
Clinical Landscape of PARP Inhibitors in Ovarian Cancer: Molecular Mechanisms and Clues to Overcome Resistance

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Two Distinct Therapeutic Mechanisms of PARPis
2.1. Enzymatic Inhibition
2.2. PARP Trapping
3. HRD as a Predictive Marker for the Efficacy and Survival Benefit of PARPis
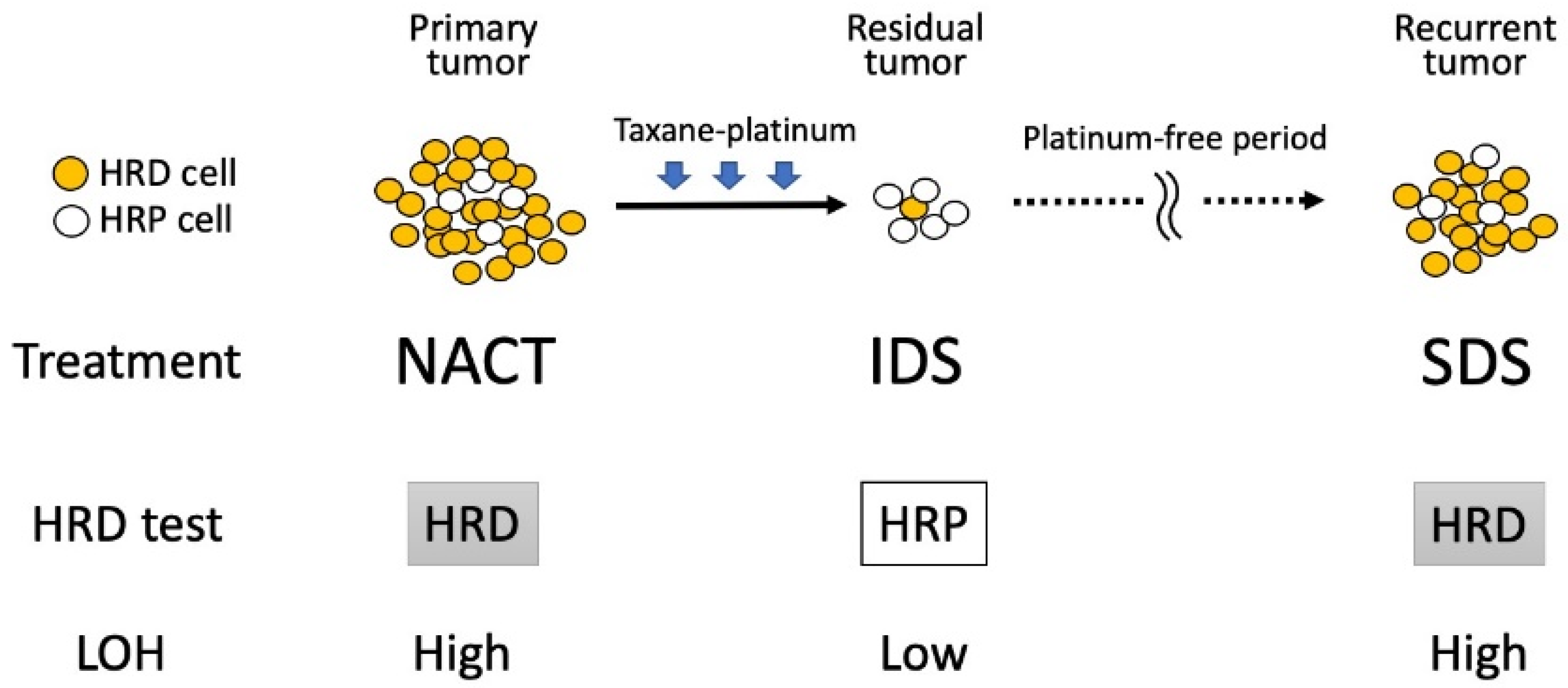
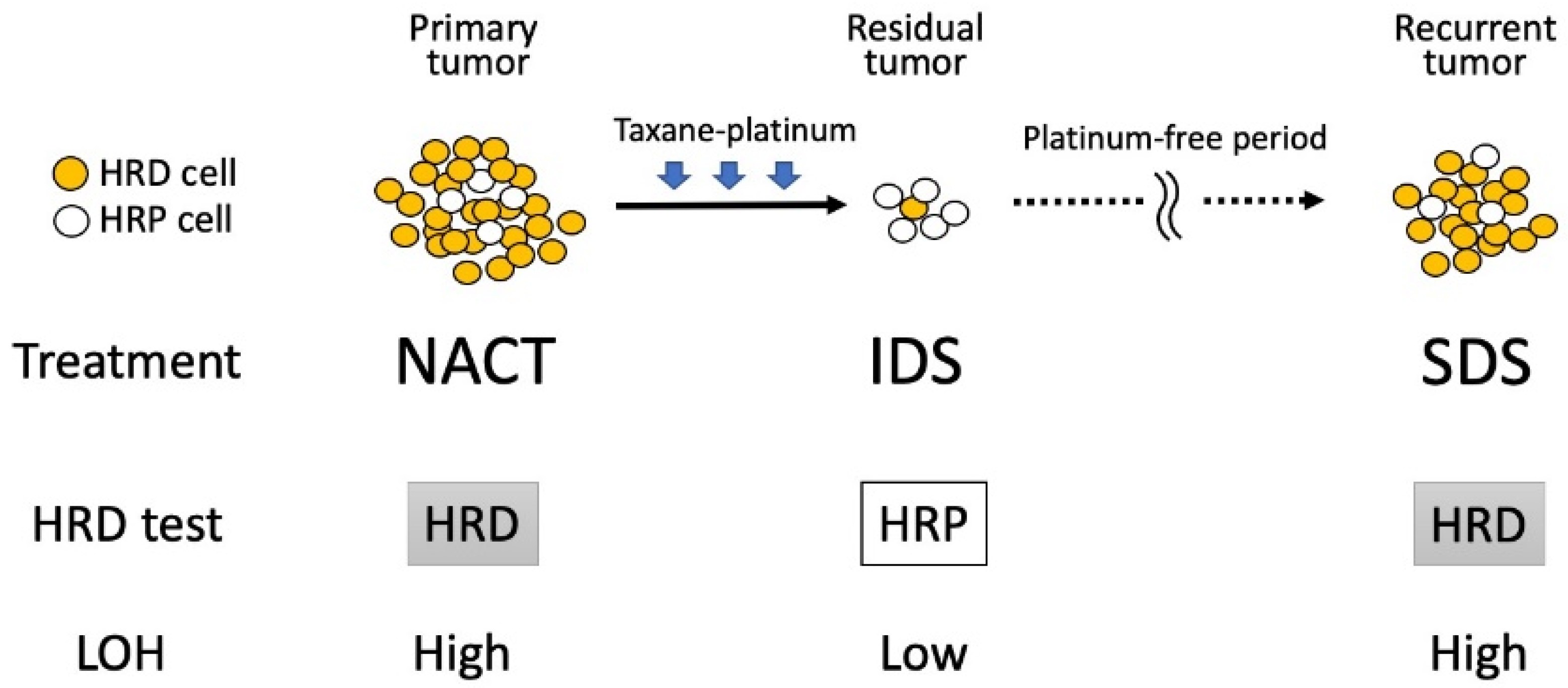
4. Dynamics of HRD Status in the Treatment of Ovarian Cancer
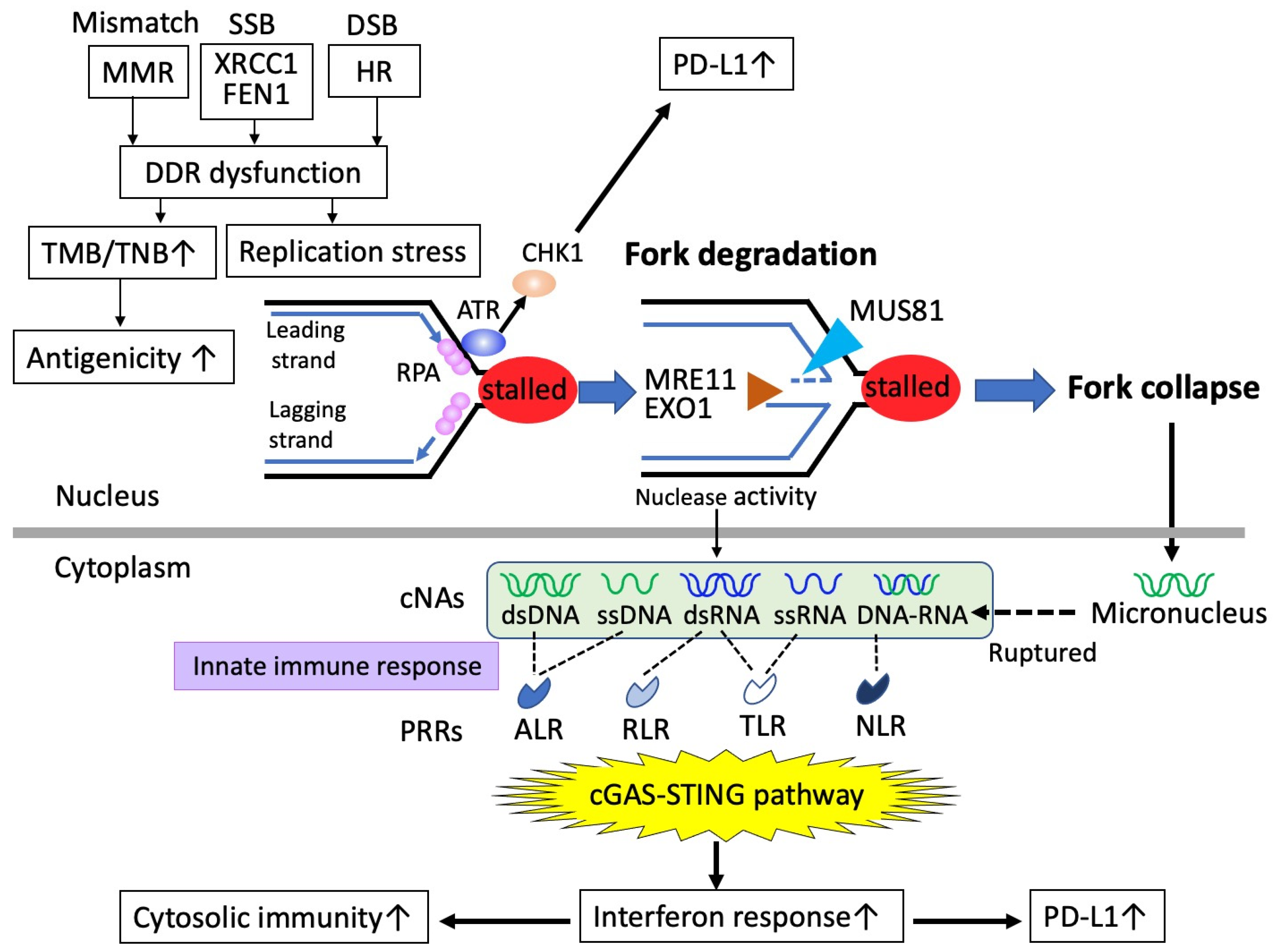
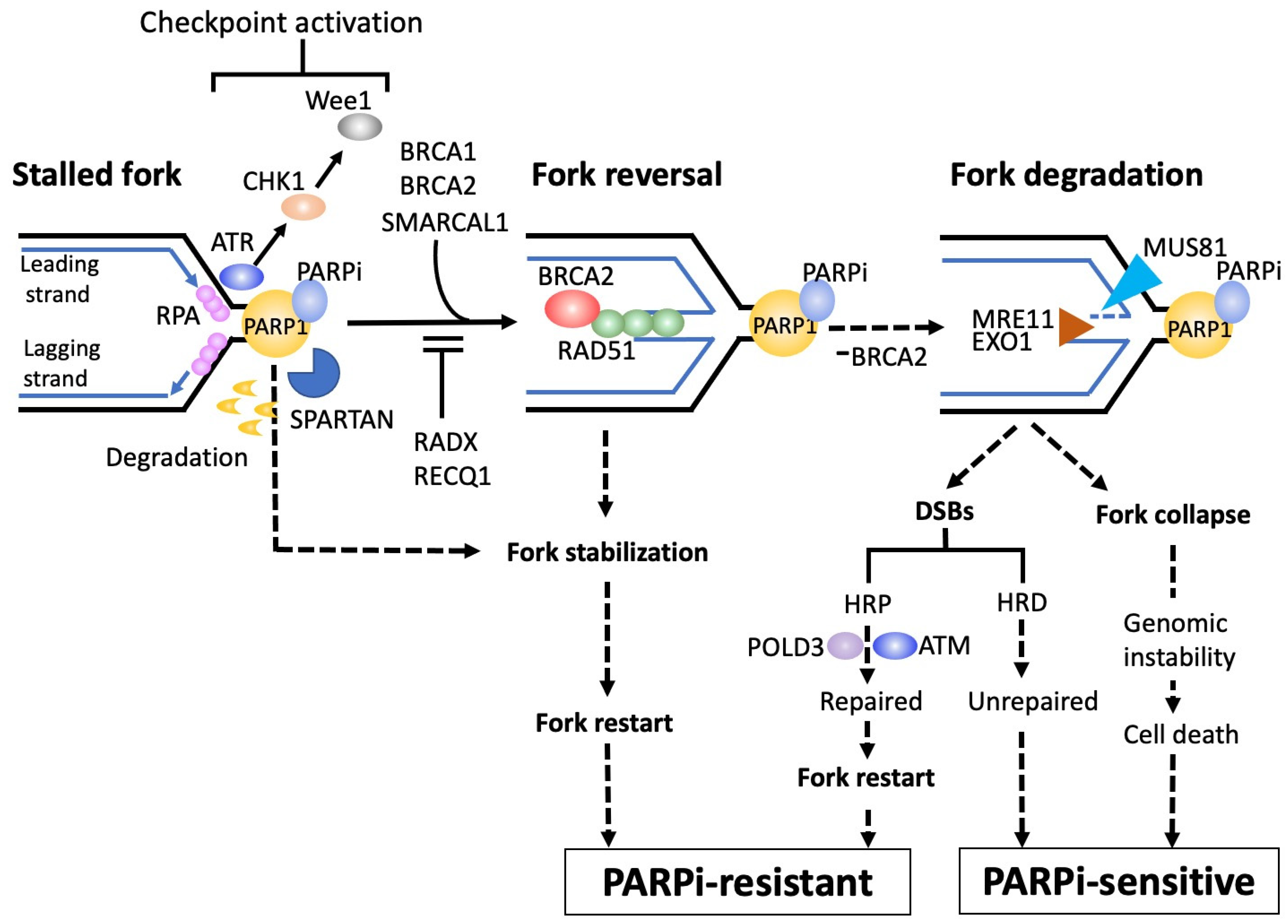
5. The Fate of PARP Trapping: Fork Stabilization or Degradation That Determines the Efficacy of PARPis
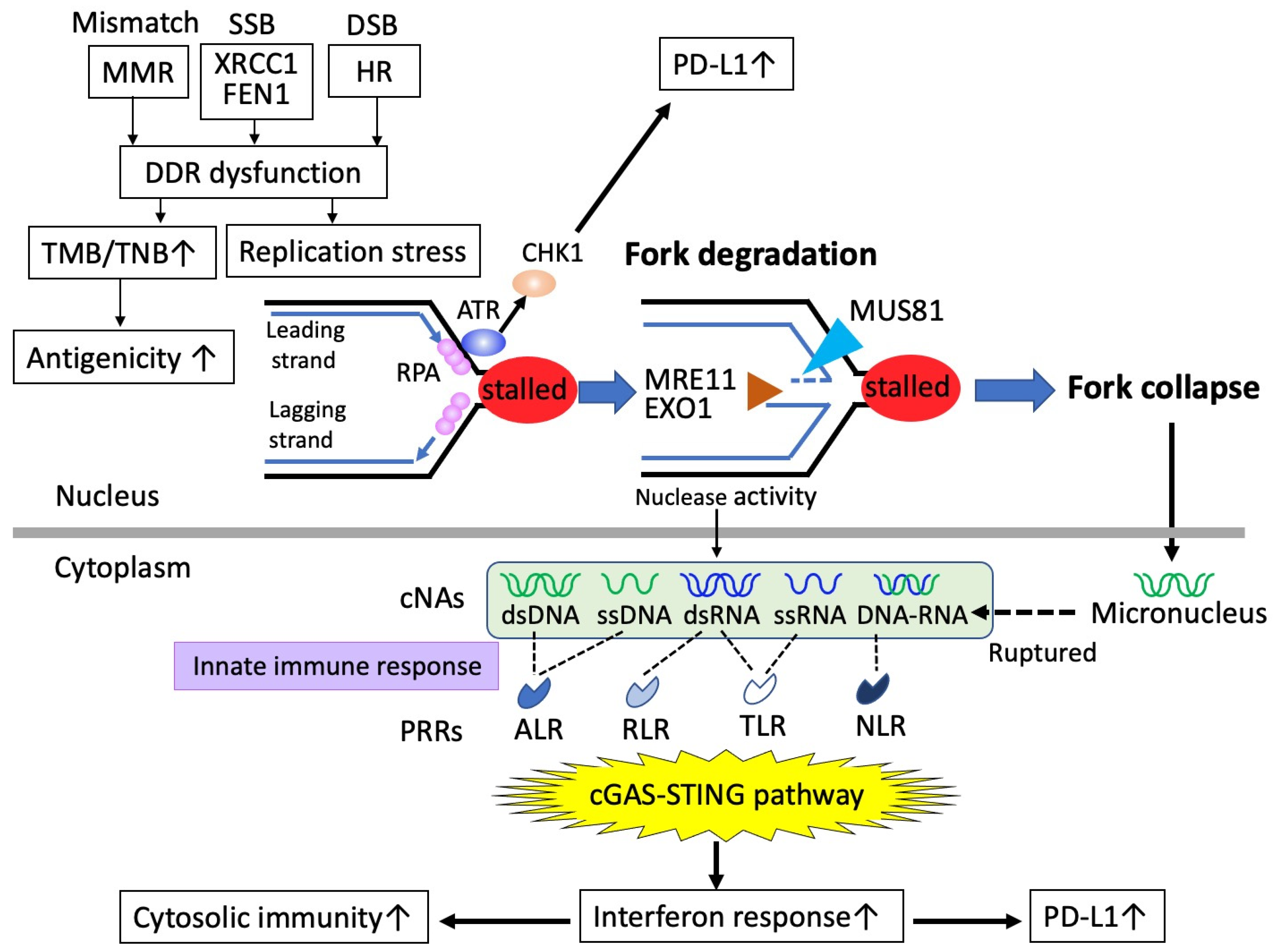
6. PARP Inhibition Triggers Tumor Immunogenicity
6.1. Defective DNA Damage Responses Enhance Tumor Antigenicity
6.2. PARPis Activate the cGAS-STING Pathway and Mediate Cytosolic Immunity
6.3. Upregulation of PD-L1 by PARP Inhibition
6.4. Clinical Application to Combine PARPi with ICIs
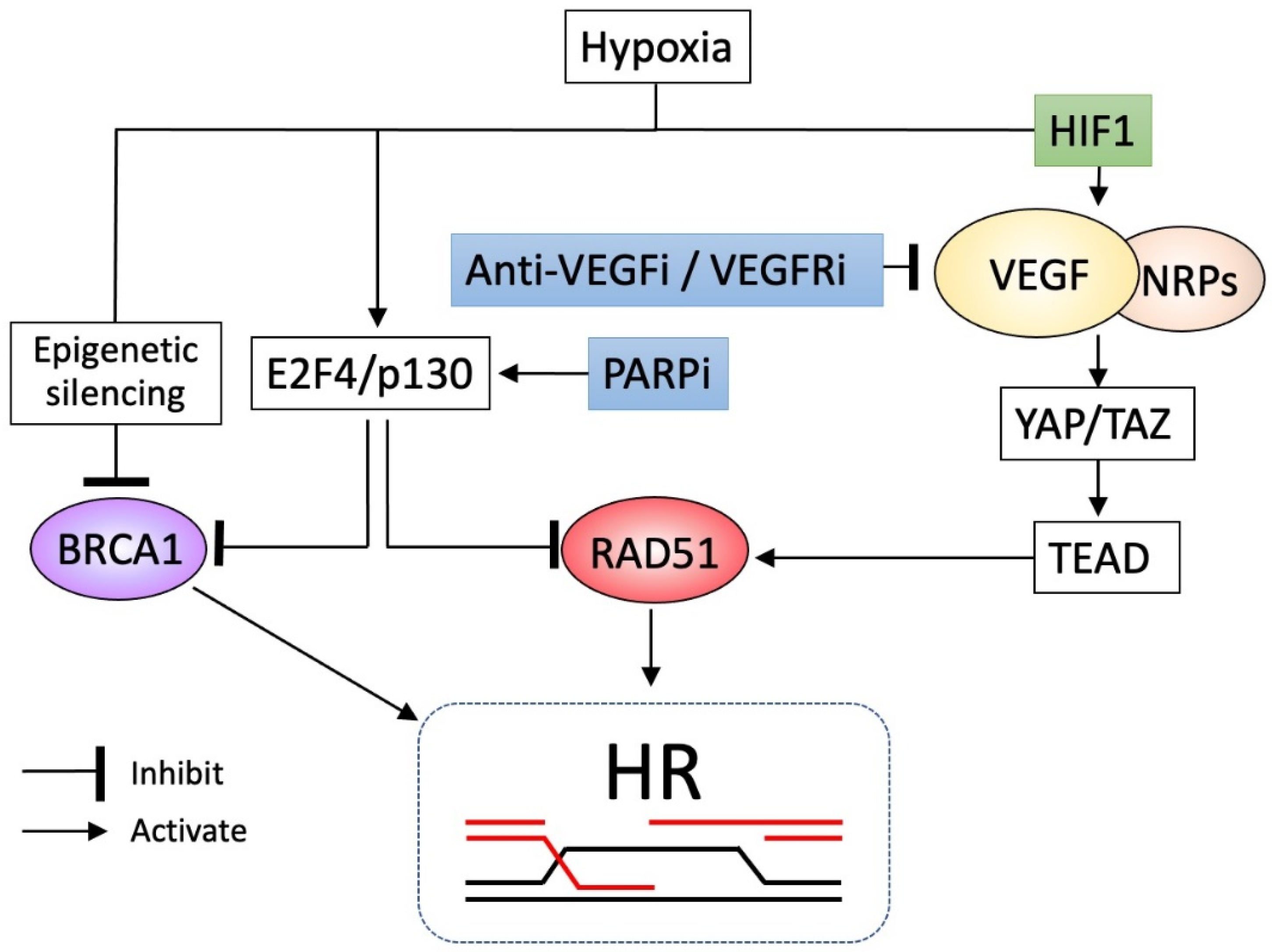
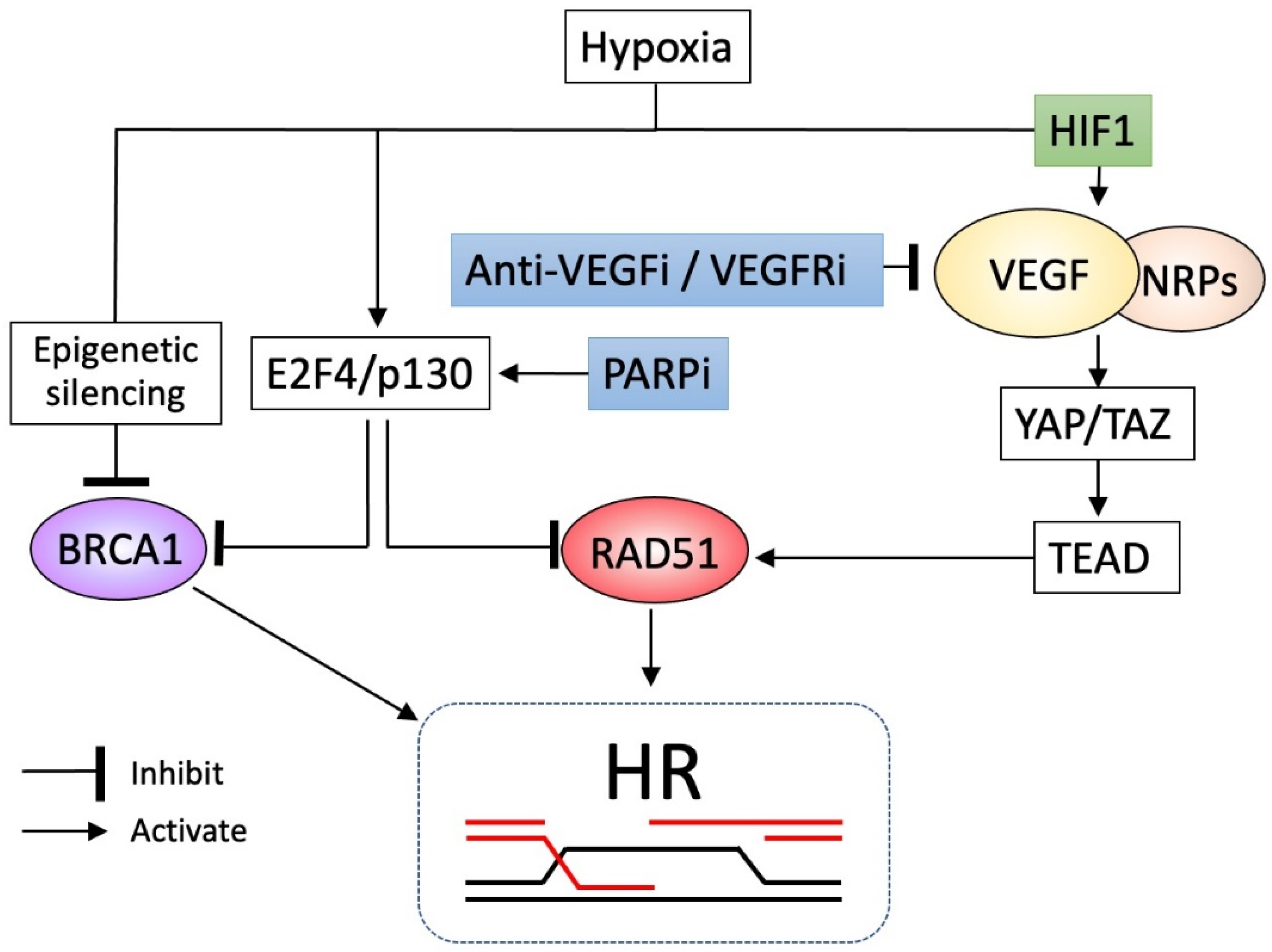
7. PARPis Can Cooperate with Antiangiogenic Agents
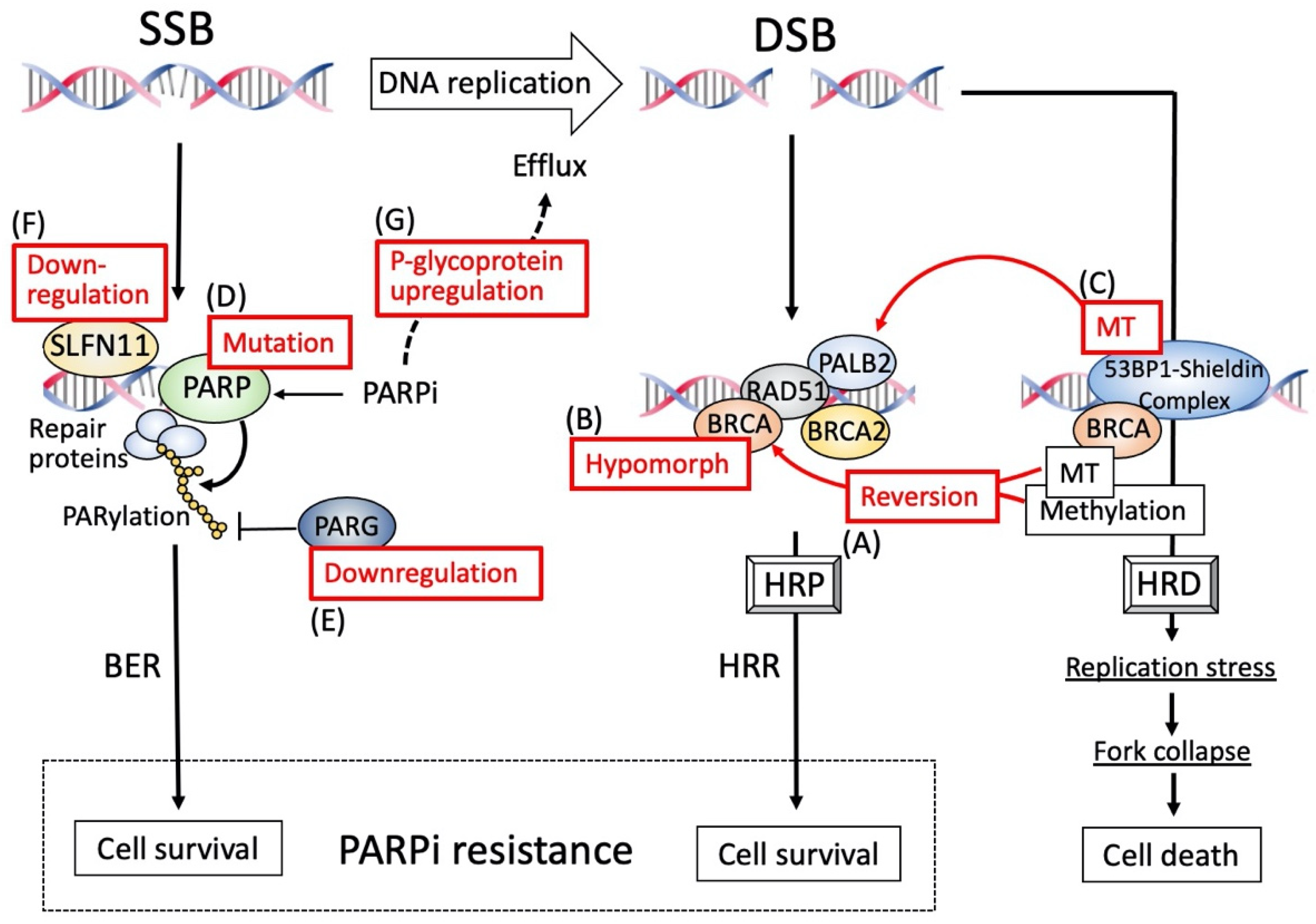
8. Molecular Factors Involved in PARPi Resistance
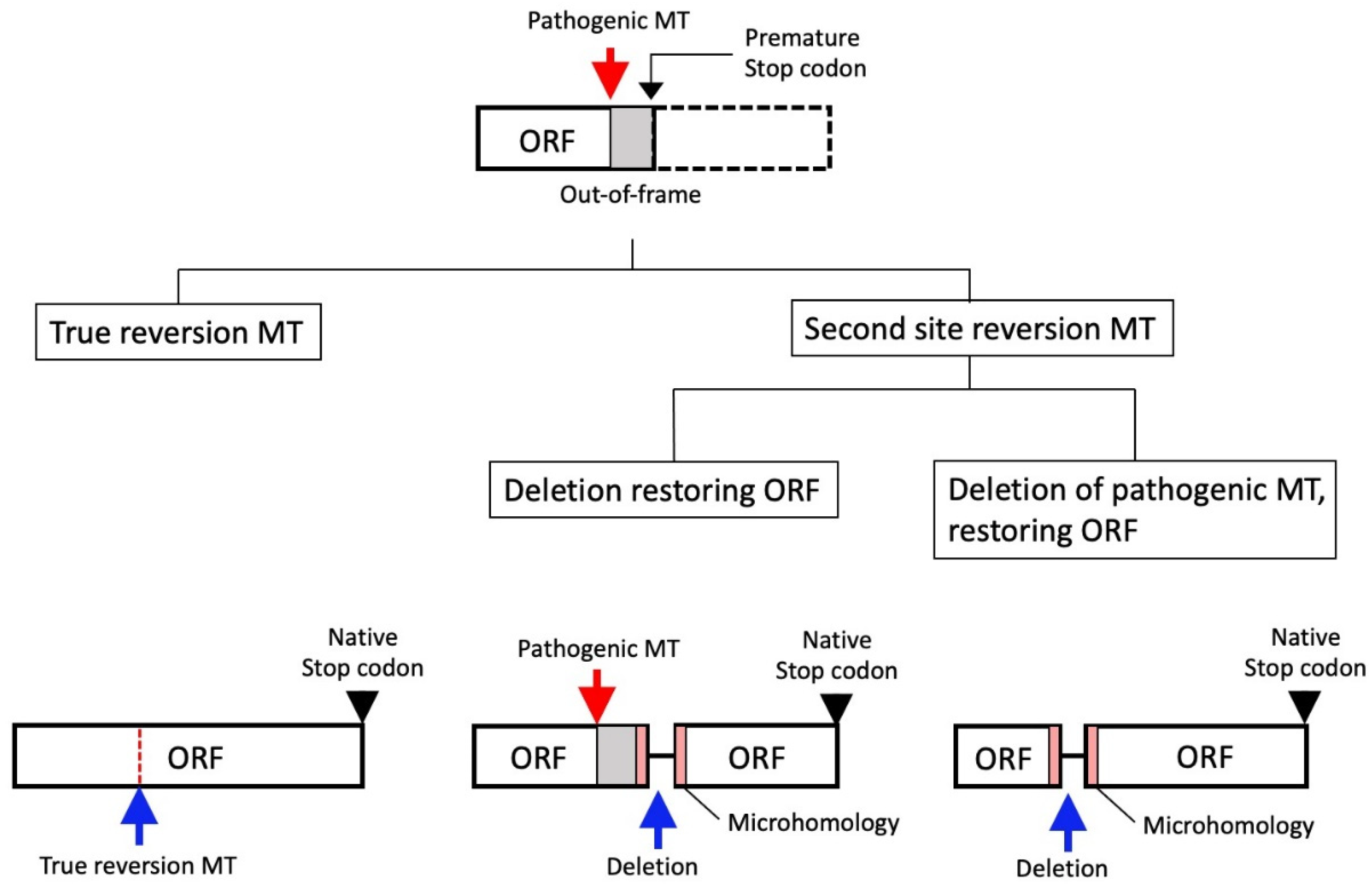
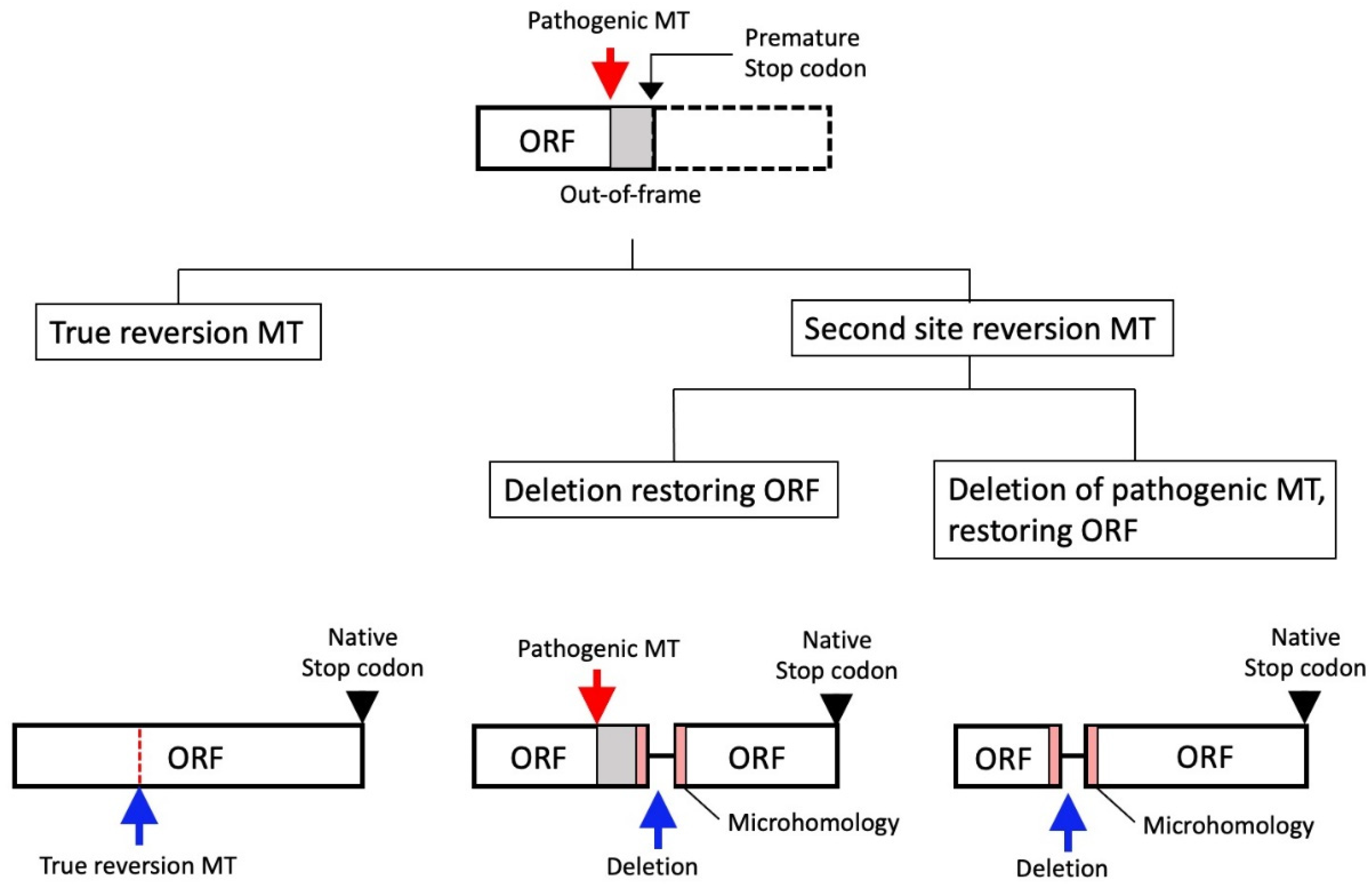
8.1. Reversion Mutation
8.2. BRCA Hypomorphic Proteins
8.3. Epigenetic Reversion
8.4. Re-Gaining HR Proficiency under BRCA Mutation
8.5. PARP1 Mutation
8.6. PARG Loss
8.7. Schlafen11 Loss
8.8. ABCB1 Overexpression
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Coleman, R.L.; Fleming, G.F.; Brady, M.F.; Swisher, E.M.; Steffensen, K.D.; Friedlander, M.; Okamoto, A.; Moore, K.N.; Efrat Ben-Baruch, N.; Werner, T.L.; et al. Veliparib with First-Line Chemotherapy and as Maintenance Therapy in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2403–2415. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.L.; Oza, A.M.; Lorusso, D.; Aghajanian, C.; Oaknin, A.; Dean, A.; Colombo, N.; Weberpals, J.I.; Clamp, A.; Scambia, G.; et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 1949–1961. [Google Scholar] [CrossRef] [Green Version]
- González-Martín, A.; Pothuri, B.; Vergote, I.; DePont Christensen, R.; Graybill, W.; Mirza, M.R.; McCormick, C.; Lorusso, D.; Hoskins, P.; Freyer, G.; et al. Niraparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2391–2402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N. Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018, 379, 2495–2505. [Google Scholar] [CrossRef]
- Pujade-Lauraine, E.; Ledermann, J.A.; Selle, F.; Gebski, V.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Poveda, A.; Pignata, S.; et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1274–1284. [Google Scholar] [CrossRef] [Green Version]
- Pujade-Lauraine, E.; Selle, F.; Scambia, G.; Asselain, B.; Marmé, F.; Lindemann, K.; Colombo, N.; Madry, R.; Glasspool, R.M.; Dubot, C.; et al. LBA33 Maintenance olaparib rechallenge in patients (pts) with ovarian carcinoma (OC) previously treated with a PARP inhibitor (PARPi): Phase IIIb OReO/ENGOT Ov-38 trial. Ann. Oncol. 2021, 32, S1308–S1309. [Google Scholar] [CrossRef]
- Amé, J.C.; Spenlehauer, C.; de Murcia, G. The PARP superfamily. Bioessays 2004, 26, 882–893. [Google Scholar] [CrossRef]
- Gibson, B.A.; Kraus, W.L. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat. Rev. Mol. Cell Biol. 2012, 13, 411–424. [Google Scholar] [CrossRef]
- Ray Chaudhuri, A.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef]
- Roy, R.; Chun, J.; Powell, S.N. BRCA1 and BRCA2: Different roles in a common pathway of genome protection. Nat. Rev. Cancer 2011, 12, 68–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Huang, S.Y.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slade, D. PARP and PARG inhibitors in cancer treatment. Genes Dev. 2020, 34, 360–394. [Google Scholar] [CrossRef] [Green Version]
- Zandarashvili, L.; Langelier, M.F.; Velagapudi, U.K.; Hancock, M.A.; Steffen, J.D.; Billur, R.; Hannan, Z.M.; Wicks, A.J.; Krastev, D.B.; Pettitt, S.J.; et al. Structural basis for allosteric PARP-1 retention on DNA breaks. Science 2020, 368, eaax6367. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef]
- Alsop, K.; Fereday, S.; Meldrum, C.; deFazio, A.; Emmanuel, C.; George, J.; Dobrovic, A.; Birrer, M.J.; Webb, P.M.; Stewart, C.; et al. BRCA mutation frequency and patterns of treatment response in BRCA mutation-positive women with ovarian cancer: A report from the Australian Ovarian Cancer Study Group. J. Clin. Oncol. 2012, 30, 2654–2663. [Google Scholar] [CrossRef] [Green Version]
- Enomoto, T.; Aoki, D.; Hattori, K.; Jinushi, M.; Kigawa, J.; Takeshima, N.; Tsuda, H.; Watanabe, Y.; Yoshihara, K.; Sugiyama, T. The first Japanese nationwide multicenter study of BRCA mutation testing in ovarian cancer: CHARacterizing the cross-sectionaL approach to Ovarian cancer geneTic TEsting of BRCA (CHARLOTTE). Int. J. Gynecol. Cancer 2019, 29, 1043–1049. [Google Scholar] [CrossRef] [Green Version]
- Frugtniet, B.; Morgan, S.; Murray, A.; Palmer-Smith, S.; White, R.; Jones, R.; Hanna, L.; Fuller, C.; Hudson, E.; Mullard, A.; et al. The detection of germline and somatic BRCA1/2 genetic variants through parallel testing of patients with high-grade serous ovarian cancer: A national retrospective audit. BJOG Int. J. Obstet. Gynaecol. 2022, 129, 433–442. [Google Scholar] [CrossRef]
- Timms, K.M.; Abkevich, V.; Hughes, E.; Neff, C.; Reid, J.; Morris, B.; Kalva, S.; Potter, J.; Tran, T.V.; Chen, J.; et al. Association of BRCA1/2 defects with genomic scores predictive of DNA damage repair deficiency among breast cancer subtypes. Breast Cancer Res. 2014, 16, 475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.L.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: A preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014, 15, 852–861. [Google Scholar] [CrossRef]
- Hodgson, D.R.; Dougherty, B.A.; Lai, Z.; Fielding, A.; Grinsted, L.; Spencer, S.; O’Connor, M.J.; Ho, T.W.; Robertson, J.D.; Lanchbury, J.S.; et al. Candidate biomarkers of PARP inhibitor sensitivity in ovarian cancer beyond the BRCA genes. Br. J. Cancer 2018, 119, 1401–1409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pujade-Lauraine, E.; Brown, J.; Barnicle, A.; Rowe, P.; Lao-Sirieix, P.; Criscione, S.; du Bois, A.; Lorusso, D.; Romero, I.; Petru, E.; et al. Homologous recombination repair mutation gene panels (excluding BRCA) are not predictive of maintenance olaparib plus bevacizumab efficacy in the first-line PAOLA-1/ENGOT-ov25 trial. Gynecol. Oncol. 2021, 162, S26–S27. [Google Scholar] [CrossRef]
- Ray-Coquard, I.; Pautier, P.; Pignata, S.; Pérol, D.; González-Martín, A.; Berger, R.; Fujiwara, K.; Vergote, I.; Colombo, N.; Mäenpää, J.; et al. Olaparib plus Bevacizumab as First-Line Maintenance in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2416–2428. [Google Scholar] [CrossRef]
- Takaya, H.; Nakai, H.; Sakai, K.; Nishio, K.; Murakami, K.; Mandai, M.; Matsumura, N. Intratumor heterogeneity and homologous recombination deficiency of high-grade serous ovarian cancer are associated with prognosis and molecular subtype and change in treatment course. Gynecol. Oncol. 2020, 156, 415–422. [Google Scholar] [CrossRef]
- Sokolenko, A.P.; Bizin, I.V.; Preobrazhenskaya, E.V.; Gorodnova, T.V.; Ivantsov, A.O.; Iyevleva, A.G.; Savonevich, E.L.; Kotiv, K.B.; Kuligina, E.S.; Imyanitov, E.N. Molecular profiles of BRCA1-associated ovarian cancer treated by platinum-based therapy: Analysis of primary, residual and relapsed tumors. Int. J. Cancer 2020, 146, 1879–1888. [Google Scholar] [CrossRef]
- Patel, J.N.; Braicu, I.; Timms, K.M.; Solimeno, C.; Tshiaba, P.; Reid, J.; Lanchbury, J.S.; Darb-Esfahani, S.; Ganapathi, M.K.; Sehouli, J.; et al. Characterisation of homologous recombination deficiency in paired primary and recurrent high-grade serous ovarian cancer. Br. J. Cancer 2018, 119, 1060–1066. [Google Scholar] [CrossRef] [Green Version]
- Cortez, D. Preventing replication fork collapse to maintain genome integrity. DNA Repair 2015, 32, 149–157. [Google Scholar] [CrossRef] [Green Version]
- Lopes, M.; Cotta-Ramusino, C.; Pellicioli, A.; Liberi, G.; Plevani, P.; Muzi-Falconi, M.; Newlon, C.S.; Foiani, M. The DNA replication checkpoint response stabilizes stalled replication forks. Nature 2001, 412, 557–561. [Google Scholar] [CrossRef]
- Sogo, J.M.; Lopes, M.; Foiani, M. Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science 2002, 297, 599–602. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.; Ji, F.; Helleday, T.; Ying, S. Mechanisms for stalled replication fork stabilization: New targets for synthetic lethality strategies in cancer treatments. EMBO Rep. 2018, 19, e46263. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Pavletich, N.P. Structure and conformational change of a replication protein A heterotrimer bound to ssDNA. Genes Dev. 2012, 26, 2337–2347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cimprich, K.A.; Cortez, D. ATR: An essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol. 2008, 9, 616–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugiyama, T.; Kowalczykowski, S.C. Rad52 protein associates with replication protein A (RPA)-single-stranded DNA to accelerate Rad51-mediated displacement of RPA and presynaptic complex formation. J. Biol. Chem. 2002, 277, 31663–31672. [Google Scholar] [CrossRef] [Green Version]
- Berti, M.; Ray Chaudhuri, A.; Thangavel, S.; Gomathinayagam, S.; Kenig, S.; Vujanovic, M.; Odreman, F.; Glatter, T.; Graziano, S.; Mendoza-Maldonado, R.; et al. Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition. Nat. Struct. Mol. Biol. 2013, 20, 347–354. [Google Scholar] [CrossRef] [Green Version]
- Couch, F.B.; Cortez, D. Fork reversal, too much of a good thing. Cell Cycle 2014, 13, 1049–1050. [Google Scholar] [CrossRef] [Green Version]
- Lemaçon, D.; Jackson, J.; Quinet, A.; Brickner, J.R.; Li, S.; Yazinski, S.; You, Z.; Ira, G.; Zou, L.; Mosammaparast, N.; et al. MRE11 and EXO1 nucleases degrade reversed forks and elicit MUS81-dependent fork rescue in BRCA2-deficient cells. Nat. Commun. 2017, 8, 860. [Google Scholar] [CrossRef]
- Jensen, R.B.; Carreira, A.; Kowalczykowski, S.C. Purified human BRCA2 stimulates RAD51-mediated recombination. Nature 2010, 467, 678–683. [Google Scholar] [CrossRef] [Green Version]
- Schlacher, K.; Wu, H.; Jasin, M. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 2012, 22, 106–116. [Google Scholar] [CrossRef] [Green Version]
- Somyajit, K.; Saxena, S.; Babu, S.; Mishra, A.; Nagaraju, G. Mammalian RAD51 paralogs protect nascent DNA at stalled forks and mediate replication restart. Nucleic Acids Res. 2015, 43, 9835–9855. [Google Scholar] [CrossRef] [PubMed]
- Dungrawala, H.; Bhat, K.P.; Le Meur, R.; Chazin, W.J.; Ding, X.; Sharan, S.K.; Wessel, S.R.; Sathe, A.A.; Zhao, R.; Cortez, D. RADX Promotes Genome Stability and Modulates Chemosensitivity by Regulating RAD51 at Replication Forks. Mol. Cell 2017, 67, 374–386.e5. [Google Scholar] [CrossRef] [PubMed]
- Schlacher, K.; Christ, N.; Siaud, N.; Egashira, A.; Wu, H.; Jasin, M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 2011, 145, 529–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saha, L.K.; Murai, Y.; Saha, S.; Jo, U.; Tsuda, M.; Takeda, S.; Pommier, Y. Replication-dependent cytotoxicity and Spartan-mediated repair of trapped PARP1-DNA complexes. Nucleic Acids Res. 2021, 49, 10493–10506. [Google Scholar] [CrossRef] [PubMed]
- Gout, J.; Perkhofer, L.; Morawe, M.; Arnold, F.; Ihle, M.; Biber, S.; Lange, S.; Roger, E.; Kraus, J.M.; Stifter, K.; et al. Synergistic targeting and resistance to PARP inhibition in DNA damage repair-deficient pancreatic cancer. Gut 2021, 70, 743–760. [Google Scholar] [CrossRef]
- Riches, L.C.; Trinidad, A.G.; Hughes, G.; Jones, G.N.; Hughes, A.M.; Thomason, A.G.; Gavine, P.; Cui, A.; Ling, S.; Stott, J.; et al. Pharmacology of the ATM Inhibitor AZD0156: Potentiation of Irradiation and Olaparib Responses Preclinically. Mol. Cancer 2020, 19, 13–25. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Xu, H.; George, E.; Hallberg, D.; Kumar, S.; Jagannathan, V.; Medvedev, S.; Kinose, Y.; Devins, K.; Verma, P.; et al. Combining PARP with ATR inhibition overcomes PARP inhibitor and platinum resistance in ovarian cancer models. Nat. Commun. 2020, 11, 3726. [Google Scholar] [CrossRef]
- Shah, P.D.; Wethington, S.L.; Pagan, C.; Latif, N.; Tanyi, J.; Martin, L.P.; Morgan, M.; Burger, R.A.; Haggerty, A.; Zarrin, H.; et al. Combination ATR and PARP Inhibitor (CAPRI): A phase 2 study of ceralasertib plus olaparib in patients with recurrent, platinum-resistant epithelial ovarian cancer. Gynecol. Oncol. 2021, 163, 246–253. [Google Scholar] [CrossRef]
- Lee, J.M.; Nair, J.; Zimmer, A.; Lipkowitz, S.; Annunziata, C.M.; Merino, M.J.; Swisher, E.M.; Harrell, M.I.; Trepel, J.B.; Lee, M.J.; et al. Prexasertib, a cell cycle checkpoint kinase 1 and 2 inhibitor, in BRCA wild-type recurrent high-grade serous ovarian cancer: A first-in-class proof-of-concept phase 2 study. Lancet Oncol. 2018, 19, 207–215. [Google Scholar] [CrossRef]
- Westin, S.N.; Coleman, R.L.; Fellman, B.M.; Yuan, Y.; Sood, A.K.; Soliman, P.T.; Wright, A.A.; Horowitz, N.S.; Campos, S.M.; Konstantinopoulos, P.A.; et al. EFFORT: EFFicacy Of adavosertib in parp ResisTance: A randomized two-arm non-comparative phase II study of adavosertib with or without olaparib in women with PARP-resistant ovarian cancer. J. Clin. Oncol. 2021, 39, 5505. [Google Scholar] [CrossRef]
- Leijen, S.; van Geel, R.M.; Sonke, G.S.; de Jong, D.; Rosenberg, E.H.; Marchetti, S.; Pluim, D.; van Werkhoven, E.; Rose, S.; Lee, M.A.; et al. Phase II Study of WEE1 Inhibitor AZD1775 Plus Carboplatin in Patients With TP53-Mutated Ovarian Cancer Refractory or Resistant to First-Line Therapy Within 3 Months. J. Clin. Oncol. 2016, 34, 4354–4361. [Google Scholar] [CrossRef] [Green Version]
- Mahdi, H.; Hafez, N.; Doroshow, D.; Sohal, D.; Keedy, V.; Do, K.T.; LoRusso, P.; Jürgensmeier, J.; Avedissian, M.; Sklar, J.; et al. Ceralasertib-Mediated ATR Inhibition Combined With Olaparib in Advanced Cancers Harboring DNA Damage Response and Repair Alterations (Olaparib Combinations). JCO Precis. Oncol. 2021, 5, 1432–1442. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Stewart, J.; Porta, N.; Toms, C.; Leary, A.; Lheureux, S.; Khalique, S.; Tai, J.; Attygalle, A.; Vroobel, K.; et al. ATARI trial: ATR inhibitor in combination with olaparib in gynecological cancers with ARID1A loss or no loss (ENGOT/GYN1/NCRI). Int. J. Gynecol. Cancer 2021, 31, 1471–1475. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. The DNA damage response and cancer therapy. Nature 2012, 481, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Imai, K. Microsatellite instability: An update. Arch. Toxicol. 2015, 89, 899–921. [Google Scholar] [CrossRef]
- Chan, T.A.; Yarchoan, M.; Jaffee, E.; Swanton, C.; Quezada, S.A.; Stenzinger, A.; Peters, S. Development of tumor mutation burden as an immunotherapy biomarker: Utility for the oncology clinic. Ann. Oncol. 2019, 30, 44–56. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [Green Version]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [Green Version]
- McAlpine, J.N.; Porter, H.; Köbel, M.; Nelson, B.H.; Prentice, L.M.; Kalloger, S.E.; Senz, J.; Milne, K.; Ding, J.; Shah, S.P.; et al. BRCA1 and BRCA2 mutations correlate with TP53 abnormalities and presence of immune cell infiltrates in ovarian high-grade serous carcinoma. Mod. Pathol. 2012, 25, 740–750. [Google Scholar] [CrossRef] [Green Version]
- Nolan, E.; Savas, P.; Policheni, A.N.; Darcy, P.K.; Vaillant, F.; Mintoff, C.P.; Dushyanthen, S.; Mansour, M.; Pang, J.B.; Fox, S.B.; et al. Combined immune checkpoint blockade as a therapeutic strategy for BRCA1-mutated breast cancer. Sci. Transl. Med. 2017, 9, eaal4922. [Google Scholar] [CrossRef]
- Viel, A.; Bruselles, A.; Meccia, E.; Fornasarig, M.; Quaia, M.; Canzonieri, V.; Policicchio, E.; Urso, E.D.; Agostini, M.; Genuardi, M.; et al. A Specific Mutational Signature Associated with DNA 8-Oxoguanine Persistence in MUTYH-defective Colorectal Cancer. EBioMedicine 2017, 20, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Kraya, A.A.; Maxwell, K.N.; Wubbenhorst, B.; Wenz, B.M.; Pluta, J.; Rech, A.J.; Dorfman, L.M.; Lunceford, N.; Barrett, A.; Mitra, N.; et al. Genomic Signatures Predict the Immunogenicity of BRCA-Deficient Breast Cancer. Clin. Cancer Res. 2019, 25, 4363–4374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solinas, C.; Marcoux, D.; Garaud, S.; Vitória, J.R.; Van den Eynden, G.; de Wind, A.; De Silva, P.; Boisson, A.; Craciun, L.; Larsimont, D.; et al. BRCA gene mutations do not shape the extent and organization of tumor infiltrating lymphocytes in triple negative breast cancer. Cancer Lett. 2019, 450, 88–97. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Matulonis, U.A.; Shapira-Frommer, R.; Santin, A.D.; Lisyanskaya, A.S.; Pignata, S.; Vergote, I.; Raspagliesi, F.; Sonke, G.S.; Birrer, M.; Provencher, D.M.; et al. Antitumor activity and safety of pembrolizumab in patients with advanced recurrent ovarian cancer: Results from the phase II KEYNOTE-100 study. Ann. Oncol. 2019, 30, 1080–1087. [Google Scholar] [CrossRef] [PubMed]
- Hamanishi, J.; Takeshima, N.; Katsumata, N.; Ushijima, K.; Kimura, T.; Takeuchi, S.; Matsumoto, K.; Ito, K.; Mandai, M.; Nakai, H.; et al. Nivolumab Versus Gemcitabine or Pegylated Liposomal Doxorubicin for Patients With Platinum-Resistant Ovarian Cancer: Open-Label, Randomized Trial in Japan (NINJA). J. Clin. Oncol. 2021, 39, 3671–3681. [Google Scholar] [CrossRef] [PubMed]
- Pujade-Lauraine, E.; Fujiwara, K.; Ledermann, J.A.; Oza, A.M.; Kristeleit, R.; Ray-Coquard, I.L.; Richardson, G.E.; Sessa, C.; Yonemori, K.; Banerjee, S.; et al. Avelumab alone or in combination with chemotherapy versus chemotherapy alone in platinum-resistant or platinum-refractory ovarian cancer (JAVELIN Ovarian 200): An open-label, three-arm, randomised, phase 3 study. Lancet Oncol. 2021, 22, 1034–1046. [Google Scholar] [CrossRef]
- Spranger, S.; Luke, J.J.; Bao, R.; Zha, Y.; Hernandez, K.M.; Li, Y.; Gajewski, A.P.; Andrade, J.; Gajewski, T.F. Density of immunogenic antigens does not explain the presence or absence of the T-cell-inflamed tumor microenvironment in melanoma. Proc. Natl. Acad. Sci. USA 2016, 113, e7759–e7768. [Google Scholar] [CrossRef] [Green Version]
- Hsiehchen, D.; Hsieh, A.; Samstein, R.M.; Lu, T.; Beg, M.S.; Gerber, D.E.; Wang, T.; Morris, L.G.T.; Zhu, H. DNA Repair Gene Mutations as Predictors of Immune Checkpoint Inhibitor Response beyond Tumor Mutation Burden. Cell Rep. Med. 2020, 1, 100034. [Google Scholar] [CrossRef]
- Chen, Q.; Sun, L.; Chen, Z.J. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immunol. 2016, 17, 1142–1149. [Google Scholar] [CrossRef]
- Ishikawa, H.; Barber, G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Barber, G.N. STING: Infection, inflammation and cancer. Nat. Rev. Immunol. 2015, 15, 760–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parkes, E.E.; Walker, S.M.; Taggart, L.E.; McCabe, N.; Knight, L.A.; Wilkinson, R.; McCloskey, K.D.; Buckley, N.E.; Savage, K.I.; Salto-Tellez, M.; et al. Activation of STING-Dependent Innate Immune Signaling By S-Phase-Specific DNA Damage in Breast Cancer. J. Natl. Cancer Inst. 2017, 109, djw199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chabanon, R.M.; Rouanne, M.; Lord, C.J.; Soria, J.C.; Pasero, P.; Postel-Vinay, S. Targeting the DNA damage response in immuno-oncology: Developments and opportunities. Nat. Rev. Cancer 2021, 21, 701–717. [Google Scholar] [CrossRef]
- Ho, S.S.; Zhang, W.Y.; Tan, N.Y.; Khatoo, M.; Suter, M.A.; Tripathi, S.; Cheung, F.S.; Lim, W.K.; Tan, P.H.; Ngeow, J.; et al. The DNA Structure-Specific Endonuclease MUS81 Mediates DNA Sensor STING-Dependent Host Rejection of Prostate Cancer Cells. Immunity 2016, 44, 1177–1189. [Google Scholar] [CrossRef] [Green Version]
- Erdal, E.; Haider, S.; Rehwinkel, J.; Harris, A.L.; McHugh, P.J. A prosurvival DNA damage-induced cytoplasmic interferon response is mediated by end resection factors and is limited by Trex1. Genes Dev. 2017, 31, 353–369. [Google Scholar] [CrossRef] [Green Version]
- Harding, S.M.; Benci, J.L.; Irianto, J.; Discher, D.E.; Minn, A.J.; Greenberg, R.A. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 2017, 548, 466–470. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.A.; Shen, Y.L.; Hsia, H.Y.; Tiang, Y.P.; Sung, T.L.; Chen, L.Y. Extrachromosomal telomere repeat DNA is linked to ALT development via cGAS-STING DNA sensing pathway. Nat. Struct. Mol. Biol. 2017, 24, 1124–1131. [Google Scholar] [CrossRef]
- Kitai, Y.; Kawasaki, T.; Sueyoshi, T.; Kobiyama, K.; Ishii, K.J.; Zou, J.; Akira, S.; Matsuda, T.; Kawai, T. DNA-Containing Exosomes Derived from Cancer Cells Treated with Topotecan Activate a STING-Dependent Pathway and Reinforce Antitumor Immunity. J. Immunol. 2017, 198, 1649–1659. [Google Scholar] [CrossRef]
- Härtlova, A.; Erttmann, S.F.; Raffi, F.A.; Schmalz, A.M.; Resch, U.; Anugula, S.; Lienenklaus, S.; Nilsson, L.M.; Kröger, A.; Nilsson, J.A.; et al. DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote anti-microbial innate immunity. Immunity 2015, 42, 332–343. [Google Scholar] [CrossRef] [Green Version]
- Mackenzie, K.J.; Carroll, P.; Martin, C.A.; Murina, O.; Fluteau, A.; Simpson, D.J.; Olova, N.; Sutcliffe, H.; Rainger, J.K.; Leitch, A.; et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 2017, 548, 461–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yum, S.; Li, M.H.; Frankel, A.E.; Chen, Z.J.J. Roles of the cGAS-STING Pathway in Cancer Immunosurveillance and Immunotherapy. Annu. Rev. Cancer Biol. 2019, 3, 323–344. [Google Scholar] [CrossRef]
- Chabanon, R.M.; Muirhead, G.; Krastev, D.B.; Adam, J.; Morel, D.; Garrido, M.; Lamb, A.; Hénon, C.; Dorvault, N.; Rouanne, M.; et al. PARP inhibition enhances tumor cell-intrinsic immunity in ERCC1-deficient non-small cell lung cancer. J. Clin. Investig. 2019, 129, 1211–1228. [Google Scholar] [CrossRef] [PubMed]
- Pantelidou, C.; Sonzogni, O.; De Oliveria Taveira, M.; Mehta, A.K.; Kothari, A.; Wang, D.; Visal, T.; Li, M.K.; Pinto, J.; Castrillon, J.A.; et al. PARP Inhibitor Efficacy Depends on CD8(+) T-cell Recruitment via Intratumoral STING Pathway Activation in BRCA-Deficient Models of Triple-Negative Breast Cancer. Cancer Discov. 2019, 9, 722–737. [Google Scholar] [CrossRef] [Green Version]
- Ding, L.; Kim, H.J.; Wang, Q.; Kearns, M.; Jiang, T.; Ohlson, C.E.; Li, B.B.; Xie, S.; Liu, J.F.; Stover, E.H.; et al. PARP Inhibition Elicits STING-Dependent Antitumor Immunity in Brca1-Deficient Ovarian Cancer. Cell Rep. 2018, 25, 2972–2980.e5. [Google Scholar] [CrossRef] [Green Version]
- Reisländer, T.; Lombardi, E.P.; Groelly, F.J.; Miar, A.; Porru, M.; Di Vito, S.; Wright, B.; Lockstone, H.; Biroccio, A.; Harris, A.; et al. BRCA2 abrogation triggers innate immune responses potentiated by treatment with PARP inhibitors. Nat. Commun. 2019, 10, 3143. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Sun, K.; Xiao, Y.; Feng, B.; Mikule, K.; Ma, X.; Feng, N.; Vellano, C.P.; Federico, L.; Marszalek, J.R.; et al. Niraparib activates interferon signaling and potentiates anti-PD-1 antibody efficacy in tumor models. Sci. Rep. 2019, 9, 1853. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.; Wang, X.D.; Yu, Y. PARP1 inhibitors trigger innate immunity via PARP1 trapping-induced DNA damage response. eLife 2020, 9, e60637. [Google Scholar] [CrossRef]
- Chopra, N.; Tovey, H.; Pearson, A.; Cutts, R.; Toms, C.; Proszek, P.; Hubank, M.; Dowsett, M.; Dodson, A.; Daley, F.; et al. Homologous recombination DNA repair deficiency and PARP inhibition activity in primary triple negative breast cancer. Nat. Commun. 2020, 11, 2662. [Google Scholar] [CrossRef]
- Jiao, S.; Xia, W.; Yamaguchi, H.; Wei, Y.; Chen, M.K.; Hsu, J.M.; Hsu, J.L.; Yu, W.H.; Du, Y.; Lee, H.H.; et al. PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clin. Cancer Res. 2017, 23, 3711–3720. [Google Scholar] [CrossRef] [Green Version]
- Xue, C.; Xu, Y.; Ye, W.; Xie, Q.; Gao, H.; Xu, B.; Zhang, D.; Jiang, J. Expression of PD-L1 in ovarian cancer and its synergistic antitumor effect with PARP inhibitor. Gynecol. Oncol. 2020, 157, 222–233. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zheng, K.; Xiong, H.; Huang, Y.; Chen, X.; Zhou, Y.; Qin, W.; Su, J.; Chen, R.; Qiu, H.; et al. PARP Inhibitor Upregulates PD-L1 Expression and Provides a New Combination Therapy in Pancreatic Cancer. Front. Immunol. 2021, 12, 762989. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Annunziata, C.M.; Houston, N.; Kohn, E.C.; Lipkowitz, S.; Minasian, L.; Nichols, E.; Trepel, J.; Trewhitt, K.; Zia, F.; et al. A phase II study of durvalumab, a PD-L1 inhibitor and olaparib in recurrent ovarian cancer (OvCa). Ann. Oncol. 2018, 29, viii334. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Waggoner, S.; Vidal, G.A.; Mita, M.; Moroney, J.W.; Holloway, R.; Van Le, L.; Sachdev, J.C.; Chapman-Davis, E.; Colon-Otero, G.; et al. Single-Arm Phases 1 and 2 Trial of Niraparib in Combination With Pembrolizumab in Patients With Recurrent Platinum-Resistant Ovarian Carcinoma. JAMA Oncol. 2019, 5, 1141–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Gaillard, S.; Hendrickson, A.W.; Moroney, J.; Yeku, O.; Diver, E.; Gunderson, C.; Arend, R.; Ratner, E.; Samnotra, V.; et al. An open-label phase II study of dostarlimab (TSR-042), bevacizumab (bev), and niraparib combination in patients (pts) with platinum-resistant ovarian cancer (PROC): Cohort A of the OPAL trial. Gynecol. Oncol. 2021, 162, S17–S18. [Google Scholar] [CrossRef]
- Drew, Y.; Kaufman, B.; Banerjee, S.; Lortholary, A.; Hong, S.H.; Park, Y.H.; Zimmermann, S.; Roxburgh, P.; Ferguson, M.; Alvarez, R.H.; et al. Phase II study of olaparib + durvalumab (MEDIOLA): Updated results in germline BRCA-mutated platinum-sensitive relapsed (PSR) ovarian cancer (OC). Ann. Oncol. 2019, 30, v485–v486. [Google Scholar] [CrossRef]
- Drew, Y.; Penson, R.T.; O’Malley, D.M.; Kim, J.W.; Zimmermann, S.; Roxburgh, P.; Sohn, J.; Stemmer, S.M.; Bastian, S.; Ferguson, M.; et al. 814MO Phase II study of olaparib (O) plus durvalumab (D) and bevacizumab (B) (MEDIOLA): Initial results in patients (pts) with non-germline BRCA-mutated (non-gBRCAm) platinum sensitive relapsed (PSR) ovarian cancer (OC). Ann. Oncol. 2020, 31, S615–S616. [Google Scholar] [CrossRef]
- Bindra, R.S.; Gibson, S.L.; Meng, A.; Westermark, U.; Jasin, M.; Pierce, A.J.; Bristow, R.G.; Classon, M.K.; Glazer, P.M. Hypoxia-induced down-regulation of BRCA1 expression by E2Fs. Cancer Res. 2005, 65, 11597–11604. [Google Scholar] [CrossRef] [Green Version]
- Bindra, R.S.; Glazer, P.M. Repression of RAD51 gene expression by E2F4/p130 complexes in hypoxia. Oncogene 2007, 26, 2048–2057. [Google Scholar] [CrossRef]
- Forsythe, J.A.; Jiang, B.H.; Iyer, N.V.; Agani, F.; Leung, S.W.; Koos, R.D.; Semenza, G.L. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell. Biol. 1996, 16, 4604–4613. [Google Scholar] [CrossRef] [Green Version]
- Elaimy, A.L.; Amante, J.J.; Zhu, L.J.; Wang, M.; Walmsley, C.S.; FitzGerald, T.J.; Goel, H.L.; Mercurio, A.M. The VEGF receptor neuropilin 2 promotes homologous recombination by stimulating YAP/TAZ-mediated Rad51 expression. Proc. Natl. Acad. Sci. USA 2019, 116, 14174–14180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegan, D.C.; Lu, Y.; Stachelek, G.C.; Crosby, M.E.; Bindra, R.S.; Glazer, P.M. Inhibition of poly(ADP-ribose) polymerase down-regulates BRCA1 and RAD51 in a pathway mediated by E2F4 and p130. Proc. Natl. Acad. Sci. USA 2010, 107, 2201–2206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.F.; Barry, W.T.; Birrer, M.; Lee, J.M.; Buckanovich, R.J.; Fleming, G.F.; Rimel, B.; Buss, M.K.; Nattam, S.; Hurteau, J.; et al. Combination cediranib and olaparib versus olaparib alone for women with recurrent platinum-sensitive ovarian cancer: A randomised phase 2 study. Lancet Oncol. 2014, 15, 1207–1214. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.F.; Barry, W.T.; Birrer, M.; Lee, J.M.; Buckanovich, R.J.; Fleming, G.F.; Rimel, B.J.; Buss, M.K.; Nattam, S.R.; Hurteau, J.; et al. Overall survival and updated progression-free survival outcomes in a randomized phase II study of combination cediranib and olaparib versus olaparib in relapsed platinum-sensitive ovarian cancer. Ann. Oncol. 2019, 30, 551–557. [Google Scholar] [CrossRef]
- Mirza, M.R.; Åvall Lundqvist, E.; Birrer, M.J.; dePont Christensen, R.; Nyvang, G.B.; Malander, S.; Anttila, M.; Werner, T.L.; Lund, B.; Lindahl, G.; et al. Niraparib plus bevacizumab versus niraparib alone for platinum-sensitive recurrent ovarian cancer (NSGO-AVANOVA2/ENGOT-ov24): A randomised, phase 2, superiority trial. Lancet Oncol. 2019, 20, 1409–1419. [Google Scholar] [CrossRef]
- Liu, J.F.; Brady, M.F.; Matulonis, U.A.; Miller, A.; Kohn, E.C.; Swisher, E.M.; Cella, D.; Tew, W.P.; Cloven, N.G.; Muller, C.Y.; et al. Olaparib With or Without Cediranib Versus Platinum-Based Chemotherapy in Recurrent Platinum-Sensitive Ovarian Cancer (NRG-GY004): A Randomized, Open-Label, Phase III Trial. J. Clin. Oncol. 2022, 40, JCO2102011. [Google Scholar] [CrossRef]
- Sakai, W.; Swisher, E.M.; Karlan, B.Y.; Agarwal, M.K.; Higgins, J.; Friedman, C.; Villegas, E.; Jacquemont, C.; Farrugia, D.J.; Couch, F.J.; et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature 2008, 451, 1116–1120. [Google Scholar] [CrossRef] [Green Version]
- Swisher, E.M.; Sakai, W.; Karlan, B.Y.; Wurz, K.; Urban, N.; Taniguchi, T. Secondary BRCA1 mutations in BRCA1-mutated ovarian carcinomas with platinum resistance. Cancer Res. 2008, 68, 2581–2586. [Google Scholar] [CrossRef] [Green Version]
- Edwards, S.L.; Brough, R.; Lord, C.J.; Natrajan, R.; Vatcheva, R.; Levine, D.A.; Boyd, J.; Reis-Filho, J.S.; Ashworth, A. Resistance to therapy caused by intragenic deletion in BRCA2. Nature 2008, 451, 1111–1115. [Google Scholar] [CrossRef]
- Tobalina, L.; Armenia, J.; Irving, E.; O’Connor, M.J.; Forment, J.V. A meta-analysis of reversion mutations in BRCA genes identifies signatures of DNA end-joining repair mechanisms driving therapy resistance. Ann. Oncol. 2021, 32, 103–112. [Google Scholar] [CrossRef]
- Sinha, S.; Villarreal, D.; Shim, E.Y.; Lee, S.E. Risky business: Microhomology-mediated end joining. Mutat. Res. Mol. Mech. Mutagen. 2016, 788, 17–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettitt, S.J.; Frankum, J.R.; Punta, M.; Lise, S.; Alexander, J.; Chen, Y.; Yap, T.A.; Haider, S.; Tutt, A.N.J.; Lord, C.J. Clinical BRCA1/2 Reversion Analysis Identifies Hotspot Mutations and Predicted Neoantigens Associated with Therapy Resistance. Cancer Discov. 2020, 10, 1475–1488. [Google Scholar] [CrossRef] [PubMed]
- Swisher, E.M.; Kristeleit, R.S.; Oza, A.M.; Tinker, A.V.; Ray-Coquard, I.; Oaknin, A.; Coleman, R.L.; Burris, H.A.; Aghajanian, C.; O’Malley, D.M.; et al. Characterization of patients with long-term responses to rucaparib treatment in recurrent ovarian cancer. Gynecol. Oncol. 2021, 163, 490–497. [Google Scholar] [CrossRef] [PubMed]
- Cruz, C.; Castroviejo-Bermejo, M.; Gutiérrez-Enríquez, S.; Llop-Guevara, A.; Ibrahim, Y.H.; Gris-Oliver, A.; Bonache, S.; Morancho, B.; Bruna, A.; Rueda, O.M.; et al. RAD51 foci as a functional biomarker of homologous recombination repair and PARP inhibitor resistance in germline BRCA-mutated breast cancer. Ann. Oncol. 2018, 29, 1203–1210. [Google Scholar] [CrossRef]
- Wang, Y.; Bernhardy, A.J.; Cruz, C.; Krais, J.J.; Nacson, J.; Nicolas, E.; Peri, S.; van der Gulden, H.; van der Heijden, I.; O’Brien, S.W.; et al. The BRCA1-Δ11q Alternative Splice Isoform Bypasses Germline Mutations and Promotes Therapeutic Resistance to PARP Inhibition and Cisplatin. Cancer Res. 2016, 76, 2778–2790. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Bernhardy, A.J.; Nacson, J.; Krais, J.J.; Tan, Y.F.; Nicolas, E.; Radke, M.R.; Handorf, E.; Llop-Guevara, A.; Balmaña, J.; et al. BRCA1 intronic Alu elements drive gene rearrangements and PARP inhibitor resistance. Nat. Commun. 2019, 10, 5661. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Krais, J.J.; Bernhardy, A.J.; Nicolas, E.; Cai, K.Q.; Harrell, M.I.; Kim, H.H.; George, E.; Swisher, E.M.; Simpkins, F.; et al. RING domain-deficient BRCA1 promotes PARP inhibitor and platinum resistance. J. Clin. Investig. 2016, 126, 3145–3157. [Google Scholar] [CrossRef] [Green Version]
- Drost, R.; Dhillon, K.K.; van der Gulden, H.; van der Heijden, I.; Brandsma, I.; Cruz, C.; Chondronasiou, D.; Castroviejo-Bermejo, M.; Boon, U.; Schut, E.; et al. BRCA1185delAG tumors may acquire therapy resistance through expression of RING-less BRCA1. J. Clin. Investig. 2016, 126, 2903–2918. [Google Scholar] [CrossRef]
- Knijnenburg, T.A.; Wang, L.; Zimmermann, M.T.; Chambwe, N.; Gao, G.F.; Cherniack, A.D.; Fan, H.; Shen, H.; Way, G.P.; Greene, C.S.; et al. Genomic and Molecular Landscape of DNA Damage Repair Deficiency across The Cancer Genome Atlas. Cell Rep. 2018, 23, 239–254.e6. [Google Scholar] [CrossRef] [Green Version]
- Patch, A.M.; Christie, E.L.; Etemadmoghadam, D.; Garsed, D.W.; George, J.; Fereday, S.; Nones, K.; Cowin, P.; Alsop, K.; Bailey, P.J.; et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature 2015, 521, 489–494. [Google Scholar] [CrossRef]
- Hurley, R.M.; McGehee, C.D.; Nesic, K.; Correia, C.; Weiskittel, T.M.; Kelly, R.L.; Venkatachalam, A.; Hou, X.; Pathoulas, N.M.; Meng, X.W.; et al. Characterization of a RAD51C-silenced high-grade serous ovarian cancer model during development of PARP inhibitor resistance. NAR Cancer 2021, 3, zcab028. [Google Scholar] [CrossRef] [PubMed]
- Swisher, E.M.; Lin, K.K.; Oza, A.M.; Scott, C.L.; Giordano, H.; Sun, J.; Konecny, G.E.; Coleman, R.L.; Tinker, A.V.; O’Malley, D.M.; et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): An international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 75–87. [Google Scholar] [CrossRef] [Green Version]
- Kondrashova, O.; Topp, M.; Nesic, K.; Lieschke, E.; Ho, G.Y.; Harrell, M.I.; Zapparoli, G.V.; Hadley, A.; Holian, R.; Boehm, E.; et al. Methylation of all BRCA1 copies predicts response to the PARP inhibitor rucaparib in ovarian carcinoma. Nat. Commun. 2018, 9, 3970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nesic, K.; Kondrashova, O.; Hurley, R.M.; McGehee, C.D.; Vandenberg, C.J.; Ho, G.Y.; Lieschke, E.; Dall, G.; Bound, N.; Shield-Artin, K.; et al. Acquired RAD51C Promoter Methylation Loss Causes PARP Inhibitor Resistance in High-Grade Serous Ovarian Carcinoma. Cancer Res. 2021, 81, 4709–4722. [Google Scholar] [CrossRef] [PubMed]
- Mirman, Z.; de Lange, T. 53BP1: A DSB escort. Genes Dev. 2020, 34, 7–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zong, D.; Adam, S.; Wang, Y.; Sasanuma, H.; Callén, E.; Murga, M.; Day, A.; Kruhlak, M.J.; Wong, N.; Munro, M.; et al. BRCA1 Haploinsufficiency Is Masked by RNF168-Mediated Chromatin Ubiquitylation. Mol. Cell 2019, 73, 1267–1281.e7. [Google Scholar] [CrossRef] [Green Version]
- Callen, E.; Zong, D.; Wu, W.; Wong, N.; Stanlie, A.; Ishikawa, M.; Pavani, R.; Dumitrache, L.C.; Byrum, A.K.; Mendez-Dorantes, C.; et al. 53BP1 Enforces Distinct Pre- and Post-resection Blocks on Homologous Recombination. Mol. Cell 2020, 77, 26–38.e7. [Google Scholar] [CrossRef]
- Belotserkovskaya, R.; Raga Gil, E.; Lawrence, N.; Butler, R.; Clifford, G.; Wilson, M.D.; Jackson, S.P. PALB2 chromatin recruitment restores homologous recombination in BRCA1-deficient cells depleted of 53BP1. Nat. Commun. 2020, 11, 819. [Google Scholar] [CrossRef] [Green Version]
- Waks, A.G.; Cohen, O.; Kochupurakkal, B.; Kim, D.; Dunn, C.E.; Buendia Buendia, J.; Wander, S.; Helvie, K.; Lloyd, M.R.; Marini, L.; et al. Reversion and non-reversion mechanisms of resistance to PARP inhibitor or platinum chemotherapy in BRCA1/2-mutant metastatic breast cancer. Ann. Oncol. 2020, 31, 590–598. [Google Scholar] [CrossRef]
- Pettitt, S.J.; Rehman, F.L.; Bajrami, I.; Brough, R.; Wallberg, F.; Kozarewa, I.; Fenwick, K.; Assiotis, I.; Chen, L.; Campbell, J.; et al. A genetic screen using the PiggyBac transposon in haploid cells identifies Parp1 as a mediator of olaparib toxicity. PLoS ONE 2013, 8, e61520. [Google Scholar] [CrossRef] [Green Version]
- Pettitt, S.J.; Krastev, D.B.; Brandsma, I.; Dréan, A.; Song, F.; Aleksandrov, R.; Harrell, M.I.; Menon, M.; Brough, R.; Campbell, J.; et al. Genome-wide and high-density CRISPR-Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance. Nat. Commun. 2018, 9, 1849. [Google Scholar] [CrossRef] [PubMed]
- Pascal, J.M.; Ellenberger, T. The rise and fall of poly(ADP-ribose): An enzymatic perspective. DNA Repair 2015, 32, 10–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gogola, E.; Duarte, A.A.; de Ruiter, J.R.; Wiegant, W.W.; Schmid, J.A.; de Bruijn, R.; James, D.I.; Guerrero Llobet, S.; Vis, D.J.; Annunziato, S.; et al. Selective Loss of PARG Restores PARylation and Counteracts PARP Inhibitor-Mediated Synthetic Lethality. Cancer Cell 2018, 33, 1078–1093.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murai, J.; Tang, S.W.; Leo, E.; Baechler, S.A.; Redon, C.E.; Zhang, H.; Al Abo, M.; Rajapakse, V.N.; Nakamura, E.; Jenkins, L.M.M.; et al. SLFN11 Blocks Stressed Replication Forks Independently of ATR. Mol. Cell 2018, 69, 371–384.e6. [Google Scholar] [CrossRef] [Green Version]
- Zoppoli, G.; Regairaz, M.; Leo, E.; Reinhold, W.C.; Varma, S.; Ballestrero, A.; Doroshow, J.H.; Pommier, Y. Putative DNA/RNA helicase Schlafen-11 (SLFN11) sensitizes cancer cells to DNA-damaging agents. Proc. Natl. Acad. Sci. USA 2012, 109, 15030–15035. [Google Scholar] [CrossRef] [Green Version]
- Murai, J.; Feng, Y.; Yu, G.K.; Ru, Y.; Tang, S.W.; Shen, Y.; Pommier, Y. Resistance to PARP inhibitors by SLFN11 inactivation can be overcome by ATR inhibition. Oncotarget 2016, 7, 76534–76550. [Google Scholar] [CrossRef] [Green Version]
- Murai, J.; Thomas, A.; Miettinen, M.; Pommier, Y. Schlafen 11 (SLFN11), a restriction factor for replicative stress induced by DNA-targeting anti-cancer therapies. Pharmacol. Ther. 2019, 201, 94–102. [Google Scholar] [CrossRef]
- Amuzu, S.; Carmona, E.; Mes-Masson, A.M.; Greenwood, C.M.T.; Tonin, P.N.; Ragoussis, J. Candidate Markers of Olaparib Response from Genomic Data Analyses of Human Cancer Cell Lines. Cancers 2021, 13, 1296. [Google Scholar] [CrossRef]
- Willis, S.E.; Winkler, C.; Roudier, M.P.; Baird, T.; Marco-Casanova, P.; Jones, E.V.; Rowe, P.; Rodriguez-Canales, J.; Angell, H.K.; Ng, F.S.L.; et al. Retrospective analysis of Schlafen11 (SLFN11) to predict the outcomes to therapies affecting the DNA damage response. Br. J. Cancer 2021, 125, 1666–1676. [Google Scholar] [CrossRef]
- Raymond, M.; Rose, E.; Housman, D.E.; Gros, P. Physical mapping, amplification, and overexpression of the mouse mdr gene family in multidrug-resistant cells. Mol. Cell. Biol. 1990, 10, 1642–1651. [Google Scholar] [CrossRef]
- Wu, C.P.; Calcagno, A.M.; Ambudkar, S.V. Reversal of ABC drug transporter-mediated multidrug resistance in cancer cells: Evaluation of current strategies. Curr. Mol. Pharm. 2008, 1, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Rottenberg, S.; Jaspers, J.E.; Kersbergen, A.; van der Burg, E.; Nygren, A.O.; Zander, S.A.; Derksen, P.W.; de Bruin, M.; Zevenhoven, J.; Lau, A.; et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc. Natl. Acad. Sci. USA 2008, 105, 17079–17084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lheureux, S.; Oaknin, A.; Garg, S.; Bruce, J.P.; Madariaga, A.; Dhani, N.C.; Bowering, V.; White, J.; Accardi, S.; Tan, Q.; et al. EVOLVE: A Multicenter Open-Label Single-Arm Clinical and Translational Phase II Trial of Cediranib Plus Olaparib for Ovarian Cancer after PARP Inhibition Progression. Clin. Cancer Res. 2020, 26, 4206–4215. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kyo, S.; Kanno, K.; Takakura, M.; Yamashita, H.; Ishikawa, M.; Ishibashi, T.; Sato, S.; Nakayama, K. Clinical Landscape of PARP Inhibitors in Ovarian Cancer: Molecular Mechanisms and Clues to Overcome Resistance. Cancers 2022, 14, 2504. https://doi.org/10.3390/cancers14102504
Kyo S, Kanno K, Takakura M, Yamashita H, Ishikawa M, Ishibashi T, Sato S, Nakayama K. Clinical Landscape of PARP Inhibitors in Ovarian Cancer: Molecular Mechanisms and Clues to Overcome Resistance. Cancers. 2022; 14(10):2504. https://doi.org/10.3390/cancers14102504
Chicago/Turabian StyleKyo, Satoru, Kosuke Kanno, Masahiro Takakura, Hitomi Yamashita, Masako Ishikawa, Tomoka Ishibashi, Seiya Sato, and Kentaro Nakayama. 2022. "Clinical Landscape of PARP Inhibitors in Ovarian Cancer: Molecular Mechanisms and Clues to Overcome Resistance" Cancers 14, no. 10: 2504. https://doi.org/10.3390/cancers14102504
APA StyleKyo, S., Kanno, K., Takakura, M., Yamashita, H., Ishikawa, M., Ishibashi, T., Sato, S., & Nakayama, K. (2022). Clinical Landscape of PARP Inhibitors in Ovarian Cancer: Molecular Mechanisms and Clues to Overcome Resistance. Cancers, 14(10), 2504. https://doi.org/10.3390/cancers14102504