
Safety and Efficacy of Crizotinib in Combination with Temozolomide and Radiotherapy in Patients with Newly Diagnosed Glioblastoma: Phase Ib GEINO 1402 Trial


 , , ,
, , , 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Material and Methods
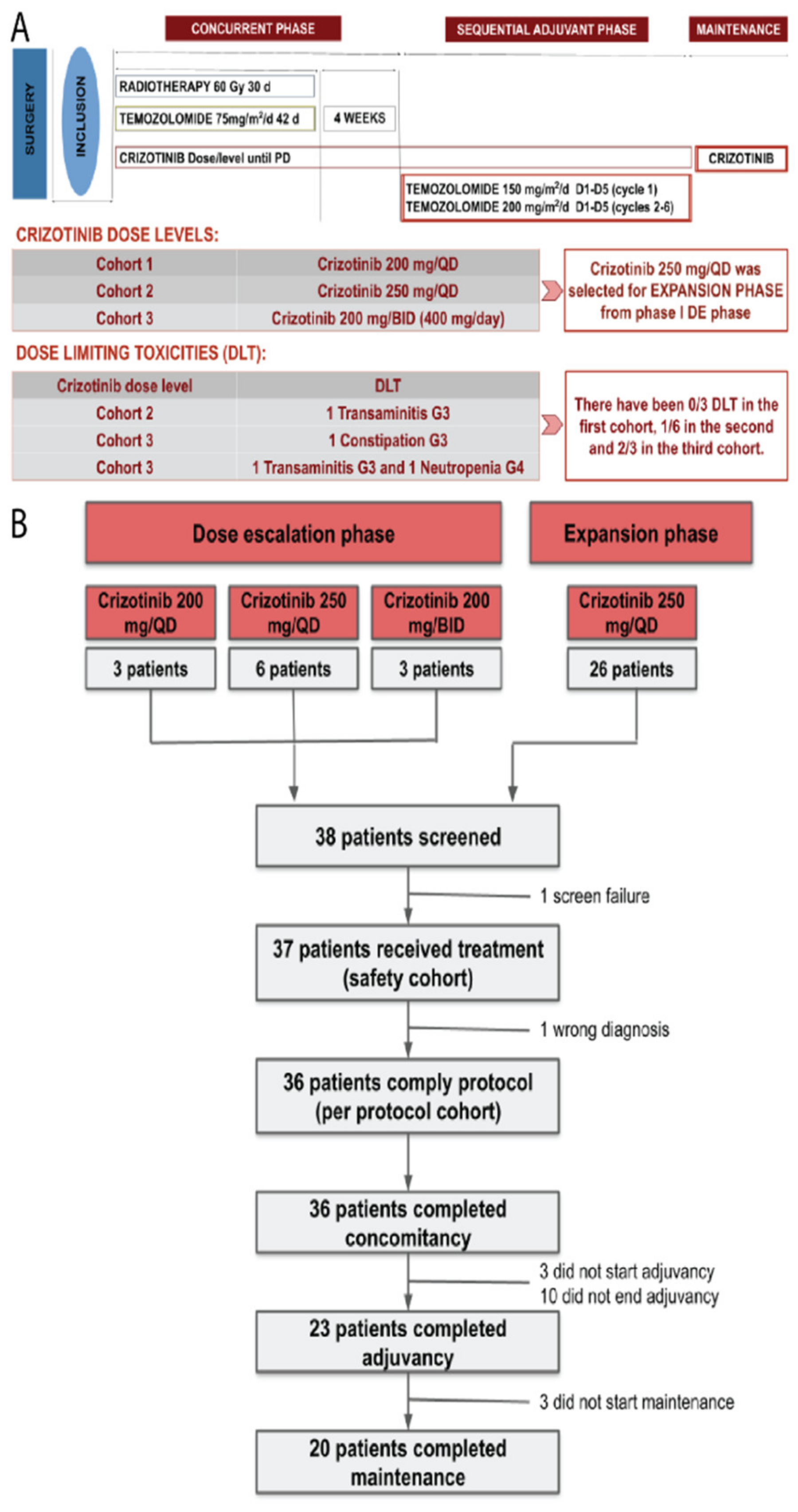
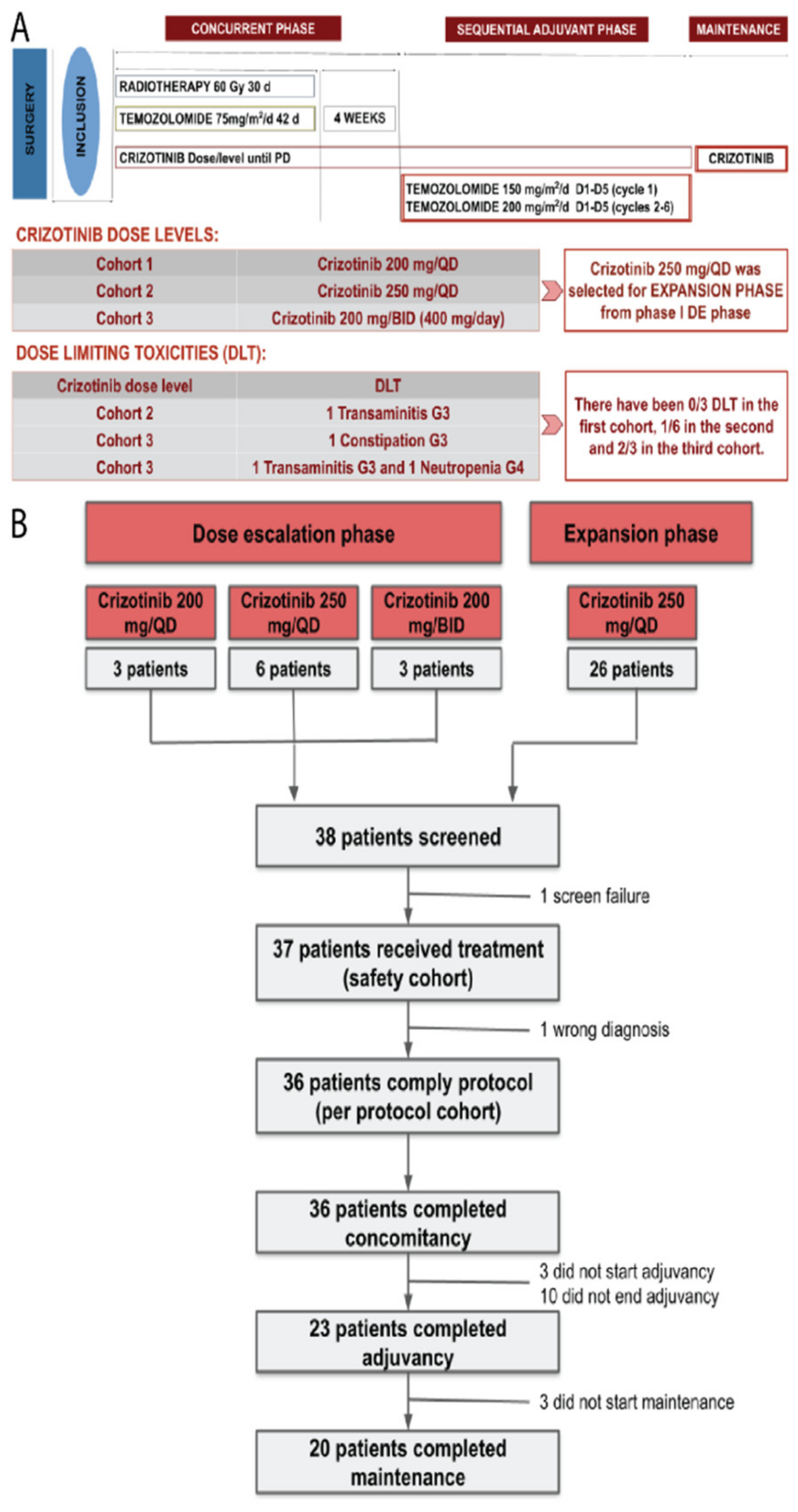
2.1. Study Design
2.2. Study Population
2.3. Treatment Plan
2.4. Objectives and Assessments
2.5. Statistics
2.6. Molecular Biomarker Analysis
3. Results
3.1. Dose-Escalation Phase
3.2. Dose-Expansion Phase
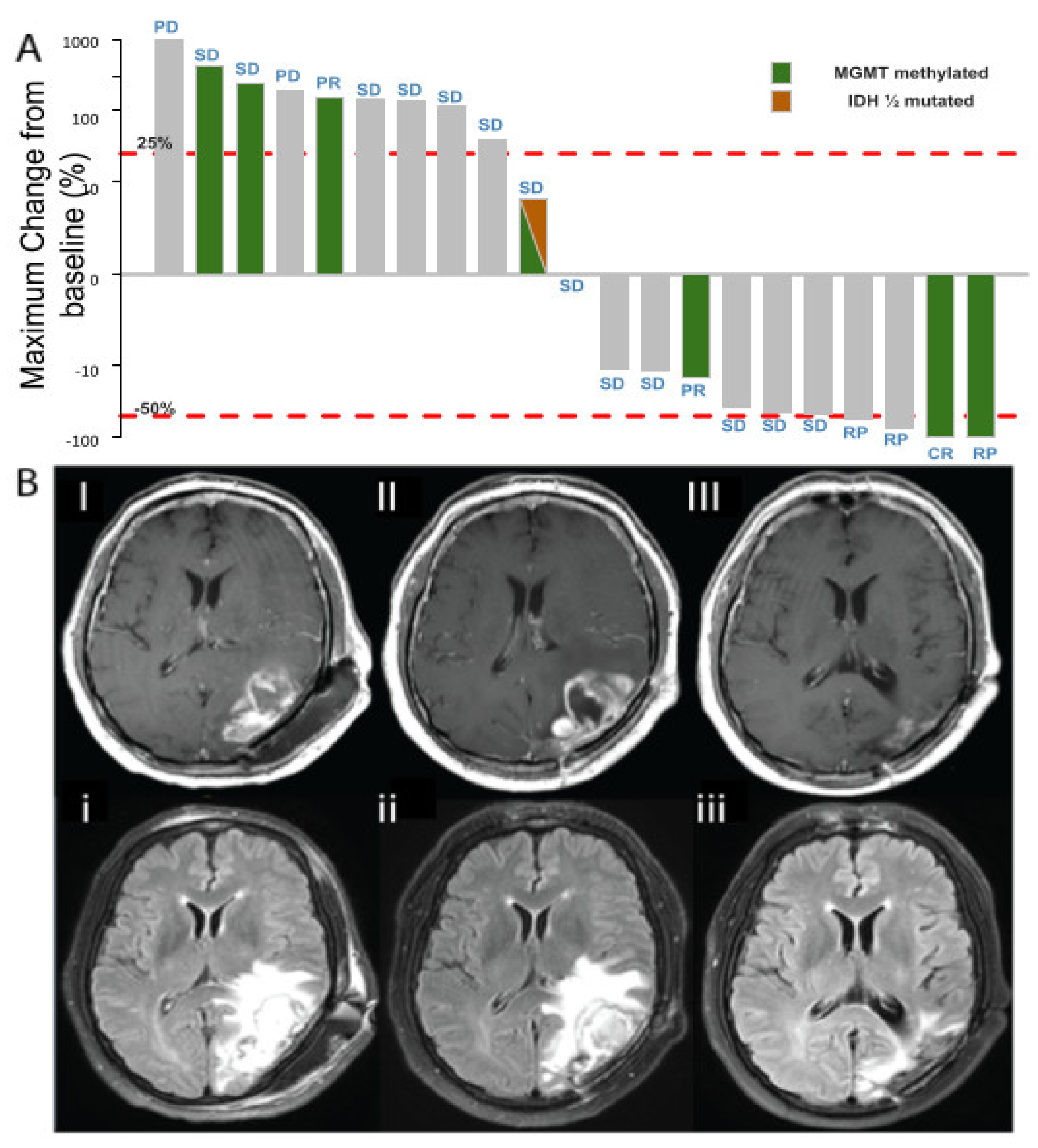
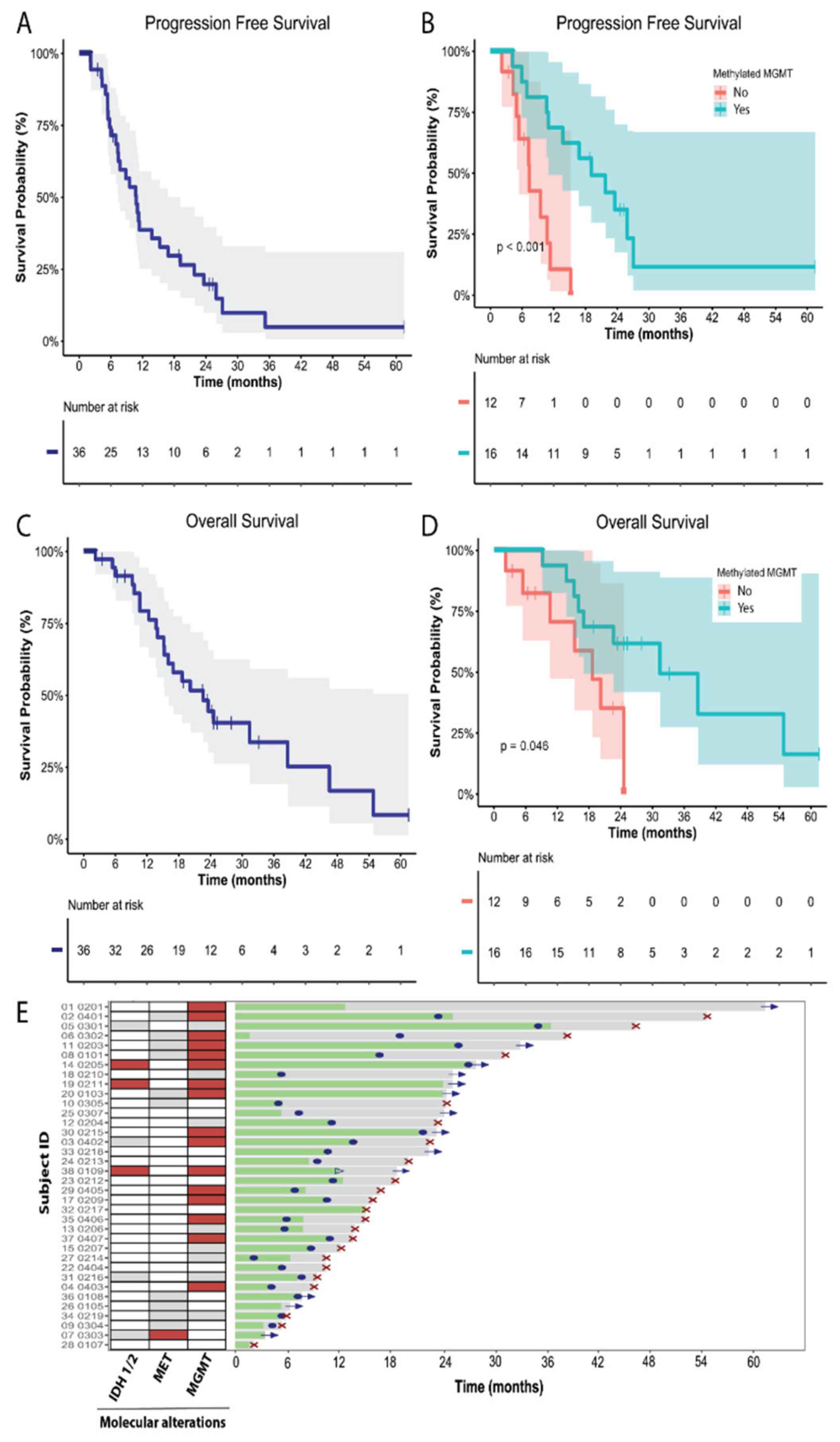
3.3. Efficacy
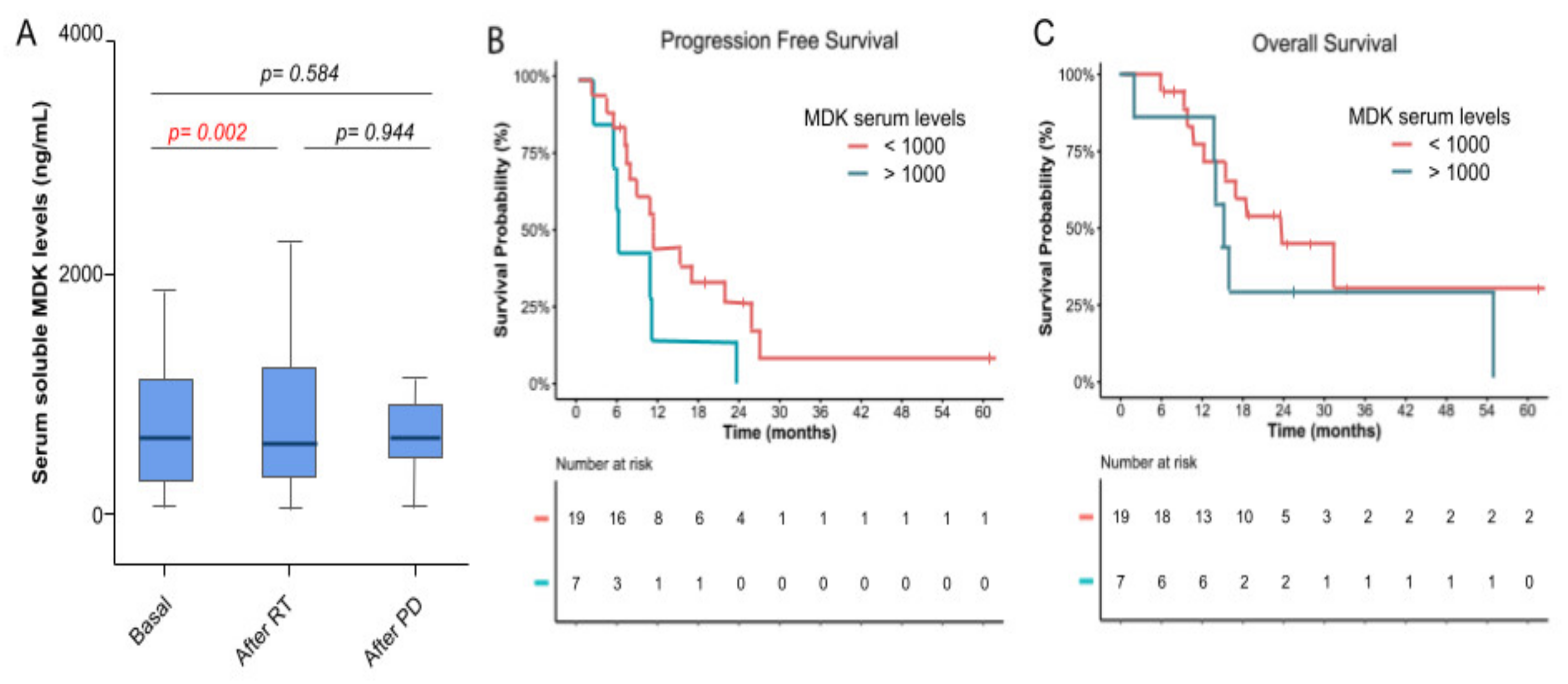
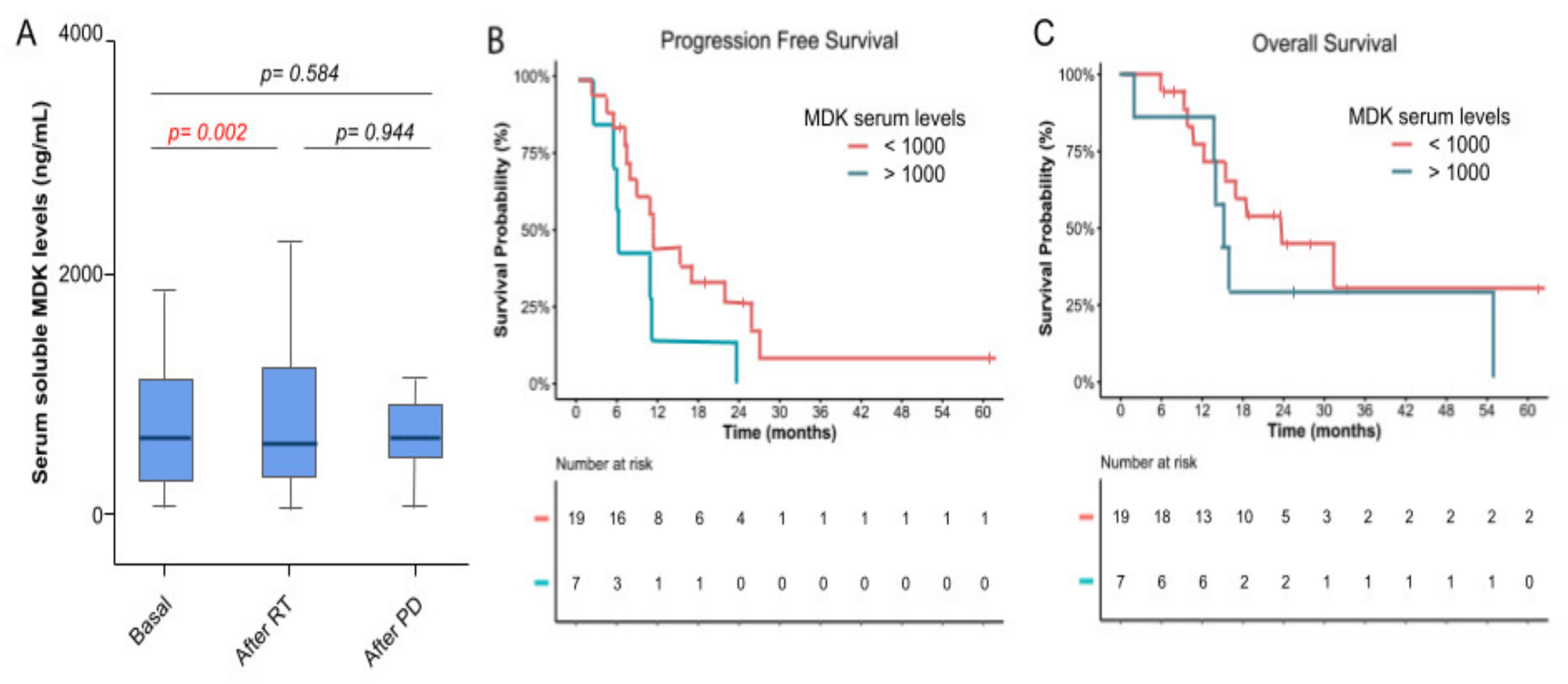
3.4. Molecular Biomarkers and Correlation with Efficacy
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegi, M.E.; Diserens, A.-C.; Gorlia, T.; Hamou, M.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsons, D.W.; Jones, S.; Zhang, X.; Cheng-Ho Lin, J.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.-M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 Mutations in Gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef]
- Blakeley, J.O.; Grossman, S.A.; Chi, A.S.; Mikkelsen, T.; Rosenfeld, M.R.; Ahluwalia, M.S.; Nabors, L.B.; Eichler, A.; Ribas, I.G.; Desideri, S.; et al. Phase II Study of Iniparib with Concurrent Chemoradiation in Patients with Newly Diagnosed Glioblastoma. Clin. Cancer. Res. 2019, 25, 73–79. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Chen, Y.; Lai, S.; Jiang, F.; Liu, X.; Tao, C.; Wang, L.; Liu, G.; Huang, J.; Tang, Y.; et al. A phase II study of anlotinib combined with STUPP regimen in the treatment of patients with newly diagnosed glioblastoma (GBM). J. Clin. Oncol. 2021, 39, 15. [Google Scholar] [CrossRef]
- Roth, P.; Gorlia, T.; Reijneveld, J.C.; Leon de Vos, F.Y.F.; Idbaih, A.; Frenel, J.-S.; le Rhun, E.; Sánchez, J.M.S.; Perry, J.R.; Masucci, L.; et al. EORTC 1709/CCTG CE.8: A phase III trial of marizomib in combination with temozolomide-based radiochemotherapy versus temozolomide-based radiochemotherapy alone in patients with newly diagnosed glioblastoma. J. Clin. Oncol. 2021, 39, 15. [Google Scholar] [CrossRef]
- Galanis, E.; Anderson, K.; Miller, R.; Sarkaria, J.N.; Jaeckle, K.; Buckner, J.C.; Ligon, K.L.; Ballman, K.V.; Moore, D.F.M., Jr.; Nebozhyn, M.; et al. Phase I/II trial of vorinostat combined with temozolomide and radiation therapy for newly diagnosed glioblastoma: Results of Alliance N0874/ABTC 02. Neuro Oncol. 2018, 20, 546–556. [Google Scholar] [CrossRef] [Green Version]
- Nayak, L.; de Groot, J.; Wefel, J.S.; Cloughesy, T.F.; Lieberman, F.; Chang, S.M.; Omuro, A.; Drappatz, J.; Batchelor, T.T.; DeAngelis, L.M.; et al. Phase I trial of aflibercept (VEGF trap) with radiation therapy and concomitant and adjuvant temozolomide in patients with high-grade gliomas. J. Neurooncol. 2017, 132, 181–188. [Google Scholar] [CrossRef]
- Awad, A.J.; Burns, T.C.; Zhang, Y.; Abounader, R. Targeting MET for glioma therapy. Neurosurg. Focus 2014, 37, E10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joo, K.M.; Jin, J.; Kim, E.; Kim, K.H.; Kim, Y.; Kang, B.G.; Youn-Jung Kang, J.D.L.; Lathia, J.D.; Cheong, K.H.; Song, P.H.; et al. MET signaling regulates glioblastoma stem cells. Cancer Res. 2012, 72, 3828–3838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Organ, S.L.; Tsao, M.-S. An overview of the c-MET signaling pathway. Ther. Adv. Med. Oncol. 2011, 3, S7–S19. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Li, A.; Glas, M.; Lal, B.; Ying, M.; Sang, Y.; Xia, S.; Trageser, D.; Guerrero-Cázares, H.; Eberhart, C.G.; et al. c-Met signaling induces a reprogramming network and supports the glioblastoma stem-like phenotype. Proc. Natl. Acad. Sci. USA 2011, 108, 9951–9956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorente, M.; Torres, S.; Salazar, M.; Carracedo, A.; Hernández-Tiedra, S.; Rodríguez-Fornés, F.; Elena García-Taboada, B.M.; Meléndez, B.; Mollejo, M.; Campos-Martín, Y.; et al. Stimulation of ALK by the growth factor midkine renders glioma cells resistant to autophagy-mediated cell death. Autophagy 2011, 7, 1071–1073. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Lang, B.; Wang, X.; Wang, L.; Dong, Y.; Hu, H. Co-expression of midkine and pleiotrophin predicts poor survival in human glioma. J. Clin. Neurosci. 2014, 21, 1885–1890. [Google Scholar] [CrossRef] [PubMed]
- Stylianou, D.C.; Auf der Maur, A.; Kodack, D.P.; Henke, R.T.; Hohn, S.; Toretsky, J.A.; Riegel, A.T.; Wellstein, A. Effect of single-chain antibody targeting of the ligand-binding domain in the anaplastic lymphoma kinase receptor. Oncogene 2019, 28, 3296–3306. [Google Scholar] [CrossRef] [Green Version]
- López-Valero, I.; Dávila, D.; González-Martínez, J.; Salvador-Tormo, N.; Lorente, M.; Saiz-Ladera, C.; Torres, S.; Gabicagogeascoa, E.; Hernández-Tiedra, S.; García-Taboada, E.; et al. Midkine signaling maintains the self-renewal and tumorigenic capacity of glioma initiating cells. Theranostics 2020, 10, 5120–5136. [Google Scholar] [CrossRef]
- Chi, A.S.; Batchelor, T.T.; Kwak, E.L.; Clark, J.W.; Wang, D.L.; Wilner, K.D.; Louis, D.N.; Iafrate, A.J. Rapid radiographic and clinical improvement after treatment of a MET-amplified recurrent glioblastoma with a mesenchymal-epithelial transition inhibitor. J. Clin. Oncol. 2012, 30, e30–e33. [Google Scholar] [CrossRef]
- Solomon, B.; Cappuzzo, F.; Felip, E.; Blackhall, F.H.; Costa, D.B.; Kim, D.-W.; Nakagawa, K.; Wu, Y.-L.; Mekhail, T.; Paolini, J.; et al. Intracranial Efficacy of Crizotinib Versus Chemotherapy in Patients With Advanced ALK-Positive Non-Small-Cell Lung Cancer: Results From PROFILE 1014. J. Clin. Oncol. 2016, 34, 2858–2865. [Google Scholar] [CrossRef]
- Camidge, D.R.; Bang, Y.-J.; Kwak, E.L.; Iafrate, A.J.; Varella-Garcia, M.; Fox, S.B.; Riely, G.J.; Solomon, B.; Ou, S.I.; Kim, D.W.; et al. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: Updated results from a phase 1 study. Lancet Oncol. 2012, 13, 1011–1019. [Google Scholar] [CrossRef] [Green Version]
- Solomon, B.J.; Kim, D.W.; Wu, Y.L.; Nakagawa, K.; Mekhail, T.; Felip, E.; Cappuzzo, F.; Paolini, J.; Usari, T.; Tang, Y.; et al. Final Overall Survival Analysis From a Study Comparing First-Line Crizotinib Versus Chemotherapy in ALK-Mutation-Positive Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2018, 36, 2251–2258. [Google Scholar] [CrossRef] [PubMed]
- Blackhall, F.; Camidge, D.R.; Shaw, A.T.; Soria, J.-C.; Solomon, B.J.; Mok, T.; Hirsh, V.; Jänne, P.A.; Shi, Y.; Yang, P.; et al. Final results of the large-scale multinational trial PROFILE 1005: Efficacy and safety of crizotinib in previously treated patients with advanced/metastatic ALK-positive non-small-cell lung cancer. ESMO Open 2017, 2, e000219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, A.; Holmen, S.; Colman, H. IDH1 and IDH2 Mutations in Gliomas. Curr. Neurol. Neurosci. Rep. 2013, 13, 345. [Google Scholar] [CrossRef] [Green Version]
- Buccarelli, M.; Marconi, M.; Pacioni, S.; De Pascalis, I.; D’Alessandris, Q.G.; Martini, M.; Ascione, B.; Malorni, W.; Larocca, L.M.; Pallini, R.; et al. Inhibition of autophagy increases susceptibility of glioblastoma stem cells to temozolomide by igniting ferroptosis. Cell Death Dis. 2018, 9, 841. [Google Scholar] [CrossRef] [PubMed]
- Binabaj, M.M.; Bahrami, A.; ShahidSales, S.; Joodi, M.; Mashhad, M.J.; Hassanian, S.M.; Anvari, K.; Avan, A. The prognostic value of MGMT promoter methylation in glioblastoma: A meta-analysis of clinical trials. J. Cell Physiol. 2018, 233, 378–386. [Google Scholar] [CrossRef]
- Chu, F.; Naiditch, J.A.; Clark, S.; Qiu, Y.-Y.; Zheng, X.; Lautz, T.B.; Holub, J.L.; Chou, P.M.; Czurylo, M.; Madonna, M.B. Midkine mediates intercellular crosstalk between drug-resistant and drug-sensitive neuroblastoma cells in vitro and in vivo. ISRN Oncol. 2013, 2013, 518637. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.C.; Kim, I.J.; Park, J.H.; Shin, Y.; Ku, J.-L.; Jung, M.S.; Yoo, B.C.; Kim, H.K.; Park, J.-G. Identification of genes with differential expression in acquired drug-resistant gastric cancer cells using high-density oligonucleotide microarrays. Clin. Cancer Res. 2004, 10, 272–284. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient Quantitative Characteristics | Median (Range) | |
|---|---|---|
| Age (years) | 51.4 (33.3–76.5) | |
| KPS (%) | 90 (70–100) | |
| Barthel (%) | 100 (75–100) | |
| Mini-mental | 29 (20–30) | |
| Time from surgery to study treatment (weeks) | 5.4 (4–8) | |
| Patient Qualitative Characteristics | n (%) | |
| Gender | Male | 16 (44.4) |
| Female | 20 (55.6) | |
| Histological diagnosis | Glioblastoma | 34 (94.4) |
| Astrocytoma | 1 (2.8) | |
| NE | 1 (2.8) | |
| MGMT methylation | Yes | 16 (44.4) |
| No | 12(33.3) | |
| NE/UK | 8 (22.2) | |
| IDH1/2 mutations | Yes | 3 (8.3) |
| No | 29 (80.6) | |
| NE/UK | 4 (11.1) | |
| EGFR amplification | Yes | 12 (33.3) |
| No | 14 (38.9) | |
| NE/UK | 10 (27.8) | |
| ALK alterations | Yes | 0 (0) |
| No | 26 (72.2) | |
| NE/UK | 10 (27.8) | |
| MET alterations | Yes | 1 (2.8) |
| No | 25 (69.4) | |
| NE/UK | 10 (27.8) | |
| ROS alterations | Yes | 0 (0) |
| No | 26 (72.2) | |
| NE/UK | 10 (27.8) | |
| Phase | Dose Escalation Phase | ||||
|---|---|---|---|---|---|
| Crizotinib dose | 200 mg/QD | 250 mg/QD | 200 mg/BID | 250 mg/QD | Any |
| Grade | Grade ≥3 | Grade ≥3 | Grade ≥3 | Grade ≥3 | Grade ≥3 |
| Number of patients n (%) | 3 (100%) | 6 (100%) | 3 (100%) | 25 (100%) | 37 (100%) |
| Fatigue | 1 (33.3) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (2.7) |
| Transaminitis | 1 (33.3) | 1 (16.7) * | 1 (33.3) * | 5 (20.0) | 8 (21.6) |
| Neutrophil count decreased | 2 (66.7) | 2 (33.3) | 1 (33.3) * | 1 (4.0) | 6 (16.2) |
| Platelet count decreased | 1 (33.3) | 1 (16.7) | 0 (0.0) | 2 (8.0) | 4 (10.8) |
| Constipation | 0 (0.0) | 0 (0.0) | 1 (33.3) * | 0 (0.0) | 1 (2.7) |
| Lymphocyte count decreased | 1 (33.3) | 0 (0.0) | 0 (0.0) | 1 (4.0) | 2 (5.4) |
| White blood cell decreased | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (4.0) | 1 (2.7) |
| Alanine aminotransferase increased | 0 (0.0) | 0 (0.0) | 0 (0.0) | 2 (8.0) | 2 (5.4) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-García, M.; Velasco, G.; Pineda, E.; Gil-Gil, M.; Alameda, F.; Capellades, J.; Martín-Soberón, M.C.; López-Valero, I.; Tovar Ambel, E.; Foro, P.; et al. Safety and Efficacy of Crizotinib in Combination with Temozolomide and Radiotherapy in Patients with Newly Diagnosed Glioblastoma: Phase Ib GEINO 1402 Trial. Cancers 2022, 14, 2393. https://doi.org/10.3390/cancers14102393
Martínez-García M, Velasco G, Pineda E, Gil-Gil M, Alameda F, Capellades J, Martín-Soberón MC, López-Valero I, Tovar Ambel E, Foro P, et al. Safety and Efficacy of Crizotinib in Combination with Temozolomide and Radiotherapy in Patients with Newly Diagnosed Glioblastoma: Phase Ib GEINO 1402 Trial. Cancers. 2022; 14(10):2393. https://doi.org/10.3390/cancers14102393
Chicago/Turabian StyleMartínez-García, María, Guillermo Velasco, Estela Pineda, Miguel Gil-Gil, Francesc Alameda, Jaume Capellades, Mari Cruz Martín-Soberón, Israel López-Valero, Elena Tovar Ambel, Palmira Foro, and et al. 2022. "Safety and Efficacy of Crizotinib in Combination with Temozolomide and Radiotherapy in Patients with Newly Diagnosed Glioblastoma: Phase Ib GEINO 1402 Trial" Cancers 14, no. 10: 2393. https://doi.org/10.3390/cancers14102393
APA StyleMartínez-García, M., Velasco, G., Pineda, E., Gil-Gil, M., Alameda, F., Capellades, J., Martín-Soberón, M. C., López-Valero, I., Tovar Ambel, E., Foro, P., Taus, Á., Arumi, M., Hernández-Laín, A., & Sepúlveda-Sánchez, J. M. (2022). Safety and Efficacy of Crizotinib in Combination with Temozolomide and Radiotherapy in Patients with Newly Diagnosed Glioblastoma: Phase Ib GEINO 1402 Trial. Cancers, 14(10), 2393. https://doi.org/10.3390/cancers14102393