
Bottom-Up Approach to the Discovery of Clinically Relevant Biomarker Genes: The Case of Colorectal Cancer



Abstract
Simple Summary
Abstract
1. Introduction
1.1. Detection and Screening in Colorectal Cancer
1.2. The Top-Down Approach Utilized in ‘Omics-Based Marker Discovery
1.3. Research Hypothesis and Implementation
1.4. Brief Justification and Explanation of the Experimental Approach Used
1.4.1. Transcription Factors
1.4.2. Biological Pathways
1.4.3. Gene Co-Expression
2. Materials and Methods
2.1. Biomarkers: Training and Validation Sets
2.2. Transcription Factors
2.3. Biological Pathways
2.4. Validation of the Gene Selection Procedure and Further Ranking of the Predicted Genes
2.5. Experimental Validation of the Predicted Biomarkers Using Human Tissues
2.6. RNA Extraction
2.7. cDNA Synthesis
2.8. RT-PCR Primer Selection
2.9. RT-PCR
2.10. Real-Time PCR
3. Results
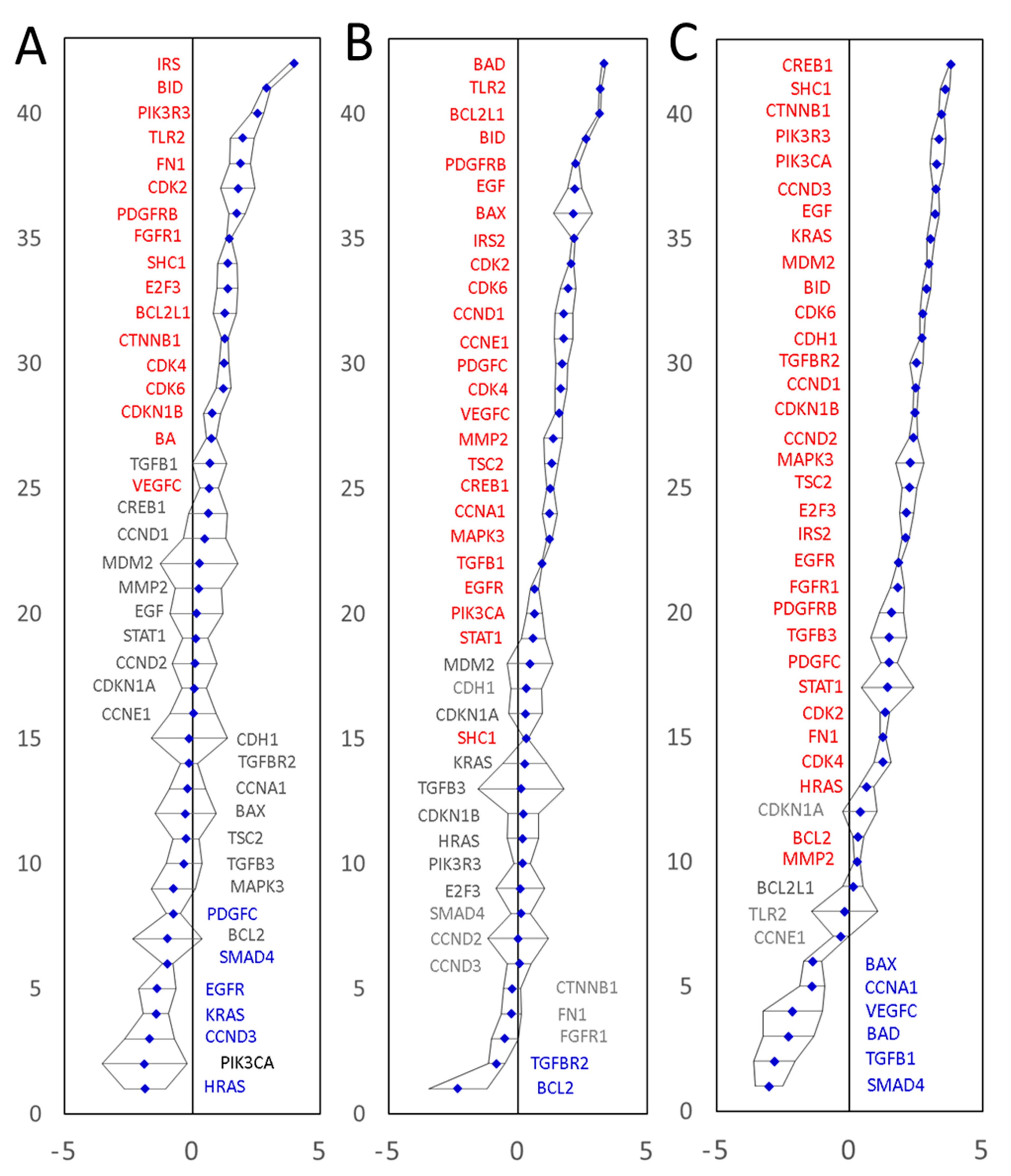
3.1. Prediction of Novel CRC Biomarkers and Validation of the Data Analysis Approach
3.2. Experimental Validation of the Predicted Putative Biomarkers
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- UK Cancer Research. Cancer Research UK Bowel Cancer Statistics. Available online: https://www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/bowel-cancer (accessed on 31 October 2021).
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Grady, W.M.; Carethers, J.M. Genomic and epigenetic instability in colorectal cancer pathogenesis. Gastroenterology 2008, 135, 1079–1099. [Google Scholar] [CrossRef]
- Marmol, I.; Sanchez-de-Diego, C.; Pradilla Dieste, A.; Cerrada, E.; Rodriguez Yoldi, M.J. Colorectal Carcinoma: A General Overview and Future Perspectives in Colorectal Cancer. Int. J. Mol. Sci. 2017, 18, 197. [Google Scholar] [CrossRef]
- Boland, C.R.; Goel, A. Microsatellite instability in colorectal cancer. Gastroenterology 2010, 138, 2073–2087.e3. [Google Scholar] [CrossRef]
- Kamel, F.; Eltarhoni, K.; Nisar, P.; Soloviev, M. Colorectal Cancer Diagnosis: The Obstacles We Face in Determining a Non-Invasive Test and Current Advances in Biomarker Detection. Cancers 2022, 14, 1889. [Google Scholar] [CrossRef]
- UK Cancer Research. Bowel Cancer Survial Statistics. Available online: https://www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/bowel-cancer/survival#heading-Three (accessed on 10 February 2022).
- Ladabaum, U.; Mannalithara, A. Comparative Effectiveness and Cost Effectiveness of a Multitarget Stool DNA Test to Screen for Colorectal Neoplasia. Gastroenterology 2016, 151, 427–439.e6. [Google Scholar] [CrossRef]
- Au, F.C.; Stein, B.; Tang, C.K. Carcinoembryonic antigen levels in colonic lesions. Am. J. Surg. 1986, 151, 61–64. [Google Scholar] [CrossRef]
- Tejpar, S.; Celik, I.; Schlichting, M.; Sartorius, U.; Bokemeyer, C.; Van Cutsem, E. Association of KRAS G13D tumor mutations with outcome in patients with metastatic colorectal cancer treated with first-line chemotherapy with or without cetuximab. J. Clin. Oncol. 2012, 30, 3570–3577. [Google Scholar] [CrossRef]
- Oh, T.; Kim, N.; Moon, Y.; Kim, M.S.; Hoehn, B.D.; Park, C.H.; Kim, T.S.; Kim, N.K.; Chung, H.C.; An, S. Genome-wide identification and validation of a novel methylation biomarker, SDC2, for blood-based detection of colorectal cancer. J. Mol. Diagn. 2013, 15, 498–507. [Google Scholar] [CrossRef]
- Zhang, L.; Dong, L.; Lu, C.; Huang, W.; Yang, C.; Wang, Q.; Wang, Q.; Lei, R.; Sun, R.; Wan, K.; et al. Methylation of SDC2/TFPI2 and Its Diagnostic Value in Colorectal Tumorous Lesions. Front. Mol. Biosci. 2021, 8, 706754. [Google Scholar] [CrossRef]
- Zhang, W.; Yang, C.; Wang, S.; Xiang, Z.; Dou, R.; Lin, Z.; Zheng, J.; Xiong, B. SDC2 and TFPI2 Methylation in Stool Samples as an Integrated Biomarker for Early Detection of Colorectal Cancer. Cancer Manag. Res. 2021, 13, 3601–3617. [Google Scholar] [CrossRef] [PubMed]
- Müller, H.M.; Oberwalder, M.; Fiegl, H.; Morandell, M.; Goebel, G.; Zitt, M.; Mühlthaler, M.; Ofner, D.; Margreiter, R.; Widschwendter, M. Methylation changes in faecal DNA: A marker for colorectal cancer screening? Lancet 2004, 363, 1283–1285. [Google Scholar] [CrossRef]
- Shirahata, A.; Hibi, K. Serum vimentin methylation as a potential marker for colorectal cancer. Anticancer Res. 2014, 34, 4121–4125. [Google Scholar] [PubMed]
- Shirahata, A.; Sakuraba, K.; Goto, T.; Saito, M.; Ishibashi, K.; Kigawa, G.; Nemoto, H.; Hibi, K. Detection of vimentin (VIM) methylation in the serum of colorectal cancer patients. Anticancer Res. 2010, 30, 5015–5018. [Google Scholar]
- Yi, J.M.; Dhir, M.; Guzzetta, A.A.; Iacobuzio-Donahue, C.A.; Heo, K.; Yang, K.M.; Suzuki, H.; Toyota, M.; Kim, H.M.; Ahuja, N. DNA methylation biomarker candidates for early detection of colon cancer. Tumour Biol. 2012, 33, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Yang, I.P.; Tsai, H.L.; Miao, Z.F.; Huang, C.W.; Kuo, C.H.; Wu, J.Y.; Wang, W.M.; Juo, S.H.; Wang, J.Y. Development of a deregulating microRNA panel for the detection of early relapse in postoperative colorectal cancer patients. J. Transl. Med. 2016, 14, 108. [Google Scholar] [CrossRef]
- Durán-Vinet, B.; Araya-Castro, K.; Calderón, J.; Vergara, L.; Weber, H.; Retamales, J.; Araya-Castro, P.; Leal-Rojas, P. CRISPR/Cas13-Based Platforms for a Potential Next-Generation Diagnosis of Colorectal Cancer through Exosomes Micro-RNA Detection: A Review. Cancers 2021, 13, 4640. [Google Scholar] [CrossRef]
- Bradner, J.E.; Hnisz, D.; Young, R.A. Transcriptional Addiction in Cancer. Cell 2017, 168, 629–643. [Google Scholar] [CrossRef]
- Rohr, M.; Beardsley, J.; Nakkina, S.P.; Zhu, X.; Aljabban, J.; Hadley, D.; Altomare, D. A merged microarray meta-dataset for transcriptionally profiling colorectal neoplasm formation and progression. Sci. Data 2021, 8, 214. [Google Scholar] [CrossRef]
- Alves Martins, B.A.; de Bulhões, G.F.; Cavalcanti, I.N.; Martins, M.M.; de Oliveira, P.G.; Martins, A.M.A. Biomarkers in Colorectal Cancer: The Role of Translational Proteomics Research. Front. Oncol. 2019, 9, 1284. [Google Scholar] [CrossRef]
- Mueller, C.; Haymond, A.; Davis, J.B.; Williams, A.; Espina, V. Protein biomarkers for subtyping breast cancer and implications for future research. Expert Rev. Proteom. 2018, 15, 131–152. [Google Scholar] [CrossRef] [PubMed]
- Cheung CH, Y.; Juan, H.F. Quantitative proteomics in lung cancer. J. Biomed. Sci. 2017, 24, 37. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Ray, S.; Ghantasala, S.; Srivastava, S. An Integrated Quantitative Proteomics Workflow for Cancer Biomarker Discovery and Validation in Plasma. Front. Oncol. 2020, 10, 543997. [Google Scholar] [CrossRef]
- Yihang, Y.; Ji, C.; Jue, W.; Ming, X.; Yunpeng, Z.; Peng, S.; Leilei, L. Identification Hub Genes in Colorectal Cancer by Integrating Weighted Gene Co-Expression Network Analysis and Clinical Validation in vivo and vitro. Front. Oncol. 2020, 10, 638. [Google Scholar] [CrossRef]
- Sun, Y.; Mironova, V.; Chen, Y.; Lundh EP, F.; Zhang, Q.; Cai, Y.; Vasiliou, V.; Zhang, Y.; Garcia-Milian, R.; Khan, S.A.; et al. Molecular Pathway Analysis Indicates a Distinct Metabolic Phenotype in Women with Right-Sided Colon Cancer. Transl. Oncol. 2020, 13, 42–56. [Google Scholar] [CrossRef]
- Ding, X.; Duan, H.; Luo, H. Identification of Core Gene Expression Signature and Key Pathways in Colorectal Cancer. Front. Genet. 2020, 11, 45. [Google Scholar] [CrossRef]
- Wilson, S.; Filipp, F.V. A network of epigenomic and transcriptional cooperation encompassing an epigenomic master regulator in cancer. NPJ Syst. Biol. Appl. 2018, 4, 24. [Google Scholar] [CrossRef]
- Netbiolab. Transcriptional Regulatory Relationships Unraveled by Sentence-Based Text Mining. Available online: https://www.grnpedia.org/trrust/ (accessed on 1 December 2019).
- Planqué, R.; Hulshof, J.; Teusink, B.; Hendriks, J.C.; Bruggeman, F.J. Maintaining maximal metabolic flux by gene expression control. PLoS Comput. Biol. 2018, 14, e1006412. [Google Scholar] [CrossRef]
- Alliance of Genome Resources. The Gene Ontology Resource. Available online: http://geneontology.org/ (accessed on 20 March 2022).
- KEGG. Kyoto Encyclopedia of Genes and Genomes. Available online: https://www.genome.jp/kegg/ (accessed on 1 December 2019).
- Khatri, P.; Sirota, M.; Butte, A.J. Ten years of pathway analysis: Current approaches and outstanding challenges. PLoS Comput. Biol. 2012, 8, e1002375. [Google Scholar] [CrossRef]
- Berlanda, N.; Somigliana, E.; Frattaruolo, M.P.; Buggio, L.; Dridi, D.; Vercellini, P. Surgery versus hormonal therapy for deep endometriosis: Is it a choice of the physician? Eur. J. Obstet. Gynecol. Reprod. Biol. 2017, 209, 67–71. [Google Scholar] [CrossRef]
- Creixell, P.; Reimand, J.; Haider, S.; Wu, G.; Shibata, T.; Vazquez, M.; Mustonen, V.; Gonzalez-Perez, A.; Pearson, J.; Sander, C.; et al. Pathway and network analysis of cancer genomes. Nat. Methods 2015, 12, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Eisen, M.B.; Spellman, P.T.; Brown, P.O.; Botstein, D. Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. USA 1998, 95, 14863–14868. [Google Scholar] [CrossRef] [PubMed]
- Cramer, P. Organization and regulation of gene transcription. Nature 2019, 573, 45–54. [Google Scholar] [CrossRef] [PubMed]
- DeRisi, J.L.; Iyer, V.R.; Brown, P.O. Exploring the metabolic and genetic control of gene expression on a genomic scale. Science 1997, 278, 680–686. [Google Scholar] [CrossRef]
- Hughes, T.R.; Marton, M.J.; Jones, A.R.; Roberts, C.J.; Stoughton, R.; Armour, C.D.; Bennett, H.A.; Coffey, E.; Dai, H.; He, Y.D.; et al. Functional discovery via a compendium of expression profiles. Cell 2000, 102, 109–126. [Google Scholar] [CrossRef]
- Reel, P.S.; Reel, S.; Pearson, E.; Trucco, E.; Jefferson, E. Using machine learning approaches for multi-omics data analysis: A review. Biotechnol. Adv. 2021, 49, 107739. [Google Scholar] [CrossRef]
- Joshi, A.; Rienks, M.; Theofilatos, K.; Mayr, M. Systems biology in cardiovascular disease: A multiomics approach. Nat. Rev. Cardiol. 2021, 18, 313–330. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Li, Y.; Bie, R.; Teran Hidalgo, S.J.; Qin, Y.; Wu, M.; Ma, S. Assisted gene expression-based clustering with AWNCut. Stat. Med. 2018, 37, 4386–4403. [Google Scholar] [CrossRef]
- Ontario Genomics Institute Genome Canada. GeneMania. Available online: https://genemania.org/ (accessed on 1 December 2019).
- Obayashi, T.; Kagaya, Y.; Aoki, Y.; Tadaka, S.; Kinoshita, K. COXPRESdb v7: A gene coexpression database for 11 animal species supported by 23 coexpression platforms for technical evaluation and evolutionary inference. Nucleic Acids Res. 2019, 47, D55–D62. [Google Scholar] [CrossRef]
- Van Dam, S.; Vosa, U.; van der Graaf, A.; Franke, L.; de Magalhaes, J.P. Gene co-expression analysis for functional classification and gene-disease predictions. Brief Bioinform. 2018, 19, 575–592. [Google Scholar] [CrossRef] [PubMed]
- PubMed. NCBI Pub Med. Available online: https://pubmed.ncbi.nlm.nih.gov/ (accessed on 6 May 2022).
- Biosystems. NCBI Biosystems. Available online: https://www.ncbi.nlm.nih.gov/biosystems/ (accessed on 31 October 2019).
- bioDBnet. Biological Database Network. Available online: https://biodbnet-abcc.ncifcrf.gov/db/db2db.php (accessed on 25 May 2022).
- NCBI. NCBI Primer Blast Tool. Available online: https://www.ncbi.nlm.nih.gov/tools/primer-blast/ (accessed on 31 October 2019).
- EMBL-EBI. Clusta lOmega. Available online: https://www.ebi.ac.uk/Tools/msa/clustalo/ (accessed on 1 May 2019).
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Huang, C.; Nice, E.C. Proteomics, genomics and transcriptomics: Their emerging roles in the discovery and validation of colorectal cancer biomarkers. Expert Rev. Proteom. 2014, 11, 179–205. [Google Scholar] [CrossRef] [PubMed]
- Chauvin, A.; Boisvert, F.M. Clinical Proteomics in Colorectal Cancer, a Promising Tool for Improving Personalised Medicine. Proteomes 2018, 6, 49. [Google Scholar] [CrossRef]
- Li, J.; Ma, X.; Chakravarti, D.; Shalapour, S.; DePinho, R.A. Genetic and biological hallmarks of colorectal cancer. Genes Dev. 2021, 35, 787–820. [Google Scholar] [CrossRef] [PubMed]
- Lindhorst, P.H.; Hummon, A.B. Proteomics of Colorectal Cancer: Tumors, Organoids, and Cell Cultures—A Minireview. Front. Mol. Biosci. 2020, 7, 604492. [Google Scholar] [CrossRef]
- Casamassimi, A.; Ciccodicola, A. Transcriptional Regulation: Molecules, Involved Mechanisms, and Misregulation. Int. J. Mol. Sci. 2019, 20, 1281. [Google Scholar] [CrossRef]
- Lambert, S.A.; Jolma, A.; Campitelli, L.F.; Das, P.K.; Yin, Y.; Albu, M.; Chen, X.; Taipale, J.; Hughes, T.R.; Weirauch, M.T. The Human Transcription Factors. Cell 2018, 172, 650–665. [Google Scholar] [CrossRef]
- Chang, Y.Y.; Lin, J.K.; Lin, T.C.; Chen, W.S.; Jeng, K.J.; Yang, S.H.; Wang, H.S.; Lan, Y.T.; Lin, C.C.; Liang, W.Y.; et al. Impact of KRAS mutation on outcome of patients with metastatic colorectal cancer. Hepatogastroenterology 2014, 61, 1946–1953. [Google Scholar]
- Ogino, S.; Nosho, K.; Kirkner, G.J.; Shima, K.; Irahara, N.; Kure, S.; Chan, A.T.; Engelman, J.A.; Kraft, P.; Cantley, L.C.; et al. PIK3CA mutation is associated with poor prognosis among patients with curatively resected colon cancer. J. Clin. Oncol. 2009, 27, 1477–1484. [Google Scholar] [CrossRef]
- Watanabe, T.; Wu, T.T.; Catalano, P.J.; Ueki, T.; Satriano, R.; Haller, D.G.; Benson, A.B., 3rd; Hamilton, S.R. Molecular predictors of survival after adjuvant chemotherapy for colon cancer. N. Engl. J. Med. 2001, 344, 1196–1206. [Google Scholar] [CrossRef] [PubMed]
- Belt, E.J.; Brosens, R.P.; Delis-van Diemen, P.M.; Bril, H.; Tijssen, M.; van Essen, D.F.; Heymans, M.W.; Beliën, J.A.; Stockmann, H.B.; Meijer, S.; et al. Cell cycle proteins predict recurrence in stage II and III colon cancer. Ann. Surg. Oncol. 2012, 19, S682–S692. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cacina, C.; Yaylim-Eraltan, I.; Arikan, S.; Saglam, E.K.; Zeybek, U.; Isbir, T. Association between CDKN1A Ser31Arg and C20T gene polymorphisms and colorectal cancer risk and prognosis. In Vivo 2010, 24, 179–183. [Google Scholar] [PubMed]
- Shen, W.; Xi, H.; Wei, B.; Chen, L. The prognostic role of matrix metalloproteinase 2 in gastric cancer: A systematic review with meta-analysis. J. Cancer Res. Clin. Oncol. 2014, 140, 1003–1009. [Google Scholar] [CrossRef] [PubMed]
- Yokota, T.; Serizawa, M.; Hosokawa, A.; Kusafuka, K.; Mori, K.; Sugiyama, T.; Tsubosa, Y.; Koh, Y. PIK3CA mutation is a favorable prognostic factor in esophageal cancer: Molecular profile by next-generation sequencing using surgically resected formalin-fixed, paraffin-embedded tissue. BMC Cancer 2018, 18, 826. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Li, X.; Wang, H.; Shi, Y.; Xu, C.; Lu, S.; Huang, J.; Xu, Y.; Zeng, H.; Su, J.; et al. The prognostic value of EGFR overexpression and amplification in Esophageal squamous cell Carcinoma. BMC Cancer 2015, 15, 377. [Google Scholar] [CrossRef]
- Zheng, L.; Zhan, Y.; Lu, J.; Hu, J.; Kong, D. A prognostic predictive model constituted with gene mutations of APC, BRCA2, CDH1, SMO, and TSC2 in colorectal cancer. Ann. Transl. Med. 2021, 9, 680. [Google Scholar] [CrossRef]
- Slattery, M.L.; Herrick, J.S.; Lundgreen, A.; Fitzpatrick, F.A.; Curtin, K.; Wolff, R.K. Genetic variation in a metabolic signaling pathway and colon and rectal cancer risk: mTOR, PTEN, STK11, RPKAA1, PRKAG2, TSC1, TSC2, PI3K and Akt1. Carcinogenesis 2010, 31, 1604–1611. [Google Scholar] [CrossRef]
- Zlobec, I.; Baker, K.; Terracciano, L.M.; Lugli, A. RHAMM, p21 combined phenotype identifies microsatellite instability-high colorectal cancers with a highly adverse prognosis. Clin. Cancer Res. 2008, 14, 3798–3806. [Google Scholar] [CrossRef][Green Version]
- Mitomi, H.; Ohkura, Y.; Fukui, N.; Kanazawa, H.; Kishimoto, I.; Nakamura, T.; Yokoyama, K.; Sada, M.; Kobayashi, K.; Tanabe, S.; et al. P21WAF1/CIP1 expression in colorectal carcinomas is related to Kras mutations and prognosis. Eur. J. Gastroenterol. Hepatol. 2007, 19, 883–889. [Google Scholar] [CrossRef]
- Bukholm, I.K.; Nesland, J.M. Protein expression of p53, p21 (WAF1/CIP1), bcl-2, Bax, cyclin D1 and pRb in human colon carcinomas. Virchows Arch. 2000, 436, 224–228. [Google Scholar] [CrossRef] [PubMed]
- Rau, B.; Sturm, I.; Lage, H.; Berger, S.; Schneider, U.; Hauptmann, S.; Wust, P.; Riess, H.; Schlag, P.M.; Dörken, B.; et al. Dynamic expression profile of p21WAF1/CIP1 and Ki-67 predicts survival in rectal carcinoma treated with preoperative radiochemotherapy. J. Clin. Oncol. 2003, 21, 3391–3401. [Google Scholar] [CrossRef]
- Ogino, S.; Nosho, K.; Shima, K.; Baba, Y.; Irahara, N.; Kirkner, G.J.; Hazra, A.; De Vivo, I.; Giovannucci, E.L.; Meyerhardt, J.A.; et al. p21 expression in colon cancer and modifying effects of patient age and body mass index on prognosis. Cancer Epidemiol. Biomark. Prev. 2009, 18, 2513–2521. [Google Scholar] [CrossRef] [PubMed]
- Bednarz-Misa, I.; Fortuna, P.; Diakowska, D.; Jamrozik, N.; Krzystek-Korpacka, M. Distinct Local and Systemic Molecular Signatures in the Esophageal and Gastric Cancers: Possible Therapy Targets and Biomarkers for Gastric Cancer. Int. J. Mol. Sci. 2020, 21, 4509. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Blatt, A.; Feld, S.; Zohar, Y.; Saadi, E.; Barki-Harrington, L.; Hammond, E.; Ilan, N.; Vlodavsky, I.; Chowers, Y.; et al. The Heparanase Inhibitor PG545 Attenuates Colon Cancer Initiation and Growth, Associating with Increased p21 Expression. Neoplasia 2017, 19, 175–184. [Google Scholar] [CrossRef]
- Li, Z.; Qiu, R.; Qiu, X.; Tian, T. SNHG6 Promotes Tumor Growth via Repression of P21 in Colorectal Cancer. Cell Physiol. Biochem. 2018, 49, 463–478. [Google Scholar] [CrossRef]
- Kang, B.W.; Jeon, H.S.; Chae, Y.S.; Lee, S.J.; Park, J.Y.; Choi, J.E.; Park, J.S.; Choi, G.S.; Kim, J.G. Association between GWAS-identified genetic variations and disease prognosis for patients with colorectal cancer. PLoS ONE 2015, 10, e0119649. [Google Scholar] [CrossRef]
- Kreis, N.N.; Louwen, F.; Yuan, J. The Multifaceted p21 (Cip1/Waf1/CDKN1A) in Cell Differentiation, Migration and Cancer Therapy. Cancers 2019, 11, 1220. [Google Scholar] [CrossRef]
- Al Bitar, S.; Gali-Muhtasib, H. The Role of the Cyclin Dependent Kinase Inhibitor p21(cip1/waf1) in Targeting Cancer: Molecular Mechanisms and Novel Therapeutics. Cancers 2019, 11, 1475. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Training Set | Validation Set |
|---|---|
| BAG1 | BAX |
| BCL-2 | CDH1 |
| CDKN1A | CDKN1B |
| CXCR4 | EGFR |
| ERBB2 | ESR1 |
| KRAS | MK167 |
| PIK3CA | PLAU |
| PTEN | TERT |
| TFGBRII | TP53 |
| TYMS | VEGF |
| Predicted Genes | Main Functions or Relevant Molecular Phenomena |
|---|---|
| CDK2, CDK4, CDK6, CDKN1A, CDKN1B, CCNA1, CCND1, CCND2, CCND3, CCNE1 | Regulation of cell cycle |
| EGF, EGFR, FGFR1, HRAS, KDR *, KRAS, PIK3CA, PIK3R3, TGFB1, TGFB3, TGFBR2 | Cell growth, proliferation, differentiation or embryogenesis, wound healing |
| BAD, BAX, BCL2, BCL2L1, BID | Regulation of apoptosis and cell death |
| CREB1, E2F3, SMAD4, STAT1 | Transcription factors |
| CDH1, CTNNB1, FN1 | Cell adhesion, motility and/or shape |
| PDGFC, PDGFRB, VEGFC | Growth factors and their receptors |
| MAPK3, SHC1, IRS2 | Cellular signaling, signal transduction |
| PTEN *, TSC2 | Tumor suppressor genes |
| MMP2 | Extracellular metalloproteinase |
| TLR2 | Immune system regulation |
| MDM2 | Ubiquitin-protein ligase |
| Gene 1 | Patient 1 | Patient 2 | Patient 3 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Expression 2 | p-Value | Expression 2 | p-Value | Expression 2 | p-Value | ||||
| CCNA1 | 0.890 | 0.4366 | 2.343 | ↑↑ | 0.0046 | 0.380 | ↓↓ | 0.0088 | |
| CCND1 | 1.423 | 0.1843 | 3.433 | ↑↑ | 0.0031 | 5.627 | ↑↑↑ | 0.0001 | |
| CCND2 | 1.093 | 0.7726 | 1.060 | 0.9889 | 5.253 | ↑↑↑ | 0.0003 | ||
| CCND3 | 0.320 | ↓↓ | 0.0260 | 1.043 | 0.7505 | 9.560 | ↑↑↑↑ | 0.0001 | |
| CCNE1 | 1.050 | 0.9274 | 3.413 | ↑↑ | 0.0036 | 0.800 | 0.0602 | ||
| CDK2 | 3.480 | ↑↑ | 0.0116 | 4.187 | ↑↑↑ | 0.0002 | 2.527 | ↑↑ | 0.0014 |
| CDK4 | 2.360 | ↑↑ | 0.0017 | 3.170 | ↑↑ | 0.0015 | 2.380 | ↑↑ | 0.0056 |
| CDK6 | 2.330 | ↑↑ | 0.0048 | 3.870 | ↑↑ | 0.0019 | 6.717 | ↑↑↑ | 0.0001 |
| CDKN1A | 1.060 | 0.6550 | 1.250 | 0.2477 | 1.333 | 0.1679 | |||
| CDKN1B | 1.707 | ↑ | 0.0163 | 1.177 | 0.3277 | 5.533 | ↑↑↑ | 0.0001 | |
| EGF | 1.160 | 0.6528 | 4.600 | ↑↑↑ | 0.0013 | 9.340 | ↑↑↑↑ | 0.0002 | |
| EGFR | 0.390 | ↓↓ | 0.0216 | 1.563 | ↑ | 0.0047 | 3.617 | ↑↑ | 0.0001 |
| FGFR1 | 2.690 | ↑↑ | 0.0002 | 0.713 | 0.0767 | 3.490 | ↑↑ | 0.0017 | |
| HRAS | 0.280 | ↓↓ | 0.0144 | 1.157 | 0.3894 | 1.560 | ↑ | 0.0151 | |
| KRAS | 0.373 | ↓↓ | 0.0097 | 1.230 | 0.3994 | 8.313 | ↑↑↑↑ | 0.0002 | |
| PIK3CA | 0.297 | 0.0576 | 1.557 | ↑ | 0.0178 | 9.830 | ↑↑↑↑ | 0.0006 | |
| PIK3R3 | 5.847 | ↑↑↑ | 0.0008 | 1.130 | 0.1943 | 10.26 | ↑↑↑↑ | 0.0005 | |
| TGFB1 | 1.617 | 0.0728 | 1.917 | ↑ | 0.0000 | 0.143 | ↓↓↓ | 0.0060 | |
| TGFB3 | 0.807 | 0.2360 | 1.203 | 0.8063 | 2.830 | ↑↑ | 0.0165 | ||
| TGFBR2 | 0.913 | 0.2790 | 0.567 | ↓ | 0.0114 | 5.737 | ↑↑↑ | 0.0008 | |
| BAD | 1.670 | ↑ | 0.0052 | 10.07 | ↑↑↑↑ | 0.0000 | 0.210 | ↓↓↓ | 0.0135 |
| BAX | 0.870 | 0.4985 | 4.483 | ↑↑↑ | 0.0098 | 0.387 | ↓↓ | 0.0049 | |
| BCL2 | 0.543 | 0.1271 | 0.210 | ↓↓↓ | 0.0184 | 1.253 | ↑ | 0.0331 | |
| BCL2L1 | 2.430 | ↑↑ | 0.0101 | 8.863 | ↑↑↑↑ | 0.0000 | 1.097 | 0.3578 | |
| BID | 7.500 | ↑↑↑ | 0.0002 | 6.157 | ↑↑↑ | 0.0001 | 7.463 | ↑↑↑ | 0.0002 |
| CREB1 | 1.563 | 0.1042 | 2.363 | ↑↑ | 0.0008 | 13.94 | ↑↑↑↑ | 0.0000 | |
| E2F3 | 2.600 | ↑↑ | 0.0071 | 1.100 | 0.7703 | 4.443 | ↑↑↑ | 0.0012 | |
| SMAD4 | 0.507 | ↓ | 0.0035 | 1.083 | 0.4171 | 0.123 | ↓↓↓↓ | 0.0023 | |
| STAT1 | 1.097 | 0.4855 | 1.523 | ↑ | 0.0456 | 2.790 | ↑↑ | 0.0370 | |
| CDH1 | 0.993 | 0.7830 | 1.267 | 0.1972 | 6.590 | ↑↑↑ | 0.0001 | ||
| CTNNB1 | 2.403 | ↑↑ | 0.0008 | 0.853 | 0.1334 | 10.96 | ↑↑↑↑ | 0.0001 | |
| FN1 | 3.673 | ↑↑ | 0.0039 | 0.843 | 0.1443 | 2.403 | ↑↑ | 0.0005 | |
| PDGFC | 0.600 | ↓ | 0.0110 | 3.247 | ↑↑ | 0.0016 | 2.813 | ↑↑ | 0.0033 |
| PDGFRB | 3.350 | ↑↑ | 0.0028 | 4.733 | ↑↑↑ | 0.0002 | 3.010 | ↑↑ | 0.0069 |
| VEGFC | 1.583 | ↑ | 0.0249 | 2.990 | ↑↑ | 0.0007 | 0.237 | ↓↓↓ | 0.0212 |
| MAPK3 | 0.617 | 0.0947 | 2.313 | ↑↑ | 0.0006 | 4.903 | ↑↑↑ | 0.0044 | |
| SHC1 | 2.607 | ↑↑ | 0.0062 | 1.250 | ↑ | 0.0030 | 12.06 | ↑↑↑↑ | 0.0002 |
| IRS2 | 15.74 | ↑↑↑↑ | 0.0001 | 4.457 | ↑↑↑ | 0.0001 | 4.300 | ↑↑↑ | 0.0003 |
| TSC2 | 0.840 | 0.2096 | 2.453 | ↑↑ | 0.0034 | 4.810 | ↑↑↑ | 0.0012 | |
| MMP2 | 1.210 | 0.4724 | 2.573 | ↑↑ | 0.0057 | 1.217 | ↑ | 0.0097 | |
| TLR2 | 3.913 | ↑↑ | 0.0045 | 9.013 | ↑↑↑↑ | 0.0000 | 0.927 | 0.6464 | |
| MDM2 | 1.300 | 0.6030 | 1.427 | 0.1993 | 7.947 | ↑↑↑ | 0.0001 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kamel, F.; Schneider, N.; Nisar, P.; Soloviev, M. Bottom-Up Approach to the Discovery of Clinically Relevant Biomarker Genes: The Case of Colorectal Cancer. Cancers 2022, 14, 2654. https://doi.org/10.3390/cancers14112654
Kamel F, Schneider N, Nisar P, Soloviev M. Bottom-Up Approach to the Discovery of Clinically Relevant Biomarker Genes: The Case of Colorectal Cancer. Cancers. 2022; 14(11):2654. https://doi.org/10.3390/cancers14112654
Chicago/Turabian StyleKamel, Faddy, Nathalie Schneider, Pasha Nisar, and Mikhail Soloviev. 2022. "Bottom-Up Approach to the Discovery of Clinically Relevant Biomarker Genes: The Case of Colorectal Cancer" Cancers 14, no. 11: 2654. https://doi.org/10.3390/cancers14112654
APA StyleKamel, F., Schneider, N., Nisar, P., & Soloviev, M. (2022). Bottom-Up Approach to the Discovery of Clinically Relevant Biomarker Genes: The Case of Colorectal Cancer. Cancers, 14(11), 2654. https://doi.org/10.3390/cancers14112654