Simple Summary
BRCA1 and−2 are critical components of the homologous recombination pathway of DNA repair required to effectively repair DNA double strand breaks, leading to an increased cancer risk in patients with inherited BRCA mutations. An additional subset of cancers exhibit ‘BRCAness’, harboring repair defects stemming from mutations in non-BRCA DNA repair genes. Both BRCA-mutant cancers and cancers with a BRCAness phenotype are sensitive to PARP inhibitors, a class of cancer therapy drugs that inhibit the repair of DNA single strand breaks. To expand the use of PARP inhibitors to a larger group of patients, studies have focused on new combination strategies using agents that can induce BRCAness. This review focuses on the current status of drug-induced BRCAness in combination with PARP inhibitors to enhance cancer treatment.
Abstract
The poly(ADP-ribose) polymerase (PARP) family of proteins has been implicated in numerous cellular processes, including DNA repair, translation, transcription, telomere maintenance, and chromatin remodeling. Best characterized is PARP1, which plays a central role in the repair of single strand DNA damage, thus prompting the development of small molecule PARP inhibitors (PARPi) with the intent of potentiating the genotoxic effects of DNA damaging agents such as chemo- and radiotherapy. However, preclinical studies rapidly uncovered tumor-specific cytotoxicity of PARPi in a subset of cancers carrying mutations in the BReast CAncer 1 and 2 genes (BRCA1/2), which are defective in the homologous recombination (HR) DNA repair pathway, and several PARPi are now FDA-approved for single agent treatment in BRCA-mutated tumors. This phenomenon, termed synthetic lethality, has now been demonstrated in tumors harboring a number of repair gene mutations that produce a BRCA-like impairment of HR (also known as a ‘BRCAness’ phenotype). However, BRCA mutations or BRCAness is present in only a small subset of cancers, limiting PARPi therapeutic utility. Fortunately, it is now increasingly recognized that many small molecule agents, targeting a variety of molecular pathways, can induce therapeutic BRCAness as a downstream effect of activity. This review will discuss the potential for targeting a broad range of molecular pathways to therapeutically induce BRCAness and PARPi synthetic lethality.
1. Introduction
Poly(ADP-ribose) polymerase (PARP) proteins catalyze the transfer of an ADP (adenosine diphosphate)-ribose subunit of nicotinamide adenine dinucleotide (NAD+) onto a broad range of proteins, forming poly(ADP-ribose) (PAR) polymers. PARP1, which is responsible for the majority of cellular PARylation [1], is activated when its N-terminal DNA binding domain recognizes and binds to damaged DNA structures [2]. The recognition of single strand breaks (SSBs) by PARP1, and its subsequent auto-PARylation, mediates interaction with the molecular scaffold protein XRCC1 and the coordination of the transient assembly of multiprotein complexes that perform the post-recognition steps of SSB repair. Simultaneously, PARylation of histone proteins around the damage site remodels the surrounding chromatin to allow repair [3,4]. When the PARP1 protein level is reduced, either by CRISPR knockout [5] or small interfering RNA (siRNA) targeting [6], or catalytic activity is impaired, by expression of a catalytically inactive mutant [7] or targeting by small molecule inhibitors [8], a delayed repair of SSBs is observed leading to G2/M arrest, chromosomal instability, and cytotoxicity.
Observations that analogs of the PARP1 catalytic byproduct nicotinamide inhibit PAR synthesis in vitro [9] provided the template for the development of small molecule PARP inhibitors (PARPi). PARPi that have now received FDA approval include olaparib (AstraZeneca/Merck/KuDOS, Cambridge, UK; approved 2014), rucaparib (Clovis, Boulder, CO, USA; approved 2016), and niraparib (GlaxoSmithKline, Brentford, UK; approved 2017), while additional agents veliparib (AbbVie, North Chicago, IL, USA) and pamiparib (Beigene, Beijing, China) are currently being evaluated in phase III trials. In 2018, the second generation PARPi talazoparib (Pfizer, New York, NY, USA) was approved by the FDA, offering enhanced potency over its predecessors [10]. PARPi interact with the binding site of the PARP substrate NAD+, reducing PARylation activity and hence impairing single strand break repair (SSBR) capacity. It is now recognized that each of the clinically available PARPi possess differing abilities to ‘trap’ PARP at damage sites [11,12], with talazoparib possessing a trapping capacity 100-fold greater than the next most potent PARP trapper, niraparib.
PARPi selectivity for cancer cells harboring defects in DNA double strand break (DSB) repair was first described in concurrent publications by Farmer et al. [13] and Bryant et al. [14] in 2005, with both groups reporting exquisite PARPi sensitivity in BRCA-mutated tumors. BRCA1 and BRCA2 proteins play central roles in homologous recombination, the high-fidelity pathway responsible for the repair of DSBs during DNA replication [15]. Importantly, defects in HR stemming from BRCA mutations increase susceptibility to single-stranded base damage. Single-strand interruptions or adducts block replication fork progression, leading to extended fork stalling or collapse into a DSB–both of which require the proficiency of HR mechanisms for successful resolution [16]. Small molecule inhibitors of PARP1 are potent inducers of replication fork disruption, whether by the inactivation of the catalytic signaling that prevents SSBR, or by direct blocking of fork progression through the trapping of PARP into the DNA. Failure of BRCA-mutant cells to repair these PARPi-induced lesions is associated with an accumulation of cytotoxic DSB damage and the eventual activation of apoptotic mechanisms to limit the deleterious effects of error-prone repair [13,14].
The discovery that BRCA mutation and PARP inhibition, two independently non-lethal defects, combine to induce potent cell death forms the prototypical example of synthetic lethality [17]. This powerful concept allows cancer-specific defects to inform an effective therapeutic strategy with few off-target complications. However, BRCA mutations are relatively rare, associated with 5–10% of breast and ovarian cancers (and a lower percentage in other tumor types). As such, there is a growing push for the identification of tumors that exhibit ‘BRCAness’, mimicking the defective HR phenotype of BRCA mutation and potentially expanding the therapeutic utility of PARPi to a larger subset of tumors [18,19]. The BRCAness phenotype has been linked to mutations in other repair factors involved in DNA damage response (such as ATM, ATR, RAD51, or the Fanconi Anemia (FA) family), as well as altered gene expression by epigenetic silencing or cellular regulatory mechanisms [20]. To capitalize upon the potential of PARPi treatment in these settings, a number of methods have been explored to detect tumor BRCAness and predict PARPi sensitivity, including panel sequencing for DNA repair gene mutations, repair gene expression microarrays, testing for surrogate markers of HR deficiency such as loss of heterozygosity or sequence deletions associated with junctional microhomology, or functional tests of repair capacity such as RAD51 foci formation (recently reviewed in [21]). However, despite the translation of several of these methods into clinical trials, the predictive value of such tests in the context of PARPi therapy has not yet been conclusively established [22].
More recently, it has become recognized that BRCAness may be induced using therapeutic agents that modulate a variety of molecular pathways, potentially providing a novel method for inducing synthetic lethality in cancers that are otherwise HR-proficient. This review will focus on the therapeutic targeting of major molecular pathways that have been implicated in BRCAness, including epigenetic mechanisms, cell cycle checkpoints, and receptor kinase activity.
2. Modulation of Epigenetic Pathways
Epigenetics refers to heritable factors that influence cellular phenotypes other than DNA sequence, including DNA methylation, histone modification, and gene silencing by non-coding RNAs [23]. Inhibitors of the first two of these mechanisms have been linked to the induction of BRCAness (Figure 1).
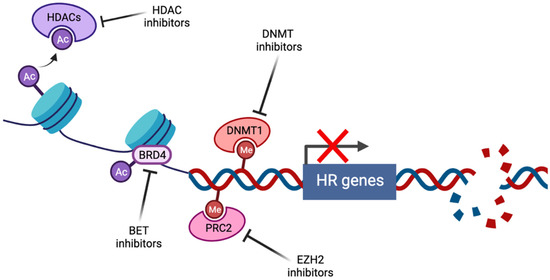
Figure 1.
Induction of BRCAness by pharmacological targeting of epigenetic pathways (created with Biorender.com, accessed 15 May 2022). Histone deacetylase inhibitors (HDACi) prevent the histone deacetylation activity of HDACs, thus maintaining chromatin in a condensed state associated with transcriptional repression. Bromodomain and extraterminal (BET) family members such as BRD4 act as transcriptional cofactors at acetylated gene promoters, including the homologous recombination (HR) genes RAD51 and BRCA1, whose expression is suppressed by BET inhibition. DNA methyltransferase (DNMT) and enhancer of zeste homolog 2 (EZH2) inhibitors alter genome-wide methylation patterns and have been linked to altered DNA double strand break repair gene expression. In each case, repression of HR gene expression and activity has been described, contributing to induction of the BRCAness phenotype.
2.1. Inhibition of DNA Methylation
DNA methyltransferases (DNMTs) are critical mediators of epigenetic gene regulation, responsible for genome-wide de novo and maintenance methylation. Aberrations in methylation have been widely implicated in cancer development, progression, and response to treatment [24,25], and consequently, DNMT inhibitors (DNMTi) including decitabine and 5-azacytidine have been developed and are now FDA-approved for the treatment of myelodysplastic syndrome [26,27]. These agents are cytosine analogs that become incorporated into replicating DNA, where they are targeted for methylation by DNMTs. Due to their altered structure, they cannot be released by DNMT by β-elimination, leading to the covalent entrapment of DNMT into the DNA [28,29].
There is evidence of a biological interplay between DNMT1, the enzyme responsible for maintenance methylation, and PARP1, that provides a rationale for combination DNMTi-PARPi therapy. DNMT1 and PARP1 are members of a multiprotein complex that localizes to sites of oxidative DNA damage [30], where the presence of PARylated PARP1 inhibits methylation activity by DNMT1 [31], possibly to maintain an open chromatin structure to permit repair. Our group has demonstrated that combining low-dose DNMTi treatment with the potent PARP-trapping PARPi talazoparib enhances PARP1-DNA binding, synergistically enhancing cytotoxicity across a number of BRCA-wildtype cancer types with minimal toxicity in in vivo models [32,33,34] or human subjects [35,36]. Similar synergism has also been observed when talazoparib is combined with the second-generation DNMTi guadecitabine [37].
In addition to a direct reduction in the free enzyme pool, DNMT entrapment also induces ubiquitin-E3 ligase-mediated proteasomal degradation of free DNMT1 [38,39]. Accordingly, low doses of DNMTi are sufficient to alter methylation patterns across the genome, leading to widespread alterations in multiple molecular pathways including the DNA damage response (DDR) and apoptosis [40]. Among the myriad of pathways contributing to the Hanahan and Weinberg ‘hallmarks of cancer’ [41] that are altered by DNMTi, our recent examination of the DNA repair reactome in non-small cell lung [33], breast, and ovarian cancers [34] demonstrated a significant reduction in DSB repair, particularly involving the FA pathway. Of note was the downregulation of FANCD2, which is mono-ubiquitinated by other FA pathway members in response to DNA damage, leading to colocalization with BRCA1 and BRCA2 during homologous recombination repair of DSBs, and resulting in it being ascribed a role as a BRCAness gene [42]. Furthermore, FANCD2 monoubiquitination is required for interactions with FANCD2/FANCI-associated nuclease 1 (FAN1), which mediates the canonical FA roles of interstrand crosslink repair and the resolution of stalled replication forks, potentially including those induced by trapped PARP1 and/or DNMT1 [43]. In keeping with the loss of these repair roles, DNMTi-induced FANCD2 downregulation was associated with a BRCAness phenotype, including increased replication fork stalling, DSB accumulation as measured by γH2AX foci accumulation, and a reduction in RAD51-mediated DSB repair capacity. Accordingly, in several human cancer cell lines and murine xenograft models, combining a low dose DNMTi with the PARPi talazoparib produces a significant and synergistic increase in tumor cell cytotoxicity [33,34]. These results have led to a dose-finding Phase 1 trial in untreated or relapsed/refractory AML using DNMTi decitabine and PARPi talazoparib [44], and a Phase 1 trial in BRCA-proficient breast cancer treated using oral decitabine and talazoparib (Table 1).

Table 1.
Clinical trials evaluating epigenetic therapy in combination with PARPi.
DNMTi-induced reversal of cancer-associated methylation abnormalities can reactivate abnormally methylated tumor suppressor gene promoters. One emerging example is Schlafen 11 (SLFN11), which irreversibly inhibits replication in cells undergoing replication stress such as DNA-damaging chemotherapy [45]. High levels of SLFN11 destabilizes the interaction between single-stranded DNA and replication protein A (RPA) at the sites of DNA damage, inhibiting downstream DSB repair and producing cell cycle checkpoint activation [46]. The suppression of SLFN11 expression is observed in ~50% of cancer cell lines and is correlated with a resistance to DNA-damaging agents including PARP inhibitors [47]. SLFN11 suppression appears to be primarily epigenetic in origin, linked to promoter methylation, histone deacetylation, and PRC-mediated histone methylation [45]. Decitabine can reverse SLFN11 promoter methylation, leading to the re-expression and re-sensitization to DNA-damaging agents, and similar results have also been observed following EZH2 [48] or HDAC inhibitors [49] (see below), providing a further rationale for future studies combining epigenetic agents with PARP inhibitors.
2.2. Maintenance of Chromatin Repressive States
Histone deacetylases (HDACs) remove acetyl groups from ϵ-N-acetyl lysine residues on histones, leading to chromatin condensation and transcriptional repression. Abnormal acetylation resulting from HDAC overexpression can downregulate the expression of various tumor suppressive mechanisms, including cyclin-dependent kinases, differentiation factors, and proapoptotic signals, leading to the uncontrolled proliferation, de-differentiation, and survival that is characteristic of oncogenesis and metastasis [50]. The eighteen identified members of the HDAC family have been classified into four groups (class I, IIa/b, V, and III/sirtuins) based on homology to yeast HDACs. Compounds with anti-HDAC activity are numerous, and can be divided into pan-HDAC inhibitors (HDACi), which exhibit activity against all non-sirtuin HDACs, or selective HDACi, which target specific HDACs [51]. FDA approval has been granted for the treatment of various hematological malignancies for three pan-HDACi (vorinostat, belinostat, and panobinostat) and one HDAC1/2-selective HDACi (romidepsin) [52].
Acetylation exerts effects over the chromatin structure that impacts the recognition and repair of DNA damage [53], and accordingly, HDACi have been reported to alter DSB repair capacity [54]. While deacetylation activity of HDAC1/2 has been shown to both directly and indirectly decrease c-NHEJ activity [55,56], the role of HDACs in HR, and hence the therapeutic potential of HDACi for the induction of BRCAness, is less clearly defined. Of note, HR proteins including BRCA1, BRCA2, and RAD51 have been reported to be suppressed by HDACi in a variety of cancers [57,58], sensitizing to PARPi [59,60,61,62,63,64,65]. Based on these results, phase I trials combining olaparib with vorinostat are underway in advanced lymphoma and breast cancer (Table 1).
Notably, the inhibition of the deacetylation activity following HDACi exposure leads to PARP1 hyperacetylation and enrichment in chromatin that resembles PARPi-induced PARP trapping. When combined with PARPi, HDACi treatment further increases PARP trapping, synergistically sensitizing to the PARP-trapping PARPi talazoparib [66]. Synergism has also been observed when HDACi are combined with DNMTi, specifically by enhancing the re-expression of genes silenced by abnormal promoter methylation [35,67]. Valdez et al. have reported synergistic inhibition of AML and lymphoma cell proliferation by the triple combination of PARPi niraparib, DNMTi decitabine, and HDACi romidepsin or pabinostat, associated with the activation of ATM-mediated DDR, increased ROS production, and the induction of apoptosis [68]. These effects were hypothesized to be the sequelae of DSB accumulation induced by triple combination through three mechanisms: significantly enhanced PARP trapping; acetylation and inhibition of DNA repair proteins including Ku70/80 and PARP1; and the downregulation of the nucleosome-remodeling deacetylase complex, a transcriptional repressor with chromatin remodeling activity that is functionally linked to efficient DNA repair [69]. While further preclinical study is required, these results provide a rationale for the future development of combination therapy using PARPi, HDACi, and DNMTi.
2.3. Polycomb Repressive Complex 2
An enhancer of the zeste homolog 2 (EZH2) is the histone methyltransferase subunit of polycomb repressive complex 2 (PRC2), which methylates histone H3 on lysine 27 (H3K27me3) to mark chromatin as transcriptionally silent. PRC2 plays an important oncogenic role through the modulation of the DDR [70]. EZH2 overexpression, which is common in many cancers [71], induces the downregulation of RAD51 homolog expression [72], cytoplasmic BRCA1 retention [73], and impaired HR that is associated with increased genomic instability. PRC2 appears to play a role in the DSB repair pathway choice, being recruited to DSBs in a Ku-dependent mechanism to promote efficient NHEJ [74], and accordingly, EZH2 depletion favors HR, impairs NHEJ, and sensitizes to irradiation damage [75]. Recent evidence indicates that this DSB repair pathway switch can be therapeutically targeted by PARPi in a subset of HR-proficient tumors overexpressing the oncogene coactivator-associated arginine methyltransferase 1 (CARM1). The overexpression of CARM1 promotes the EZH2 silencing of MAD2L2, a member of the shieldin complex that limits DNA end resection to favor NHEJ. Accordingly, in CARM1-high cells, EZH2 inhibition upregulates MAD2L2, increasing error-prone NHEJ activity and associated chromosomal abnormalities, and producing mitotic catastrophe in combination with PARPi treatment [76]. An ongoing phase II clinical trial, evaluating multiple targeted therapies in a biomarker-guided precision therapy approach, includes the novel agents SHR2554 (EZH2 inhibitor) and SHR3162 (PARPi) (Table 1).
2.4. BET Proteins
The conserved bromodomain and extraterminal (BET) family of proteins are characterized by two tandem bromodomains that bind to activated lysine residues on target proteins [77,78]. BET members preferentially interact with hyperacetylated histones, leading to an accumulation at the transcriptionally active regulatory elements [79]. BET family member BRD4 acts as a transcriptional cofactor, influencing the expression of a wide range of genes involved in cell fate determination. In cancer, BRD4 has been implicated in the activation of a multitude of oncogenes, co-occupying a set of promoter super-enhancers associated with prominent oncogenic drivers such as c-MYC [80,81]. High affinity small molecules targeting the BET bromodomains demonstrate preclinical efficacy in a wide range of cancers associated with transcriptional suppression of key proto-oncogenes including c-MYC, N-MYC, FOSL1, and BLC2 (reviewed in [79].
The first report of potential synergism between BET inhibition and PARPi was based on a drug combination screen testing PARPi olaparib in BRCA-wildtype triple negative breast (TNBC), ovarian, and prostate cancer in combination with 20 well-characterized epigenetic modulators across seven classes, demonstrating synergism for all tested BET inhibitors (BETi) [82]. BETi treatment significantly enhanced PARPi-induced DSB accumulation independent of PARP-trapping, associated with the repression of BRCA1 and RAD51 transcription, which is suggestive of induced BRCAness. Notably, BETi treatment could disrupt the enrichment of BRD2/3/4 at the BRCA1 and RAD51 promoter regions, in addition to the putative super-enhancer region downstream of the BRCA1 promoter that exerted a stronger transcriptional enhancing activity than the promoter region alone [82]. Validation of these results, with similar BETi-induced repression of BRCA1 and RAD51, induction of an HR defect, and sensitivity to PARPi, has since been reported in TNBC [83].
A subsequent study used publicly available transcriptional profiling data to demonstrate that BRD4 inhibition modulates a previously validated HR defect gene signature [84], though finding minimal impact on BRCA1 or RAD51 expression in cell lines of multiple cancer types. Instead, a consistent and marked downregulation of CtIP was observed, in keeping with ChIP-seq analysis that indicates that both the CtIP promoter and an associated enhancer region are directly targeted by BRD4. BETi induced PARPi sensitivity in 40 of 55 cancer cell lines and five in vivo models spanning breast, ovarian, and pancreatic cancer, as well as resensitizing PARPi-resistant cells [85]. Despite the mechanistic discrepancies between the studies, these results indicate the therapeutic potential of the PARPi-BETi combination that warrants further investigation.
3. Cell Cycle Checkpoints and the DNA Damage Response
Cell cycle progression is directed by the activity of cyclin-dependent kinases (CDKs), which phosphorylate targets that include transcription factors and regulatory elements such as retinoblastoma (Rb), promoting checkpoint transit. Cell cycle arrest occurs in response to DNA damage, initiated by the activation of ataxia-telangiectasia mutated (ATM; by DSBs) and ataxia-telangiectasia and Rad3-related (ATR; by single strand breaks and stalled replication forks), leading to the phosphorylation of checkpoint kinases (CHK) 2 and 1. Activated CHK1 and CHK2 antagonize the function of the Cdc25 phosphatase family, allowing the accumulation of inhibitory phosphorylation on CDKs, thus delaying cell cycle progression so that DNA repair can occur [87] (Figure 2).
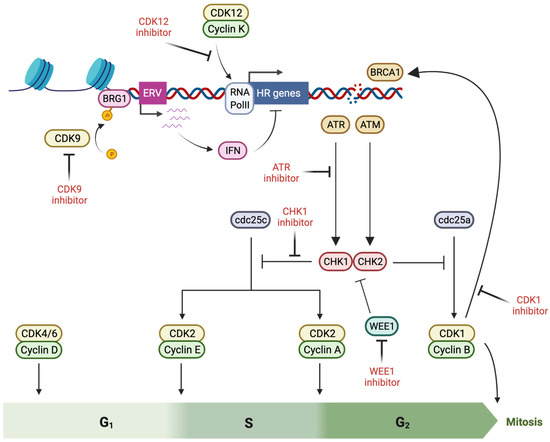
Figure 2.
Induction of BRCAness by pharmacological targeting of cell cycle checkpoint proteins (created with Biorender.com, accessed 15 May 2022). Cell cycle arrest is initiated as a component of the response to DNA damage, initiated by activation of ATM (ataxia-telangiectasia mutated) and ATR (ataxia-telangiectasia and Rad3-related), which lead to CHK (checkpoint kinase) 2 and −1 phosphorylation and Cdc25 antagonism, producing inhibitory phosphorylation of cyclin-dependent kinases (CDK) that prevents cell cycle progression. Multiple factors in this cell cycle checkpoint response directly interact with double strand break repair proteins, promoting repair activity–and thus offering a potential for BRCAness induction via inhibitory molecules.
3.1. Cyclin-Dependent Kinases
CDKs are a family of proline-directed Ser/Thr kinases, first characterized for their highly evolutionarily conserved function in the cell cycle, although now recognized to also have an important role in the modulation of transcription [88]. CDK activity is promoted by conformational changes induced by binding to cyclin subunits, while Thr14/Tyr15 phosphorylation by regulatory proteins such as WEE1 inhibit CDK kinase function until removed by Cdc25 family phosphatases [88]. CDK1, which has roles in S and G2 phase transit, is required for the efficient recruitment of BRCA1 to DNA damage sites, with depletion or small molecule inhibition abrogating S phase checkpoint arrest [89], reducing BRCA1 recruitment [90], and impairing RPA and RAD51 loading [91]. Accordingly, CDK1 inhibition sensitizes to PARPi, both in vitro [92] and in BRCA-wildtype xenograft models [90].
Disruption of transcription-related CDK activity may also impact HR activity. CDK12, in complex with cyclin K, phosphorylates Ser2 at the C-terminal domain of RNA PolII, stimulating productive transcriptional elongation. Additionally, CDK12 phosphorylates and regulates pre-mRNA processing factors such as the U1 snRNP complex, which recognizes and inhibits intronic polyadenylation sites. The mutation of CDK12 or inhibition using the CDK12/13 inhibitor THZ531 produces premature cleavage and polyadenylation (PCPA), leading to truncated gene products, particularly in long (>45 kb) genes [93,94]. Notably, several DNA damage response genes are susceptible to PCPA due to their relatively long length and high number of cryptic polyadenylation sites, including BRCA1, ATR, and the FA genes FANCI and FANCD2, and hence CDK12 has been ascribed a role in the promotion of HR activity and genomic stability [95]. SR-4835, a novel selective CDK12/13 inhibitor, triggers PCPA at polyadenylation sites, impairing HR gene expression and efficiency, and sensitizing to PARPi [96]. Similarly, the multi-CDK targeting inhibitor dinaciclib reduces HR gene expression in TNBC specifically through the inhibition of CDK12, sensitizing the BRCA-wildtype in vitro and in vivo models to PARPi, and reversing de novo and acquired PARPi resistance in BRCA-mutated cells [97]. However, this preclinical promise has yet been translated into clinical trials.
A novel mechanism by which CDKs with transcriptional regulatory activity may impact gene expression is through interplay with epigenetic mechanisms. In euchromatin, CDK9 promotes transcriptional elongation by promoting RNA PolII promoter-proximal pause release [98]. Recent evidence suggests that it also plays a contrasting role in gene silencing, mediated through the regulatory phosphorylation of the SWI/SNF member BRG1, an ATP-dependent helicase that maintains heterochromatic DNA in a condensed conformation. Inhibition of CDK9 by the novel targeted agent MC180295 promotes dephosphorylation of BRG1 leading to chromatin opening and gene reactivation [99]. Amongst the epigenetically silenced genes reactivated by CDK9 inhibition are endogenous retrovirus (ERV) elements, which form cytoplasmic double-stranded RNA aggregations that trigger cellular immune signaling—the basis for the ‘pathogen mimicry’ immune response that has also been described following DNMTi treatment [35,36]. Our work in breast and ovarian cancer has recently linked ERV re-expression and immune signaling directly to the induction of HR defects, providing a mechanistic basis for the observed PARPi sensitivity both in DNMTi treatment [34] and following CDK9 inhibition [100].
3.2. ATR Inhibitors
ATR plays an important role in DNA damage sensing and the cellular response, particularly during S phase. As a member of the phosphatidylinositol-3-kinase-like kinase (PIKK) family, ATR directly phosphorylates Ser/Thr-Glu motifs on hundreds of target proteins across multiple processes involved in the DDR, in addition to activating CHK1-mediated kinase activity and cell cycle arrest [87]. ATR interacts with replication protein A (RPA), which coats single-stranded DNA during replication, allowing it to sense stalled replication forks [101], and it also interacts with ATM on resected ends of DSBs to promote repair [102,103]. Accordingly, ATR inhibition attenuates G2/M arrest following DNA damage, limits RAD51 focus formation, promotes early mitotic entry, and sensitizes to both DNA-damaging cytotoxic agents and PARPi [104].
ATR has been ascribed a role in the development of PARPi resistance in BRCA1-mutated cells, which can become ‘rewired’ to reverse the BRCAness phenotype. In BRCA1-mutant cells, the absence of BRCA1 recruitment to DSB ends is associated with the suppression of end resection, and the impaired recruitment of PALB2-BRCA2, which is required to load RAD51 onto resected single-stranded DNA ends to enact repair. Phosphorylation of RPA by ATR can bypass this block to HR by promoting PALB2-BRCA2 recruitment and subsequent RAD51 loading, reversing BRCA1 mutation-associated HR deficiency [105]. Furthermore, ATR-mediated RAD51 recruitment plays an important role in the stabilization of stalled replication forks in the absence of BRCA1 [105,106], which ordinarily functions to prevent extensive resection by MRE11 [107]. These ATR-dependent pathways of reactivated HR and fork protection produce PARPi resistance that can be abrogated by ATR knockdown or small molecule inhibitors (ATRi) [108]. Several clinical trials combining the ATRi AZD6738 (ceralasertib, AstraZeneca, Cambridge, UK) and olaparib are underway in PARPi-naïve and PARPi-treated cancers (Table 2).
Table 2.
Clinical trials evaluating cell cycle inhibitors in combination with PARPi.
3.3. CHK1 Inhibitors
Activated by direct interaction with ATR, CHK1 signaling plays an important role in the cell cycle signaling pathway in response to replication stress. Notably, potent CHK1 activation is observed in BRCA-mutant cells treated with PARPi, reflecting an increased dependence upon ATR-mediated fork protection [108]. Akin to ATRi treatment, CHK1 inhibitors (CHK1i) induce unrepaired DSB DNA damage while releasing cells from cell cycle arrest into early mitosis [109]. Furthermore, CHK1 directly phosphorylates RAD51 at Thr309, stimulating recruitment after DNA damage [110] and suppressing proteasomal degradation [111,112]. However, despite inducing early mitotic entry, CHK1 inhibition does not produce the marked accumulation of DNA damage and robust PARPi sensitization observed with ATRi [108,113,114]. Accordingly, clinical trials of a CHK1i and PARPi combination have not been initiated at the same rate, with a single phase I of the CHK1i prexasertib in combination with olaparib being recorded on ClinicalTrials.gov (Table 2).
3.4. WEE1 Inhibitors
WEE1 is a member of the serine-threonine (Ser/Thr) kinase family that limits G2 progression by inactivating the phosphorylation of CDK1, thus cooperating with the inhibitory phosphorylation of the Cdc25 phosphatase by ATR/CHK1 [115]. Furthermore, activated WEE1 sustains ATR and CHK1 phosphorylation during the DDR to delay mitotic entry [116]. The inhibition of WEE1 results in the forced activation of CDK1, leading to the phosphorylation of BRCA2 that limits HR [117,118]. Similar to ATR, WEE1 has also been implicated in replication fork protection through direct interaction and negative regulation of DNA cleavage by the endonuclease MUS81 [119], which has structure specific activity against Holliday junctions formed during HR [120]. In keeping with these functions, the inhibition of WEE1 slows replication fork progression, limits HR, activates DDR, and forces mitotic entry, particularly in response to genotoxic agents [121]. Accordingly, the WEE1 inhibitor AZD1775 sensitizes gastric [122], non-small cell lung cancer (NSCLC) [123], and pancreatic cancer [124] to olaparib, particularly when combined with genotoxic stress via ionizing radiation, and a small number of clinical trials are currently underway (Table 2).
4. Tyrosine Kinase Signaling Pathways
4.1. FMS-Like Tyrosine Kinase 3 (FLT3)
FLT3 is a transmembrane receptor that plays a role in hematopoietic cell differentiation, proliferation, and survival via the phosphorylation of downstream targets including phosphoinositide 3-kinase (PI3K), protein kinase B (also known as AKT), Ras, mitogen-activated protein kinase (MAPK), and signal transducer and activator of transcription 5 (STAT5) [125] (Figure 3). FLT3 is predominantly expressed on CD34-positive hematopoietic stem or progenitor cells, where it drives early myeloid and lymphoid lineage development. Approximately 30% of acute myeloid leukemias (AML) carry poor prognosis FLT3 mutations, most commonly internal tandem duplications (ITD) that constitutively activate kinase activity and aberrant signaling via STAT5 to promote proliferation and survival [125,126]. Multitargeted tyrosine kinase inhibitors (TKIs) such as sunitinib and sorafenib possess activity against FLT3, but have demonstrated limited antileukemic activity in clinical trials, prompting the development of inhibitors with greater FLT3 specificity and potency such as gilteritinib and quizartinib [125]. Remissions associated with FLT3 inhibition are usually short-lived, commonly due to the persistence of therapy-refractory leukemia stem cells (LSCs), despite clearance of the bulk of leukemia progenitor cells (LPCs) [127]. Therefore, new approaches are required to enhance therapeutic targeting of LSCs and prolong the efficacy of these agents.
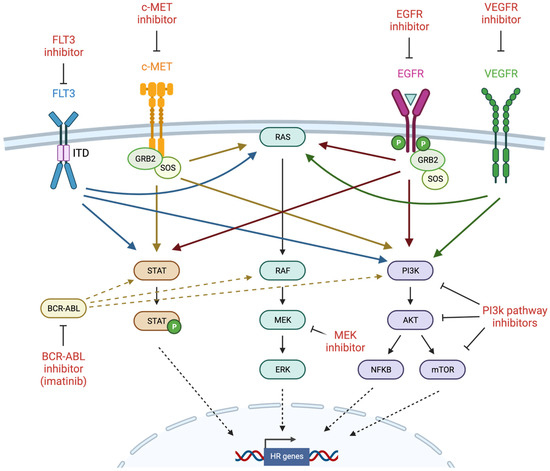
Figure 3.
Induction of BRCAness by tyrosine kinase inhibitors (created with Biorender.com, accessed 15 May 2022). Receptor tyrosine kinases (RTKs) such as FLT3, c-MET, EGFR, and VEGFR are frequently mutated and constitutively activated in cancer, and thus are important targets for therapeutic inhibition. RTKs activate several major intracellular signaling pathways such as JAK/STAT, MAPK, and PI3K, which have also been the target of inhibitor development. Homologous recombination genes have been identified among the transcriptional targets of these signaling pathways, leading to investigation of TKIs as potential inducers of BRCAness. GRB2 = growth factor receptor bound protein 2; SOS = son of sevenless.
AML cells generate an elevated level of reactive oxygen species (ROS) that has been linked to STAT5 activation of RAC1, a critical member of the NADPH oxidase (NOX) pathway [128,129]. This ROS generation is associated with an increased DNA damage burden, including DSBs [129], that is frequently accompanied by the functional dysregulation of DSB repair [130]. In certain subsets of AML, such as those driven by the fusion oncoproteins AML1-ETO or PML-RARα, the expression of key HR proteins (including RAD51, ATM, BRCA1, and BRCA2) is suppressed, producing sensitivity to PARP inhibition [131]. In contrast, DSB repair is proficient in FLT3-ITDhigh leukemic cells [132,133], rendering resistance to PARPi [131]. Treating FLT3-ITDhigh cells with quizartinib suppresses downstream signaling by Janus kinase 2 (JAK2) and PI3K to produce a rapid downregulation of DSB repair proteins (including BRCA1, BRCA2, PALB2, and RAD51) that sensitizes to PARP inhibition in preclinical and in vivo models [134]. Importantly, this effect is observed in both LPCs and in proliferating and quiescent LSCs, including those cultured under conditions mimicking the bone marrow microenvironment, an important reservoir of treatment-resistant cells [134]. Of note, these effects may be mediated in part by the effects of PARP1 inhibition outside of DNA repair, such as STAT5 protein stabilization through PARylation [135]. This approach may therefore offer a potential to improve long-term outcomes by targeting the disease-initiating stem cell population without losing sensitivity in the presence of resistance mutations.
4.2. c-MET
The c-MET transmembrane receptor tyrosine kinase is expressed on stem and progenitor cells, where activation by its ligand hepatocyte growth factor (HGF) activates an invasive growth program during embryogenesis and organ regeneration [136]. C-MET activation is a common event in numerous cancers, driving a number of proliferation, differentiation, and survival signaling pathways, including sustained Ras/MAPK activity, as well as PI3K, STAT, β-catenin, NFκB, and Notch activation [137] (Figure 3). Several small molecules and antibodies targeting the HGF-MET pathway have been developed, categorized as ligand inhibitors (blocking pro-HGF cleavage to the active form or preventing ligand-receptor binding) or MET receptor inhibitors (competitively antagonizing receptor binding or inhibiting MET tyrosine kinase activity). Of these, TKIs with a specific activity against MET kinase (capmatinib) or non-specific global kinase inhibition (crizotinib, cabozantinib) have progressed farthest in clinical development, with FDA approval for indications such as NSCLC, renal cell, hepatocellular, and medullary thyroid cancers [137].
In addition to the activation of signaling cascades, c-MET interacts and phosphorylates a wide range of target proteins. Recent evidence indicates that c-MET becomes activated following oxidative stress, inducing an antiapoptotic cytoprotective response that includes the phosphorylation of PARP1 at Tyr907 located within the catalytic domain [138]. Phospho-Tyr907 not only enhances PARylation activity, but also reduces PARPi binding, and may be a predictive marker of PARPi resistance [139]. The inhibition of c-MET using the non-specific pan-kinase inhibitor crizotinib [139] or the MET-specific HS-10,241 [140] abolishes Tyr907 phosphorylation, sensitizing to PARPi in in vitro and xenograft models of TNBC, NSCLC, and high-grade serous ovarian cancer (HGSOC), independent of BRCA status. Notably, elevated c-MET expression in BRCA-mutant TNBC cell lines correlates to PARPi resistance that can be reversed by c-MET inhibition. These results highlight the potential of a therapeutic strategy to combine PARPi with c-MET inhibitors in PARPi-resistant cancers.
4.3. EGFR
The epidermal growth factor receptor (EGFR) is a transmembrane receptor tyrosine kinase that binds to a variety of ligands including epidermal growth factor (EGF) and transforming growth factor α (TGFα) [141]. Upon ligand binding, inactive EGFR monomers dimerize to an active form, either as homodimers or as heterodimers with other members of the ErbB receptor family such as HER2. Autophosphorylation induces the activation of target proteins via interaction at phosphotyrosine SH2 domains, resulting in the stimulation of signal transduction cascades including MAPK (see below), AKT, and JNK (Figure 3). Ultimately, EGFR activation stimulates a phenotype that promotes proliferation, cell adhesion, and migration; hence, constitutive activation is an oncogenic driver in multiple human cancers. TKIs with specific activity against EGFR (erlotinib, gefitinib, and the EGFR/HER2-targeting lapatinib) and monoclonal antibodies that prevent EGFR-ligand binding (cetuximab and panitumumab) have been approved for use in a variety of EGFR-expressing malignancies, including NSCLC, head and neck squamous cell carcinoma (HNSCC), and colorectal cancer.
In response to EGFR inhibition, physical interaction between EGFR and components of the classical NHEJ pathway of DSB repair (particularly DNA-PKCS) have been reported [142], leading to globally reduced levels of DNA-PK [143,144] and/or subcellular relocalization away from the nucleus [145] that reduces NHEJ repair capacity and sensitizes to radiation [146]. EGFR inhibition has also been linked to the transient downregulation of mismatch repair (MLH1, MSH2, and MSH6) and HR (BRCA2, RAD51) genes in cetuximab-sensitive colorectal cancer cell lines, increasing the mutability that leads to the development of permanent resistance with prolonged exposure [147]. Although a potential strategy to target this repair downregulation has not yet been extensively explored, the Hung lab have built upon their work combining MET inhibitors with PARPi (see above) to demonstrate that EGFR cooperates with MET in subsets of hepatocellular cancers [148] and TNBCs [149] to phosphorylate PARP1 Tyr907 in response to DNA damage, demonstrating that dual EGFR/MET inhibition is required in this group to block phosphorylation and sensitize resistant cells to PARPi. This may further broaden the therapeutic potential of MET inhibition to overcome PARPi resistance in certain cancers.
4.4. VEGFR
Vascular endothelial growth factor receptors (VEGFRs) are tyrosine kinase receptors that play critical roles in signal transduction during vasculogenesis and angiogenesis [150]. The abnormal expression of VEGFR ligands (VEGFs) by tumor-associated macrophages contributes to tumor neoangiogenesis, an observation that led to the development of targeted anti-angiogenic therapies. An important consequence of VEGFR inhibition is tumor hypoxia [151], a state that has been linked to HR defects via the downregulation of BRCA1, BRCA2, and RAD51 [152,153]. Accordingly, in preclinical models, VEGFR inhibition is reported to sensitize to PARPi, and a phase 2 clinical trial in platinum-sensitive HGSOC indicates that the combination prolongs progression-free survival over single agent treatment [154]. Several early-stage clinical trials combining the VEGFR inhibitor cediranib with olaparib are currently underway (Table 3).
Table 3.
Clinical trials evaluating TKI in combination with PARPi.
4.5. MAPK Pathway
The mitogen-activated protein kinase (MAPK) signaling pathway regulates a diverse range of cellular processes, including proliferation, differentiation, and survival [155]. In response to extracellular stimuli including growth factors or cytokines, a transmembrane receptor-linked tyrosine kinase (such as EGFR, fibroblast growth factor receptor [FGFR], or FLT3) activates a member of the Ras subfamily. This triggers a kinase signaling cascade through RAF, MEK1/2, and ERK (also known as MAPK) (Figure 3), leading to the phosphorylation of a range of target proteins including transcription factors (such as c-MYC, c-Jun, and c-Fos), cell cycle proteins (such as CDK4/6 for S-phase entry), apoptotic factors (inactivating pro-apoptotic proteins such as Bad, Bim, and caspase 9), and regulators of the translational machinery such as the 90 kDa ribosomal S6 kinase (p90Rsk) [155]. Activated Ras can also interact with other signaling pathways, such as the Pi3K-AKT-mTOR (mammalian target of rapamycin) pathway (see below) and RAS-like (RAL) GTPases. Unsurprisingly, members of the MAPK pathway are proto-oncogenes, with constitutive activation of Ras observed in ~30% of cancers, and BRAF in ~7% [156]. While Ras has not proven to be an effectively druggable target, inhibitors of downstream MAPK members such as MEK are under development.
Recent evidence indicates that constitutive Ras activation produces a PARPi-resistant phenotype. Using a reverse phase protein assay to determine changes in signaling pathway expression, Sun et al. demonstrated that transient PARPi treatment induces Ras/MAPK activation, producing a downregulation of the pro-apoptotic targets that induced PARPi resistance, and furthermore, recapitulated the PARPi resistance observed in Ras-mutant cell lines. MEK (or ERK) inhibition in KRAS-mutant or KRAS-induced cell lines enforced the expression of the Ras-regulated FOXO3a transcription factor, leading to increased expression of pro-apoptotic factors such as Bim. Phosphorylation patterns of multiple DNA repair proteins were also found to be altered by MEKi, associated with altered expression levels of DSB repair proteins that reversed an enhanced level of DSBR observed in KRAS-mutant cells. This reduced MEKi-induced reduction in DSBR sensitized KRAS-mutant cells to talazoparib, compounded by increased PARP1 expression that enhanced the accumulation of cytotoxic PARP-trapping lesions [157]. These results were subsequently confirmed by a second group [158], suggesting a combinatorial role for PARPi and MEKi in the treatment of PARPi-resistant and/or KRAS-mutant tumors that is now being tested in a phase I/II trial (Table 3). Interestingly, PARPi synergism is not recapitulated by BRAF inhibition, likely because other RAF homologs bypass the effects of therapeutic inhibition [157].
4.6. PI3K Pathway
The phosphoinositide 3-kinase pathway is a major effector of receptor tyrosine kinase activation, transducing signals via phospholipid generation to protein kinase B (also known as AKT), mammalian target of rapamycin (mTOR), and other downstream targets [159]. Loss of function mutations in the negative regulator phosphatase and tensin homolog (PTEN) are a common occurrence in human cancers, as are activating mutations of other components of the PI3K pathway, producing an accelerated growth and proliferation phenotype. PI3K has long been ascribed a role in promoting the DDR [160], including regulating the binding of the NBS1 damage sensor to DNA [161], and the control of RAD51 recruitment to DSBs [162]. The downstream effector mTOR also modulates the DDR, maintaining HR and NHEJ [163,164], at least in part by the stimulation of FANCD2 expression [165,166]. The PI3K pathway also exerts transcriptional control over repair gene expression, including BRCA1/2, RAD51 [167,168], PRKDC (DNA-PKCS), and ATM [169]. Several studies have therefore considered the potential for PI3K pathway inhibitors to induce DSB repair defects. In both in vitro and in vivo models of BRCA-proficient TNBC, BRCA1/2 downregulation induced by the PI3K inhibitor (PI3Ki) BKM120 [167], mTOR inhibitors everolimus or KU0063794 [170], or the dual PI3K/mTOR inhibitor GDC-0980 [171] impairs HR and sensitizes to PARPi. Similar results have been observed in PTEN-mutant, PI3K-activated endometrial cancer [172], and in PI3K-wildtype [173,174] or mutant [175] ovarian cancer. Phase I/II clinical trials examining PARPi in combination with inhibitors of PI3K, AKT, or mTOR are underway (Table 3).
5. Other Targets
5.1. BCR-ABL
c-ABL tyrosine kinase is constitutively activated in most chronic myeloid leukemias (CML) following translocation adjacent to the BCR gene, forming the BCR-ABL ‘Philadelphia chromosome’ [176]. Activated c-ABL interacts with multiple proliferative and survival pathways, including MAPK, PI3K, and JAK/STAT. Following activation induced by IR, c-ABL phosphorylates RAD51 at Tyr315, enhancing complex formation with RAD52 [177,178] and stabilizing chromatin binding [179]. In the presence of BCR-ABL, RAD51 and RAD51 paralog expression is significantly enhanced, mediated via JAK/STAT signaling [180]. Imatinib is a multi-kinase inhibitor that possesses selectivity for BCR-ABL, along with c-kit and PDGFR, and is FDA-approved in hematological malignancies and gastrointestinal stromal tumors. Imatinib reduces RAD51 nuclear expression and chromatin binding, and inhibits HR-mediated repair [181], thus sensitizing to PARPi in ovarian cancer [174]. Further study, particularly in the BCR-ABL fusion setting, is required to evaluate the clinical potential of this combination.
5.2. NAMPT Inhibition
Nicotinamide phosphoribosyl transferase (NAMPT) is a rate-limiting enzyme required for the generation of the PARP substrate β-NAD+. Small molecule inhibition of NAMPT suppresses β-NAD+ synthesis, preventing PARP1 PARylation activity. Synthetic lethality between an experimental NAMPT inhibitor and olaparib has been observed in different tumor models independent of BRCA status, producing a synergistic and non-redundant NAD+ depletion, a reduction in PARylation, an increase in DNA damage, and an induction of apoptosis [182,183]. While this combination does not induce a BRCAness phenotype, it may offer an opportunity to further optimize therapeutic strategies by maximizing PARP inactivation.
5.3. Pharmacological Ascorbate
High doses of vitamin C (ascorbate) have been evaluated as an anticancer therapy in a range of malignancies. Cytotoxicity is mediated in part through DNA damage accumulation resulting from the generation of hydrogen peroxide, which activates PARP1 and subsequently depletes the PARP1 substrate nicotinamide adenine dinucleotide (NAD+) leading to ATP depletion and cell death [184]. Although PARPi treatment prevents NAD+/ATP depletion, cell death still ensues secondary to DSB accumulation linked to the ascorbate-induced downregulation of BRCA1, BRCA2, and RAD51 [185].
Additionally, low doses of vitamin C, particularly in the context of vitamin C deficiency, may synergistically enhance the effects of DNMTi in hematological malignancy [186]. Vitamin C acts as a cofactor for ten-eleven translocation (TET) enzymes that convert 5-methylcytosine (5mc) to 5-hydroxymethylcytosine (5 hmc), thus producing active demethylation in concert with the passive demethylation induced by DNMT inhibition. Notably, these demethylation effects are enriched at the long terminal repeat (LTR) regions of ERVs, leading to ERV re-expression and pathogen mimicry immune responses [187] that induce HR defects as previously described in Section 3.1. An early clinical trial has demonstrated an enhanced 5 hmc/5 mc ratio following oral vitamin C supplementation compared to placebos in patients with myeloid cancers treated with DNMTi [186], although the impact upon HR capacity and PARPi efficacy has not yet been considered.
6. Conclusions
PARP inhibitor sensitivity in BRCA-mutated breast and ovarian cancers is the prototypical example of synthetic lethality but represents only a small number of total cancer diagnoses. To expand PARPi utility into a wider setting, research has primarily focused on the identification of other genetic and epigenetic determinants of BRCAness. With the advent of increasing numbers and a widening scope of targeted therapies, it has become apparent that BRCAness—and hence PARPi sensitivity—may also be pharmaceutically induced. With the exploration of the additional roles of PARP in the regulation of gene expression and protein translation, this may increase the targets for the induction of BRCAness. This review has summarized the current status of therapeutic BRCAness induction, focusing on epigenetic agents, drugs targeting cell cycle checkpoints and the DNA damage response, and tyrosine kinase inhibitors. The translation of promising preclinical results to clinical trials and beyond is critical to maximizing PARPi therapeutic scope and optimizing treatment outcomes.
Author Contributions
Writing—Original Draft Preparation, R.A., A.J.D.; Writing—Review & Editing, R.A., A.J.D., F.V.R.; Supervision—F.V.R. All authors have read and agreed to the published version of the manuscript.
Funding
Our studies were supported by funding from the National Cancer Institute–Cancer Center Support Grant P30 CA134274 University of Maryland Marlene and Stewart Greenebaum Comprehensive Cancer Center (F.V.R.); the Molecular Medicine Graduate Program (A.D.), University of Maryland Department of Health’s Cigarette Restitution Fund Program (F.V.R., R.A.); and the Van Andel Research Institute–Stand Up to Cancer Epigenetics Dream Team (F.V.R., R.A.). Stand Up to Cancer is a program of the Entertainment Industry Foundation. Specialized Program of Research Excellence (SPORE) program, through National Cancer Institute (NCI) grant P50CA254897 (F.V.R., A.D.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Acknowledgments
We thank Stephen Bruce Baylin, M.D. (JHU) for careful reading of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bock, F.; Chang, P. New directions in poly(ADP-ribose) polymerase biology. FEBS J. 2016, 283, 4017–4031. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.Y.; Zhang, T.; Kraus, W.L. Poly(ADP-ribosyl)ation by PARP-1: PAR-laying NAD+ into a nuclear signal. Genes Dev. 2005, 19, 1951–1967. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.Y.; Mauro, S.; Gévry, N.; Lis, J.T.; Kraus, W. NAD+-Dependent Modulation of Chromatin Structure and Transcription by Nucleosome Binding Properties of PARP-1. Cell 2004, 119, 803–814. [Google Scholar] [CrossRef]
- Timinszky, G.; Till, S.; Hassa, P.O.; Hothorn, M.; Kustatscher, G.; Nijmeijer-Winter, B.; Colombelli, J.; Altmeyer, M.; Stelzer, E.H.; Scheffzek, K.; et al. A macrodomain-containing histone rearranges chromatin upon sensing PARP1 activation. Nat. Struct. Mol. Biol. 2009, 16, 923–929. [Google Scholar] [CrossRef]
- Trucco, C.; Oliver, F.J.; de Murcia, G.; Menissier-de Murcia, J. DNA repair defect in poly(ADP-ribose) polymerase-deficient cell lines. Nucleic Acids Res. 1998, 26, 2644–2649. [Google Scholar] [CrossRef] [PubMed]
- Ding, R.; Pommier, Y.; Kang, V.; Smulson, M. Depletion of poly(ADP-ribose) polymerase by antisense RNA expression results in a delay in DNA strand break rejoining. J. Biol. Chem. 1992, 267, 12804–12812. [Google Scholar] [CrossRef]
- Schreiber, V.; Hunting, D.; Trucco, C.; Gowans, B.; Grunwald, D.; De Murcia, G.; De Murcia, J.M. A dominant-negative mutant of human poly(ADP-ribose) polymerase affects cell recovery, apoptosis, and sister chromatid exchange following DNA damage. Proc. Natl. Acad. Sci. USA 1995, 92, 4753–4757. [Google Scholar] [CrossRef]
- Boulton, S.; Kyle, S.; Durkacz, B.W. Interactive effects of inhibitors of poly(ADP-ribose) polymerase and DNA-dependent protein kinase on cellular responses to DNA damage. Carcinogenesis 1999, 20, 199–203. [Google Scholar] [CrossRef][Green Version]
- Terada, M.; Fujiki, H.; Marks, P.A.; Sugimura, T. Induction of erythroid differentiation of murine erythroleukemia cells by nicotinamide and related compounds. Proc. Natl. Acad. Sci. USA 1979, 76, 6411–6414. [Google Scholar] [CrossRef]
- Shen, Y.; Rehman, F.L.; Feng, Y.; Boshuizen, J.; Bajrami, I.; Elliott, R.; Wang, B.; Lord, C.J.; Post, L.E.; Ashworth, A. BMN 673, a novel and highly potent PARP1/2 inhibitor for the treatment of human cancers with DNA repair deficiency. Clin. Cancer Res. 2013, 19, 5003–5015. [Google Scholar] [CrossRef]
- Murai, J.; Huang, S.-Y.N.; Renaud, A.; Zhang, Y.; Ji, J.; Takeda, S.; Morris, J.; Teicher, B.; Doroshow, J.H.; Pommier, Y. Stereospecific PARP Trapping by BMN 673 and Comparison with Olaparib and Rucaparib. Mol. Cancer Ther. 2013, 13, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Huang, S.-Y.N.; DAS, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [PubMed]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Prakash, R.; Zhang, Y.; Feng, W.; Jasin, M. Homologous Recombination and Human Health: The Roles of BRCA1, BRCA2, and Associated Proteins. Cold Spring Harb. Perspect. Biol. 2015, 7, a016600. [Google Scholar] [CrossRef] [PubMed]
- Saleh-Gohari, N.; Bryant, H.E.; Schultz, N.; Parker, K.M.; Cassel, T.N.; Helleday, T. Spontaneous Homologous Recombination Is Induced by Collapsed Replication Forks That Are Caused by Endogenous DNA Single-Strand Breaks. Mol. Cell. Biol. 2005, 25, 7158–7169. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G., Jr. The concept of synthetic lethality in the context of anticancer therapy. Nat. Rev. Cancer 2005, 5, 689–698. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. BRCAness revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef]
- Turner, N.; Tutt, A.; Ashworth, A. Hallmarks of BRCAness in sporadic cancers. Nat. Cancer 2004, 4, 814–819. [Google Scholar] [CrossRef]
- Byrum, A.K.; Vindigni, A.; Mosammaparast, N. Defining and Modulating BRCAness. Trends Cell Biol. 2019, 29, 740–751. [Google Scholar] [CrossRef]
- Ladan, M.; van Gent, D.; Jager, A. Homologous Recombination Deficiency Testing for BRCA-Like Tumors: The Road to Clinical Validation. Cancers 2021, 13, 1004. [Google Scholar] [CrossRef] [PubMed]
- Vergote, I.; González-Martín, A.; Ray-Coquard, I.; Harter, P.; Colombo, N.; Pujol, P.; Lorusso, D.; Mirza, M.; Brasiuniene, B.; Madry, R.; et al. European experts consensus: BRCA/homologous recombination deficiency testing in first-line ovarian cancer. Ann. Oncol. 2021, 33, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Werner, R.J.; Kelly, A.D.; Issa, J.-P. Epigenetics and Precision Oncology. Cancer J. 2017, 23, 262–269. [Google Scholar] [CrossRef]
- Issa, J.-P.J.; Kantarjian, H.M. Targeting DNA Methylation. Clin. Cancer Res. 2009, 15, 3938–3946. [Google Scholar] [CrossRef]
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome—Biological and translational implications. Nat. Cancer 2011, 11, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Issa, J.-P.J.; Garcia-Manero, G.; Giles, F.J.; Mannari, R.; Thomas, D.; Faderl, S.; Bayar, E.; Lyons, J.; Rosenfeld, C.S.; Cortes, J.; et al. Phase 1 study of low-dose prolonged exposure schedules of the hypomethylating agent 5-aza-2′-deoxycytidine (decitabine) in hematopoietic malignancies. Blood 2004, 103, 1635–1640. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.; Issa, J.P.; Rosenfeld, C.S.; Bennett, J.M.; Albitar, M.; DiPersio, J. Decitabine Improves Patient Outcomes in Myelodysplastic Syndromes: Results of a Phase III Randomized Study. Cancer 2006, 106, 1794–1803. [Google Scholar] [CrossRef]
- Gnyszka, A.; Jastrzebski, Z.; Flis, S. DNA methyltransferase inhibitors and their emerging role in epigenetic therapy of cancer. Anticancer Res. 2013, 33, 2989–2996. [Google Scholar]
- Öz, S.; Raddatz, G.; Rius, M.; Blagitko-Dorfs, N.; Lübbert, M.; Maercker, C.; Lyko, F. Quantitative Determination of Decitabine Incorporation Into DNA and Its Effect on Mutation Rates in Human Cancer Cells. Nucleic Acids Res. 2014, 42, e152. [Google Scholar] [CrossRef]
- O’Hagan, H.; Wang, W.; Sen, S.; Shields, C.D.; Lee, S.; Zhang, Y.W.; Clements, E.G.; Cai, Y.; Van Neste, L.; Easwaran, H.; et al. Oxidative Damage Targets Complexes Containing DNA Methyltransferases, SIRT1, and Polycomb Members to Promoter CpG Islands. Cancer Cell 2011, 20, 606–619. [Google Scholar] [CrossRef]
- Caiafa, P.; Guastafierro, T.; Zampieri, M. Epigenetics: Poly(ADP-ribosyl)ation of PARP-1 regulates genomic methylation patterns. FASEB J. 2008, 23, 672–678. [Google Scholar] [CrossRef] [PubMed]
- Muvarak, N.E.; Chowdhury, K.; Xia, L.; Robert, C.; Choi, E.Y.; Cai, Y. Enhancing the Cytotoxic Effects of PARP Inhibitors with DNA Demethylating Agents-A Potential Therapy for Cancer. Cancer Cell 2016, 30, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Abbotts, R.; Topper, M.J.; Biondi, C.; Fontaine, D.; Goswami, R.; Stojanovic, L.; Choi, E.Y.; McLaughlin, L.; Kogan, A.A.; Xia, L.; et al. DNA methyltransferase inhibitors induce a BRCAness phenotype that sensitizes NSCLC to PARP inhibitor and ionizing radiation. Proc. Natl. Acad. Sci. USA 2019, 116, 22609–22618. [Google Scholar] [CrossRef]
- McLaughlin, L.J.; Stojanovic, L.; Kogan, A.A.; Rutherford, J.L.; Choi, E.Y.; Yen, R.-W.C.; Xia, L.; Zou, Y.; Lapidus, R.G.; Baylin, S.B.; et al. Pharmacologic induction of innate immune signaling directly drives homologous recombination deficiency. Proc. Natl. Acad. Sci. USA 2020, 117, 17785–17795. [Google Scholar] [CrossRef]
- Topper, M.J.; Vaz, M.; Chiappinelli, K.B.; Shields, C.E.D.; Niknafs, N.; Yen, R.-W.C.; Wenzel, A.; Hicks, J.; Ballew, M.; Stone, M.; et al. Epigenetic Therapy Ties MYC Depletion to Reversing Immune Evasion and Treating Lung Cancer. Cell 2017, 171, 1284–1300.e21. [Google Scholar] [CrossRef] [PubMed]
- Chiappinelli, K.B.; Strissel, P.L.; Desrichard, A.; Li, H.; Henke, C.; Akman, B.; Hein, A.; Rote, N.S.; Cope, L.M.; Snyder, A.; et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell 2015, 162, 974–986. [Google Scholar] [CrossRef] [PubMed]
- Pulliam, N.; Fang, F.; Ozes, A.R.; Tang, J.; Adewuyi, A.; Keer, H.; Lyons, J.; Baylin, S.B.; Matei, D.; Nakshatri, H.; et al. An Effective Epigenetic-PARP Inhibitor Combination Therapy for Breast and Ovarian Cancers Independent of BRCA Mutations. Clin. Cancer Res. 2018, 24, 3163–3175. [Google Scholar] [CrossRef]
- Ghoshal, K.; Datta, J.; Majumder, S.; Bai, S.; Kutay, H.; Motiwala, T.; Jacob, S.T. 5-Aza-Deoxycytidine Induces Selective Degradation of DNA Methyltransferase 1 by a Proteasomal Pathway That Requires the KEN Box, Bromo-Adjacent Homology Domain, and Nuclear Localization Signal. Mol. Cell. Biol. 2005, 25, 4727–4741. [Google Scholar] [CrossRef]
- Patel, K.; Dickson, J.; Din, S.; Macleod, K.; Jodrell, D.; Ramsahoye, B. Targeting of 5-aza-2′-deoxycytidine residues by chromatin-associated DNMT1 induces proteasomal degradation of the free enzyme. Nucleic Acids Res. 2010, 38, 4313–4324. [Google Scholar] [CrossRef]
- Tsai, H.-C.; Li, H.; Van Neste, L.; Cai, Y.; Robert, C.; Rassool, F.V.; Shin, J.J.; Harbom, K.M.; Beaty, R.; Pappou, E.; et al. Transient Low Doses of DNA-Demethylating Agents Exert Durable Antitumor Effects on Hematological and Epithelial Tumor Cells. Cancer Cell 2012, 21, 430–446. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Bogliolo, M.; Surrallés, J. The Fanconi Anemia/BRCA Pathway: FANCD2 at the Crossroad between Repair and Checkpoint Responses to DNA Damage. In Madame Curie Bioscience Database [Internet]; Landes Bioscience: Austin, TX, USA, 2013. [Google Scholar]
- Yoshikiyo, K.; Kratz, K.; Hirota, K.; Nishihara, K.; Takata, M.; Kurumizaka, H. KIAA1018/FAN1 Nuclease Protects Cells against Genomic Instability Induced by Interstrand Cross-Linking Agents. Proc. Natl. Acad. Sci. USA 2010, 107, 21553–21557. [Google Scholar] [CrossRef] [PubMed]
- Baer, M.R.; Kogan, A.A.; Bentzen, S.M.; Mi, T.; Lapidus, R.G.; Duong, V.H.; Emadi, A.; Niyongere, S.; O’Connell, C.L.; Youngblood, B.A.; et al. Phase I clinical trial of DNA methyltransferase inhibitor decitabine and PARP inhibitor talazoparib combination therapy in relapsed/refractory acute myeloid leukemia. Clin. Cancer Res. 2022, 28, 1313–1322. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Thomas, A.; Miettinen, M.; Pommier, Y. Schlafen 11 (SLFN11), a restriction factor for replicative stress induced by DNA-targeting anti-cancer therapies. Pharmacol. Ther. 2019, 201, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Mu, Y.; Lou, J.; Srivastava, M.; Zhao, B.; Feng, X.; Liu, T.; Chen, J.; Huang, J. SLFN 11 inhibits checkpoint maintenance and homologous recombination repair. EMBO Rep. 2015, 17, 94–109. [Google Scholar] [CrossRef]
- Lok, B.H.; Gardner, E.E.; Schneeberger, V.E.; Ni, A.; Desmeules, P.; Rekhtman, N.; De Stanchina, E.; Teicher, B.A.; Riaz, N.; Powell, S.N.; et al. PARP Inhibitor Activity Correlates with SLFN11 Expression and Demonstrates Synergy with Temozolomide in Small Cell Lung Cancer. Clin. Cancer Res. 2017, 23, 523–535. [Google Scholar] [CrossRef]
- Gardner, E.; Lok, B.; Schneeberger, V.E.; Desmeules, P.; Miles, L.; Arnold, P.; Ni, A.; Khodos, I.; De Stanchina, E.; Nguyen, T.; et al. Chemosensitive Relapse in Small Cell Lung Cancer Proceeds through an EZH2-SLFN11 Axis. Cancer Cell 2017, 31, 286–299. [Google Scholar] [CrossRef]
- Tang, S.-W.; Thomas, A.; Murai, J.; Trepel, J.B.; Bates, S.E.; Rajapakse, V.N.; Pommier, Y. Overcoming Resistance to DNA-Targeted Agents by Epigenetic Activation of Schlafen 11 (SLFN11) Expression with Class I Histone Deacetylase Inhibitors. Clin. Cancer Res. 2018, 24, 1944–1953. [Google Scholar] [CrossRef]
- Glozak, M.A.; Seto, E. Histone Deacetylases and Cancer. Oncogene 2007, 26, 5420–5432. [Google Scholar] [CrossRef]
- Hontecillas-Prieto, L.; Flores-Campos, R.; Silver, A.; De Álava, E.; Hajji, N.; García-Domínguez, D.J. Synergistic Enhancement of Cancer Therapy Using HDAC Inhibitors: Opportunity for Clinical Trials. Front. Genet. 2020, 11, 1113. [Google Scholar] [CrossRef]
- Ho, T.C.S.; Chan, A.H.Y.; Ganesan, A. Thirty Years of HDAC Inhibitors: 2020 Insight and Hindsight. J. Med. Chem. 2020, 63, 12460–12484. [Google Scholar] [CrossRef] [PubMed]
- Misteli, T.; Soutoglou, E. The emerging role of nuclear architecture in DNA repair and genome maintenance. Nat. Rev. Mol. Cell Biol. 2009, 10, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Koprinarova, M.; Botev, P.; Russev, G. Histone deacetylase inhibitor sodium butyrate enhances cellular radiosensitivity by inhibiting both DNA nonhomologous end joining and homologous recombination. DNA Repair 2011, 10, 970–977. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.M.; Tjeertes, J.V.; Coates, J.; Legube, G.; Polo, S.E.; Britton, S.; Jackson, S.P. Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nat. Struct. Mol. Biol. 2010, 17, 1144–1151. [Google Scholar] [CrossRef]
- Mladenov, E.; Iliakis, G. Induction and repair of DNA double strand breaks: The increasing spectrum of non-homologous end joining pathways. Mutat. Res. Mol. Mech. Mutagen. 2011, 711, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Ladd, B.; Ackroyd, J.J.; Hicks, J.K.; Canman, C.E.; Flanagan, S.A.; Shewach, D.S. Inhibition of homologous recombination with vorinostat synergistically enhances ganciclovir cytotoxicity. DNA Repair 2013, 12, 1114–1121. [Google Scholar] [CrossRef]
- Xiao, W.; Graham, P.H.; Hao, J.; Chang, L.; Ni, J.; Power, C.A.; Dong, Q.; Kearsley, J.H.; Li, Y. Combination Therapy with the Histone Deacetylase Inhibitor LBH589 and Radiation Is an Effective Regimen for Prostate Cancer Cells. PLoS ONE 2013, 8, e74253. [Google Scholar] [CrossRef]
- Ha, K.; Fiskus, W.; Choi, D.S.; Bhaskara, S.; Cerchietti, L.; Devaraj, S.G.T.; Shah, B.; Sharma, S.; Chang, J.C.; Melnick, A.M.; et al. Histone deacetylase inhibitor treatment induces ‘BRCAness’ and synergistic lethality with PARP inhibitor and cisplatin against human triple negative breast cancer cells. Oncotarget 2014, 5, 5637–5650. [Google Scholar] [CrossRef]
- Wiegmans, A.P.; Yap, P.-Y.; Ward, A.; Lim, Y.C.; Khanna, K.K. Differences in Expression of Key DNA Damage Repair Genes after Epigenetic-Induced BRCAness Dictate Synthetic Lethality with PARP1 Inhibition. Mol. Cancer Ther. 2015, 14, 2321–2331. [Google Scholar] [CrossRef]
- Yin, L.; Liu, Y.; Peng, Y.; Peng, Y.; Yu, X.; Gao, Y. PARP Inhibitor Veliparib and HDAC Inhibitor SAHA Synergistically Co-Target the UHRF1/BRCA1 DNA Damage Repair Complex in Prostate Cancer Cells. J. Exp. Clin. Cancer Res. CR 2018, 37, 153. [Google Scholar] [CrossRef]
- Baldan, F.; Mio, C.; Allegri, L.; Puppin, C.; Russo, D.; Filetti, S.; Damante, G. Synergy between HDAC and PARP Inhibitors on Proliferation of a Human Anaplastic Thyroid Cancer-Derived Cell Line. Int. J. Endocrinol. 2015, 2015, 978371. [Google Scholar] [CrossRef] [PubMed]
- Liang, B.Y.; Xiong, M.; Ji, G.B.; Zhang, E.L.; Zhang, Z.Y.; Dong, K.S. Synergistic Suppressive Effect of PARP-1 Inhibitor PJ34 and HDAC Inhibitor SAHA on Proliferation of Liver Cancer Cells. J. Huazhong Univ. Sci. Technol. 2015, 35, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Hegde, M.; Mantelingu, K.; Pandey, M.; Pavankumar, C.S.; Rangappa, K.S.; Raghavan, S.C. Combinatorial Study of a Novel Poly (ADP-ribose) Polymerase Inhibitor and an HDAC Inhibitor, SAHA, in Leukemic Cell Lines. Target. Oncol. 2016, 11, 655–665. [Google Scholar] [CrossRef]
- Rasmussen, R.D.; Gajjar, M.K.; Jensen, K.E.; Hamerlik, P. Enhanced efficacy of combined HDAC and PARP targeting in glioblastoma. Mol. Oncol. 2016, 10, 751–763. [Google Scholar] [CrossRef]
- Robert, C.; Nagaria, P.K.; Pawar, N.; Adewuyi, A.; Gojo, I.; Meyers, D.J.; Cole, P.A.; Rassool, F.V. Histone deacetylase inhibitors decrease NHEJ both by acetylation of repair factors and trapping of PARP1 at DNA double-strand breaks in chromatin. Leuk. Res. 2016, 45, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Cameron, E.E.; Bachman, K.E.; Myöhänen, S.; Herman, J.G.; Baylin, S.B. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat. Genet. 1999, 21, 103–107. [Google Scholar] [CrossRef]
- Valdez, B.C.; Li, Y.; Murray, D.; Liu, Y.; Nieto, Y.; Champlin, R.E.; Andersson, B.S. Combination of a hypomethylating agent and inhibitors of PARP and HDAC traps PARP1 and DNMT1 to chromatin, acetylates DNA repair proteins, down-regulates NuRD and induces apoptosis in human leukemia and lymphoma cells. Oncotarget 2017, 9, 3908–3921. [Google Scholar] [CrossRef]
- Li, D.-Q.; Kumar, R. Mi-2/NuRD Complex Making Inroads Into DNA-damage Response Pathway. Cell Cycle 2010, 9, 2071–2079. [Google Scholar] [CrossRef]
- Veneti, Z.; Gkouskou, K.K.; Eliopoulos, A. Polycomb Repressor Complex 2 in Genomic Instability and Cancer. Int. J. Mol. Sci. 2017, 18, 1657. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Hung, M.-C. Regulation and Role of EZH2 in Cancer. Cancer Res. Treat. 2014, 46, 209–222. [Google Scholar] [CrossRef]
- Zeidler, M.; Varambally, S.; Cao, Q.; Chinnaiyan, A.M.; Ferguson, D.O.; Merajver, S.D.; Kleer, C.G. The Polycomb Group Protein EZH2 Impairs DNA Repair in Breast Epithelial Cells. Neoplasia 2005, 7, 1011–1019. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, M.E.; DuPrie, M.L.; Krueger, H.; Merajver, S.D.; Ventura, A.C.; Toy, K.A.; Kleer, C.G. Histone Methyltransferase EZH2 Induces Akt-Dependent Genomic Instability and BRCA1 Inhibition in Breast Cancer. Cancer Res. 2011, 71, 2360–2370. [Google Scholar] [CrossRef] [PubMed]
- Hong, Z.; Jiang, J.; Lan, L.; Nakajima, S.; Kanno, S.-I.; Koseki, H.; Yasui, A. A polycomb group protein, PHF1, is involved in the response to DNA double-strand breaks in human cell. Nucleic Acids Res. 2008, 36, 2939–2947. [Google Scholar] [CrossRef] [PubMed]
- Campbell, S.; Ismail, I.H.; Young, L.C.; Poirier, G.G.; Hendzel, M.J. Polycomb repressive complex 2 contributes to DNA double-strand break repair. Cell Cycle 2013, 12, 2675–2683. [Google Scholar] [CrossRef]
- Karakashev, S.; Fukumoto, T.; Zhao, B.; Lin, J.; Wu, S.; Fatkhutdinov, N.; Park, P.-H.; Semenova, G.; Jean, S.; Cadungog, M.G.; et al. EZH2 Inhibition Sensitizes CARM1-High, Homologous Recombination Proficient Ovarian Cancers to PARP Inhibition. Cancer Cell 2020, 37, 157–167.e6. [Google Scholar] [CrossRef]
- Dhalluin, C.; Carlson, J.E.; Zeng, L.; He, C.; Aggarwal, A.K.; Zhou, M.-M. Structure and ligand of a histone acetyltransferase bromodomain. Nature 1999, 399, 491–496. [Google Scholar] [CrossRef]
- Wu, S.-Y.; Chiang, C.-M. The Double Bromodomain-containing Chromatin Adaptor Brd4 and Transcriptional Regulation. J. Biol. Chem. 2007, 282, 13141–13145. [Google Scholar] [CrossRef]
- Shi, J.; Vakoc, C.R. The Mechanisms behind the Therapeutic Activity of BET Bromodomain Inhibition. Mol. Cell 2014, 54, 728–736. [Google Scholar] [CrossRef]
- Lovén, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective Inhibition of Tumor Oncogenes by Disruption of Super-Enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef]
- Hnisz, D.; Schuijers, J.; Lin, C.Y.; Weintraub, A.S.; Abraham, B.; Lee, T.I.; Bradner, J.E.; Young, R.A. Convergence of Developmental and Oncogenic Signaling Pathways at Transcriptional Super-Enhancers. Mol. Cell 2015, 58, 362–370. [Google Scholar] [CrossRef]
- Yang, L.; Zhang, Y.; Shan, W.; Hu, Z.; Yuan, J.; Pi, J.; Wang, Y.; Fan, L.; Tang, Z.; Li, C.; et al. Repression of BET activity sensitizes homologous recombination–proficient cancers to PARP inhibition. Sci. Transl. Med. 2017, 9, eaal1645. [Google Scholar] [CrossRef] [PubMed]
- Mio, C.; Gerratana, L.; Bolis, M.; Caponnetto, F.; Zanello, A.; Barbina, M.; Di Loreto, C.; Garattini, E.; Damante, G.; Puglisi, F. BET proteins regulate homologous recombination-mediated DNA repair: BRCAness and implications for cancer therapy. Int. J. Cancer 2019, 144, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Peng, G.; Lin, C.C.-J.; Mo, W.; Dai, H.; Park, Y.-Y.; Kim, S.M.; Peng, Y.; Mo, Q.; Siwko, S.; Hu, R.; et al. Genome-wide transcriptome profiling of homologous recombination DNA repair. Nat. Commun. 2014, 5, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Yin, J.; Fang, Y.; Chen, J.; Jeong, K.J.; Chen, X.; Vellano, C.P.; Ju, Z.; Zhao, W.; Zhang, D.; et al. BRD4 Inhibition Is Synthetic Lethal with PARP Inhibitors through the Induction of Homologous Recombination Deficiency. Cancer Cell 2018, 33, 401–416.e8. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Available online: https://www.clinicaltrials.gov (accessed on 15 May 2022).
- Maréchal, A.; Zou, L. DNA Damage Sensing by the ATM and ATR Kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a012716. [Google Scholar] [CrossRef]
- Malumbres, M. Cyclin-dependent Kinases. Genome Biol. 2014, 15, 122. [Google Scholar] [CrossRef]
- Johnson, N.; Cai, D.; Kennedy, R.D.; Pathania, S.; Arora, M.; Li, Y.-C.; D’Andrea, A.D.; Parvin, J.D.; Shapiro, G.I. Cdk1 Participates in BRCA1-Dependent S Phase Checkpoint Control in Response to DNA Damage. Mol. Cell 2009, 35, 327–339. [Google Scholar] [CrossRef]
- Johnson, N.; Li, Y.C.; Walton, Z.E.; Cheng, K.A.; Li, D.; Rodig, S.J.; Moreau, L.A.; Unitt, C.; Bronson, R.T.; Thomas, H.D.; et al. Compromised CDK1 activity sensitizes BRCA-proficient cancers to PARP inhibition. Nat. Med. 2011, 17, 875–882. [Google Scholar] [CrossRef]
- Ira, G.; Pellicioli, A.; Balijja, A.; Wang, X.; Fiorani, S.; Carotenuto, W.; Liberi, G.; Bressan, D.; Wan, L.; Hollingsworth, N.M.; et al. DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature 2004, 431, 1011–1017. [Google Scholar] [CrossRef]
- Turner, N.C.; Lord, C.; Iorns, E.; Brough, R.; Swift, S.; Elliott, R.; Rayter, S.; Tutt, A.N.; Ashworth, A. A synthetic lethal siRNA screen identifying genes mediating sensitivity to a PARP inhibitor. EMBO J. 2008, 27, 1368–1377. [Google Scholar] [CrossRef]
- Krajewska, M.; Dries, R.; Grassetti, A.V.; Dust, S.; Gao, Y.; Huang, H.; Sharma, B.; Day, D.S.; Kwiatkowski, N.; Pomaville, M.; et al. CDK12 loss in cancer cells affects DNA damage response genes through premature cleavage and polyadenylation. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Iniguez, A.B.; Stolte, B.; Wang, E.J.; Conway, A.S.; Alexe, G.; Dharia, N.; Kwiatkowski, N.; Zhang, T.; Abraham, B.; Mora, J.; et al. EWS/FLI Confers Tumor Cell Synthetic Lethality to CDK12 Inhibition in Ewing Sarcoma. Cancer Cell 2018, 33, 202–216.e6. [Google Scholar] [CrossRef] [PubMed]
- Blazek, D.; Kohoutek, J.; Bartholomeeusen, K.; Johansen, E.; Hulinkova, P.; Luo, Z.; Cimermancic, P.; Ule, J.; Peterlin, B.M. The Cyclin K/Cdk12 complex maintains genomic stability via regulation of expression of DNA damage response genes. Genes Dev. 2011, 25, 2158–2172. [Google Scholar] [CrossRef] [PubMed]
- Quereda, V.; Bayle, S.; Vena, F.; Frydman, S.M.; Monastyrskyi, A.; Roush, W.R. Therapeutic Targeting of CDK12/CDK13 in Triple-Negative Breast Cancer. Cancer Cell 2019, 36, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.F.; Cruz, C.; Greifenberg, A.K.; Dust, S.; Stover, D.; Chi, D.; Primack, B.; Cao, S.; Bernhardy, A.J.; Coulson, R.; et al. CDK12 Inhibition Reverses De Novo and Acquired PARP Inhibitor Resistance in BRCA Wild-Type and Mutated Models of Triple-Negative Breast Cancer. Cell Rep. 2016, 17, 2367–2381. [Google Scholar] [CrossRef]
- Adelman, K.; Lis, J.T. Promoter-proximal pausing of RNA polymerase II: Emerging roles in metazoans. Nat. Rev. Genet. 2012, 13, 720–731. [Google Scholar] [CrossRef]
- Zhang, H.; Pandey, S.; Travers, M.; Sun, H.; Morton, G.; Madzo, J.; Chung, W.; Khowsathit, J.; Perez-Leal, O.; Barrero, C.; et al. Targeting CDK9 Reactivates Epigenetically Silenced Genes in Cancer. Cell 2018, 175, 1244–1258.e26. [Google Scholar] [CrossRef]
- Li, J.; Zhi, X.; Chen, S.; Shen, X.; Chen, C.; Yuan, L.; Guo, J.; Meng, D.; Chen, M.; Yao, L. CDK9 inhibitor CDKI-73 is synergetic lethal with PARP inhibitor olaparib in BRCA1 wide-type ovarian cancer. Am. J. Cancer Res. 2020, 10, 1140–1155. [Google Scholar]
- Zou, L.; Elledge, S.J. Sensing DNA Damage Through ATRIP Recognition of RPA-ssDNA Complexes. Science 2003, 300, 1542–1548. [Google Scholar] [CrossRef]
- Jazayeri, A.; Falck, J.; Lukas, C.; Bartek, J.; Smith, G.C.M.; Lukas, J.; Jackson, S.P. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat. Cell Biol. 2006, 8, 37–45. [Google Scholar] [CrossRef]
- Shiotani, B.; Zou, L. Single-Stranded DNA Orchestrates an ATM-to-ATR Switch at DNA Breaks. Mol. Cell 2009, 33, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Peasland, A.; Wang, L.-Z.; Rowling, E.; Kyle, S.; Chen, T.; Hopkins, A.; Cliby, W.A.; Sarkaria, J.; Beale, G.; Edmondson, R.J.; et al. Identification and evaluation of a potent novel ATR inhibitor, NU6027, in breast and ovarian cancer cell lines. Br. J. Cancer 2011, 105, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Yazinski, S.A.; Comaills, V.; Buisson, R.; Genois, M.-M.; Nguyen, H.D.; Ho, C.K.; Kwan, T.T.; Morris, R.; Lauffer, S.; Nussenzweig, A.; et al. ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor-resistant BRCA-deficient cancer cells. Genes Dev. 2017, 31, 318–332. [Google Scholar] [CrossRef] [PubMed]
- Haynes, B.; Murai, J.; Lee, J.-M. Restored replication fork stabilization, a mechanism of PARP inhibitor resistance, can be overcome by cell cycle checkpoint inhibition. Cancer Treat. Rev. 2018, 71, 1–7. [Google Scholar] [CrossRef]
- Schlacher, K.; Christ, N.; Siaud, N.; Egashira, A.; Wu, H.; Jasin, M. Double-strand Break Repair-Independent Role for BRCA2 in Blocking Stalled Replication Fork Degradation by MRE11. Cell 2011, 145, 529–542. [Google Scholar] [CrossRef]
- Kim, H.; George, E.; Ragland, R.L.; Rafail, S.; Zhang, R.; Krepler, C.; Morgan, M.A.; Herlyn, M.; Brown, E.J.; Simpkins, F. Targeting the ATR/CHK1 Axis with PARP Inhibition Results in Tumor Regression in BRCA-Mutant Ovarian Cancer Models. Clin. Cancer Res. 2017, 23, 3097–3108. [Google Scholar] [CrossRef]
- King, C.; Diaz, H.B.; McNeely, S.; Barnard, D.; Dempsey, J.; Blosser, W.; Beckmann, R.P.; Barda, D.; Marshall, M.S. LY2606368 Causes Replication Catastrophe and Antitumor Effects through CHK1-Dependent Mechanisms. Mol. Cancer Ther. 2015, 14, 2004–2013. [Google Scholar] [CrossRef]
- Sorensen, C.; Hansen, L.T.; Dziegielewski, J.; Syljuåsen, R.G.; Lundin, C.; Bartek, J.; Helleday, T. The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat. Cell Biol. 2005, 7, 195–201. [Google Scholar] [CrossRef]
- Mani, C.; Pai, S.; Papke, C.M.; Palle, K.; Gmeiner, W.H. Thymineless Death by the Fluoropyrimidine Polymer F10 Involves Replication Fork Collapse and Is Enhanced by Chk1 Inhibition. Neoplasia 2018, 20, 1236–1245. [Google Scholar] [CrossRef]
- Mani, C.; Jonnalagadda, S.; Lingareddy, J.; Awasthi, S.; Gmeiner, W.H.; Palle, K. Prexasertib treatment induces homologous recombination deficiency and synergizes with olaparib in triple-negative breast cancer cells. Breast Cancer Res. 2019, 21, 1–14. [Google Scholar] [CrossRef]
- Huntoon, C.J.; Flatten, K.S.; Hendrickson, A.E.W.; Huehls, A.M.; Sutor, S.L.; Kaufmann, S.H.; Karnitz, L.M. ATR Inhibition Broadly Sensitizes Ovarian Cancer Cells to Chemotherapy Independent of BRCA Status. Cancer Res. 2013, 73, 3683–3691. [Google Scholar] [CrossRef] [PubMed]
- Brill, E.; Yokoyama, T.; Nair, J.; Yu, M.; Ahn, Y.R.; Lee, J.M. Prexasertib, a Cell Cycle Checkpoint Kinases 1 and 2 Inhibitor, Increases in vitro Toxicity of PARP Inhibition by Preventing Rad51 Foci Formation in BRCA Wild Type High-Grade Serous Ovarian Cancer. Oncotarget 2017, 8, 111026. [Google Scholar] [CrossRef] [PubMed]
- Lindqvist, A.; Rodriguez-Bravo, V.; Medema, R.H. The decision to enter mitosis: Feedback and redundancy in the mitotic entry network. J. Cell Biol. 2009, 185, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Saini, P.; Li, Y.; Dobbelstein, M. Wee1 is required to sustain ATR/Chk1 signaling upon replicative stress. Oncotarget 2015, 6, 13072–13087. [Google Scholar] [CrossRef]
- Krajewska, M.; Heijink, A.M.; Bisselink, Y.J.; Seinstra, R.I.; Silljé, H.H.; de Vries, E.G.; van Vugt, M.A. Forced Activation of Cdk1 via wee1 Inhibition Impairs Homologous Recombination. Oncogene 2013, 32, 3001–3008. [Google Scholar] [CrossRef] [PubMed]
- Kausar, T.; Schreiber, J.S.; Karnak, D.; Parsels, L.A.; Parsels, J.D.; Davis, M.A.; Zhao, L.; Maybaum, J.; Lawrence, T.S.; Morgan, M.A. Sensitization of Pancreatic Cancers to Gemcitabine Chemoradiation by WEE1 Kinase Inhibition Depends on Homologous Recombination Repair. Neoplasia 2015, 17, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Domínguez-Kelly, R.; Martín, Y.; Koundrioukoff, S.; Tanenbaum, M.E.; Smits, V.A.; Medema, R.; Debatisse, M.; Freire, R. Wee1 controls genomic stability during replication by regulating the Mus81-Eme1 endonuclease. J. Cell Biol. 2011, 194, 567–579. [Google Scholar] [CrossRef]
- Taylor, E.R.; McGowan, C.H. Cleavage mechanism of human Mus81–Eme1 acting on Holliday-junction structures. Proc. Natl. Acad. Sci. USA 2008, 105, 3757–3762. [Google Scholar] [CrossRef]
- Hirai, H.; Arai, T.; Okada, M.; Nishibata, T.; Kobayashi, M.; Sakai, N.; Imagaki, K.; Ohtani, J.; Sakai, T.; Yoshizumi, T.; et al. MK-1775, a small molecule Wee1 inhibitor, enhances anti-tumor efficacy of various DNA-damaging agents, including 5-fluorouracil. Cancer Biol. Ther. 2010, 9, 514–522. [Google Scholar] [CrossRef]
- Lin, X.; Chen, D.; Zhang, C.; Zhang, X.; Li, Z.; Dong, B.; Gao, J.; Shen, L. Augmented antitumor activity by olaparib plus AZD1775 in gastric cancer through disrupting DNA damage repair pathways and DNA damage checkpoint. J. Exp. Clin. Cancer Res. 2018, 37, 129. [Google Scholar] [CrossRef]
- Parsels, L.A.; Karnak, D.; Parsels, J.D.; Zhang, Q.; Vélez-Padilla, J.; Reichert, Z.R.; Wahl, D.R.; Maybaum, J.; O’Connor, M.J.; Lawrence, T.S.; et al. PARP1 Trapping and DNA Replication Stress Enhance Radiosensitization with Combined WEE1 and PARP Inhibitors. Mol. Cancer Res. 2017, 16, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Karnak, D.; Engelke, C.G.; Parsels, L.A.; Kausar, T.; Wei, D.; Robertson, J.R. Combined Inhibition of Wee1 and PARP1/2 for Radiosensitization in Pancreatic Cancer. Clin. Cancer Res. 2014, 20, 5085–5096. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.; Schlenk, R.F.; Russell, N.H.; Levis, M.J. Targeting FLT3 mutations in AML: Review of current knowledge and evidence. Leukemia 2019, 33, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Grafone, T.; Palmisano, M.; Nicci, C.; Storti, S. An overview on the role of FLT3-tyrosine kinase receptor in acute myeloid leukemia: Biology and treatment. Oncol. Rev. 2012, 6, e8. [Google Scholar] [CrossRef]
- Ho, T.-C.; LaMere, M.; Stevens, B.M.; Ashton, J.M.; Myers, J.R.; O’Dwyer, K.M.; Liesveld, J.L.; Mendler, J.H.; Guzman, M.; Morrissette, J.D.; et al. Evolution of acute myelogenous leukemia stem cell properties after treatment and progression. Blood 2016, 128, 1671–1678. [Google Scholar] [CrossRef]
- Sallmyr, A.; Fan, J.; Datta, K.; Kim, K.T.; Grosu, D.; Shapiro, P. Internal Tandem Duplication of FLT3 (FLT3/ITD) Induces Increased ROS Production, DNA Damage, and Misrepair: Implications for Poor Prognosis in AML. Blood 2008, 111, 3173–3182. [Google Scholar] [CrossRef]
- Hole, P.S.; Zabkiewicz, J.; Munje, C.; Newton, Z.; Pearn, L.; White, P.; Marquez, N.; Hills, R.; Burnett, A.K.; Tonks, A.; et al. Overproduction of NOX-derived ROS in AML promotes proliferation and is associated with defective oxidative stress signaling. Blood 2013, 122, 3322–3330. [Google Scholar] [CrossRef]
- Jacoby, M.A.; Pizarro, R.D.J.; Shao, J.; Koboldt, D.C.; Fulton, R.S.; Zhou, G.; Wilson, R.K.; Walter, M.J. The DNA double-strand break response is abnormal in myeloblasts from patients with therapy-related acute myeloid leukemia. Leukemia 2013, 28, 1242–1251. [Google Scholar] [CrossRef]
- Esposito, M.T.; Zhao, L.; Fung, T.K.; Rane, J.K.; Wilson, A.; Martin, N.; Gil, J.; Leung, A.Y.; Ashworth, A.; So, C.W.E. Synthetic lethal targeting of oncogenic transcription factors in acute leukemia by PARP inhibitors. Nat. Med. 2015, 21, 1481–1490. [Google Scholar] [CrossRef]
- Gaymes, T.J.; Mohamedali, A.; Eiliazadeh, A.L.; Darling, D.; Mufti, G.J. FLT3 and JAK2 Mutations in Acute Myeloid Leukemia Promote Interchromosomal Homologous Recombination and the Potential for Copy Neutral Loss of Heterozygosity. Cancer Res. 2017, 77, 1697–1708. [Google Scholar] [CrossRef][Green Version]
- Seedhouse, C.H.; Hunter, H.M.; Lloyd-Lewis, B.; Massip, A.-M.; Pallis, M.; Carter, G.I.; Grundy, M.; Shang, S.; Russell, N.H. DNA repair contributes to the drug-resistant phenotype of primary acute myeloid leukaemia cells with FLT3 internal tandem duplications and is reversed by the FLT3 inhibitor PKC412. Leukemia 2006, 20, 2130–2136. [Google Scholar] [CrossRef] [PubMed]
- Maifrede, S.; Nieborowska-Skorska, M.; Sullivan-Reed, K.; Dasgupta, Y.; Podszywalow-Bartnicka, P.; Le, B.V.; Solecka, M.; Lian, Z.; Belyaeva, E.A.; Nersesyan, A.; et al. Tyrosine kinase inhibitor–induced defects in DNA repair sensitize FLT3(ITD)-positive leukemia cells to PARP1 inhibitors. Blood 2018, 132, 67–77. [Google Scholar] [CrossRef]
- Dellomo, A.J.; Abbotts, R.; Eberly, C.L.; Karbowski, M.; Baer, M.R.; Kingsbury, T.J.; Rassool, F.V. PARP1 PARylates and stabilizes STAT5 in FLT3-ITD acute myeloid leukemia and other STAT5-activated cancers. Transl. Oncol. 2021, 15, 101283. [Google Scholar] [CrossRef] [PubMed]
- Boccaccio, C.; Comoglio, P.M. Invasive growth: A MET-driven genetic programme for cancer and stem cells. Nat. Cancer 2006, 6, 637–645. [Google Scholar] [CrossRef]
- Park, K.C.; Richardson, D.R. The c-MET oncoprotein: Function, mechanisms of degradation and its targeting by novel anti-cancer agents. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2020, 1864, 129650. [Google Scholar] [CrossRef]
- Chakraborty, S.; Balan, M.; Flynn, E.; Zurakowski, D.; Choueiri, T.K.; Pal, S. Activation of c-Met in cancer cells mediates growth-promoting signals against oxidative stress through Nrf2-HO-1. Oncogenesis 2019, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Yamaguchi, H.; Wei-Chao, C.; Hsu, J.L.; Wang, H.-L.; Hsu, Y.-H.; Lin, W.-C.; Yu, W.-H.; Leonard, P.G.; Lee, G.R.; et al. Blocking c-Met–mediated PARP1 phosphorylation enhances anti-tumor effects of PARP inhibitors. Nat. Med. 2016, 22, 194–201. [Google Scholar] [CrossRef]
- Han, Y.; Chen, M.-K.; Wang, H.-L.; Hsu, J.L.; Li, C.-W.; Chu, Y.-Y.; Liu, C.-X.; Nie, L.; Chan, L.-C.; Yam, C.; et al. Synergism of PARP inhibitor fluzoparib (HS10160) and MET inhibitor HS10241 in breast and ovarian cancer cells. Am. J. Cancer Res. 2019, 9, 608–618. [Google Scholar]
- Hajjo, R.; Sweidan, K. Review on Epidermal Growth Factor Receptor (EGFR) Structure, Signaling Pathways, Interactions, and Recent Updates of EGFR Inhibitors. Curr. Top. Med. Chem. 2020, 20, 815–834. [Google Scholar]
- Bandyopadhyay, D.; Mandal, M.; Adam, L.; Mendelsohn, J.; Kumar, R. Physical Interaction between Epidermal Growth Factor Receptor and DNA-dependent Protein Kinase in Mammalian Cells. J. Biol. Chem. 1998, 273, 1568–1573. [Google Scholar] [CrossRef]
- Magné, N.; Fischel, J.-L.; Tiffon, C.; Formento, P.; Dubreuil, A.; Renée, N.; Formento, J.-L.; Francoual, M.; Ciccolini, J.; Etienne, M.-C.; et al. Molecular mechanisms underlying the interaction between ZD1839 (‘Iressa’) and cisplatin/5-fluorouracil. Br. J. Cancer 2003, 89, 585–592. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chu, P.L.; Shihabuddeen, W.A.; Low, K.P.; Poon, D.J.; Ramaswamy, B.; Liang, Z.-G.; Nei, W.L.; Chua, K.L.; Thong, P.S.; Soo, K.C.; et al. Vandetanib sensitizes head and neck squamous cell carcinoma to photodynamic therapy through modulation of EGFR-dependent DNA repair and the tumour microenvironment. Photodiagnosis Photodyn. Ther. 2019, 27, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.M.; Harari, P.M. Modulation of radiation response after epidermal growth factor receptor blockade in squamous cell carcinomas: Inhibition of damage repair, cell cycle kinetics, and tumor angiogenesis. Clin. Cancer Res. 2000, 6, 2166–2174. [Google Scholar] [PubMed]
- Friedmann, B.; Caplin, M.; Hartley, J.A.; Hochhauser, D. Modulation of DNA Repair In vitro after Treatment with Chemotherapeutic Agents by the Epidermal Growth Factor Receptor Inhibitor Gefitinib (ZD1839). Clin. Cancer Res. 2004, 10, 6476–6486. [Google Scholar] [CrossRef]
- Russo, M.; Crisafulli, G.; Sogari, A.; Reilly, N.M.; Arena, S.; Lamba, S.; Bartolini, A.; Amodio, V.; Magrì, A.; Novara, L.; et al. Adaptive mutability of colorectal cancers in response to targeted therapies. Science 2019, 366, 1473–1480. [Google Scholar] [CrossRef]
- Dong, Q.; Du, Y.; Li, H.; Liu, C.; Wei, Y.; Chen, M.-K.; Zhao, X.; Chu, Y.-Y.; Qiu, Y.; Qin, L.; et al. EGFR and c-MET Cooperate to Enhance Resistance to PARP Inhibitors in Hepatocellular Carcinoma. Cancer Res. 2018, 79, 819–829. [Google Scholar] [CrossRef]
- Chu, Y.-Y.; Yam, C.; Chen, M.-K.; Chan, L.-C.; Xiao, M.; Wei, Y.-K.; Yamaguchi, H.; Lee, P.-C.; Han, Y.; Nie, L.; et al. Blocking c-Met and EGFR reverses acquired resistance of PARP inhibitors in triple-negative breast cancer. Am. J. Cancer Res. 2020, 10, 648–661. [Google Scholar]
- Simons, M.; Gordon, E.; Claesson-Welsh, L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat. Rev. Mol. Cell Biol. 2016, 17, 611–625. [Google Scholar] [CrossRef]
- Lim, J.J.; Yang, K.; Taylor-Harding, B.; Wiedemeyer, W.R.; Buckanovich, R.J. VEGFR3 Inhibition Chemosensitizes Ovarian Cancer Stemlike Cells through Down-Regulation of BRCA1 and BRCA2. Neoplasia 2014, 16, 343–353.e2. [Google Scholar] [CrossRef]
- Bindra, R.S.; Gibson, S.L.; Meng, A.; Westermark, U.; Jasin, M.; Pierce, A.J.; Bristow, R.G.; Classon, M.K.; Glazer, P.M. Hypoxia-Induced Down-regulation of BRCA1 Expression by E2Fs. Cancer Res. 2005, 65, 11597–11604. [Google Scholar] [CrossRef]
- Bindra, R.S.; Schaffer, P.J.; Meng, A.; Woo, J.; Måseide, K.; Roth, M.E.; Lizardi, P.; Hedley, D.W.; Bristow, R.G.; Glazer, P.M. Down-Regulation of Rad51 and Decreased Homologous Recombination in Hypoxic Cancer Cells. Mol. Cell. Biol. 2004, 24, 8504–8518. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.F.; Barry, W.T.; Birrer, M.; Lee, J.M.; Buckanovich, R.J.; Fleming, G.F. A randomized phase 2 study of combination cediranib and olaparib versus olaparib alone as recurrence therapy in platinum-sensitive ovarian cancer. Lancet Oncol. 2014, 15, 1207–1214. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.T.; Chang, F.; Lehmann, B.; Terrian, D.M.; Milella, M.; Tafuri, A.; et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim. Biophys. Acta Mol. Cell Res. 2007, 1773, 1263–1284. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Fang, Y.; Yin, J.; Chen, J.; Ju, Z.; Zhang, D.; Chen, X.; Vellano, C.P.; Jeong, K.J.; Ng, P.K.-S.; et al. Rational combination therapy with PARP and MEK inhibitors capitalizes on therapeutic liabilities in RAS mutant cancers. Sci. Transl. Med. 2017, 9, eaal5148. [Google Scholar] [CrossRef]
- Vena, F.; Jia, R.; Esfandiari, A.; García-Gómez, J.J.; Rodríguez-Justo, M.; Ma, J.; Syed, S.; Crowley, L.; Elenbaas, B.; Goodstal, S.; et al. MEK inhibition leads to BRCA2 downregulation and sensitization to DNA damaging agents in pancreas and ovarian cancer models. Oncotarget 2018, 9, 11592–11603. [Google Scholar] [CrossRef]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef]
- Keith, C.T.; Schreiber, S.L. PIK-Related Kinases: DNA Repair, Recombination, and Cell Cycle Checkpoints. Science 1995, 270, 50. [Google Scholar] [CrossRef]
- Kumar, A.; Fernandez-Capetillo, O.; Carrera, A.C. Nuclear Phosphoinositide 3-kinase Beta Controls Double-Strand Break DNA Repair. Proc. Natl. Acad. Sci. USA 2010, 107, 7491–7496. [Google Scholar] [CrossRef]
- Juvekar, A.; Burga, L.N.; Hu, H.; Lunsford, E.P.; Ibrahim, Y.H.; Balmañà, J. Combining a PI3K Inhibitor with a PARP Inhibitor Provides an Effective Therapy for BRCA1-related Breast Cancer. Cancer Discov. 2012, 2, 1048–1063. [Google Scholar] [CrossRef]
- Chen, H.; Ma, Z.; VanderWaal, R.P.; Feng, Z.; Gonzalez-Suarez, I.; Wang, S.; Zhang, J.; Roti, J.L.R.; Gonzalo, S.; Zhang, J. The mTOR Inhibitor Rapamycin Suppresses DNA Double-Strand Break Repair. Radiat. Res. 2010, 175, 214–224. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Lancaster, C.S.; Shi, B.; Guo, H.; Thimmaiah, P.; Bjornsti, M.-A. TOR Signaling Is a Determinant of Cell Survival in Response to DNA Damage. Mol. Cell. Biol. 2007, 27, 7007–7017. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Li, J.; Du, W.; Zhang, S.; O’Connor, M.; Thomas, G. mTOR Regulates DNA Damage Response Through NF-κB-mediated FANCD2 Pathway in Hematopoietic Cells. Leukemia 2013, 27, 2040–2046. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Oswald, D.; Phelps, D.; Cam, H.; Pelloski, C.E.; Pang, Q.; Houghton, P.J. Regulation of FANCD2 by the mTOR Pathway Contributes to the Resistance of Cancer Cells to DNA Double-Strand Breaks. Cancer Res. 2013, 73, 3393–3401. [Google Scholar] [CrossRef]
- Ibrahim, Y.H.; García-García, C.; Serra, V.; He, L.; Torres-Lockhart, K. Prat A. PI3K Inhibition Impairs BRCA1/2 Expression and Sensitizes BRCA-proficient Triple-Negative Breast Cancer to PARP Inhibition. Cancer Discov. 2012, 2, 1036–1047. [Google Scholar] [CrossRef]
- Chang, L.; Graham, P.H.; Hao, J.; Ni, J.; Bucci, J.; Cozzi, P.J.; Kearsley, J.H.; Li, Y. PI3K/Akt/mTOR pathway inhibitors enhance radiosensitivity in radioresistant prostate cancer cells through inducing apoptosis, reducing autophagy, suppressing NHEJ and HR repair pathways. Cell Death Dis. 2014, 5, e1437. [Google Scholar] [CrossRef]
- Mukherjee, B.; Tomimatsu, N.; Amancherla, K.; Camacho, C.V.; Pichamoorthy, N.; Burma, S. The Dual PI3K/mTOR Inhibitor NVP-BEZ235 Is a Potent Inhibitor of ATM- and DNA-PKCs-Mediated DNA Damage Responses. Neoplasia 2012, 14, 34–43. [Google Scholar] [CrossRef]
- Mo, W.; Liu, Q.; Lin, C.C.-J.; Dai, H.; Peng, Y.; Liang, Y.; Peng, G.; Meric-Bernstam, F.; Mills, G.B.; Li, K.; et al. mTOR Inhibitors Suppress Homologous Recombination Repair and Synergize with PARP Inhibitors via Regulating SUV39H1 in BRCA-Proficient Triple-Negative Breast Cancer. Clin. Cancer Res. 2015, 22, 1699–1712. [Google Scholar] [CrossRef]
- De, P.; Sun, Y.; Carlson, J.H.; Friedman, L.S.; Leyland-Jones, B.R.; Dey, N. Doubling Down on the PI3K-AKT-mTOR Pathway Enhances the Antitumor Efficacy of PARP Inhibitor in Triple Negative Breast Cancer Model beyond BRCA-ness12. Neoplasia 2012, 162014, 43–72. [Google Scholar] [CrossRef]
- Philip, C.A.; Laskov, I.; Beauchamp, M.C.; Marques, M.; Amin, O.; Bitharas, J. Inhibition of PI3K-AKT-mTOR Pathway Sensitizes Endometrial Cancer Cell Lines to PARP Inhibitors. BMC Cancer 2017, 17, 638. [Google Scholar] [CrossRef]
- Wang, D.; Li, C.; Zhang, Y.; Wang, M.; Jiang, N.; Xiang, L.; Li, T.; Roberts, T.M.; Zhao, J.J.; Cheng, H.; et al. Combined inhibition of PI3K and PARP is effective in the treatment of ovarian cancer cells with wild-type PIK3CA genes. Gynecol. Oncol. 2016, 142, 548–556. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, A.; Drew, Y.; Matheson, E.; Salehan, M.; Gentles, L.; Pachter, J.A.; Curtin, N.J. Evaluating the potential of kinase inhibitors to suppress DNA repair and sensitise ovarian cancer cells to PARP inhibitors. Biochem. Pharmacol. 2018, 167, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Wang, M.; Jiang, N.; Zhang, Y.; Bian, X.; Wang, X.; Roberts, T.M.; Zhao, J.J.; Liu, P.; Cheng, H. Effective use of PI3K inhibitor BKM120 and PARP inhibitor Olaparib to treat PIK3CA mutant ovarian cancer. Oncotarget 2016, 7, 13153–13166. [Google Scholar] [CrossRef]
- Cilloni, D.; Saglio, G. Molecular Pathways: BCR-ABL. Clin. Cancer Res. 2011, 18, 930–937. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Yuan, S.-S.; Liu, W.; Xu, Y.; Trujillo, K.; Song, B.; Cong, F.; Goff, S.P.; Wu, Y.; Arlinghaus, R.; et al. Radiation-induced Assembly of Rad51 and Rad52 Recombination Complex Requires ATM and c-Abl. J. Biol. Chem. 1999, 274, 12748–12752. [Google Scholar] [CrossRef]
- Kitao, H.; Yuan, Z.-M. Regulation of Ionizing Radiation-induced Rad52 Nuclear Foci Formation by c-Abl-mediated Phosphorylation. J. Biol. Chem. 2002, 277, 48944–48948. [Google Scholar] [CrossRef]
- Shimizu, H.; Popova, M.; Fleury, F.; Kobayashi, M.; Hayashi, N.; Sakane, I.; Kurumizaka, H.; Venkitaraman, A.R.; Takahashi, M.; Yamamoto, K.-I. c-ABL tyrosine kinase stabilizes RAD51 chromatin association. Biochem. Biophys. Res. Commun. 2009, 382, 286–291. [Google Scholar] [CrossRef]
- Slupianek, A.; Schmutte, C.; Tombline, G.; Nieborowska-Skorska, M.; Hoser, G.; Nowicki, M.O.; Pierce, A.J.; Fishel, R.; Skorski, T. BCR/ABL Regulates Mammalian RecA Homologs, Resulting in Drug Resistance. Mol. Cell 2001, 8, 795–806. [Google Scholar] [CrossRef]
- Choudhury, A.; Zhao, H.; Jalali, F.; Al Rashid, S.; Ran, J.; Supiot, S.; Kiltie, A.; Bristow, R. Targeting homologous recombination using imatinib results in enhanced tumor cell chemosensitivity and radiosensitivity. Mol. Cancer Ther. 2009, 8, 203–213. [Google Scholar] [CrossRef]
- Bajrami, I.; Kigozi, A.; van Weverwijk, A.; Brough, R.; Frankum, J.; Lord, C.; Ashworth, A. Synthetic lethality of PARP and NAMPT inhibition in triple—negative breast cancer cells. EMBO Mol. Med. 2012, 4, 1087–1096. [Google Scholar] [CrossRef]
- Heske, C.M.; Davis, M.I.; Baumgart, J.T.; Wilson, K.; Gormally, M.V.; Chen, L.; Zhang, X.; Ceribelli, M.; Duveau, D.Y.; Guha, R.; et al. Matrix Screen Identifies Synergistic Combination of PARP Inhibitors and Nicotinamide Phosphoribosyltransferase (NAMPT) Inhibitors in Ewing Sarcoma. Clin. Cancer Res. 2017, 23, 7301–7311. [Google Scholar] [CrossRef]
- Ma, E.; Chen, P.; Wilkins, H.; Wang, T.; Swerdlow, R.H.; Chen, Q. Pharmacologic ascorbate induces neuroblastoma cell death by hydrogen peroxide mediated DNA damage and reduction in cancer cell glycolysis. Free Radic. Biol. Med. 2017, 113, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Chen, P.; Drisko, J.A.; Khabele, D.; Godwin, A.K.; Chen, Q. Pharmacological ascorbate induces ‘BRCAness’ and enhances the effects of Poly(ADP-Ribose) polymerase inhibitors against BRCA1/2 wild-type ovarian cancer. Oncol. Lett. 2020, 192020, 2629–2638. [Google Scholar] [CrossRef] [PubMed]
- Gillberg, L.; Orskov, A.D.; Nasif, A.; Ohtani, H.; Madaj, Z.; Hansen, J.W.; Rapin, N.; Mogensen, J.B.; Liu, M.; Dufva, I.H.; et al. Oral vitamin C supplementation to patients with myeloid cancer on azacitidine treatment: Normalization of plasma vitamin C induces epigenetic changes. Clin. Epigenetics 2019, 11, 143. [Google Scholar] [CrossRef]
- Liu, M.; Ohtani, H.; Zhou, W.; Orskov, A.D.; Charlet, J.; Zhang, Y.W.; Shen, H.; Baylin, S.B.; Liang, G.; Jones, P.A.; et al. Vitamin C increases viral mimicry induced by 5-aza-2′-deoxycytidine. Proc. Natl. Acad. Sci. USA 2016, 113, 10238–10244. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).