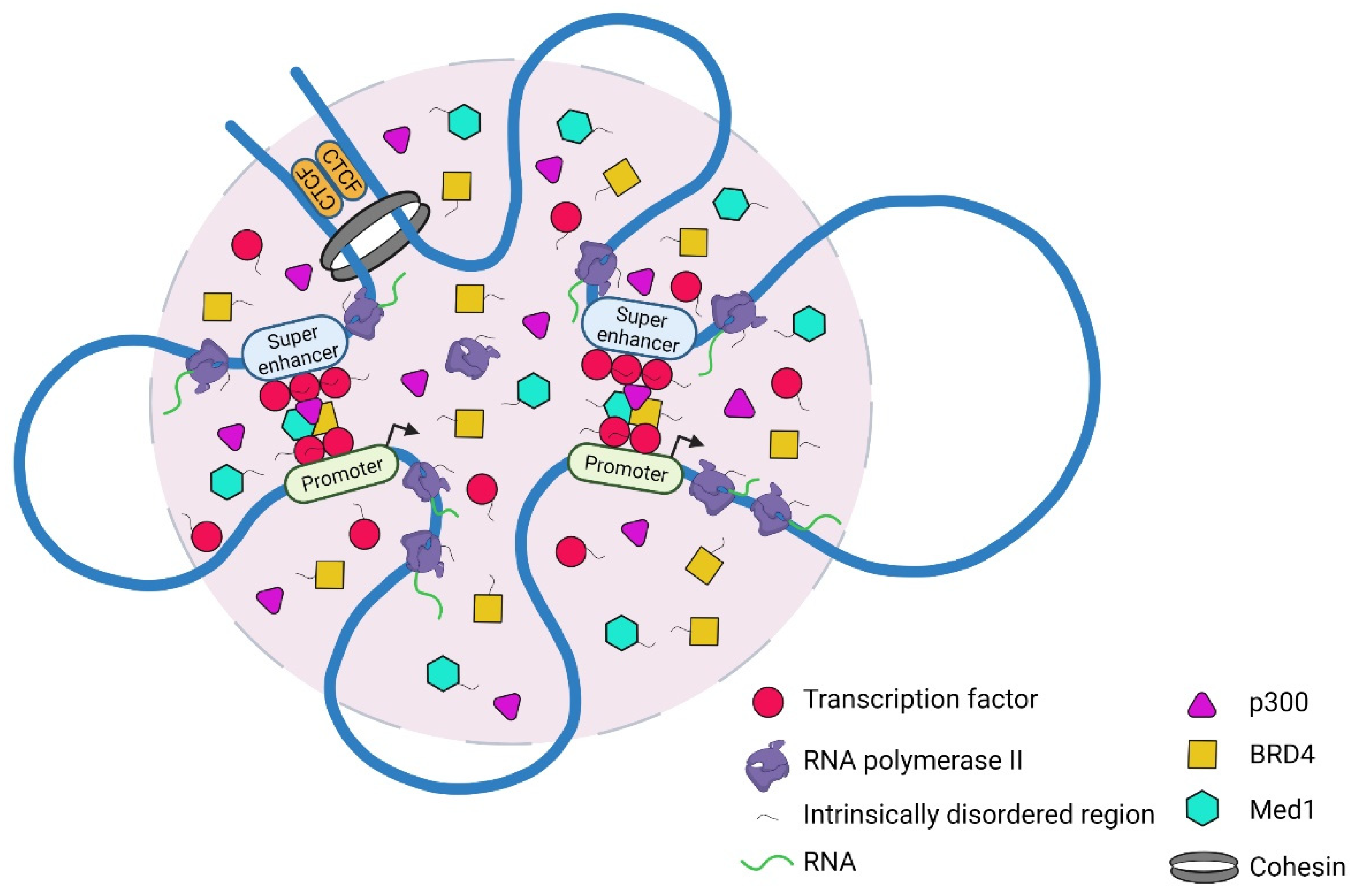{kind=link}
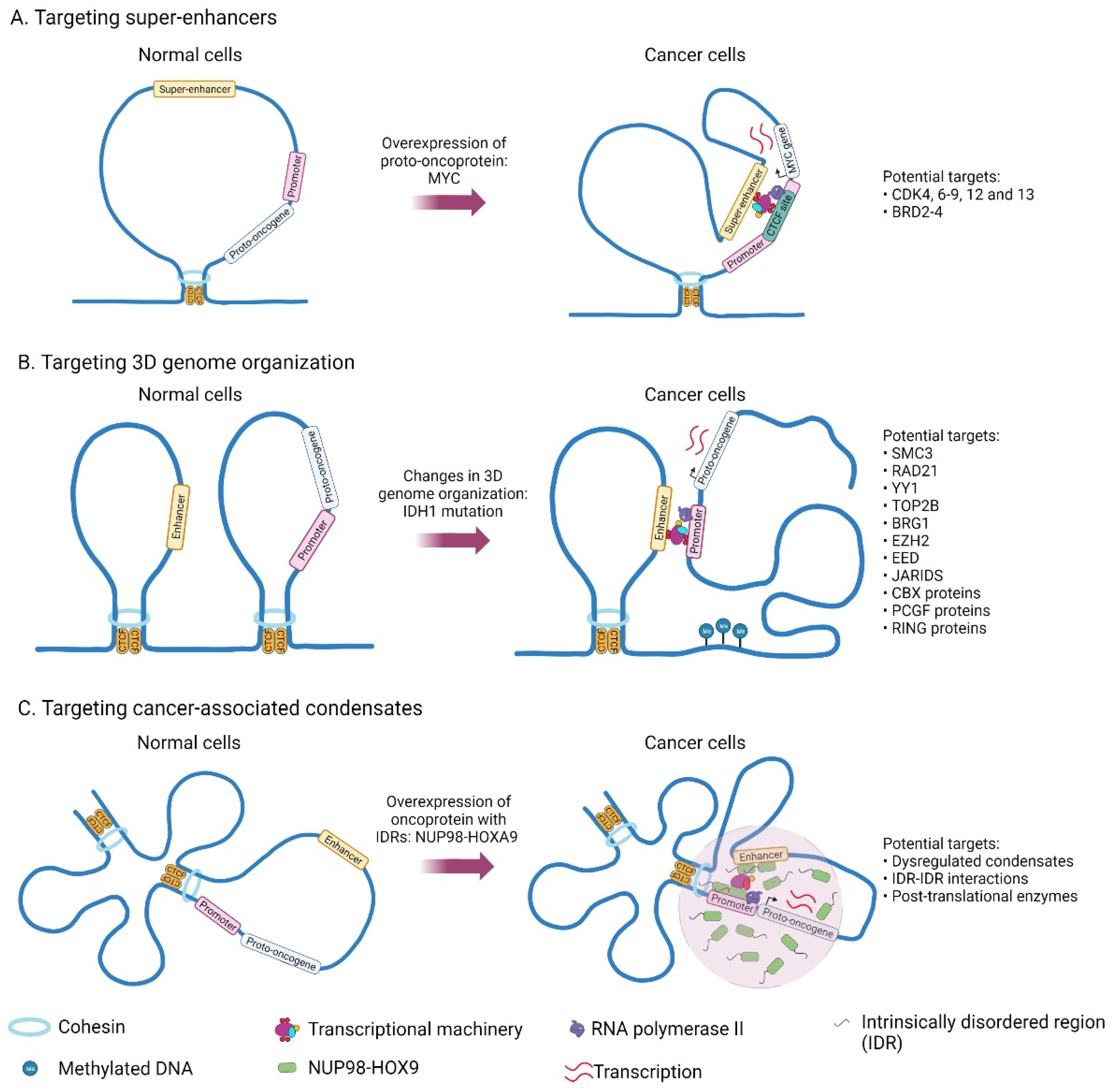{kind=link}
| Target | Potential Small-Molecule Inhibitors | Reference |
|---|---|---|
| CDK7 | THZ1, SY-1365, SY-5609, and THZ2 | [50,61,62,63,64,65] |
| CDK4 | Ribociclib (LEE011) | [6,66] |
| CDK6 | Ribociclib (LEE011) | [6,66] |
| CDK12 | THZ1, THZ531 | [64,67] |
| CDK13 | THZ1, THZ531 | [64,67] |
| CDK8 | Cortistatin A, SEL120-34A | [58,68] |
| CDK9 | NVP-2 | [64] |
| BRD2 | I-BET762, OTX015, CPI0610, and BI-89499 | [69,70,71] |
| BRD3 | I-BET762, OTX015, CPI0610, and BI-89499 | [69,70,71] |
| BRD4 | JQ1, I-BET151, and I-BET762, OTX015, CPI0610, and BI-89499 | [41,69,70,71,72] |
| Target | Potential Small-Molecule Inhibitors | Effects in Cancer Hallmarks | Reference |
|---|---|---|---|
| SMC3 | Glycyrrhizic acid, HDAC8-specific inhibitor PCI-34051 | Delay cell cycle progression in MCF7 cells | [87,88] |
| RAD21 | Glycyrrhizic acid | Inhibit Kaposi’s sarcoma-associated herpesvirus (KSHV)-infected cell growth | [87] |
| SMC1 | No | ||
| SA1 or SA2 | No | ||
| NIPBL | No | ||
| WAPL | No | ||
| YY1 | siRNA YY1, nitric oxide donors, proteasome inhibitors, and inhibitors of activated survival pathways, such as inhibitors of nuclear factor-kappa beta | Inhibit cancer cell proliferation, viability and epithelial–mesenchymal transition | [91] |
| MAZ | No | ||
| TOP2B | Doxorubicin, epirubicin, daunorubicin, idarubicin, mitoxantrone, etoposide, and mAMSA | Induce DNA damage and stop growth of cancer cells | [126] |
| ZNF143 | No | ||
| SNF2H | No | ||
| TAF3 | No | ||
| BRG1 | BRM/BRG1 ATP Inhibitor-1, PFI-3 | Deprive of stemness and deregulated lineage specification for embryonic stem cells | [127,128] |
| EZH2 | DZNep, EI1, EPZ005687, GSK343, GSK126, UNC1999, EPZ-6438, and Stabilized α-helix of EZH2 peptide (SAH-EZH2) | Inhibit cell growth, induce cell cycle arrest and apoptosis | [117] |
| EED | EED226, A-395, BR-001, EEDi-5285, EEDi-1056, MAK683, SAH-EZH2, Astemizole, Wedelolactone, and DC-PRC2in-01 | The antitumor abilities of A-395 and GSK126 were validated in the Pfeiffer xenograft animal model | [119] |
| SUZ12 | No | ||
| RbAp46/48 | No | ||
| AEBP2 | No | ||
| PCLs | No | ||
| JARIDS | Dihydroartemisinin | Inhibit cancer cell proliferation, migration, invasion, and tumor formation | [129] |
| CBX proteins | MS37452, UNC3866, MS37452, MS351, UNC4976, and SW2_152F | Block neuroendocrine differentiation and promote prostate cancer cell death | [130,131,132] |
| PCGF proteins | PTC209, QW24, PTC596, and PTC-028 | Inhibit colorectal cancer cell proliferation, migration and self-renewal; and induce mitochondrial apoptosis in AML progenitor cells | [133,134,135,136] |
| HPH proteins | No | ||
| RING proteins | RB-3, PRT4165 | Induce differentiation in leukemia cell lines and primary AML samples; and inhibit DNA double-strand break repair and cause G2/M checkpoint failure | [137,138] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, S.C.; Vijayakumar, U.; Zhang, Y.; Fullwood, M.J. Super-Enhancers, Phase-Separated Condensates, and 3D Genome Organization in Cancer. Cancers 2022, 14, 2866. https://doi.org/10.3390/cancers14122866
Tang SC, Vijayakumar U, Zhang Y, Fullwood MJ. Super-Enhancers, Phase-Separated Condensates, and 3D Genome Organization in Cancer. Cancers. 2022; 14(12):2866. https://doi.org/10.3390/cancers14122866
Chicago/Turabian StyleTang, Seng Chuan, Udhaya Vijayakumar, Ying Zhang, and Melissa Jane Fullwood. 2022. "Super-Enhancers, Phase-Separated Condensates, and 3D Genome Organization in Cancer" Cancers 14, no. 12: 2866. https://doi.org/10.3390/cancers14122866
APA StyleTang, S. C., Vijayakumar, U., Zhang, Y., & Fullwood, M. J. (2022). Super-Enhancers, Phase-Separated Condensates, and 3D Genome Organization in Cancer. Cancers, 14(12), 2866. https://doi.org/10.3390/cancers14122866