
Early Assessment of Chemotherapy Response in Advanced Non-Small Cell Lung Cancer with Circulating Tumor DNA

 , , ,
, , ,  ,
,  , , , add
Show full author list
, , , add
Show full author list
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
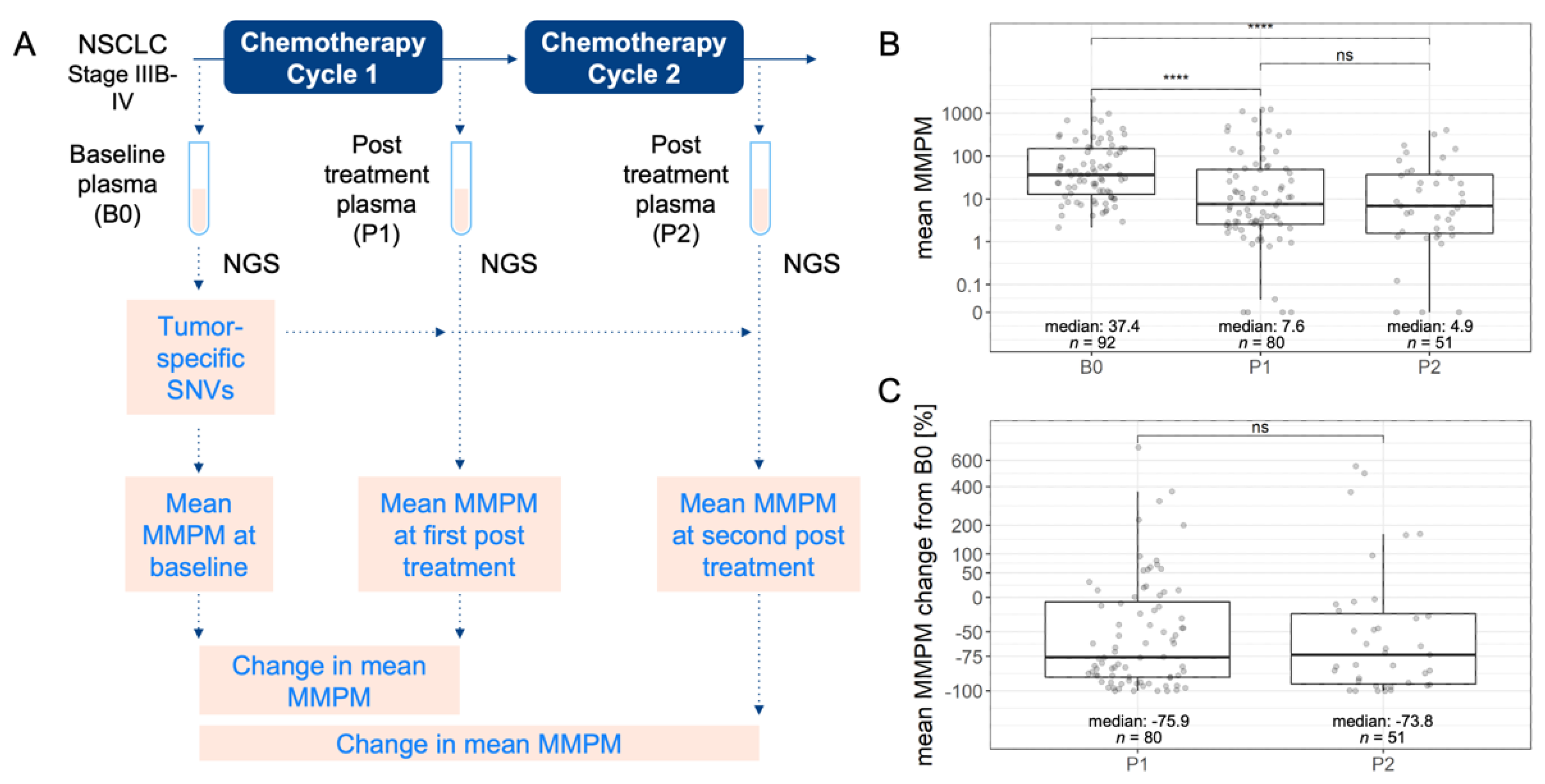
2.1. Study Design and Population
2.2. ctDNA Sequencing and Bioinformatics Analysis
2.3. ctDNA Thresholds for Molecular Response
2.4. Statistical Analysis
3. Results
3.1. Cohort Baseline Characteristics
3.2. Quantification of Circulating Tumor DNA in Plasma
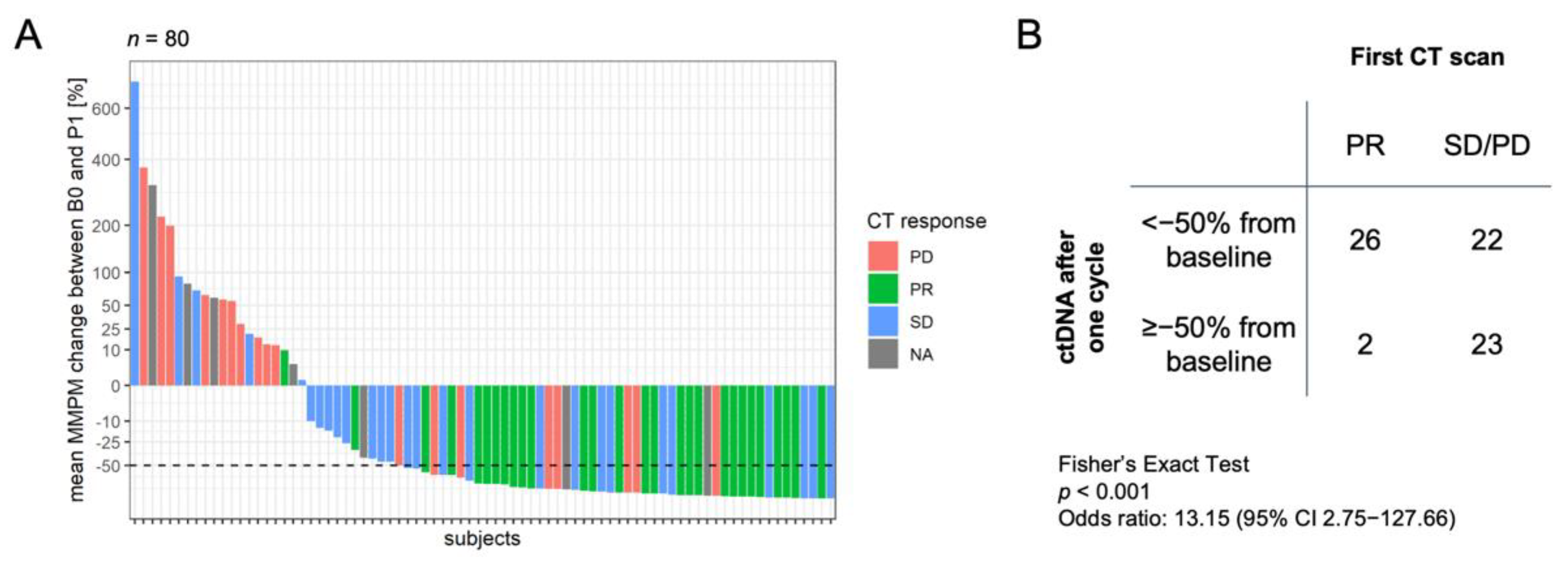
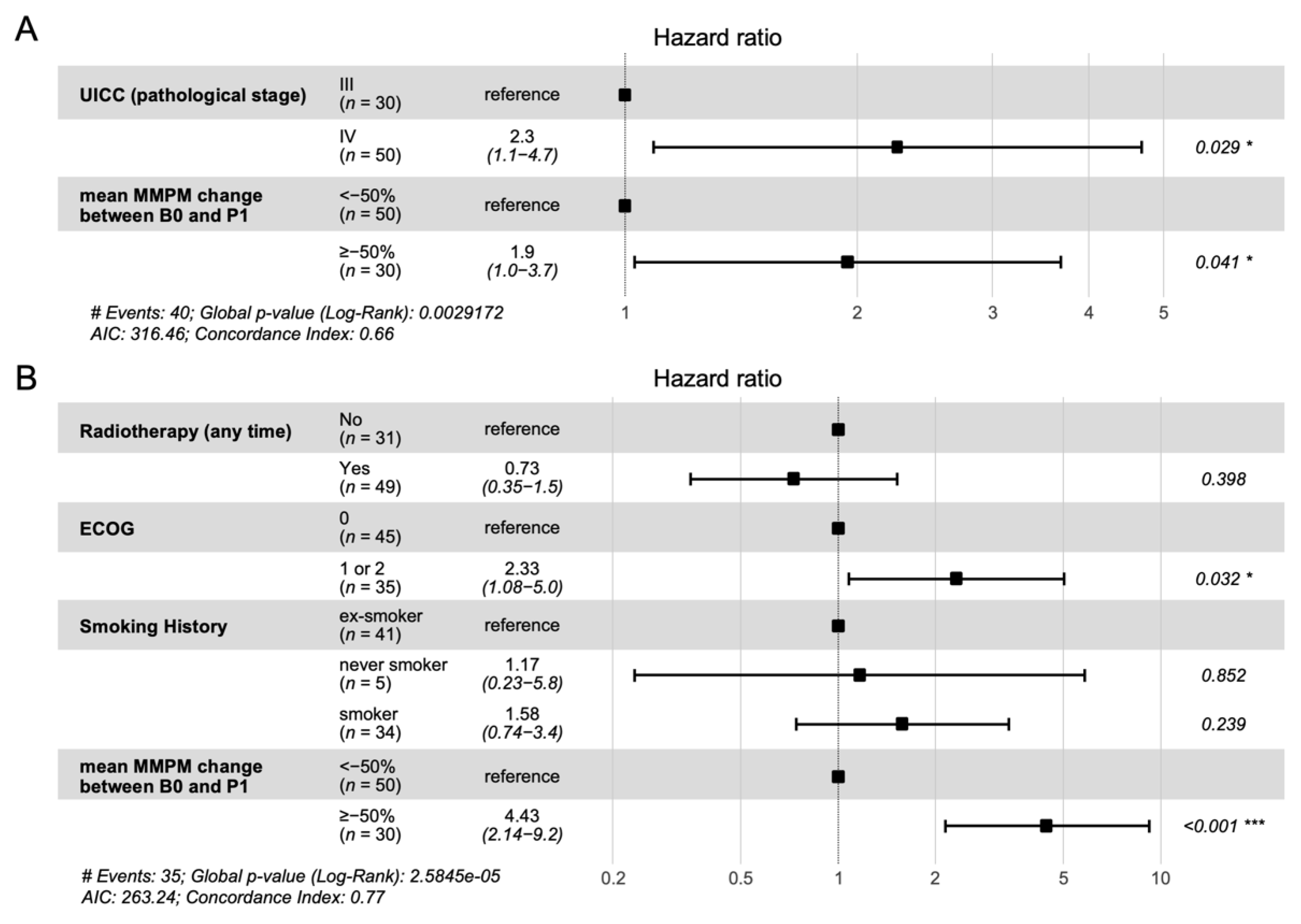
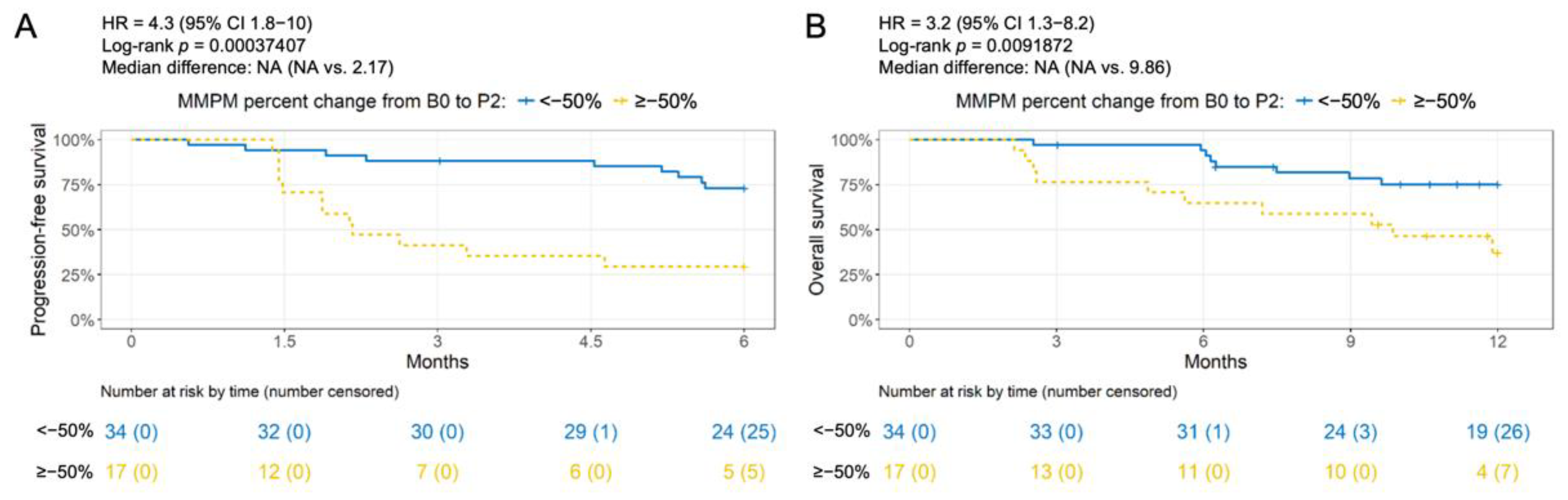
3.3. Association of Change in ctDNA Level with Clinical Outcome
3.4. Association of Single Time Point ctDNA Level with Clinical Outcome
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLO-BOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA. Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Allemani, C.; Matsuda, T.; Di Carlo, V.; Harewood, R.; Matz, M.; Nikšić, M.; Bonaventure, A.; Valkov, M.; Johnson, C.J.; Estève, J.; et al. Global Surveillance of Trends in Cancer Survival 2000–14 (CONCORD-3): Analysis of Individual Records for 37 513 025 Patients Diagnosed with One of 18 Cancers from 322 Population-Based Registries in 71 Countries. Lancet 2018, 391, 1023–1075. [Google Scholar] [CrossRef] [Green Version]
- Ganti, A.K.; Klein, A.B.; Cotarla, I.; Seal, B.; Chou, E. Update of Incidence, Prevalence, Survival, and Initial Treatment in Patients with Non-Small Cell Lung Cancer in the US. JAMA Oncol. 2021, 7, 1824–1832. [Google Scholar] [CrossRef]
- National Comprehensive Cancer Network Non-Small Cell Lung Cancer (Version 3.2022). Available online: https://www.nccn.org/professionals/physician_gls/pdf/nscl.pdf (accessed on 31 March 2022).
- Bruzzi, J.F.; Truong, M.; Zinner, R.; Erasmus, J.J.; Sabloff, B.; Munden, R. Short-Term Restaging of Patients with Non-Small Cell Lung Cancer Receiving Chemotherapy. J. Thorac. Oncol. 2006, 1, 425–429. [Google Scholar] [CrossRef] [PubMed]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New Response Evaluation Criteria in Solid Tumours: Revised RECIST Guideline (Version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef] [PubMed]
- Ko, C.C.; Yeh, L.R.; Kuo, Y.T.; Chen, J.H. Imaging Biomarkers for Evaluating Tumor Response: RECIST and Beyond. Biomark. Res. 2021, 9, 52. [Google Scholar] [CrossRef]
- Siravegna, G.; Marsoni, S.; Siena, S.; Bardelli, A. Integrating Liquid Biopsies into the Management of Cancer. Nat. Rev. Clin. Oncol. 2017, 14, 531–548. [Google Scholar] [CrossRef]
- Rolfo, C.; Mack, P.C.; Scagliotti, G.V.; Baas, P.; Barlesi, F.; Bivona, T.G.; Herbst, R.S.; Mok, T.S.; Peled, N.; Pirker, R.; et al. Liquid Biopsy for Advanced Non-Small Cell Lung Cancer (NSCLC): A Statement Paper from the IASLC. J. Thorac. Oncol. 2018, 13, 1248–1268. [Google Scholar] [CrossRef] [Green Version]
- Boonstra, P.A.; Wind, T.T.; van Kruchten, M.; Schuuring, E.; Hospers, G.A.P.; van der Wekken, A.J.; de Groot, D.J.; Schröder, C.P.; Fehrmann, R.S.N.; Reyners, A.K.L. Clinical Utility of Circulating Tumor DNA as a Response and Follow-up Marker in Cancer Therapy. Cancer Metastasis Rev. 2020, 39, 999–1013. [Google Scholar] [CrossRef]
- Tie, J.; Kinde, I.; Wang, Y.; Wong, H.L.; Roebert, J.; Christie, M.; Tacey, M.; Wong, R.; Singh, M.; Karapetis, C.S.; et al. Circu-lating Tumor DNA as an Early Marker of Therapeutic Response in Patients with Metastatic Colorectal Cancer. Ann. Oncol. 2015, 26, 1715–1722. [Google Scholar] [CrossRef]
- Garlan, F.; Laurent-Puig, P.; Sefrioui, D.; Siauve, N.; Didelot, A.; Sarafan-Vasseur, N.; Michel, P.; Perkins, G.; Mulot, C.; Blons, H.; et al. Early Evaluation of Circulating Tumor DNA as Marker of Therapeutic Efficacy in Metastatic Colorectal Cancer Pa-tients (PLACOL Study). Clin. Cancer Res. 2017, 23, 5416–5425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.M.; Bendell, J.C.; Hurwitz, H.I.; Ju, C.; Lee, J.J.; Lovejoy, A.; Mancao, C.; Nicholas, A.; Price, R.; Sommer, N.; et al. Disease Monitoring Using Post-Induction Circulating Tumor DNA Analysis Following First-Line Therapy in Patients with Metastatic Colorectal Cancer. Clin. Cancer Res. 2020, 26, 4010–4017. [Google Scholar] [CrossRef] [Green Version]
- Dawson, S.-J.; Tsui, D.W.Y.; Murtaza, M.; Biggs, H.; Rueda, O.M.; Chin, S.-F.; Dunning, M.J.; Gale, D.; Forshew, T.; Mah-ler-Araujo, B.; et al. Analysis of Circulating Tumor DNA to Monitor Metastatic Breast Cancer. N. Engl. J. Med. 2013, 368, 1199–1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hrebien, S.; Citi, V.; Garcia-Murillas, I.; Cutts, R.; Fenwick, K.; Kozarewa, I.; McEwen, R.; Ratnayake, J.; Maudsley, R.; Carr, T.H.; et al. Early CtDNA Dynamics as a Surrogate for Progression-Free Survival in Advanced Breast Cancer in the BEECH Trial. Ann. Oncol. 2019, 30, 945–952. [Google Scholar] [CrossRef]
- Goldberg, S.B.; Narayan, A.; Kole, A.J.; Decker, R.H.; Teysir, J.; Carriero, N.J.; Lee, A.; Nemati, R.; Nath, S.K.; Mane, S.M.; et al. Early Assessment of Lung Cancer Immunotherapy Response via Circulating Tumor DNA. Clin. Cancer Res. 2018, 24, 1872–1880. [Google Scholar] [CrossRef] [Green Version]
- Guibert, N.; Jones, G.; Beeler, J.F.; Plagnol, V.; Morris, C.; Mourlanette, J.; Delaunay, M.; Keller, L.; Rouquette, I.; Favre, G.; et al. Targeted Sequencing of Plasma Cell-Free DNA to Predict Response to PD1 Inhibitors in Advanced Non-Small Cell Lung Cancer. Lung Cancer 2019, 137, 1–6. [Google Scholar] [CrossRef]
- Nabet, B.Y.; Esfahani, M.S.; Moding, E.J.; Hamilton, E.G.; Chabon, J.J.; Rizvi, H.; Steen, C.B.; Chaudhuri, A.A.; Liu, C.L.; Hui, A.B.; et al. Noninvasive Early Identification of Therapeutic Benefit from Immune Checkpoint Inhibition. Cell 2020, 183, 363–376. [Google Scholar] [CrossRef]
- Van der Leest, P.; Hiddinga, B.; Miedema, A.; Azpurua, M.L.A.; Rifaela, N.; ter Elst, A.; Timens, W.; Groen, H.J.M.; van Kempen, L.C.; Hiltermann, T.J.N.; et al. Circulating Tumor DNA as a Biomarker for Monitoring Early Treatment Responses of Patients with Advanced Lung Adenocarcinoma Receiving Immune Checkpoint Inhibitors. Mol. Oncol. 2021, 15, 2910–2922. [Google Scholar] [CrossRef]
- Zou, W.; Yaung, S.J.; Fuhlbrück, F.; Ballinger, M.; Peters, E.; Palma, J.F.; Shames, D.S.; Gandara, D.; Jiang, Y.; Patil, N.S. CtDNA Predicts Overall Survival in Patients With NSCLC Treated With PD-L1 Blockade or With Chemotherapy. JCO Precis. Oncol. 2021, 5, 827–838. [Google Scholar] [CrossRef]
- Kato, K.; Uchida, J.; Kukita, Y.; Kumagai, T.; Nishino, K.; Inoue, T.; Kimura, M.; Oba, S.; Imamura, F. Numerical Indices Based on Circulating Tumor DNA for the Evaluation of Therapeutic Response and Disease Progression in Lung Cancer Patients. Sci. Rep. 2016, 6, 29093. [Google Scholar] [CrossRef] [Green Version]
- Phallen, J.; Leal, A.; Woodward, B.D.; Forde, P.M.; Naidoo, J.; Marrone, K.A.; Brahmer, J.R.; Fiksel, J.; Medina, J.E.; Cristiano, S.; et al. Early Noninvasive Detection of Response to Targeted Therapy in Non–Small Cell Lung Cancer. Cancer Res. 2019, 79, 1204–1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, M.L.; Lau, C.J.; Milan, M.S.D.; Supplee, J.G.; Riess, J.W.; Bradbury, P.A.; Jänne, P.A.; Oxnard, G.R.; Paweletz, C.P. Plasma CtDNA Response Is an Early Marker of Treatment Effect in Advanced NSCLC. JCO Precis. Oncol. 2021, 5, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Angeles, A.K.; Christopoulos, P.; Yuan, Z.; Bauer, S.; Janke, F.; Ogrodnik, S.J.; Reck, M.; Schlesner, M.; Meister, M.; Schneider, M.A.; et al. Early Identification of Disease Progression in ALK-Rearranged Lung Cancer Using Circulating Tumor DNA Analysis. NPJ Precis. Oncol. 2021, 5, 100. [Google Scholar] [CrossRef] [PubMed]
- Nygaard, A.D.; Spindler, K.L.G.; Pallisgaard, N.; Andersen, R.F.; Jakobsen, A. Levels of Cell-Free DNA and Plasma KRAS during Treatment of Advanced NSCLC. Oncol. Rep. 2014, 31, 969–974. [Google Scholar] [CrossRef] [Green Version]
- Zulato, E.; Attili, I.; Pavan, A.; Nardo, G.; Del Bianco, P.; Boscolo Bragadin, A.; Verza, M.; Pasqualini, L.; Pasello, G.; Fassan, M.; et al. Early Assessment of KRAS Mutation in CfDNA Correlates with Risk of Progression and Death in Advanced Non-Small-Cell Lung Cancer. Br. J. Cancer 2020, 123, 81–91. [Google Scholar] [CrossRef]
- Jiang, T.; Li, X.; Wang, J.; Su, C.; Han, W.; Zhao, C.; Wu, F.; Gao, G.; Li, W.; Chen, X.; et al. Mutational, Andscape of CfDNA Identifies Distinct Molecular Features Associated with Therapeutic Response to First-Line Platinum-Based Doublet Chemo-therapy in Patients with Advanced NSCLC. Theranostics 2017, 7, 4753–4762. [Google Scholar] [CrossRef]
- Pécuchet, N.; Zonta, E.; Didelot, A.; Combe, P.; Thibault, C.; Gibault, L.; Lours, C.; Rozenholc, Y.; Taly, V.; Laurent-Puig, P.; et al. Base-Position Error Rate Analysis of Next-Generation Sequencing Applied to Circulating Tumor DNA in Non-Small Cell Lung Cancer: A Prospective Study. PLoS Med. 2016, 13, 1–19. [Google Scholar] [CrossRef]
- Jiang, J.; Adams, H.-P.; Lange, M.; Siemann, S.; Feldkamp, M.; McNamara, S.; Froehler, S.; Yaung, S.J.; Yao, L.; Bal-asubramanyam, A.; et al. Plasma-Based Longitudinal Mutation Monitoring as a Potential Predictor of Disease Progression in Subjects with Adenocarcinoma in Advanced Non-Small Cell Lung Cancer. BMC Cancer 2020, 20, 885. [Google Scholar] [CrossRef]
- Sholl, L.M.; Aisner, D.L.; Allen, T.C.; Beasley, M.B.; Cagle, P.T.; Capelozzi, V.L.; Dacic, S.; Hariri, L.P.; Kerr, K.M.; Lantuejoul, S.; et al. Liquid Biopsy in Lung Cancer: A Perspective from Members of the Pulmonary Pathology Society. Arch. Pathol. Lab. Med. 2016, 140, 825–829. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez, M.E.; Choi, K.; Lanman, R.B.; Licitra, E.J.; Skrzypczak, S.M.; Pe Benito, R.; Wu, T.; Arunajadai, S.; Kaur, S.; Harper, H.; et al. Genomic Profiling of Advanced Non–Small Cell Lung Cancer in Community Settings: Gaps and Opportunities. Clin. Lung Cancer 2017, 18, 651–659. [Google Scholar] [CrossRef] [Green Version]
- Saelee, S.L.; Lovejoy, A.F.; Hinzmann, B.; Mayol, K.; Huynh, S.; Harrell, A.; Lefkowitz, J.; Deodhar, N.; Garcia-Montoya, G.; Yaung, S.J.; et al. Quantitative PCR Based Method to Assess CfDNA Quality, Adjust Input Mass, and Improve next-Generation Sequencing Assay Performance. J. Mol. Diagnostics 2022. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Bratman, S.V.; To, J.; Wynne, J.F.; Eclov, N.C.W.; Modlin, L.A.; Liu, C.L.; Neal, J.W.; Wakelee, H.A.; Merritt, R.E.; et al. An Ultrasensitive Method for Quantitating Circulating Tumor DNA with Broad Patient Coverage. Nat. Med. 2014, 20, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Deveson, I.W.; Gong, B.; Lai, K.; LoCoco, J.S.; Richmond, T.A.; Schageman, J.; Zhang, Z.; Novoradovskaya, N.; Willey, J.C.; Jones, W.; et al. Evaluating the Analytical Validity of Circulating Tumor DNA Sequencing Assays for Precision Oncology. Nat. Biotechnol. 2021, 39, 1115–1128. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Lovejoy, A.F.; Klass, D.M.; Kurtz, D.M.; Chabon, J.J.; Scherer, F.; Stehr, H.; Liu, C.L.; Bratman, S.V.; Say, C.; et al. Integrated Digital Error Suppression for Improved Detection of Circulating Tumor DNA. Nat. Biotechnol. 2016, 34, 547–555. [Google Scholar] [CrossRef]
- Yao, L.; Lal, P.; Fang, L.-T.; Lee, J.; Palma, J.; Rosenthal, A.; Hinzmann, B.; Lovejoy, A.; Lam, H.Y.K. Abstract 5293: A Method to Identify Somatic Mutations from Tumor Samples in the Absence of Matched Normal Tissue. Cancer Res. 2018, 78, 5293. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Weisburd, B.; Thomas, B.; Solomonson, M.; Ruderfer, D.M.; Kavanagh, D.; Hamamsy, T.; Lek, M.; Samocha, K.E.; Cummings, B.B.; et al. The ExAC Browser: Displaying Reference Data Information from over 60,000 Exomes. Nucleic Acids Res. 2017, 45, D840–D845. [Google Scholar] [CrossRef] [Green Version]
- Auton, A.; Abecasis, G.R.; Altshuler, D.M.; Durbin, R.M.; Bentley, D.R.; Chakravarti, A.; Clark, A.G.; Donnelly, P.; Eichler, E.E.; Flicek, P.; et al. A Global Reference for Human Genetic Variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [Green Version]
- Smigielski, E.M.; Sirotkin, K.; Ward, M.; Sherry, S.T. DbSNP: A Database of Single Nucleotide Polymorphisms. Nucleic Acids Res. 2000, 28, 352–355. [Google Scholar] [CrossRef] [Green Version]
- Izzo, B.; Gottardi, E.M.; Errichiello, S.; Daraio, F.; Baratè, C.; Galimberti, S. Monitoring Chronic Myeloid Leukemia: How Mo-lecular Tools May Drive Therapeutic Approaches. Front. Oncol. 2019, 9, 833. [Google Scholar] [CrossRef] [Green Version]
- Hochhaus, A.; Baccarani, M.; Silver, R.T.; Schiffer, C.; Apperley, J.F.; Cervantes, F.; Clark, R.E.; Cortes, J.E.; Deininger, M.W.; Guilhot, F.; et al. European LeukemiaNet 2020 Recommendations for Treating Chronic Myeloid Leukemia. Leukemia 2020, 34, 966–984. [Google Scholar] [CrossRef] [Green Version]
- Hothorn, T.; Lausen, B. On the Exact Distribution of Maximally Selected Rank Statistics. Comput. Stat. Data Anal. 2003, 43, 121–137. [Google Scholar] [CrossRef]
- Chabon, J.J.; Simmons, A.D.; Lovejoy, A.F.; Esfahani, M.S.; Newman, A.M.; Haringsma, H.J.; Kurtz, D.M.; Stehr, H.; Scherer, F.; Karlovich, C.A.; et al. Circulating Tumour DNA Profiling Reveals Heterogeneity of EGFR Inhibitor Resistance Mechanisms in Lung Cancer Patients. Nat. Commun. 2016, 7, 11815. [Google Scholar] [CrossRef] [PubMed]
- Diehl, F.; Schmidt, K.; Choti, M.A.; Romans, K.; Goodman, S.; Li, M.; Thornton, K.; Agrawal, N.; Sokoll, L.; Szabo, S.A.; et al. Circulating Mutant DNA to Assess Tumor Dynamics. Nat. Med. 2008, 14, 985–990. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, A.A.; Chabon, J.J.; Lovejoy, A.F.; Newman, A.M.; Stehr, H.; Azad, T.D.; Khodadoust, M.S.; Esfahani, M.S.; Liu, C.L.; Zhou, L.; et al. Early Detection of Molecular Residual Disease in Localized Lung Cancer by Circulating Tumor DNA Profiling. Cancer Discov. 2017, 7, 1394–1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novello, S.; Vavalà, T.; Levra, M.G.; Solitro, F.; Pelosi, E.; Veltri, A.; Scagliotti, G.V. Early Response to Chemotherapy in Patients with Non-Small-Cell Lung Cancer Assessed by [18F]-Fluoro-Deoxy-d-Glucose Positron Emission Tomography and Computed Tomography. Clin. Lung Cancer 2013, 14, 230–237. [Google Scholar] [CrossRef]
- Vollmer, R.T.; Govindan, R.; Graziano, S.L.; Gamble, G.; Garst, J.; Kelley, M.J.; Christenson, R.H. Serum CYFRA 21-1 in Ad-vanced Stage Non-Small Cell Lung Cancer: An Early Measure of Response. Clin. Cancer Res. 2003, 9, 1728–1733. [Google Scholar]
- Ardizzoni, A.; Cafferata, M.A.; Tiseo, M.; Filiberti, R.; Marroni, P.; Grossi, F.; Paganuzzi, M. Decline in Serum Carcinoem-bryonic Antigen and Cytokeratin 19 Fragment during Chemotherapy Predicts Objective Response and Survival in Patients with Advanced Nonsmall Cell Lung Cancer. Cancer 2006, 107, 2842–2849. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Subjects (n = 92) |
|---|---|
| Histology Subtype, n (%) | |
| Adenocarcinoma | 56 (61) |
| Squamous cell carcinoma | 36 (39) |
| Age, median (IQR) | 65 (59, 71) |
| Sex, n (%) | |
| Female | 23 (25) |
| Male | 69 (75) |
| Smoking History, n (%) | |
| Ex-smoker | 48 (52) |
| Never smoked | 5 (5) |
| Smoker | 39 (42) |
| UICC, n (%) | |
| IIIB | 29 (32) |
| IIIC | 9 (10) |
| IV | 34 (37) |
| IVA | 14 (15) |
| IVB | 6 (7) |
| ECOG, n (%) | |
| 0 | 55 (60) |
| 1 | 36 (39) |
| 2 | 1 (1) |
| Tumor Stage, n (%) | |
| T1 | 3 (3) |
| T2 | 15 (16) |
| T3 | 31 (34) |
| T4 | 42 (46) |
| TX | 1 (1) |
| N Stage, n (%) | |
| N0 | 4 (4) |
| N1 | 6 (7) |
| N2 | 35 (38) |
| N3 | 46 (50) |
| NX | 1 (1) |
| M Stage, n (%) | |
| M0 | 34 (37) |
| M1 | 54 (59) |
| MX | 4 (4) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yaung, S.J.; Woestmann, C.; Ju, C.; Ma, X.M.; Gattam, S.; Zhou, Y.; Xi, L.; Pal, S.; Balasubramanyam, A.; Tikoo, N.; et al. Early Assessment of Chemotherapy Response in Advanced Non-Small Cell Lung Cancer with Circulating Tumor DNA. Cancers 2022, 14, 2479. https://doi.org/10.3390/cancers14102479
Yaung SJ, Woestmann C, Ju C, Ma XM, Gattam S, Zhou Y, Xi L, Pal S, Balasubramanyam A, Tikoo N, et al. Early Assessment of Chemotherapy Response in Advanced Non-Small Cell Lung Cancer with Circulating Tumor DNA. Cancers. 2022; 14(10):2479. https://doi.org/10.3390/cancers14102479
Chicago/Turabian StyleYaung, Stephanie J., Corinna Woestmann, Christine Ju, Xiaoju Max Ma, Sandeep Gattam, Yiyong Zhou, Liu Xi, Subrata Pal, Aarthi Balasubramanyam, Nalin Tikoo, and et al. 2022. "Early Assessment of Chemotherapy Response in Advanced Non-Small Cell Lung Cancer with Circulating Tumor DNA" Cancers 14, no. 10: 2479. https://doi.org/10.3390/cancers14102479
APA StyleYaung, S. J., Woestmann, C., Ju, C., Ma, X. M., Gattam, S., Zhou, Y., Xi, L., Pal, S., Balasubramanyam, A., Tikoo, N., Heussel, C. P., Thomas, M., Kriegsmann, M., Meister, M., Schneider, M. A., Herth, F. J., Wehnl, B., Diehn, M., Alizadeh, A. A., ... Muley, T. (2022). Early Assessment of Chemotherapy Response in Advanced Non-Small Cell Lung Cancer with Circulating Tumor DNA. Cancers, 14(10), 2479. https://doi.org/10.3390/cancers14102479