
GPR87 Promotes Metastasis through the AKT-eNOS-NO Axis in Lung Adenocarcinoma

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Transfection
2.2. Invasion Assay
2.3. Proliferation Assay
2.4. Western Blot Analysis
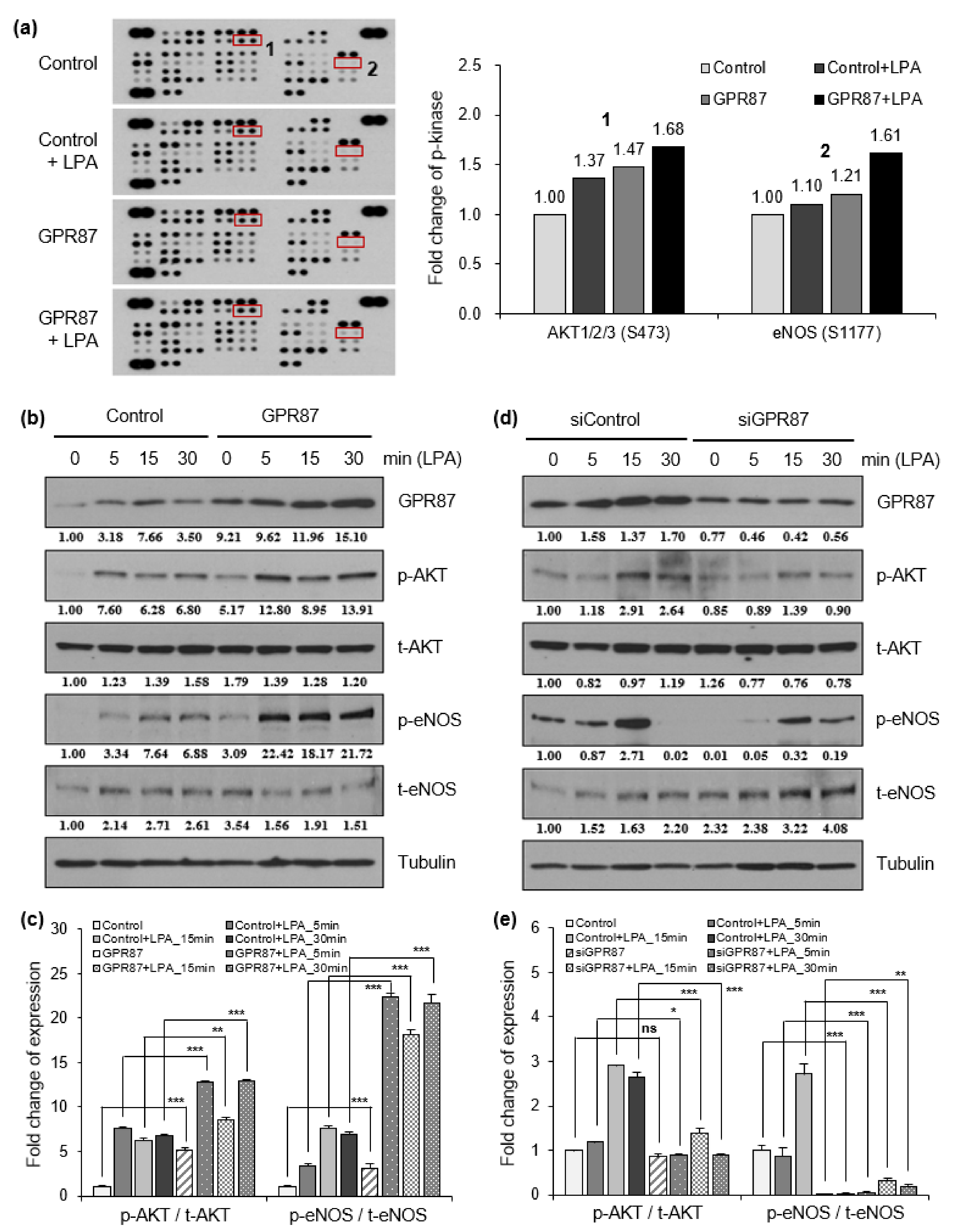
2.5. Phospho-Kinase Array
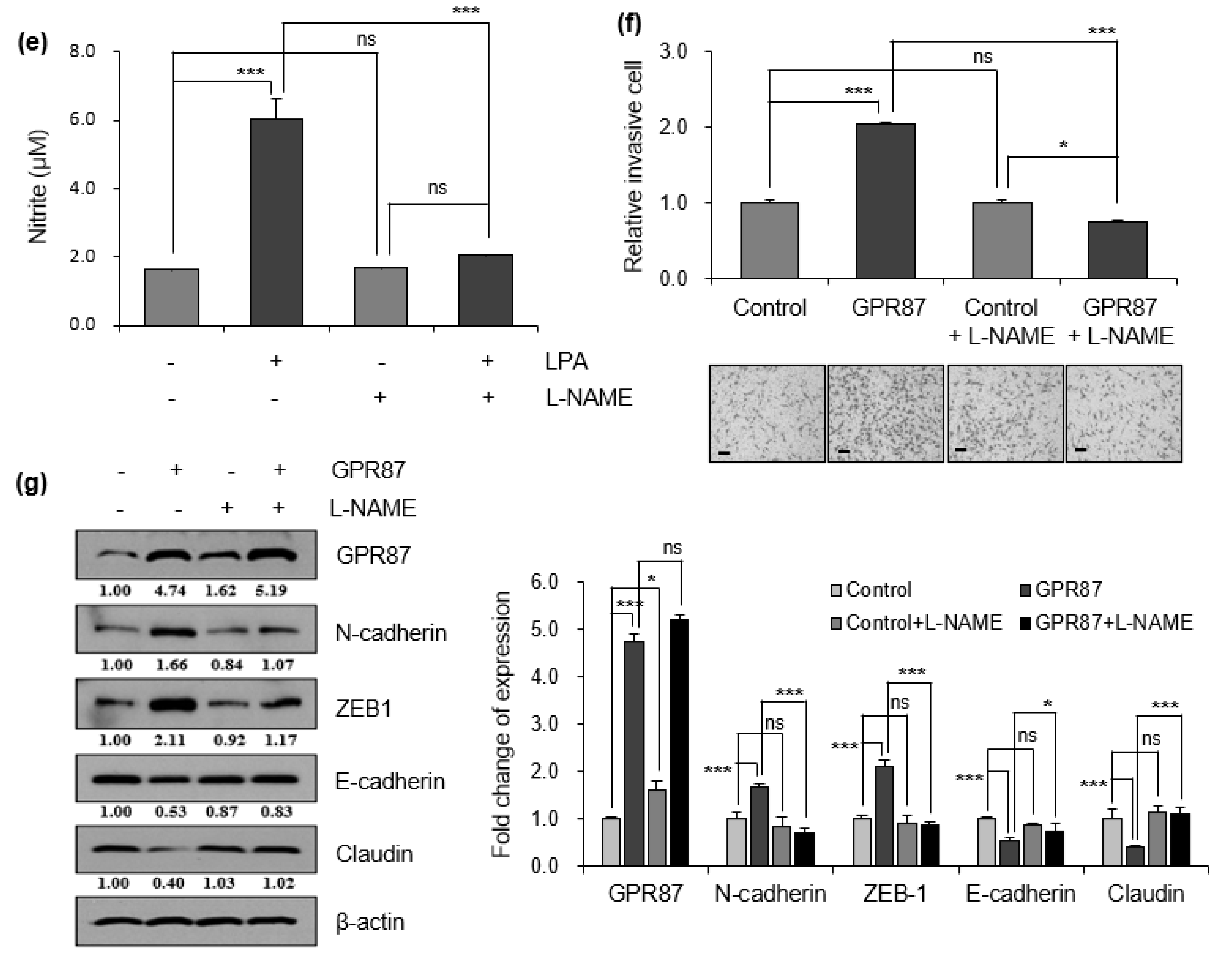
2.6. Nitrite Assay
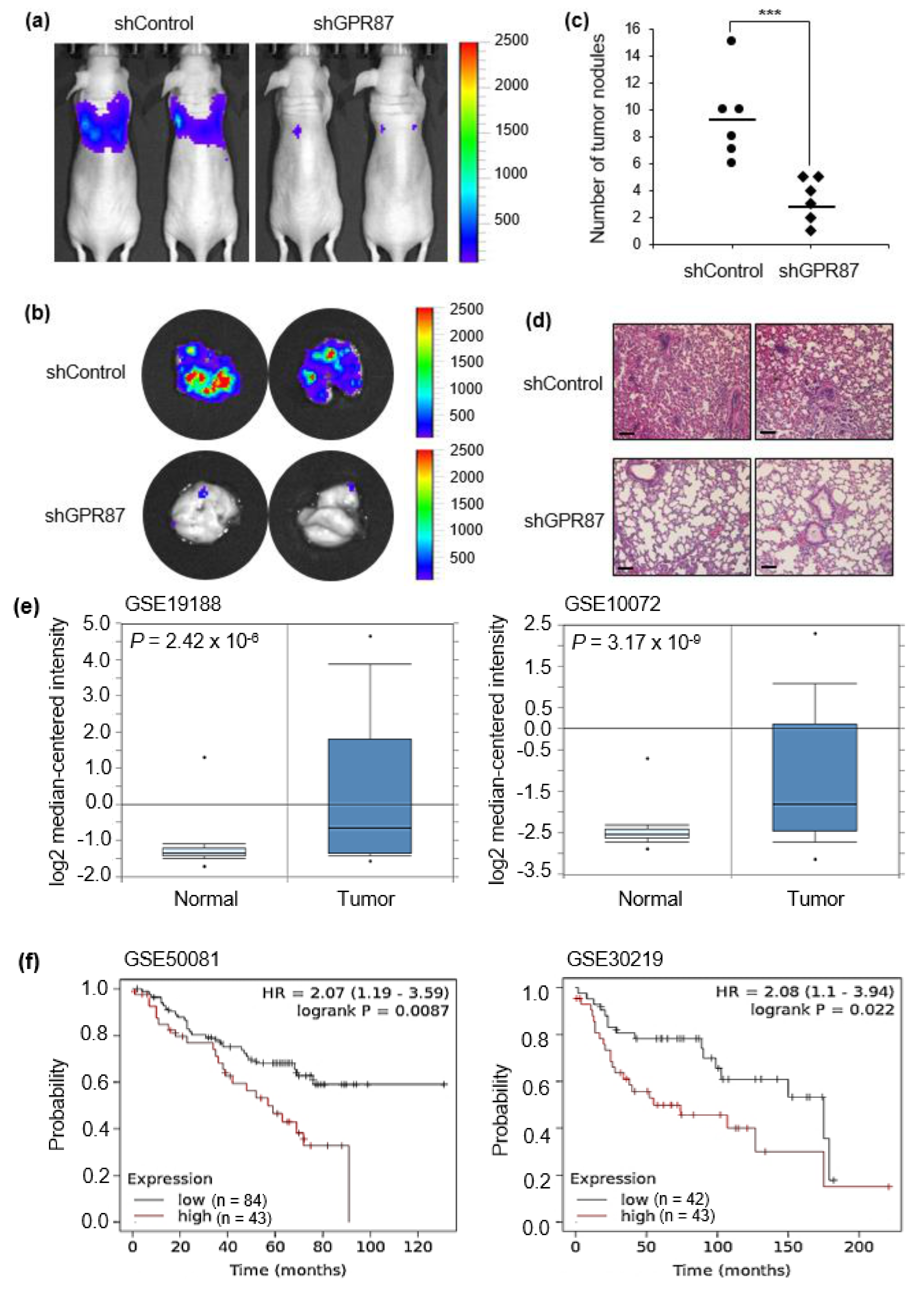
2.7. In Vivo Tumor Models
2.8. Evaluation of Lung Metastasis
2.9. Public Data Analysis
2.10. Statistical Analysis
3. Results
3.1. GPR87 Promotes Invasiveness of Lung Adenocarcinoma Cells
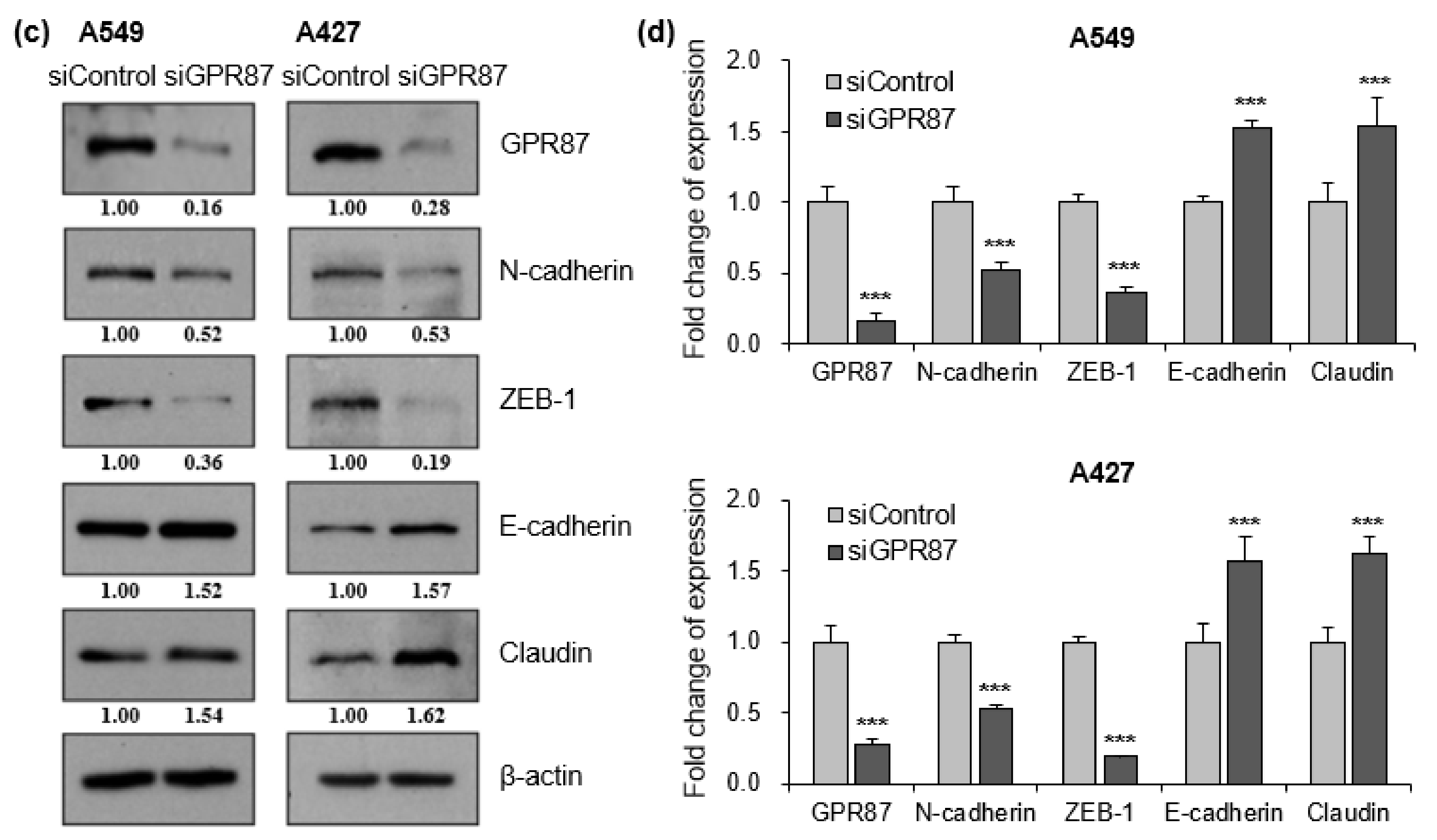
3.2. GPR87 Regulates Mesenchymal Phenotype in Lung Adenocarcinoma Cells
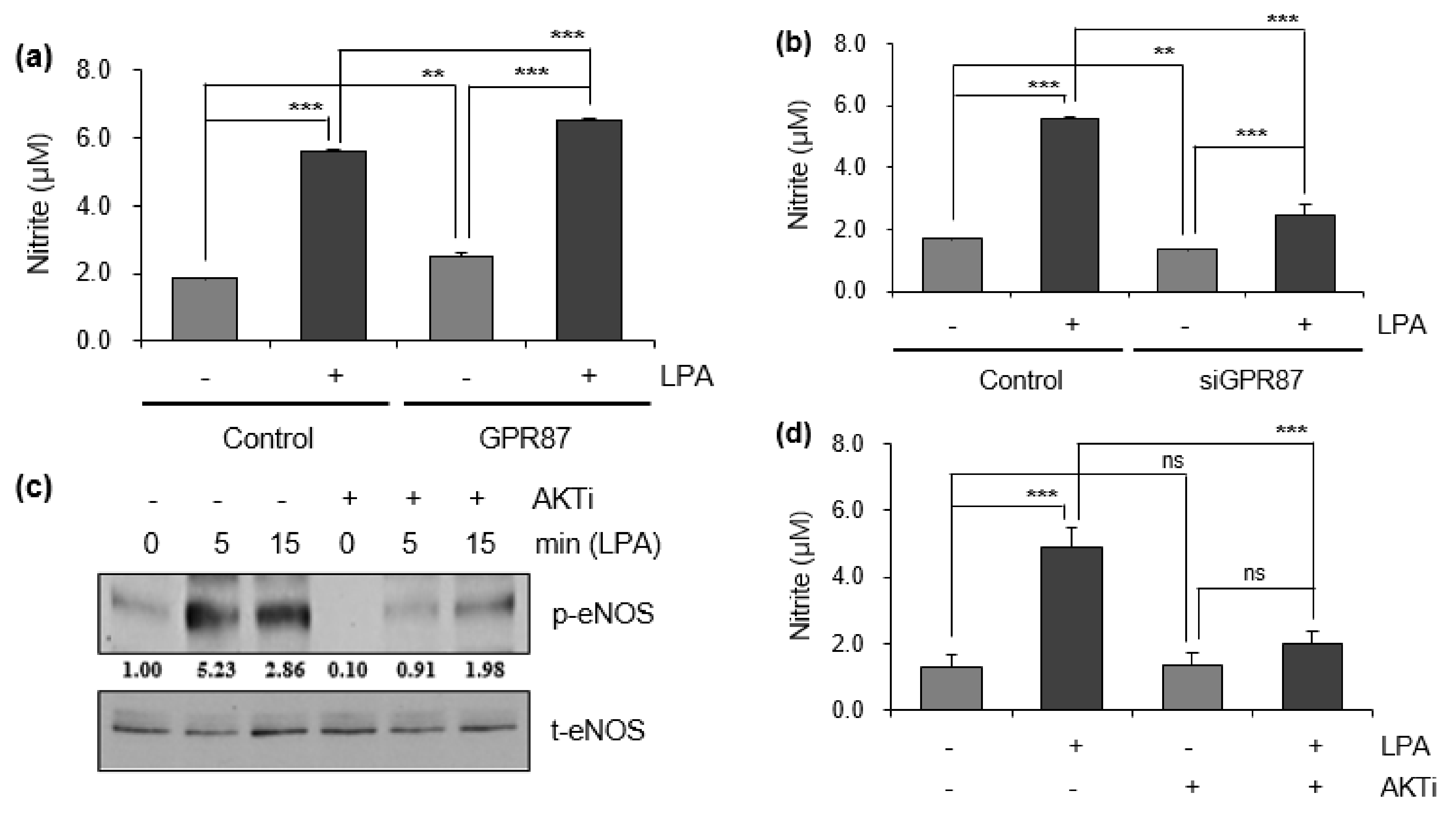
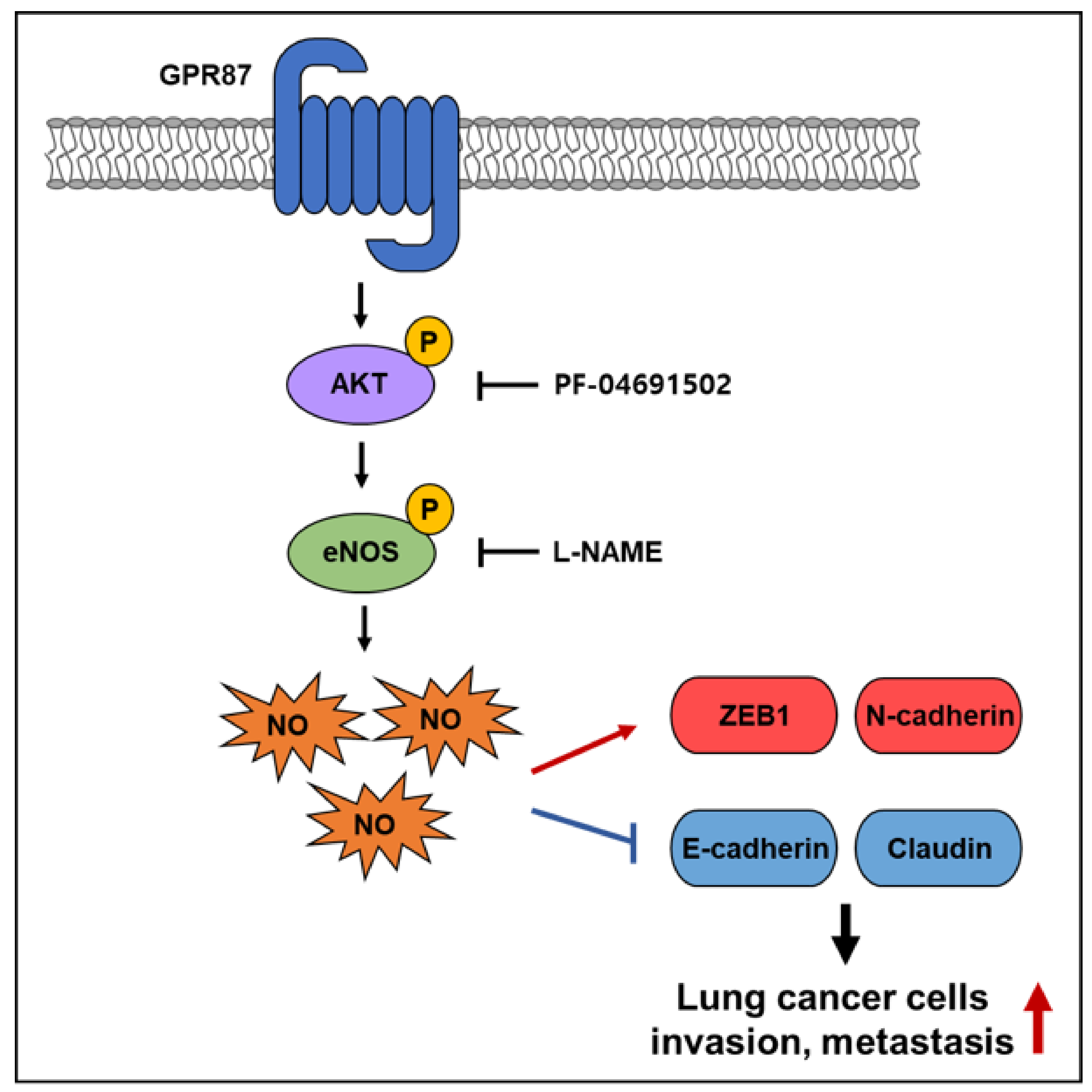
3.3. GPR87 Promotes Malignant Features by Activating AKT-eNOS Signaling
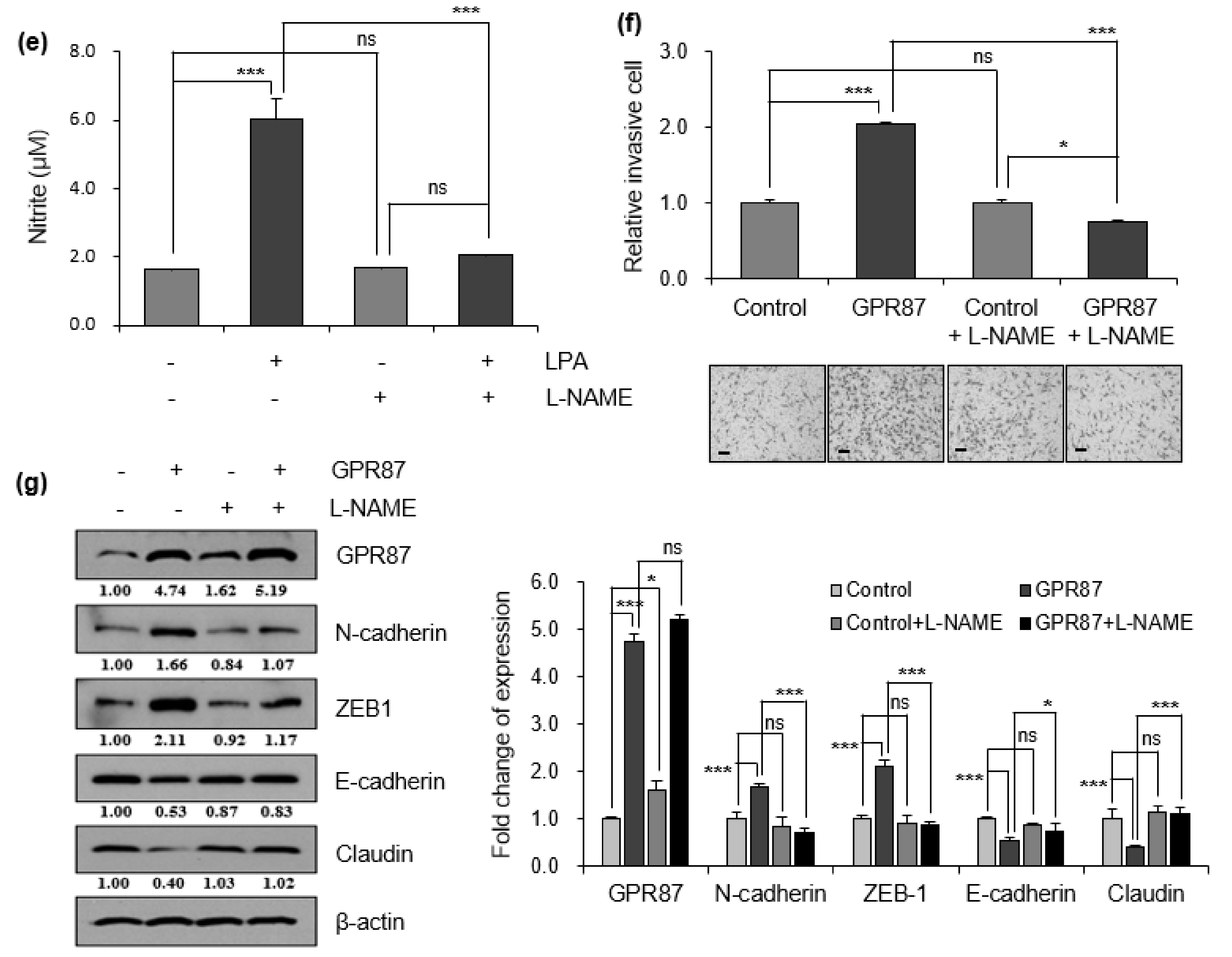
3.4. eNOS Is a Key Mediator for GPR87 in Regulating Metastatic Properties in Lung Adenocarcinoma Cells
3.5. GPR87 Promotes Lung Adenocarcinoma Metastasis and Is Correlated with Poor Prognosis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.-E.; Yin, J.-Y.; Chen, D.-X.; He, S.; Chen, H. Agmatinase promotes the lung adenocarcinoma tumorigenesis by activating the NO-MAPKs-PI3K/Akt pathway. Cell Death Dis. 2019, 10, 854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giuliani, J.; Bonetti, A. Financial Toxicity and Non-small Cell Lung Cancer Treatment: The Optimization in the Choice of Immune Check Point Inhibitors. Anticancer Res. 2019, 39, 3961–3965. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Uchida, N.; Yamamoto, Y.; Kanai, O.; Okamura, M.; Nakatani, K.; Sawai, S.; Mio, T. Retreatment with anti-PD-L1 antibody in advanced non-small cell lung cancer previously treated with anti-PD-1 antibodies. Anticancer Res. 2019, 39, 3917–3921. [Google Scholar] [CrossRef] [PubMed]
- Crystal, A.S.; Shaw, A.T.; Sequist, L.V.; Friboulet, L.; Niederst, M.J.; Lockerman, E.L.; Frias, R.L.; Gainor, J.F.; Amzallag, A.; Greninger, P.; et al. Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science 2014, 346, 1480–1486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellone, M.; Elia, A.R. Constitutive and acquired mechanisms of resistance to immune checkpoint blockade in human cancer. Cytokine Growth Factor Rev. 2017, 36, 17–24. [Google Scholar] [CrossRef]
- Zhao, Q.; Wu, B.-L. Ice breaking in GPCR structural biology. Acta Pharmacol. Sin. 2012, 33, 324–334. [Google Scholar] [CrossRef] [Green Version]
- Fidom, K.; Isberg, V.; Hauser, A.S.; Mordalski, S.; Lehto, T.; Bojarski, A.J.; Gloriam, D.E. A new crystal structure fragment-based pharmacophore method for G protein-coupled receptors. Methods 2015, 71, 104–112. [Google Scholar] [CrossRef]
- Lappano, R.; Maggiolini, M. G protein-coupled receptors: Novel targets for drug discovery in cancer. Nat. Rev. Drug Discov. 2011, 10, 47–60. [Google Scholar] [CrossRef]
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schioth, H.B.; Gloriam, D.E. Trends in GPCR drug discovery: New agents, targets and indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef]
- Dorsam, R.T.; Gutkind, J.S. G-protein-coupled receptors and cancer. Nat. Rev. Cancer 2007, 7, 79–94. [Google Scholar] [CrossRef]
- Kita, Y.; Go, T.; Nakashima, N.; Liu, D.; Tokunaga, Y.; Zhang, X.; Nakano, T.; Nii, K.; Chang, S.S.; Yokomise, H. Inhibition of cell-surface molecular GPR87 with GPR87-suppressing adenoviral vector disturb tumor proliferation in lung cancer cells. Anticancer Res. 2020, 40, 733–741. [Google Scholar] [CrossRef]
- Nii, K.; Tokunaga, Y.; Liu, D.; Zhang, X.; Nakano, J.; Ishikawa, S.; Kakehi, Y.; Haba, R.; Yokomise, H. Overexpression of G protein-coupled receptor 87 correlates with poorer tumor differentiation and higher tumor proliferation in non-small-cell lung cancer. Mol. Clin. Oncol. 2014, 2, 539–544. [Google Scholar] [CrossRef] [Green Version]
- Glatt, S.; Halbauer, D.; Heindl, S.; Wernitznig, A.; Kozina, D.; Su, K.-C.; Puri, C.; Garin-Chesa, P.; Sommergruber, W. hGPR87 contributes to viability of human tumor cells. Int. J. Cancer 2008, 122, 2008–2016. [Google Scholar] [CrossRef]
- Zhang, Y.; Qian, Y.; Lu, W.; Chen, X. The G protein–coupled receptor 87 is necessary for p53-dependent cell survival in response to genotoxic stress. Cancer Res. 2009, 69, 6049–6056. [Google Scholar] [CrossRef] [Green Version]
- Arfelt, K.N.; Fares, S.; Sparre-Ulrich, A.H.; Hjorto, G.M.; Gasbjerg, L.S.; Molleskov-Jensen, A.-S.; Benned-Jensen, T.; Rosenkilde, M.M. Signaling via G proteins mediates tumorigenic effects of GPR87. Cell. Signal. 2017, 30, 9–18. [Google Scholar] [CrossRef]
- Wang, L.; Zhou, W.; Zhong, Y.; Huo, Y.; Fan, P.; Zhan, S.; Xiao, J.; Jin, X.; Gou, S.; Yin, T.; et al. Overexpression of G protein-coupled receptor GPR87 promotes pancreatic cancer aggressiveness and activates NF-kappaB signaling pathway. Mol. Cancer 2017, 16, 61. [Google Scholar] [CrossRef] [Green Version]
- Yan, M.; Li, H.; Zhu, M.; Zhao, F.; Zhang, L.; Chen, T.; Jiang, G.; Xie, H.; Cui, Y.; Yao, M.; et al. G protein-coupled receptor 87 (GPR87) promotes the growth and metastasis of CD133(+) cancer stem-like cells in hepatocellular Carcinoma. PLoS ONE 2013, 8, e61056. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.; Yu, C.; Guo, X.; Zhang, H.; Tian, S.; Cai, K.; He, Z.; Sun, C. G protein-coupled receptor GPR87 promotes the expansion of PDA stem cells through activating JAK2/STAT3. Mol. Ther. Oncolytics 2020, 17, 384–393. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.-J.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Marinissen, M.J.; Gutkind, J. G-protein-coupled receptors and signaling networks: Emerging paradigms. Trends Pharmacol. Sci. 2001, 22, 368–376. [Google Scholar] [CrossRef]
- Nonaka, Y.; Hiramoto, T.; Fujita, N. Identification of endogenous surrogate ligands for human P2Y12 receptors by in silico and in vitro methods. Biochem. Biophys. Res. Commun. 2005, 337, 281–288. [Google Scholar] [CrossRef]
- Tabata, K.-I.; Baba, K.; Shiraishi, A.; Ito, M.; Fujita, N. The orphan GPCR GPR87 was deorphanized and shown to be a lysophosphatidic acid receptor. Biochem. Biophys. Res. Commun. 2007, 363, 861–866. [Google Scholar] [CrossRef]
- Ochiai, S.; Furuta, D.; Sugita, K.; Taniura, H.; Fujita, N. GPR87 mediates lysophosphatidic acid-induced colony dispersal in A431 cells. Eur. J. Pharmacol. 2013, 715, 15–20. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, D.; Hayashida, Y.; Okazoe, H.; Hashimoto, T.; Ueda, N.; Sugimoto, M.; Kakehi, Y. G protein-coupled receptor 87 (GPR87) promotes cell proliferation in human bladder cancer cells. Int. J. Mol. Sci. 2015, 16, 24319–24331. [Google Scholar] [CrossRef]
- Hu, Y.; Xiang, J.; Su, L.; Tang, X. The regulation of nitric oxide in tumor progression and therapy. J. Int. Med. Res. 2020, 48, 27. [Google Scholar] [CrossRef] [Green Version]
- Bogdan, C. Nitric oxide synthase in innate and adaptive immunity: An update. Trends Immunol. 2015, 36, 161–178. [Google Scholar] [CrossRef]
- Palmer, R.M.J.; Ferrige, A.G.; Moncada, S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature 1987, 327, 524–526. [Google Scholar] [CrossRef]
- Seabra, A. Nitric Oxide Donors: Novel Biomedical Applications and Perspectives; Academic Press: Cambridge, MA, USA, 2017. [Google Scholar]
- Fukumura, D.; Kashiwagi, S.; Jain, R.K. The role of nitric oxide in tumour progression. Nat. Rev. Cancer 2006, 6, 521–534. [Google Scholar] [CrossRef]
- Seabra, A.B.; Durán, N.J.E.J.O.P. Nitric oxide donors for prostate and bladder cancers: Current state and challenges. Eur. J. Pharmacol. 2018, 826, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, L.L.; Miles, D.W.; Happerfield, L.; Bobrow, L.G.; Knowles, R.G.; Moncada, S. Nitric oxide synthase activity in human breast cancer. Br. J. Cancer 1995, 72, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Naidu, M.S.K.; Suryakar, A.N.; Swami, S.C.; Katkam, R.V.; Kumbar, K.M. Oxidative stress and antioxidant status in cervical cancer patients. Indian J. Clin. Biochem. 2007, 22, 140–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yagihashi, N.; Kasajima, H.; Sugai, S.; Matsumoto, K.; Ebina, Y.; Morita, T.; Murakami, T.; Yagihashi, S. Increased in situ expression of nitric oxide synthase in human colorectal cancer. Virchows Arch. 2000, 436, 109–114. [Google Scholar] [CrossRef]
- Fulton, D.; Gratton, J.-P.; McCabe, T.J.; Fontana, J.; Fujio, Y.; Walsh, K.; Franke, T.F.; Papapetropoulos, A.; Sessa, W.C. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature 1999, 399, 597–601. [Google Scholar] [CrossRef]
- Dimmeler, S.; Fleming, I.; Fisslthaler, B.; Hermann, C.; Busse, R.; Zeiher, A.M. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature 1999, 399, 601–605. [Google Scholar] [CrossRef]
- Michel, J.B.; Feron, O.; Sacks, D.; Michel, T. Reciprocal regulation of endothelial nitric-oxide synthase by Ca2+-calmodulin and caveolin. J. Biol. Chem. 1997, 272, 15583–15586. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Zhang, J.; Feng, G.; Shen, J.; Kong, D.; Zhao, Q. Nitric oxide-releasing biomaterials for biomedical applications. Curr. Med. Chem. 2016, 23, 2579–2601. [Google Scholar] [CrossRef]
- Lim, K.-H.; Ancrile, B.B.; Kashatus, D.; Counter, C.M. Tumour maintenance is mediated by eNOS. Nature 2008, 452, 646–649. [Google Scholar] [CrossRef] [Green Version]
- Lampson, B.L.; Kendall, S.D.; Ancrile, B.B.; Morrison, M.M.; Shealy, M.J.; Barrientos, K.S.; Crowe, M.; Kashatus, D.; White, R.R.; Gurley, S.B.; et al. Targeting eNOS in pancreatic cancer. Cancer Res. 2012, 72, 4472–4482. [Google Scholar] [CrossRef] [Green Version]
- Suksawat, M.; Techasen, A.; Namwat, N.; Boonsong, T.; Titapun, A.; Ungarreevittaya, P.; Yongvanit, P.; Loilome, W. Inhibition of endothelial nitric oxide synthase in cholangiocarcinoma cell lines—A new strategy for therapy. FEBS Open Bio 2018, 8, 513–522. [Google Scholar] [CrossRef] [Green Version]
- Tuveson, D.; Clevers, H. Cancer modeling meets human organoid technology. Science 2019, 364, 952–955. [Google Scholar] [CrossRef]
- De Vita, A.; Recine, F.; Mercatali, L.; Miserocchi, G.; Spadazzi, C.; Liverani, C.; Bongiovanni, A.; Pieri, F.; Casadei, R.; Riva, N.; et al. Primary culture of undifferentiated pleomorphic sarcoma: Molecular characterization and response to anticancer agents. Int. J. Mol. Sci. 2017, 18, 2662. [Google Scholar] [CrossRef] [Green Version]
- Yasui, H.; Nishinaga, Y.; Taki, S.; Takahashi, K.; Isobe, Y.; Shimizu, M.; Koike, C.; Taki, T.; Sakamoto, A.; Katsumi, K.; et al. Near-infrared photoimmunotherapy targeting GPR87: Development of a humanised anti-GPR87 mAb and therapeutic efficacy on a lung cancer mouse model. EBioMedicine 2021, 67, 103372. [Google Scholar] [CrossRef]
- Fujita, M.; Somasundaram, V.; Basudhar, D.; Cheng, R.Y.; Ridnour, L.A.; Higuchi, H.; Imadome, K.; No, J.H.; Bharadwaj, G.; Wink, D.A. Role of nitric oxide in pancreatic cancer cells exhibiting the invasive phenotype. Redox Biol. 2019, 22, 101158. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahn, H.-M.; Choi, E.-Y.; Kim, Y.-J. GPR87 Promotes Metastasis through the AKT-eNOS-NO Axis in Lung Adenocarcinoma. Cancers 2022, 14, 19. https://doi.org/10.3390/cancers14010019
Ahn H-M, Choi E-Y, Kim Y-J. GPR87 Promotes Metastasis through the AKT-eNOS-NO Axis in Lung Adenocarcinoma. Cancers. 2022; 14(1):19. https://doi.org/10.3390/cancers14010019
Chicago/Turabian StyleAhn, Hye-Mi, Eun-Young Choi, and Youn-Jae Kim. 2022. "GPR87 Promotes Metastasis through the AKT-eNOS-NO Axis in Lung Adenocarcinoma" Cancers 14, no. 1: 19. https://doi.org/10.3390/cancers14010019
APA StyleAhn, H.-M., Choi, E.-Y., & Kim, Y.-J. (2022). GPR87 Promotes Metastasis through the AKT-eNOS-NO Axis in Lung Adenocarcinoma. Cancers, 14(1), 19. https://doi.org/10.3390/cancers14010019