
Tipping Growth Inhibition into Apoptosis by Combining Treatment with MDM2 and WIP1 Inhibitors in p53WT Uterine Leiomyosarcoma


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Reagents
2.2. Growth Inhibition Assay
2.3. Clonogenic Assay
2.4. Western Immunoblotting
2.5. Quantitative Reverse Transcriptase PCR
2.6. Caspase-Glo® 3/7 Assay
2.7. Statistical Analysis
3. Results
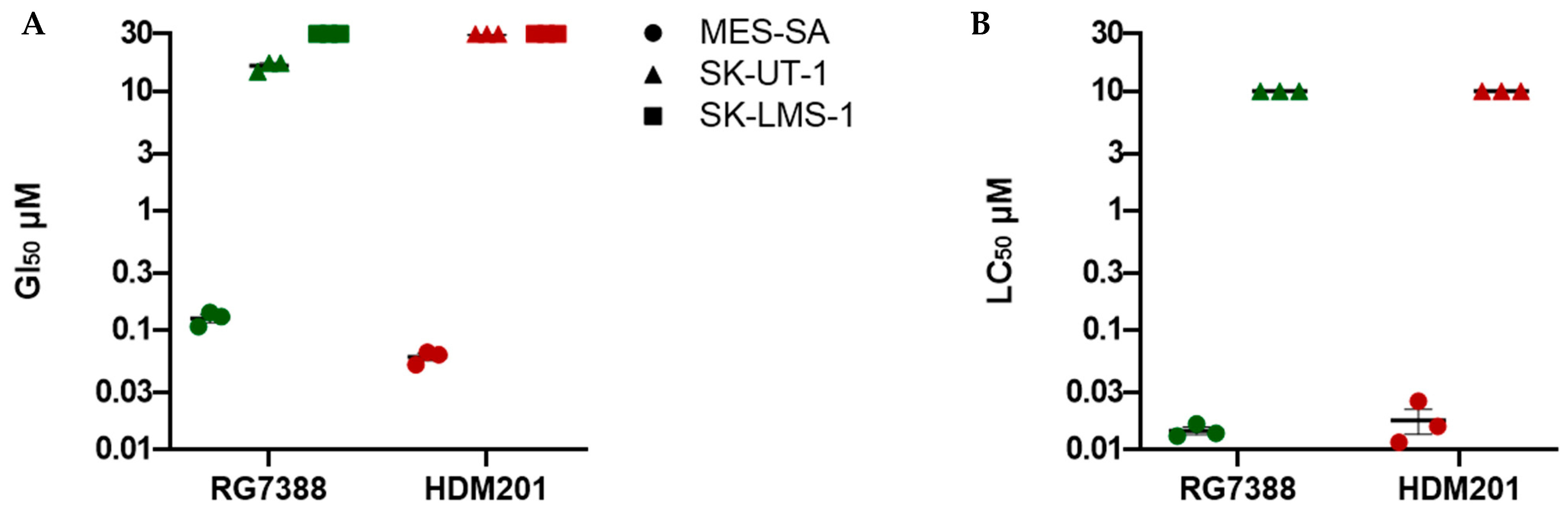
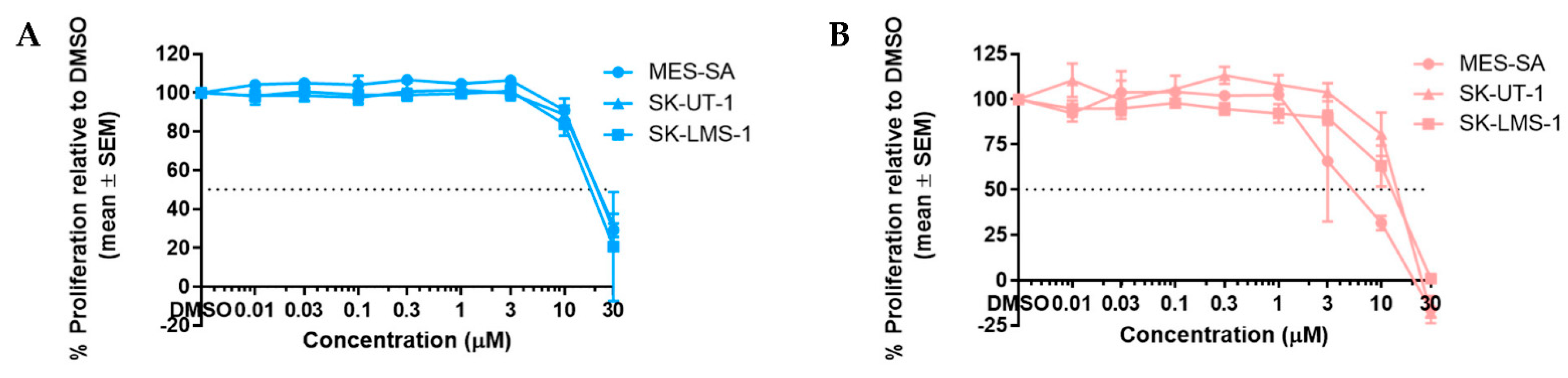
3.1. RG7388 and HDM201 Are Potent Inhibitors in p53WT uLMS Cells but Not p53MUT
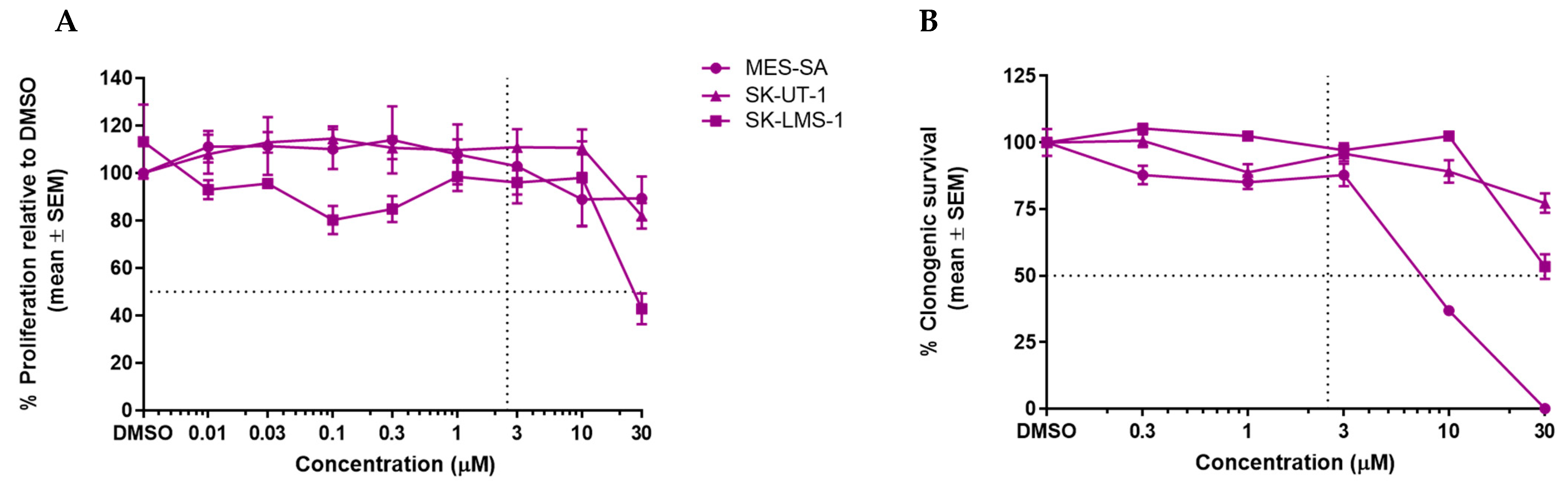
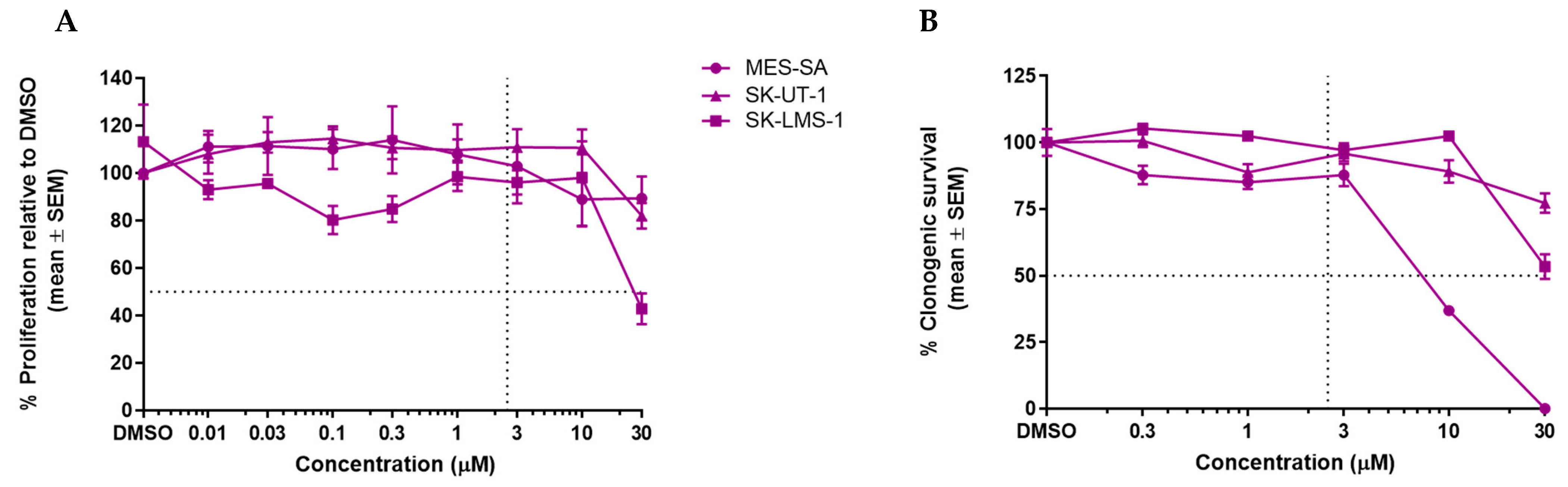
3.2. GSK2830371 Has No Growth Inhibitory Effects as a Single Agent
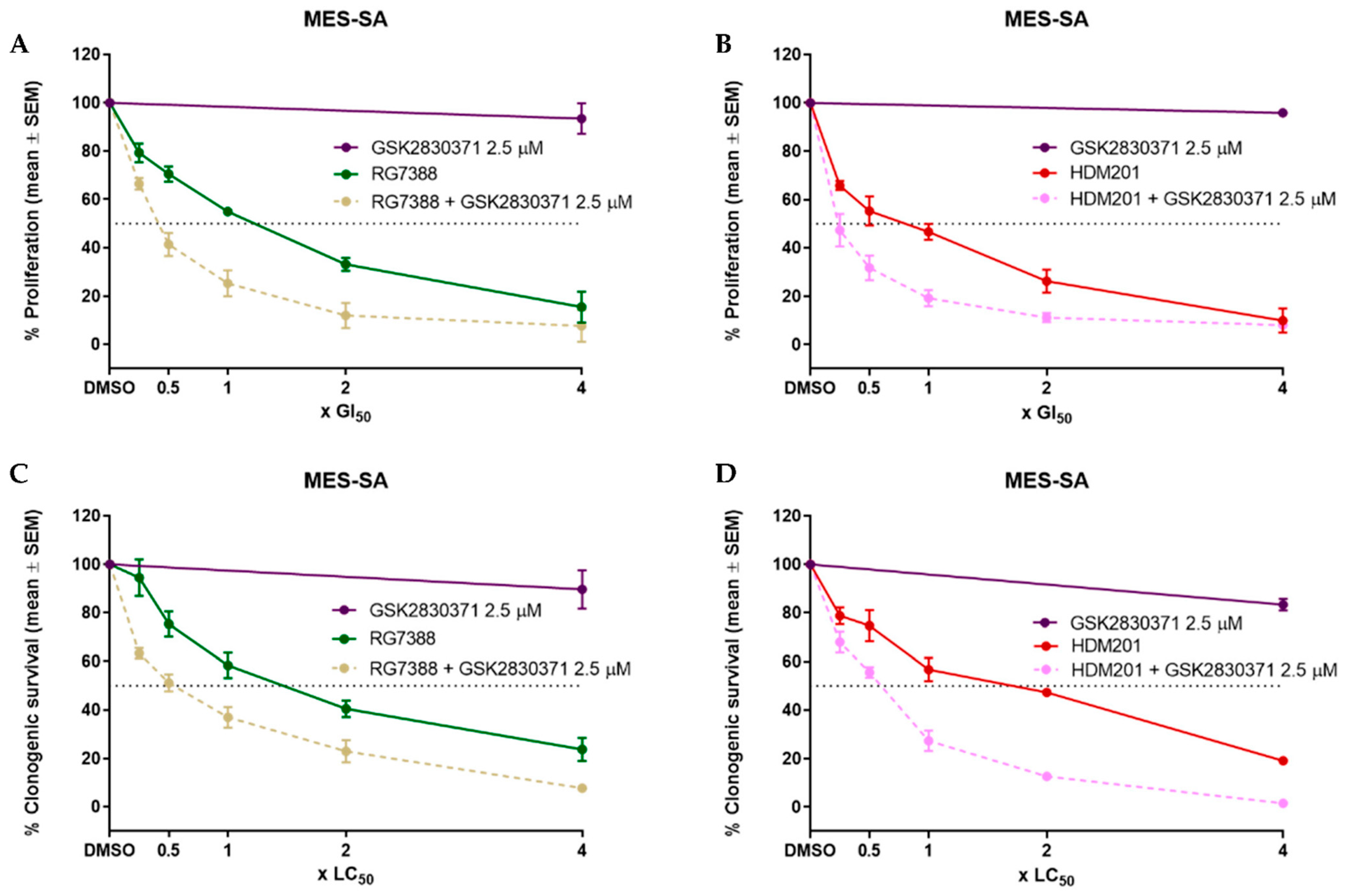
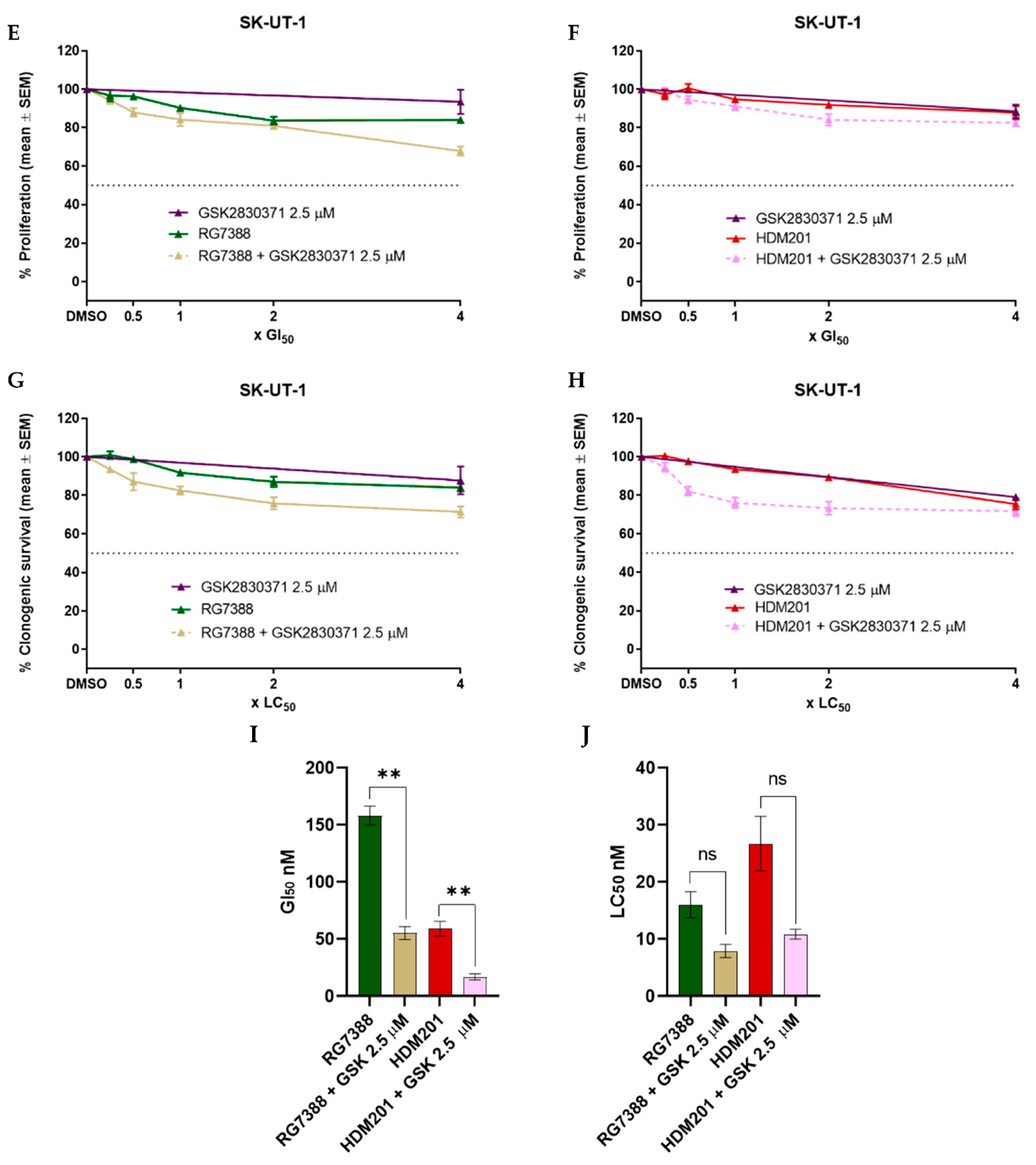
3.3. WIP1 Inhibitor GSK2830371 Potentiates the Growth Inhibitory and Cytotoxic Effects of MDM2 Inhibitors on uLMS p53WT Cells
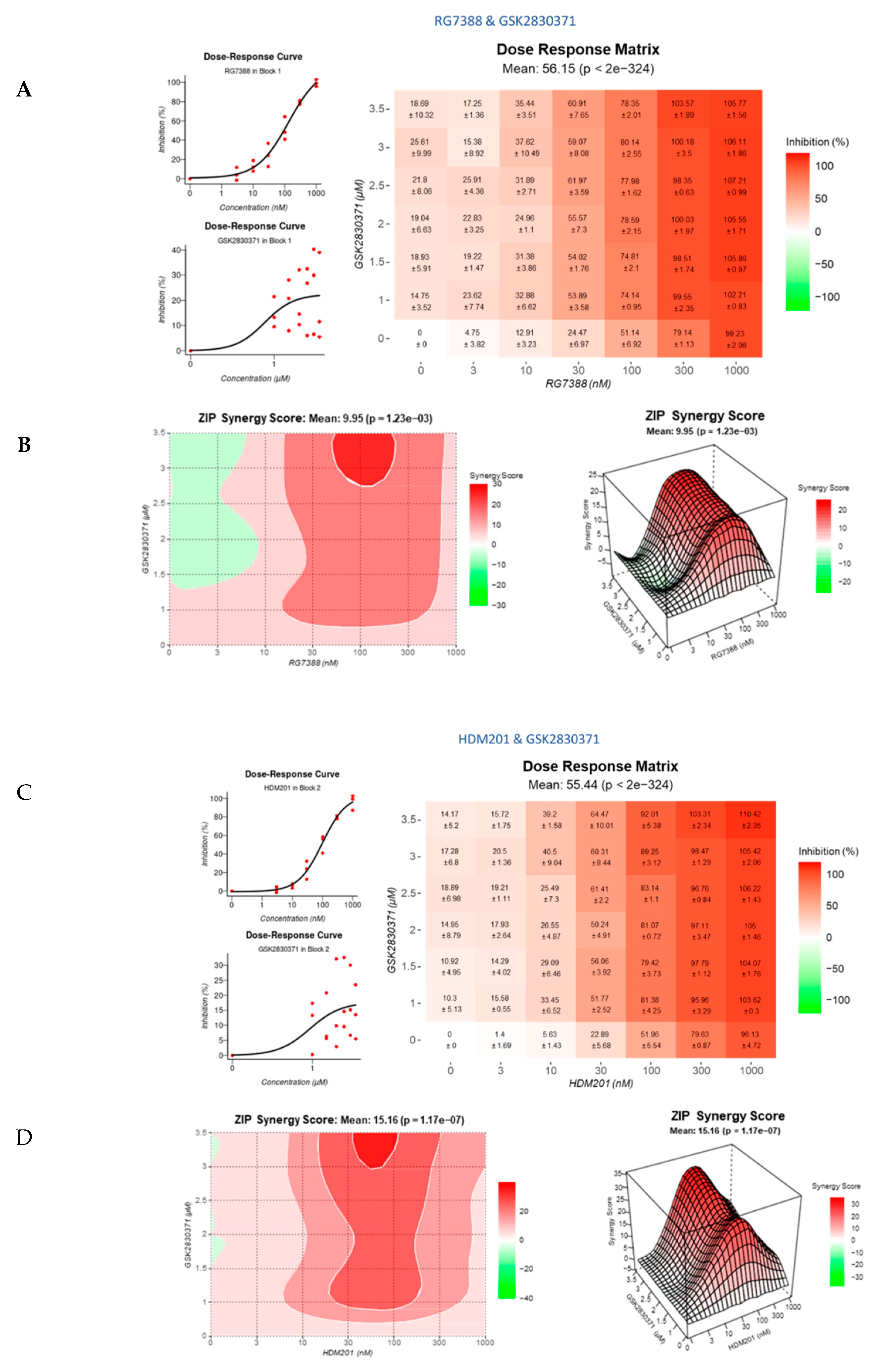
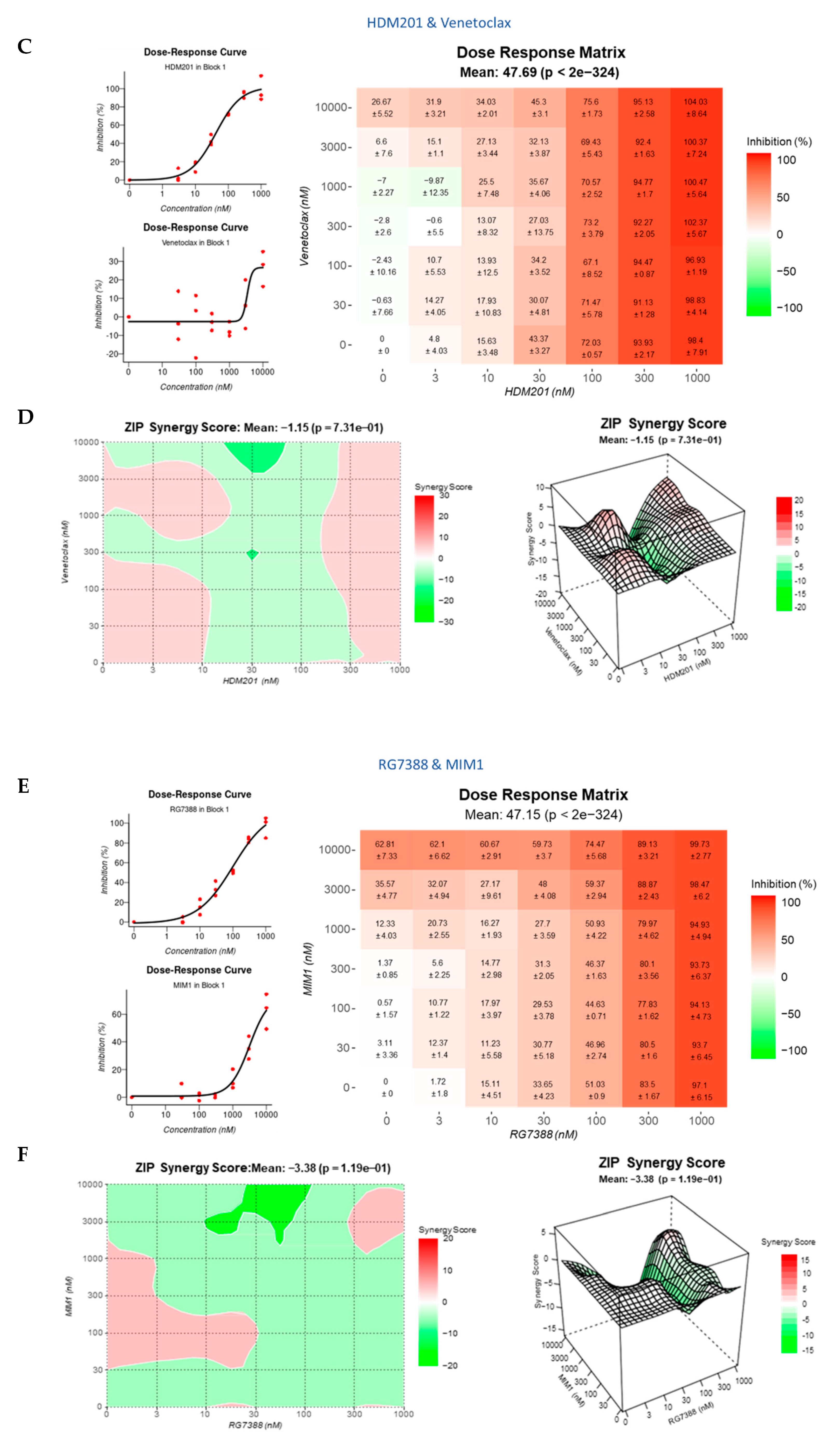
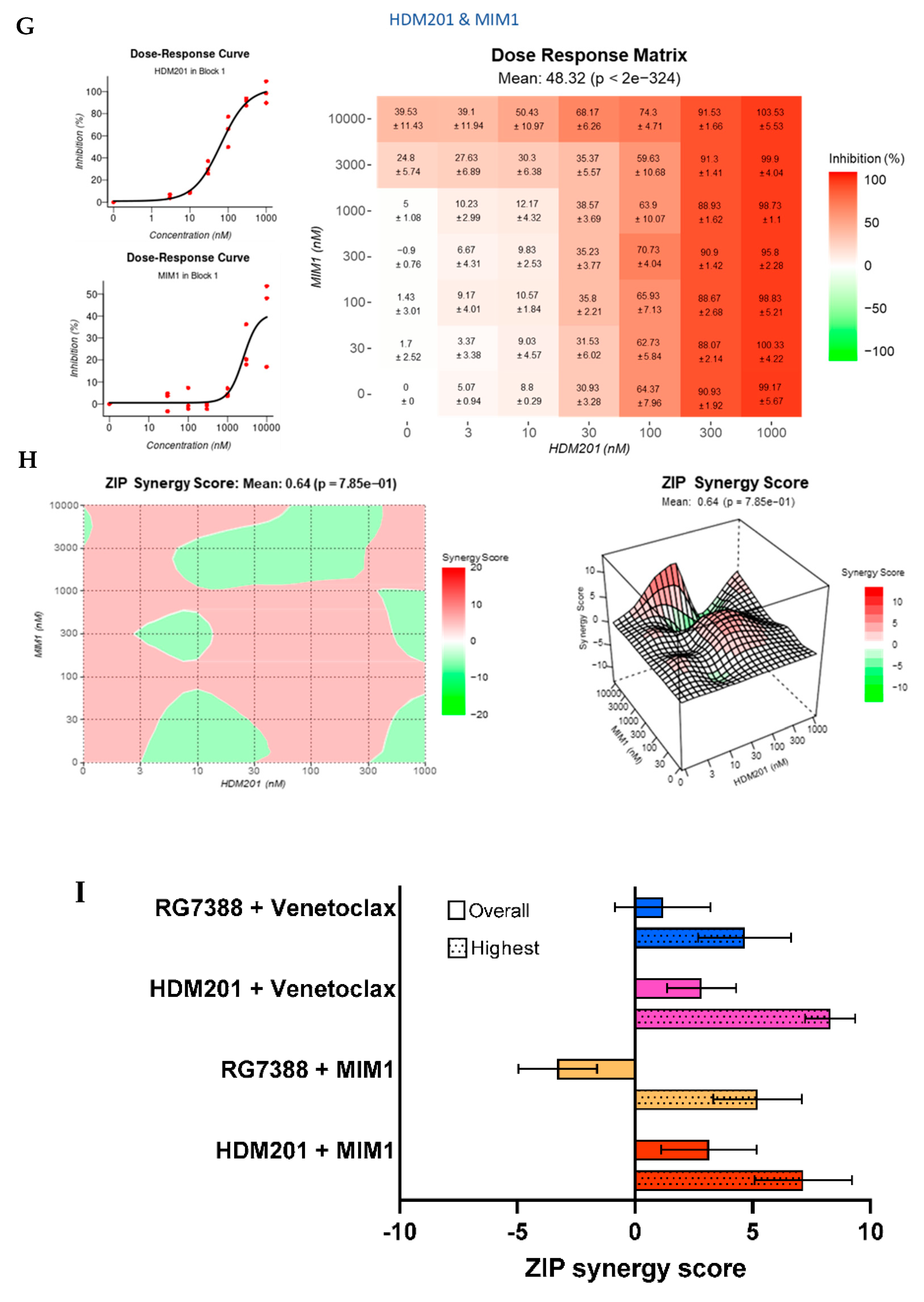
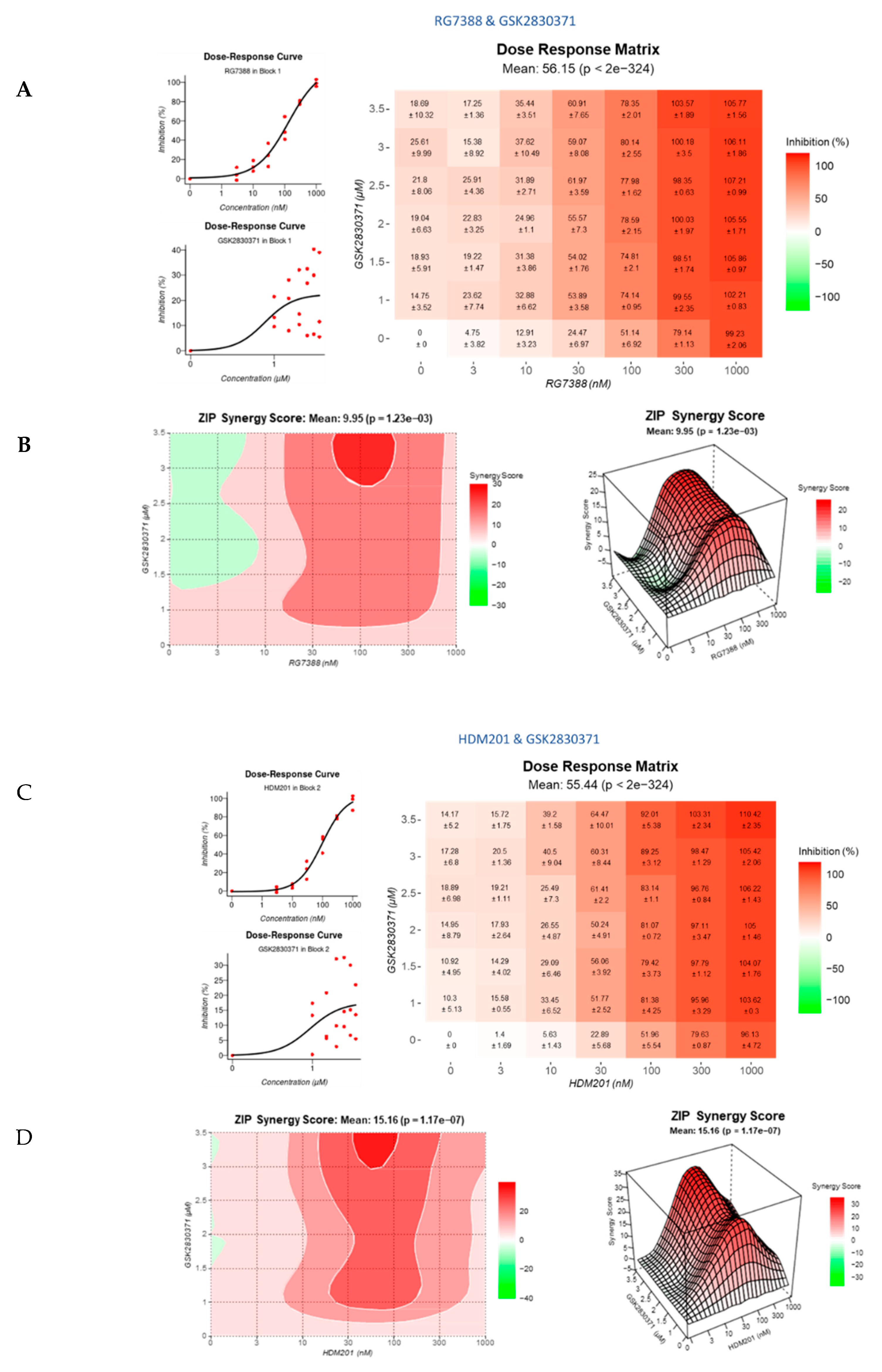
3.4. Strong Synergy Observed with MDM2 Inhibitors in Combination with GSK2830371
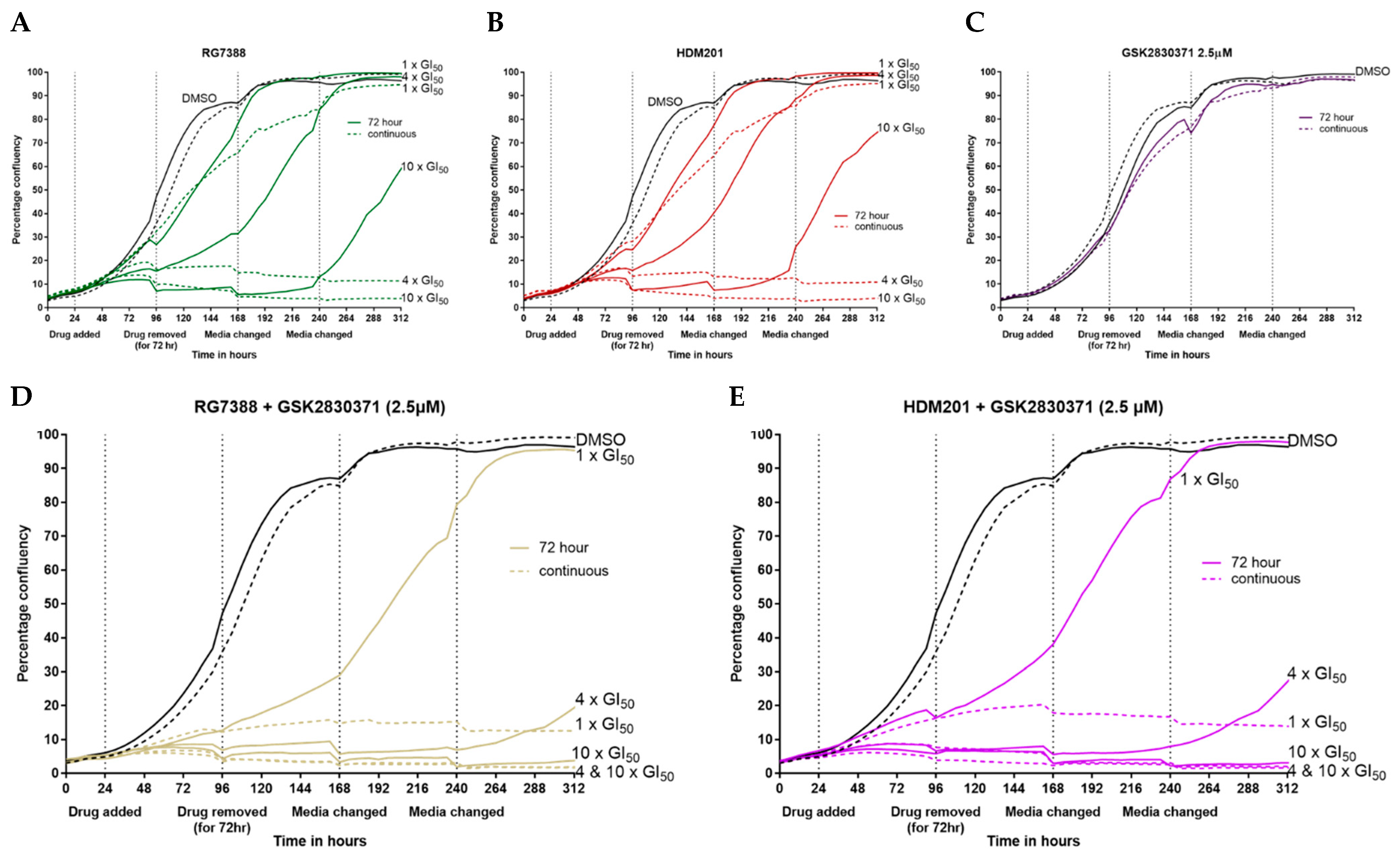
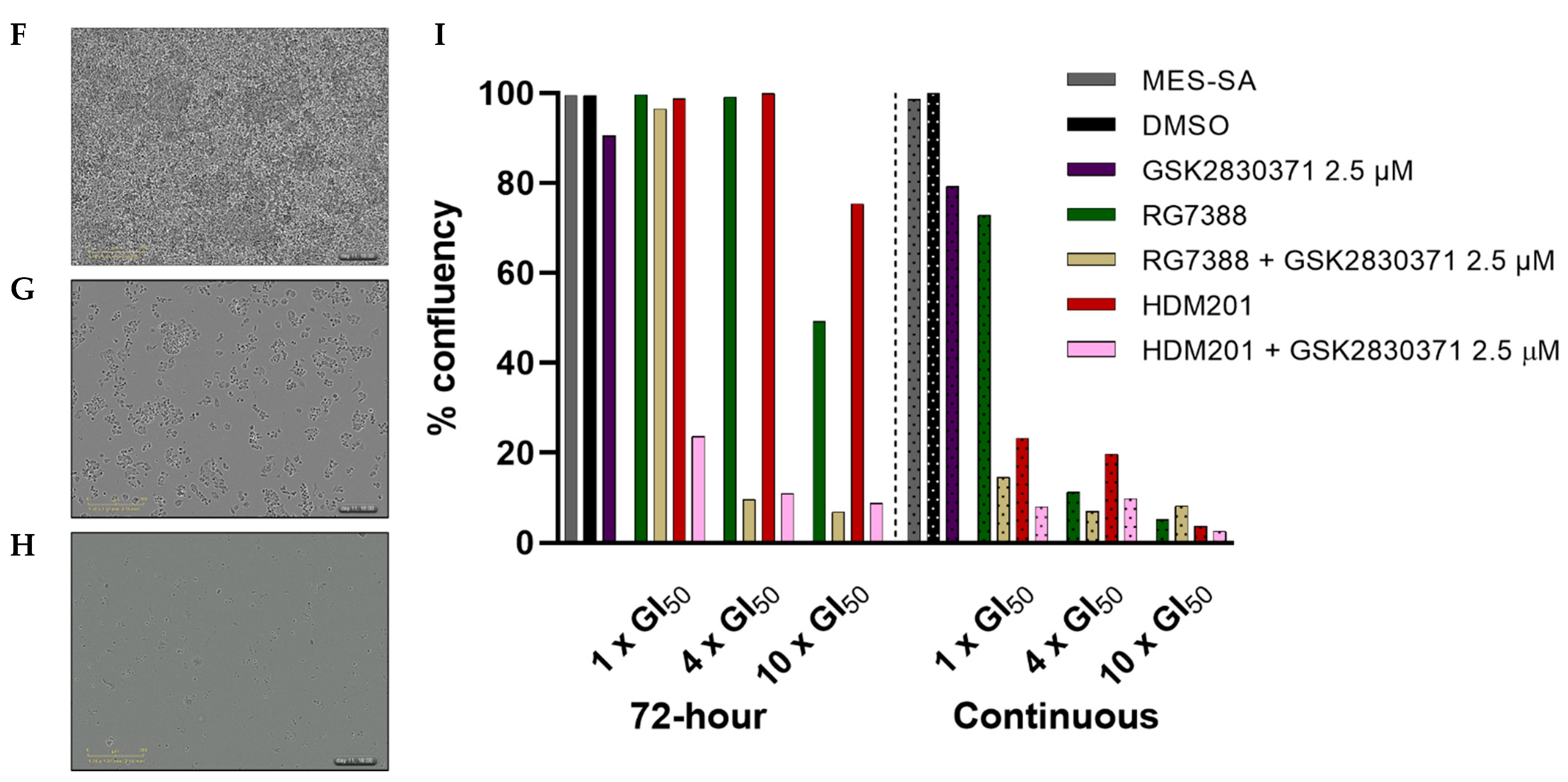
3.5. Combination with GSK2830371 Induces Irreversible Growth Arrest Following 72 h Treatment of p53WT uLMS Cells with nM Concentrations of MDM2 Inhibitors
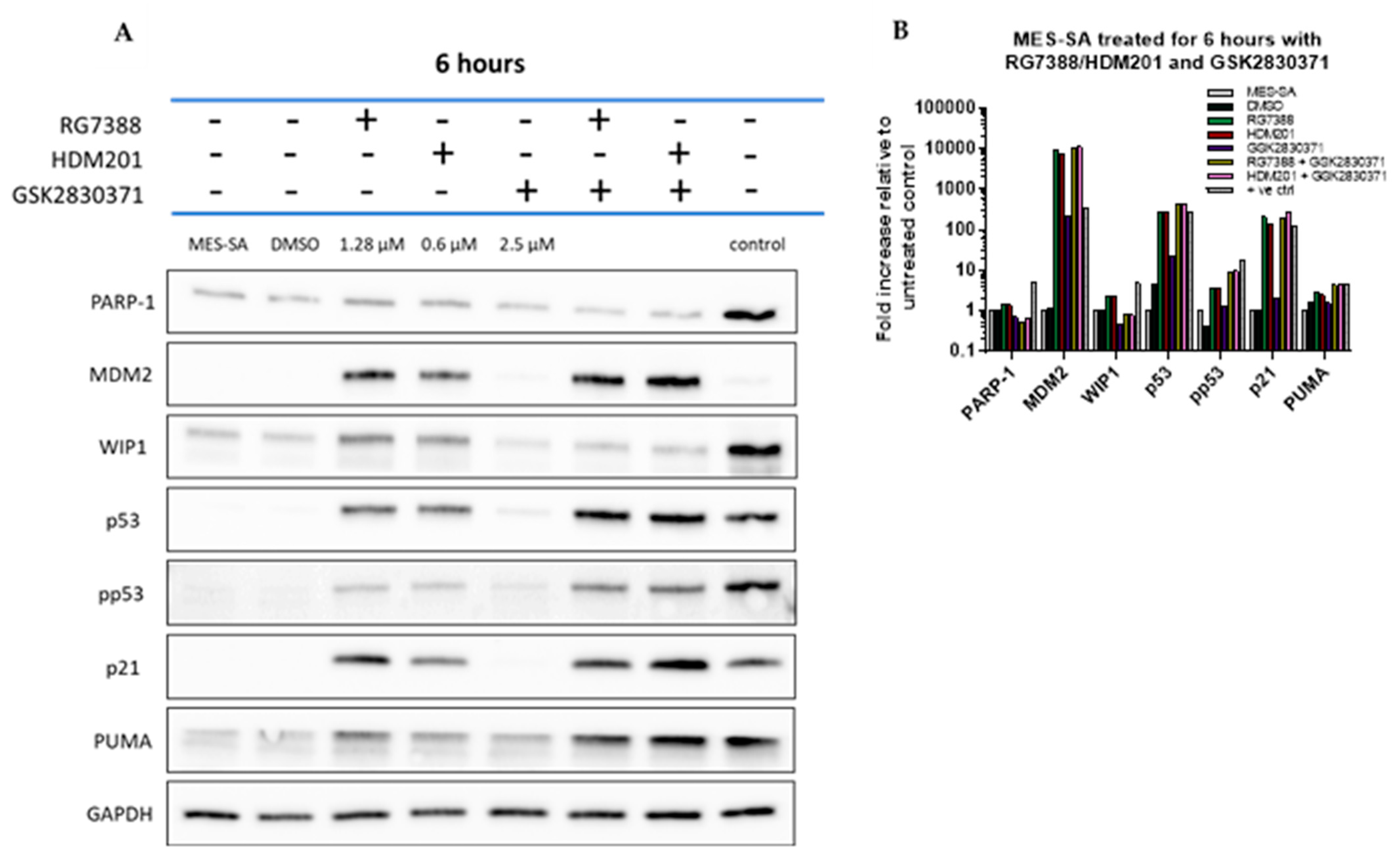
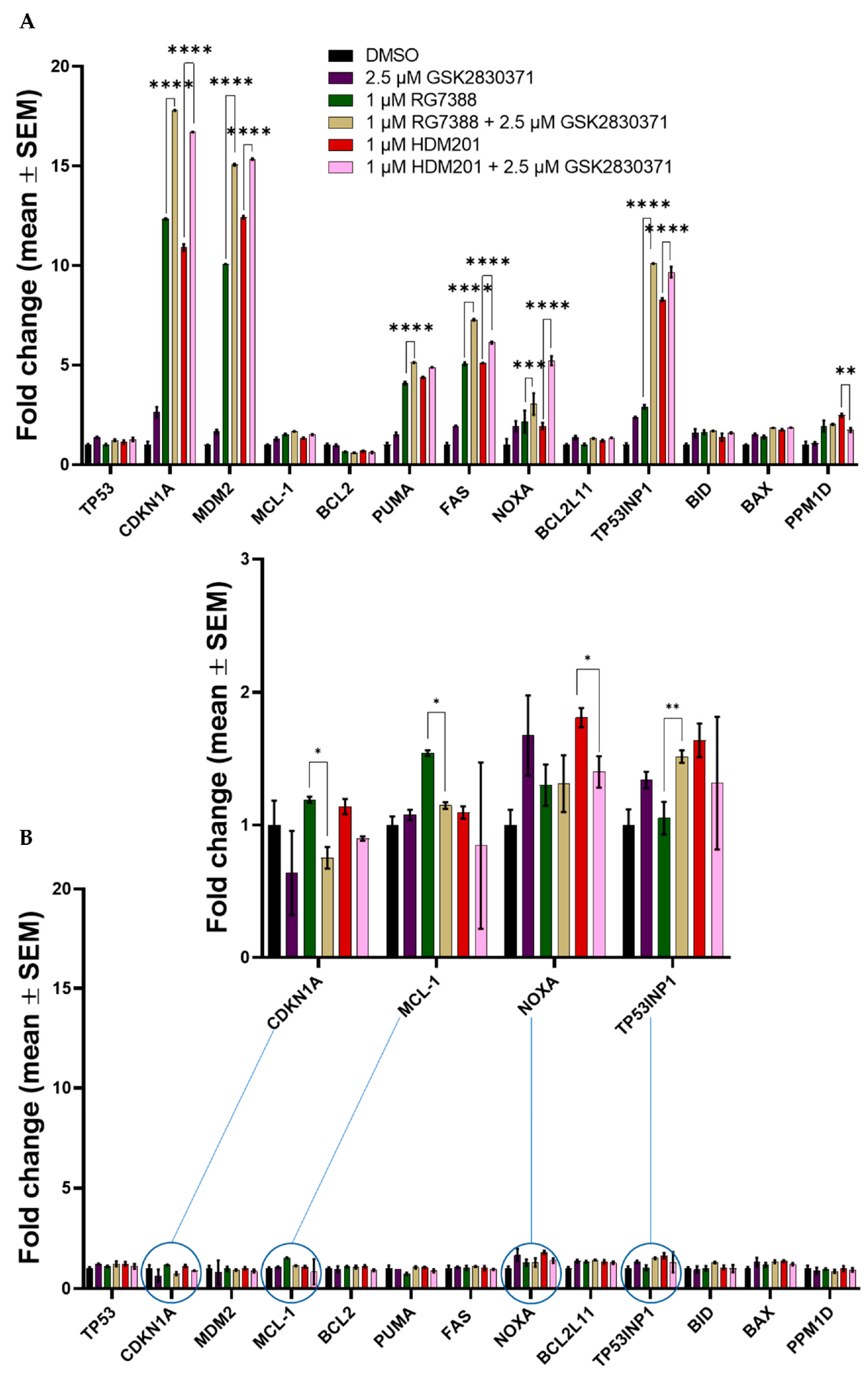
3.6. Combined MDM2 and WIP1 Inhibition Significantly Increases Transcript Levels of Pro-Apoptotic Genes in p53WT uLMS Cells
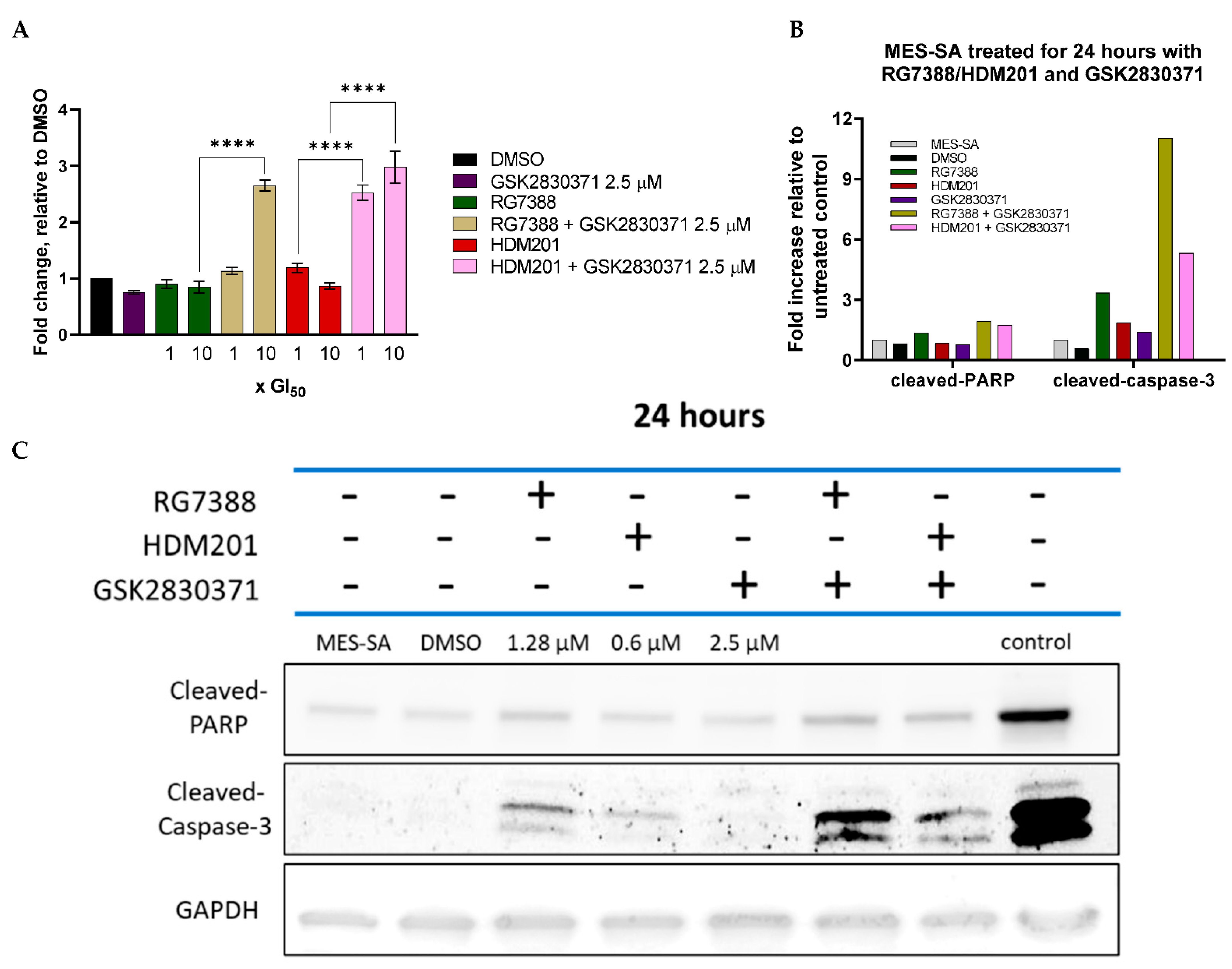
3.7. GSK2830371 Combination with MDM2 Inhibitors Induces Apoptosis
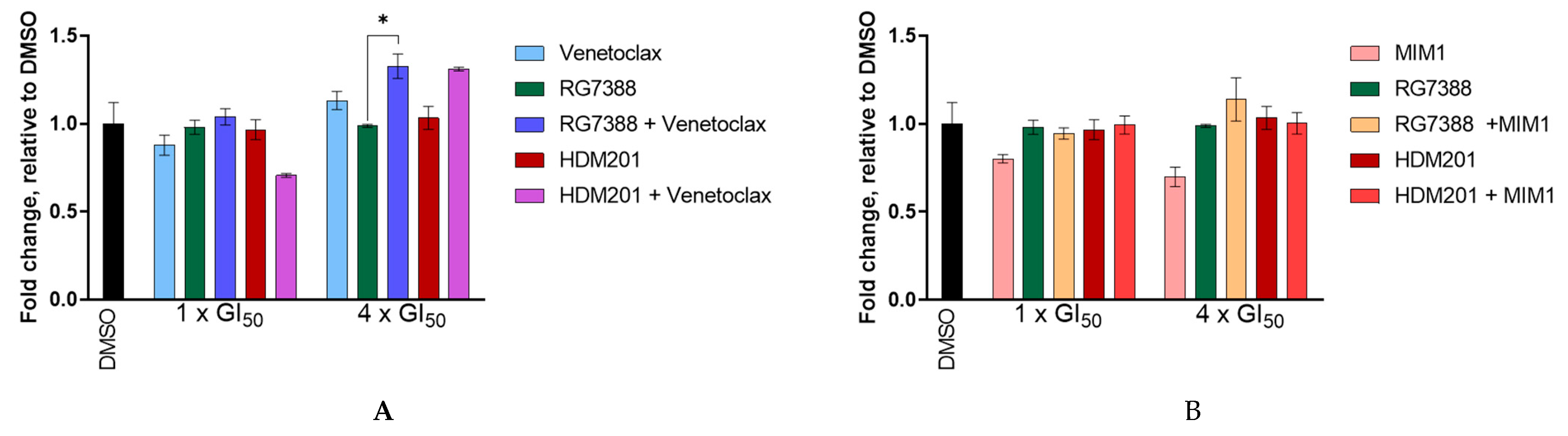
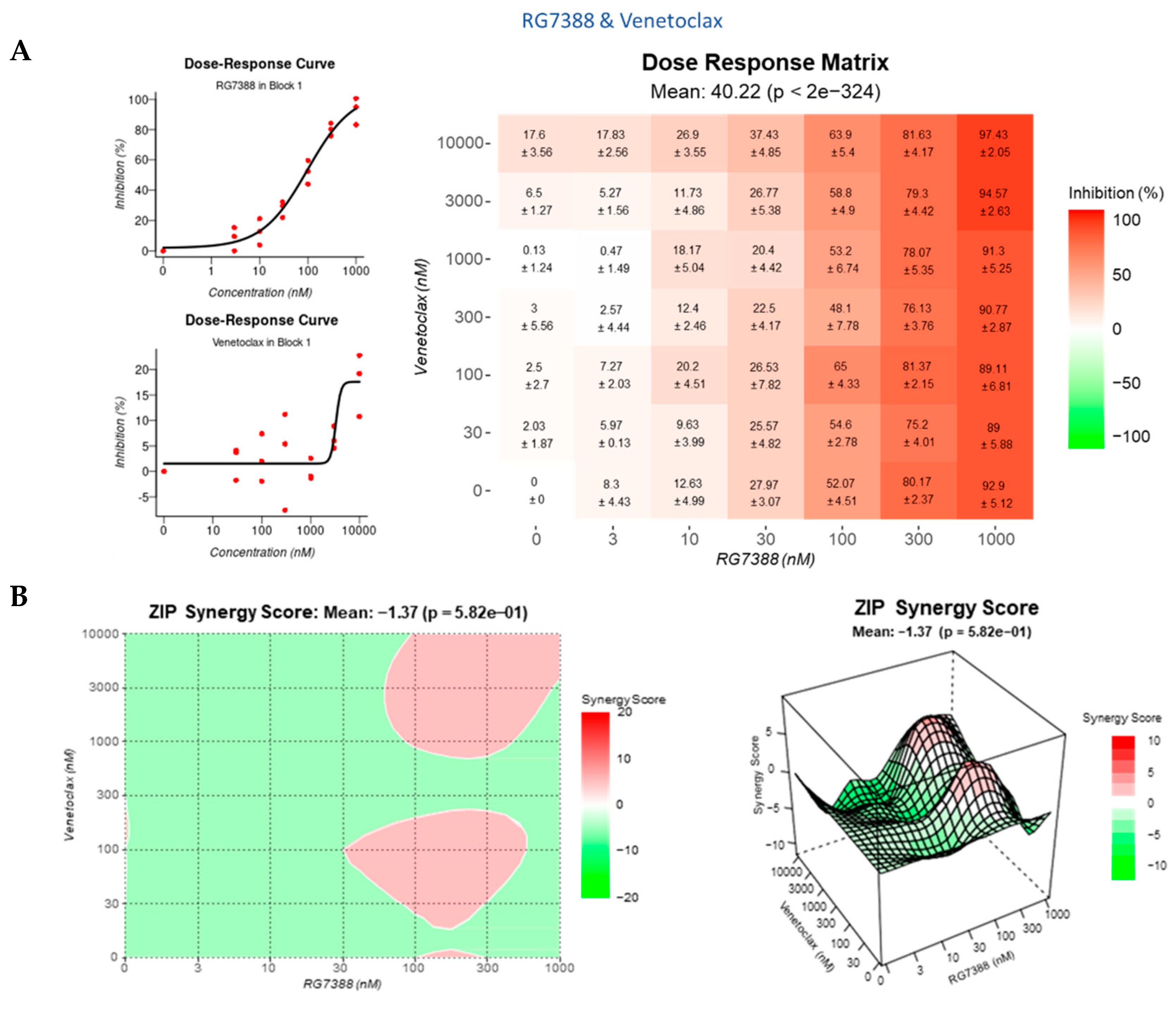
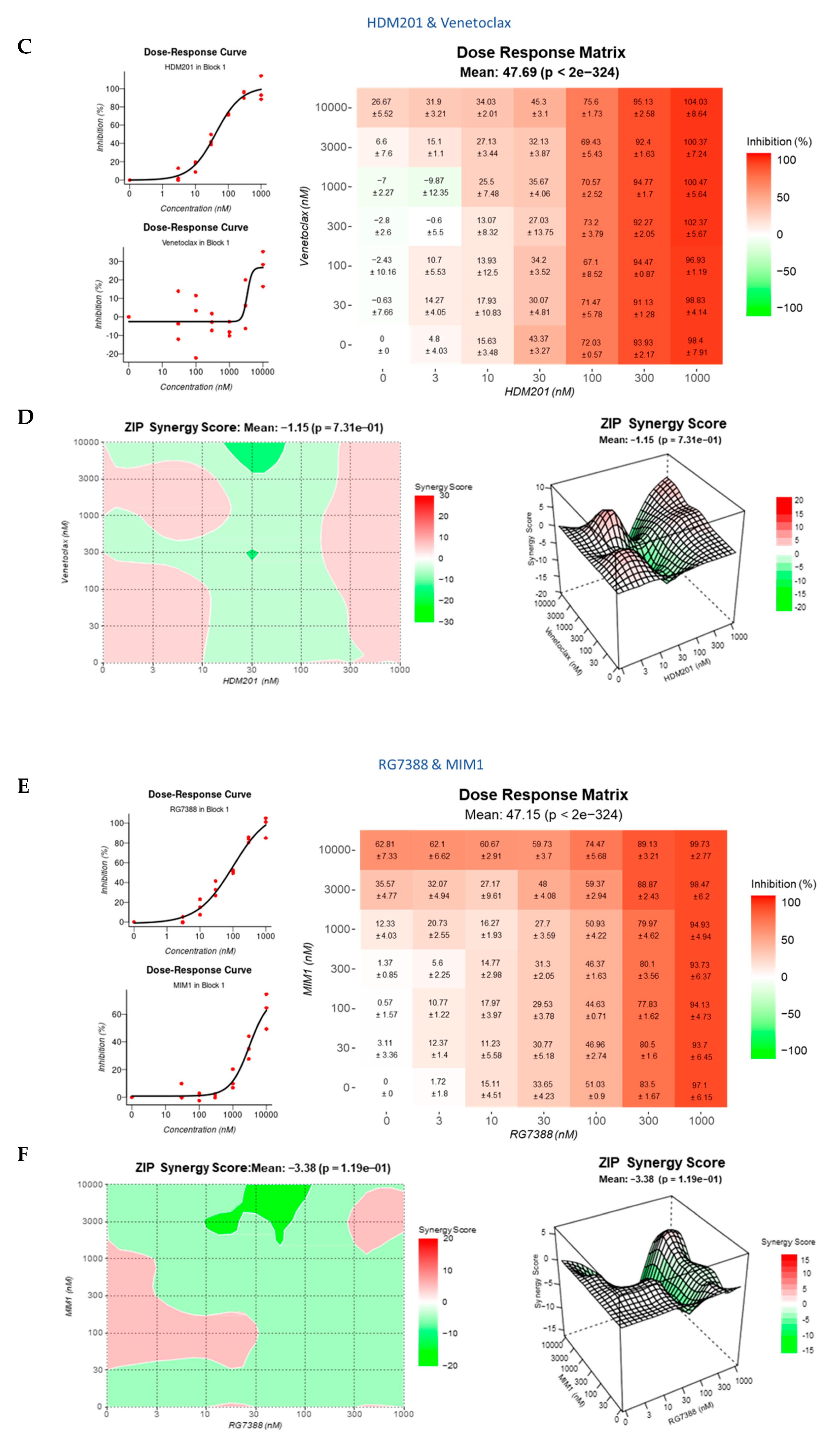
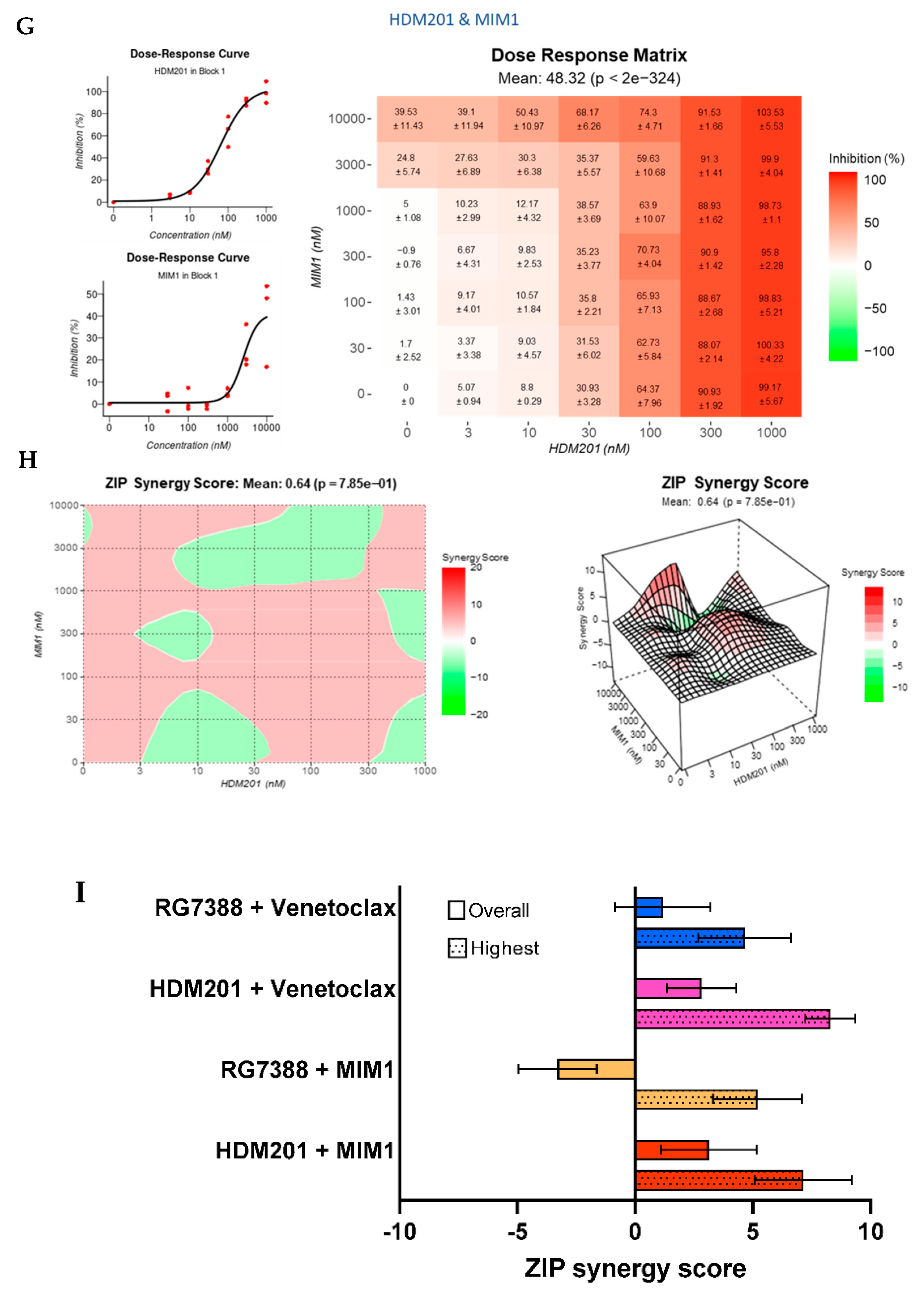
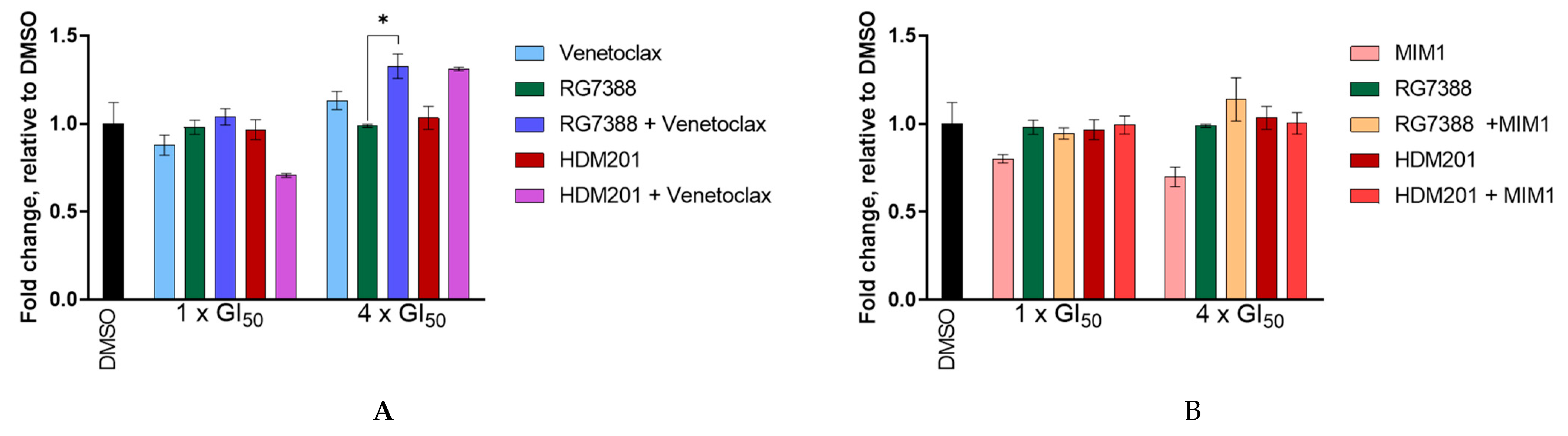
3.8. BCL2 and MCL-1 Inhibitors in Combination with MDM2 Inhibitors Fail to Lower the Apoptotic Threshold Required to Tip p53WT Cells into Apoptosis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Product | Source | Company | Dilution | Diluent |
|---|---|---|---|---|---|
| GAPDH | (14C10) #2118 | Rabbit | Cell Signalling Technologies | 1:1000 | Milk |
| PARP-1 | (C2-10) #4338-MC | Mouse | Trevigen | 1:1000 | Milk |
| Cleaved-PARP | (Asp 214) #9541 | Rabbit | Cell Signalling Technologies | 1:1000 | Milk |
| MDM2 | OP46 | Mouse | EMD Millipore Corporation | 1:300 | Milk |
| WIP1 | (F-10) sc-376257 | Mouse | Santa Cruz | 1:1000 | Milk |
| p53 | (DO-1) sc-126 | Mouse | Santa Cruz | 1:500 | Milk |
| pp53 | (E9Y4U) #82530 | Rabbit | Cell Signalling Technologies | 1:1000 | BSA |
| p21 | (EA10) OP64 | Mouse | EMD Millipore Corporation | 1:100 | Milk |
| PUMA | (D30C10) #12450 | Rabbit | Cell Signalling Technologies | 1:1000 | BSA |
| Cleaved-caspase-3 | (Asp175) (5A1E) #9664 | Rabbit | Cell Signalling Technologies | 1:1000 | Milk |
| Gene Name | Primer Sequence |
|---|---|
| GAPDH | F-5′-CAATGACCCCTTCATTGACC-3′ R-5′-GATCTCGCTCCTGGAAGAT-3′ |
| MCL-1 | F-5′-GTGCCTTTGTGGCTAAACACT-3′ R-5′-AGTCCCGTTTTGTCCTTACGA-3′ |
| BCL2 | F-5′-GGTGGGGTCATGTGTGTGG-3′ R-5′-CGGTTCAGGTACTCAGTCATCC-3′ |
| TP53 | F-5′-CAGCACATGACGGAGGTTGT-3′ R-5′-TCATCCAAATACTCCACACGC-3′ |
| P21 | F-5′-TGTCCGTCAGAACCCATGC-3′ R-5′-AAAGTCGAAGTTCCATCGCTC-3′ |
| MDM2 | F-5′-AGTAGCAGTGAATCTACAGGGA-3′ R-5′-CTGATCCAACCAATCACCTGAAT-3′ |
| PUMA | F-5′-ACCTCAACGCACAGTACGA-3′ R-5′-CTGGGTAAGGGCAGGAGTC-3′ |
| FAS | F-5′-AGATTGTGTGATGAAGGACATGG-3′ R-5′-TGTTGCTGGTGAGTGTGCATT-3′ |
| NOXA | F-5′-TGCTACACAATGTGGCGTC-3′ R-5′-ACTTGGACATGGCCTCCCTTA-3′ |
| BCL2L11 | F-5′-TAAGTTCTGAGTGTGACCGAGA-3′ R-5′-GCTCTGTCTGTAGGGAGGTAGG-3′ |
| TP53INP1 | F-5′-TCTTGAGTGCTTGGCTGATACA-3′ R-5′-GGTGGGGTGATAAACCAGCTC-3′ |
| BID | F-5′-ATGGACCGTAGCATCCCTCC-3′ R-5′-GTAGGTGCGTAGGTTCTGGT-3′ |
| BAX | F-5′-CCCGAGAGGTCTTTTTCCGAG-3′ R-5′-CCAGCCCATGATGGTTCTGAT-3′ |
| PPM1D | F-5′-TTGTCAGAGCTGTGGAGGTG-3′ R-5′-AGTGAGTCGAGGTCGTTTCC-3′ |
| MES-SA | Treatment | p-Value |
|---|---|---|
| CDKN1A | DMSO vs. 2.5 µM GSK2830371 | <0.0001 |
| DMSO vs. 1 µM RG7388 | <0.0001 | |
| DMSO vs. 1 µM HDM201 | <0.0001 | |
| DMSO vs. 1 µM HDM201 + 2.5 µM GSK2830371 | <0.0001 | |
| DMSO vs. 1 µM RG7388 + 2.5 µM GSK2830371 | <0.0001 | |
| 2.5 µM GSK2830371 vs. 1 µM RG7388 | <0.0001 | |
| 2.5 µM GSK2830371 vs. 1 µM HDM201 | <0.0001 | |
| 2.5 µM GSK2830371 vs. 1 µM HDM201 + 2.5 µM GSK2830371 | <0.0001 | |
| 2.5 µM GSK2830371 vs. 1 µM RG7388 + 2.5 µM GSK2830371 | <0.0001 | |
| 1 µM RG7388 vs. 1 µM HDM201 | <0.0001 | |
| 1 µM RG7388 vs. 1 µM HDM201 + 2.5 µM GSK2830371 | <0.0001 | |
| 1 µM RG7388 vs. 1 µM RG7388 + 2.5 µM GSK2830371 | <0.0001 | |
| 1 µM HDM201 vs. 1 µM HDM201 + 2.5 µM GSK2830371 | <0.0001 | |
| 1 µM HDM201 vs. 1 µM RG7388 + 2.5 µM GSK2830371 | <0.0001 | |
| 1 µM HDM201 + 2.5 µM GSK2830371 vs. 1 µM RG7388 + 2.5 µM GSK2830371 | <0.0001 | |
| MDM2 | DMSO vs. 2.5 µM GSK2830371 | 0.0088 |
| DMSO vs. 1 µM RG7388 | <0.0001 | |
| DMSO vs. 1 µM HDM201 | <0.0001 | |
| DMSO vs. 1 µM HDM201 + 2.5 µM GSK2830371 | <0.0001 | |
| DMSO vs. 1 µM RG7388 + 2.5 µM GSK2830371 | <0.0001 | |
| 2.5 µM GSK2830371 vs. 1 µM RG7388 | <0.0001 | |
| 2.5 µM GSK2830371 vs. 1 µM HDM201 | <0.0001 | |
| 2.5 µM GSK2830371 vs. 1 µM HDM201 + 2.5 µM GSK2830371 | <0.0001 | |
| 2.5 µM GSK2830371 vs. 1 µM RG7388 + 2.5 µM GSK2830371 | <0.0001 | |
| 1 µM RG7388 vs. 1 µM HDM201 | <0.0001 | |
| 1 µM RG7388 vs. 1 µM HDM201 + 2.5 µM GSK2830371 | <0.0001 | |
| 1 µM RG7388 vs. 1 µM RG7388 + 2.5 µM GSK2830371 | <0.0001 | |
| 1 µM HDM201 vs. 1 µM HDM201 + 2.5 µM GSK2830371 | <0.0001 | |
| 1 µM HDM201 vs. 1 µM RG7388 + 2.5 µM GSK2830371 | <0.0001 | |
| MCL-1 | DMSO vs. 1 µM RG7388 + 2.5 µM GSK2830371 | 0.0060 |
| PUMA | DMSO vs. 1 µM RG7388 | <0.0001 |
| DMSO vs. 1 µM HDM201 | <0.0001 | |
| DMSO vs. 1 µM HDM201 + 2.5 µM GSK2830371 | <0.0001 | |
| DMSO vs. 1 µM RG7388 + 2.5 µM GSK2830371 | <0.0001 | |
| 2.5 µM GSK2830371 vs. 1 µM RG7388 | <0.0001 | |
| 2.5 µM GSK2830371 vs. 1 µM HDM201 | <0.0001 | |
| 2.5 µM GSK2830371 vs. 1 µM HDM201 + 2.5 µM GSK2830371 | <0.0001 | |
| 2.5 µM GSK2830371 vs. 1 µM RG7388 + 2.5 µM GSK2830371 | <0.0001 | |
| 1 µM RG7388 vs. 1 µM HDM201 + 2.5 µM GSK2830371 | 0.0008 | |
| 1 µM RG7388 vs. 1 µM RG7388 + 2.5 µM GSK2830371 | <0.0001 | |
| 1 µM HDM201 vs. 1 µM RG7388 + 2.5 µM GSK2830371 | 0.0018 | |
| FAS | DMSO vs. 2.5 µM GSK2830371 | <0.0001 |
| DMSO vs. 1 µM RG7388 | <0.0001 | |
| DMSO vs. 1 µM HDM201 | <0.0001 | |
| DMSO vs. 1 µM HDM201 + 2.5 µM GSK2830371 | <0.0001 | |
| DMSO vs. 1 µM RG7388 + 2.5 µM GSK2830371 | <0.0001 | |
| 2.5 µM GSK2830371 vs. 1 µM RG7388 | <0.0001 | |
| 2.5 µM GSK2830371 vs. 1 µM HDM201 | <0.0001 | |
| 2.5 µM GSK2830371 vs. 1 µM HDM201 + 2.5 µM GSK2830371 | <0.0001 | |
| 2.5 µM GSK2830371 vs. 1 µM RG7388 + 2.5 µM GSK2830371 | <0.0001 | |
| 1 µM RG7388 vs. 1 µM HDM201 + 2.5 µM GSK2830371 | <0.0001 | |
| 1 µM RG7388 vs. 1 µM RG7388 + 2.5 µM GSK2830371 | <0.0001 | |
| 1 µM HDM201 vs. 1 µM HDM201 + 2.5 µM GSK2830371 | <0.0001 | |
| 1 µM HDM201 vs. 1 µM RG7388 + 2.5 µM GSK2830371 | <0.0001 | |
| 1 µM HDM201 + 2.5 µM GSK2830371 vs. 1 µM RG7388 + 2.5 µM GSK2830371 | <0.0001 | |
| NOXA | DMSO vs. 2.5 µM GSK2830371 | <0.0001 |
| DMSO vs. 1 µM RG7388 | <0.0001 | |
| DMSO vs. 1 µM HDM201 | <0.0001 | |
| DMSO vs. 1 µM HDM201 + 2.5 µM GSK2830371 | <0.0001 | |
| DMSO vs. 1 µM RG7388 + 2.5 µM GSK2830371 | <0.0001 | |
| 2.5 µM GSK2830371 vs. 1 µM HDM201 + 2.5 µM GSK2830371 | <0.0001 | |
| 2.5 µM GSK2830371 vs. 1 µM RG7388 + 2.5 µM GSK2830371 | <0.0001 | |
| 1 µM RG7388 vs. 1 µM HDM201 + 2.5 µM GSK2830371 | <0.0001 | |
| 1 µM RG7388 vs. 1 µM RG7388 + 2.5 µM GSK2830371 | 0.0001 | |
| 1 µM HDM201 vs. 1 µM HDM201 + 2.5 µM GSK2830371 | <0.0001 | |
| 1 µM HDM201 vs. 1 µM RG7388 + 2.5 µM GSK2830371 | <0.0001 | |
| 1 µM HDM201 + 2.5 µM GSK2830371 vs. 1 µM RG7388 + 2.5 µM GSK2830371 | <0.0001 | |
| TP53INP1 | DMSO vs. 2.5 µM GSK2830371 | <0.0001 |
| DMSO vs. 1 µM RG7388 | <0.0001 | |
| DMSO vs. 1 µM HDM201 | <0.0001 | |
| DMSO vs. 1 µM HDM201 + 2.5 µM GSK2830371 | <0.0001 | |
| DMSO vs. 1 µM RG7388 + 2.5 µM GSK2830371 | <0.0001 | |
| 2.5 µM GSK2830371 vs. 1 µM HDM201 | <0.0001 | |
| 2.5 µM GSK2830371 vs. 1 µM HDM201 + 2.5 µM GSK2830371 | <0.0001 | |
| 2.5 µM GSK2830371 vs. 1 µM RG7388 + 2.5 µM GSK2830371 | <0.0001 | |
| 1 µM RG7388 vs. 1 µM HDM201 | <0.0001 | |
| 1 µM RG7388 vs. 1 µM HDM201 + 2.5 µM GSK2830371 | <0.0001 | |
| 1 µM RG7388 vs. 1 µM RG7388 + 2.5 µM GSK2830371 | <0.0001 | |
| 1 µM HDM201 vs. 1 µM HDM201 + 2.5 µM GSK2830371 | <0.0001 | |
| 1 µM HDM201 vs. 1 µM RG7388 + 2.5 µM GSK2830371 | <0.0001 | |
| BID | DMSO vs. 2.5 µM GSK2830371 | 0.0242 |
| DMSO vs. 1 µM RG7388 | 0.0119 | |
| DMSO vs. 1 µM HDM201 + 2.5 µM GSK2830371 | 0.0220 | |
| DMSO vs. 1 µM RG7388 + 2.5 µM GSK2830371 | 0.0039 | |
| BAX | DMSO vs. 1 µM HDM201 | 0.0013 |
| DMSO vs. 1 µM HDM201 + 2.5 µM GSK2830371 | 0.0001 | |
| DMSO vs. 1 µM RG7388 + 2.5 µM GSK2830371 | 0.0002 | |
| PPM1D | DMSO vs. 1 µM RG7388 | <0.0001 |
| DMSO vs. 1 µM HDM201 | <0.0001 | |
| DMSO vs. 1 µM HDM201 + 2.5 µM GSK2830371 | 0.0020 | |
| DMSO vs. 1 µM RG7388 + 2.5 µM GSK2830371 | <0.0001 | |
| 2.5 µM GSK2830371 vs. 1 µM RG7388 | 0.0002 | |
| 2.5 µM GSK2830371 vs. 1 µM HDM201 | <0.0001 | |
| 2.5 µM GSK2830371 vs. 1 µM HDM201 + 2.5 µM GSK2830371 | 0.0088 | |
| 2.5 µM GSK2830371 vs. 1 µM RG7388 + 2.5 µM GSK2830371 | <0.0001 | |
| 1 µM HDM201 vs. 1 µM HDM201 + 2.5 µM GSK2830371 | 0.0019 |
| SK-UT-1 | Treatment | p-Value |
|---|---|---|
| CDKN1A | 2.5 µM GSK2830371 vs. 1 µM RG7388 | 0.0005 |
| 2.5 µM GSK2830371 vs. 1 µM HDM201 | 0.0022 | |
| 1 µM RG7388 vs. 1 µM RG7388 + 2.5 µM GSK2830371 | 0.0114 | |
| 1 µM HDM201 vs. 1 µM RG7388 + 2.5 µM GSK2830371 | 0.0381 | |
| MCL-1 | DMSO vs. 1 µM RG7388 | 0.0006 |
| 2.5 µM GSK2830371 vs. 1 µM RG7388 | 0.0053 | |
| 1 µM RG7388 vs. 1 µM HDM201 | 0.0085 | |
| 1 µM RG7388 vs. 1 µM HDM201 + 2.5 µM GSK2830371 | <0.0001 | |
| 1 µM RG7388 vs. 1 µM RG7388 + 2.5 µM GSK2830371 | 0.0300 | |
| NOXA | DMSO vs. 2.5 µM GSK2830371 | <0.0001 |
| DMSO vs. 1 µM HDM201 | <0.0001 | |
| DMSO vs. 1 µM HDM201 + 2.5 µM GSK2830371 | 0.0280 | |
| 2.5 µM GSK2830371 vs. 1 µM RG7388 | 0.0492 | |
| 1 µM RG7388 vs. 1 µM HDM201 | 0.0017 | |
| 1 µM HDM201 vs. 1 µM HDM201 + 2.5 µM GSK2830371 | 0.0228 | |
| 1 µM HDM201 vs. 1 µM RG7388 + 2.5 µM GSK2830371 | 0.0024 | |
| BCL2L11 | DMSO vs. 2.5 µM GSK2830371 | 0.0462 |
| DMSO vs. 1 µM RG7388 + 2.5 µM GSK2830371 | 0.0136 | |
| TP53INP1 | DMSO vs. 1 µM HDM201 | <0.0001 |
| DMSO vs. 1 µM RG7388 + 2.5 µM GSK2830371 | 0.0014 | |
| 1 µM RG7388 vs. 1 µM HDM201 | 0.0002 | |
| 1 µM RG7388 vs. 1 µM RG7388 + 2.5 µM GSK2830371 | 0.0058 | |
| BAX | DMSO vs. 1 µM HDM201 | 0.0453 |
References
- Zhao, Y.; Aguilar, A.; Bernard, D.; Wang, S. Small-Molecule Inhibitors of the MDM2–p53 Protein–Protein Interaction (MDM2 Inhibitors) in Clinical Trials for Cancer Treatment. J. Med. Chem. 2014, 58, 1038–1052. [Google Scholar] [CrossRef] [PubMed]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In Vivo Activation of the p53 Pathway by Small-Molecule Antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef] [Green Version]
- Ray-Coquard, I.; Blay, J.-Y.; Italiano, A.; Le Cesne, A.; Penel, N.; Zhi, J.; Heil, F.; Rueger, R.; Graves, B.; Ding, M.; et al. Effect of the MDM2 antagonist RG7112 on the P53 pathway in patients with MDM2-amplified, well-differentiated or dedifferentiated liposarcoma: An exploratory proof-of-mechanism study. Lancet Oncol. 2012, 13, 1133–1140. [Google Scholar] [CrossRef]
- Levine, A.J. p53, the Cellular Gatekeeper for Growth and Division. Cell 1997, 88, 323–331. [Google Scholar] [CrossRef] [Green Version]
- Kern, S.E.; Kinzler, K.W.; Bruskin, A.; Jarosz, D.; Friedman, P.; Prives, C.; Vogelstein, B. Identification of p53 as a Sequence-Specific DNA-Binding Protein. Science 1991, 252, 1708–1711. [Google Scholar] [CrossRef]
- Pietenpol, J.A.; Tokino, T.; Thiagalingam, S.; El-Deiry, W.S.; Kinzler, K.W.; Vogelstein, B. Sequence-specific transcriptional activation is essential for growth suppression by p53. Proc. Natl. Acad. Sci. USA 1994, 91, 1998–2002. [Google Scholar] [CrossRef] [Green Version]
- Rotter, V. p53, a transformation-related cellular-encoded protein, can be used as a biochemical marker for the detection of primary mouse tumor cells. Proc. Natl. Acad. Sci. USA 1983, 80, 2613–2617. [Google Scholar] [CrossRef] [Green Version]
- Shi, D.; Gu, W. Dual Roles of MDM2 in the Regulation of p53: Ubiquitination Dependent and Ubiquitination Independent Mechanisms of MDM2 Repression of p53 Activity. Genes Cancer 2012, 3, 240–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowe, S.W.; Cepero, E.; Evan, G. Intrinsic tumour suppression. Nat. Cell Biol. 2004, 432, 307–315. [Google Scholar] [CrossRef]
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ventura, A.; Kirsch, D.G.; McLaughlin, M.E.; Tuveson, D.A.; Grimm, J.; Lintault, L.; Newman, J.; Reczek, E.E.; Weissleder, R.; Jacks, T. Restoration of p53 function leads to tumour regression in vivo. Nature 2007, 445, 661–665. [Google Scholar] [CrossRef]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Kubbutat, M.H.; Vousden, K.H. Proteolytic cleavage of human p53 by calpain: A potential regulator of protein stability. Mol. Cell. Biol. 1997, 17, 460–468. [Google Scholar] [CrossRef] [Green Version]
- Moll, U.M.; Petrenko, O. The MDM2-p53 interaction. Mol. Cancer Res. 2003, 1, 1001–1008. [Google Scholar]
- Michaelis, M.; Rothweiler, F.; Barth, S.; Cinatl, J.; Van Rikxoort, M.; Löschmann, N.; Voges, Y.; Breitling, R.; Von Deimling, A.; Rödel, F.; et al. Adaptation of cancer cells from different entities to the MDM2 inhibitor nutlin-3 results in the emergence of p53-mutated multi-drug-resistant cancer cells. Cell Death Dis. 2011, 2, e243. [Google Scholar] [CrossRef]
- Ruark, E.; Snape, K.; Humburg, P.; Loveday, C.; Bajrami, I.; Brough, R.; Rodrigues, D.N.; Renwick, A.; Seal, S.; Ramsay, E.; et al. Mosaic PPM1D mutations are associated with predisposition to breast and ovarian cancer. Nature 2013, 493, 406–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rauta, J.; Alarmo, E.-L.; Kauraniemi, P.; Karhu, R.; Kuukasjärvi, T.; Kallioniemi, A. The serine-threonine protein phosphatase PPM1D is frequently activated through amplification in aggressive primary breast tumours. Breast Cancer Res. Treat. 2005, 95, 257–263. [Google Scholar] [CrossRef]
- Tan, D.S.P.; Lambros, M.B.K.; Rayter, S.; Natrajan, R.; Vatcheva, R.; Gao, Q.; Marchiò, C.; Geyer, F.C.; Savage, K.; Parry, S.; et al. PPM1D Is a Potential Therapeutic Target in Ovarian Clear Cell Carcinomas. Clin. Cancer Res. 2009, 15, 2269–2280. [Google Scholar] [CrossRef] [Green Version]
- Lowe, J.; Cha, H.; Lee, M.O.; Mazur, S.J.; Appella, E.; Fornace, A.J., Jr. Regulation of the Wip1 phosphatase and its effects on the stress response. Front. Biosci. 2012, 17, 1480–1498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, M.; Shi, J.; Jung, S.H.; Chen, X.; Cho, K.-H. Attractor Landscape Analysis Reveals Feedback Loops in the p53 Network That Control the Cellular Response to DNA Damage. Sci. Signal. 2012, 5, ra83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilmartin, A.G.; Faitg, T.H.; Richter, M.; Groy, A.; Seefeld, M.A.; Darcy, M.G.; Peng, X.; Federowicz, K.; Yang, J.; Zhang, S.-Y.; et al. Allosteric Wip1 phosphatase inhibition through flap-subdomain interaction. Nat. Chem. Biol. 2014, 10, 181–187. [Google Scholar] [CrossRef]
- Pechackova, S.; Burdova, K.; Benada, J.; Kleiblova, P.; Jenikova, G.; Macurek, L. Inhibition of WIP1 phosphatase sensitizes breast cancer cells to genotoxic stress and to MDM2 antagonist nutlin-3. Oncotarget 2016, 7, 14458–14475. [Google Scholar] [CrossRef] [Green Version]
- Esfandiari, A.; Hawthorne, T.A.; Nakjang, S.; Lunec, J. Chemical Inhibition of Wild-Type p53-Induced Phosphatase 1 (WIP1/PPM1D) by GSK2830371 Potentiates the Sensitivity to MDM2 Inhibitors in a p53-Dependent Manner. Mol. Cancer Ther. 2016, 15, 379–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sriraman, A.; Radovanović, M.; Wienken, M.; Najafova, Z.; Li, Y.; Dobbelstein, M. Cooperation of Nutlin-3a and a Wip1 inhibitor to induce p53 activity. Oncotarget 2016, 7, 31623–31638. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.-E.; Esfandiari, A.; Ho, Y.-H.; Wang, N.; Mahdi, A.; Aptullahoglu, E.; Lovat, P.; Lunec, J. Targeting negative regulation of p53 by MDM2 and WIP1 as a therapeutic strategy in cutaneous melanoma. Br. J. Cancer 2017, 118, 495–508. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Tsujimoto, Y.; Cossman, J.; Jaffe, E.; Croce, C.M. Involvement of the bcl-2 Gene in Human Follicular Lymphoma. Science 1985, 228, 1440–1443. [Google Scholar] [CrossRef] [PubMed]
- Delia, D.; Aiello, A.; Soligo, D.; Fontanella, E.; Melani, C.; Pezzella, F.; Pierotti, M.A.; Della Porta, G. bcl-2 proto-oncogene expression in normal and neoplastic human myeloid cells. Blood 1992, 79, 1291–1298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aisenberg, A.C.; Wilkes, B.M.; Jacobson, J.O. The bcl-2 gene is rearranged in many diffuse B-cell lymphomas. Blood 1988, 71, 969–972. [Google Scholar] [CrossRef] [Green Version]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef]
- Hoffman-Luca, C.G.; Ziazadeh, D.; McEachern, N.; Zhao, Y.; Sun, W.; Debussche, L.; Wang, S. Elucidation of Acquired Resistance to Bcl-2 and MDM2 Inhibitors in Acute Leukemia In Vitro and In Vivo. Clin. Cancer Res. 2015, 21, 2558–2568. [Google Scholar] [CrossRef] [Green Version]
- Carter, B.Z.; Mak, P.Y.; Mak, D.H.; Ruvolo, V.R.; Schober, W.; McQueen, T.; Cortes, J.; Kantarjian, H.M.; Champlin, R.E.; Konopleva, M.; et al. Synergistic effects of p53 activation via MDM2 inhibition in combination with inhibition of Bcl-2 or Bcr-Abl in CD34+ proliferating and quiescent chronic myeloid leukemia blast crisis cells. Oncotarget 2015, 6, 30487–30499. [Google Scholar] [CrossRef] [Green Version]
- Pan, R.; Ruvolo, V.; Mu, H.; Leverson, J.D.; Nichols, G.; Reed, J.C.; Konopleva, M.; Andreeff, M. Synthetic Lethality of Combined Bcl-2 Inhibition and p53 Activation in AML: Mechanisms and Superior Antileukemic Efficacy. Cancer Cell 2017, 32, 748–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decaudin, D.; Leitz, E.F.D.; Nemati, F.; Tarin, M.; Naguez, A.; Zerara, M.; Marande, B.; Vivet-Noguer, R.; Halilovic, E.; Fabre, C.; et al. Preclinical evaluation of drug combinations identifies co-inhibition of Bcl-2/XL/W and MDM2 as a potential therapy in uveal melanoma. Eur. J. Cancer 2020, 126, 93–103. [Google Scholar] [CrossRef] [Green Version]
- Mallick, D.; Soderquist, R.S.; Bates, D.; Eastman, A. Confounding off-target effects of BH3 mimetics at commonly used concentrations: MIM1, UMI-77, and A-1210477. Cell Death Dis. 2019, 10, 185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, N.A.; Stewart, M.L.; Gavathiotis, E.; Tepper, J.L.; Bruekner, S.R.; Koss, B.; Opferman, J.T.; Walensky, L.D. A Competitive Stapled Peptide Screen Identifies a Selective Small Molecule that Overcomes MCL-1-Dependent Leukemia Cell Survival. Chem. Biol. 2012, 19, 1175–1186. [Google Scholar] [CrossRef] [Green Version]
- Respondek, M.; Beberok, A.; Rok, J.; Rzepka, Z.; Wrześniok, D.; Buszman, E. MIM1, the Mcl-1—specific BH3 mimetic induces apoptosis in human U87MG glioblastoma cells. Toxicol. In Vitro 2018, 53, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Respondek, M.; Beberok, A.; Rzepka, Z.; Rok, J.; Wrześniok, D. MIM1 induces COLO829 melanoma cell death through mitochondrial membrane breakdown, GSH depletion, and DNA damage. Fundam. Clin. Pharmacol. 2019, 34, 20–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ianevski, A.; He, L.; Aittokallio, T.; Tang, J. SynergyFinder: A web application for analyzing drug combination dose–response matrix data. Bioinformatics 2017, 33, 2413–2415. [Google Scholar] [CrossRef]
- Italiano, A.; Miller, W.H.; Blay, J.-Y.; Gietema, J.A.; Bang, Y.-J.; Mileshkin, L.R.; Hirte, H.W.; Higgins, B.; Blotner, S.; Nichols, G.L.; et al. Phase I study of daily and weekly regimens of the orally administered MDM2 antagonist idasanutlin in patients with advanced tumors. Investig. New Drugs 2021, 39, 1587–1597. [Google Scholar] [CrossRef]
- Zanjirband, M.; Edmondson, R.; Lunec, J. Pre-clinical efficacy and synergistic potential of the MDM2-p53 antagonists, Nutlin-3 and RG7388, as single agents and in combined treatment with cisplatin in ovarian cancer. Oncotarget 2016, 7, 40115–40134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Deiry, W.S.; Tokino, T.; Velculescu, V.E.; Levy, D.B.; Parsons, R.; Trent, J.M.; Lin, D.; Mercer, W.E.; Kinzler, K.W.; Vogelstein, B. WAF1, a potential mediator of p53 tumor suppression. Cell 1993, 75, 817–825. [Google Scholar] [CrossRef]
- Khoo, K.H.; Verma, C.S.; Lane, D.P. Drugging the p53 pathway: Understanding the route to clinical efficacy. Nat. Rev. Drug Discov. 2014, 13, 217–236. [Google Scholar] [CrossRef] [PubMed]
- Kracikova, M.; Akiri, G.; George, A.; Sachidanandam, R.; Aaronson, S.A. A threshold mechanism mediates p53 cell fate decision between growth arrest and apoptosis. Cell Death Differ. 2013, 20, 576–588. [Google Scholar] [CrossRef] [Green Version]
- Lu, M.; Breyssens, H.; Salter, V.; Zhong, S.; Hu, Y.; Baer, C.; Ratnayaka, I.; Sullivan, A.; Brown, N.R.; Endicott, J.; et al. Restoring p53 function in human melanoma cells by inhibiting MDM2 and cyclin B1/CDK1-phosphorylated nuclear iASPP. Cancer Cell 2013, 23, 618–633. [Google Scholar] [CrossRef] [Green Version]
- Ciardullo, C.; Aptullahoglu, E.; Woodhouse, L.; Lin, W.-Y.; Wallis, J.P.; Marr, H.; Marshall, S.; Bown, N.; Willmore, E.; Lunec, J. Non-genotoxic MDM2 inhibition selectively induces a pro-apoptotic p53 gene signature in chronic lymphocytic leukemia cells. Haematologica 2019, 104, 2429–2442. [Google Scholar] [CrossRef] [Green Version]
- Tracz-Gaszewska, Z.; Klimczak, M.; Biecek, P.; Herok, M.; Kosinski, M.; Olszewski, M.; Czerwińska, P.; Wiech, M.; Wiznerowicz, M.; Zylicz, A.; et al. Molecular chaperones in the acquisition of cancer cell chemoresistance with mutated TP53 and MDM2 up-regulation. Oncotarget 2017, 8, 82123–82143. [Google Scholar] [CrossRef] [Green Version]
- Maganti, H.B.; Jrade, H.; Cafariello, C.; Rothberg, J.L.M.; Porter, C.J.; Yockell-Lelièvre, J.; Battaion, H.L.; Khan, S.T.; Howard, J.P.; Li, Y.; et al. Targeting the MTF2–MDM2 Axis Sensitizes Refractory Acute Myeloid Leukemia to Chemotherapy. Cancer Discov. 2018, 8, 1376–1389. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Hu, B.; Qin, J.; Cheng, J.-W.; Li, X.; Rajaei, M.; Fan, J.; Yang, X.-R.; Zhang, R. A novel inhibitor of MDM2 oncogene blocks metastasis of hepatocellular carcinoma and overcomes chemoresistance. Genes Dis. 2019, 6, 419–430. [Google Scholar] [CrossRef]
- Giordana, M.T.; Duó, D.; Gasverde, S.; Trevisan, E.; Boghi, A.; Morra, I.; Pradotto, L.; Mauro, A.; Chió, A. MDM2 overexpression is associated with short survival in adults with medulloblastoma. Neuro Oncol. 2002, 4, 115–122. [Google Scholar] [CrossRef]
- Gu, L.; Zhu, N.; Findley, H.W.; Zhou, M. MDM2 antagonist nutlin-3 is a potent inducer of apoptosis in pediatric acute lymphoblastic leukemia cells with wild-type p53 and overexpression of MDM2. Leukemia 2008, 22, 730–739. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Gu, L.; Li, J.; Shah, N.; He, J.; Yang, L.; Hu, Q.; Zhou, M. Degradation of MDM2 by the Interaction between Berberine and DAXX Leads to Potent Apoptosis in MDM2-Overexpressing Cancer Cells. Cancer Res. 2010, 70, 9895–9904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barretina, J.; Taylor, B.S.; Banerji, S.; Ramos, A.H.; Lagos-Quintana, M.; DeCarolis, P.L.; Shah, K.; Socci, N.D.; Weir, B.A.; Ho, A.; et al. Subtype-specific genomic alterations define new targets for soft-tissue sarcoma therapy. Nat. Genet. 2010, 42, 715–721. [Google Scholar] [CrossRef] [Green Version]
- Hensley, M.L.; Chavan, S.S.; Solit, D.B.; Murali, R.; Soslow, R.; Chiang, S.; Jungbluth, A.A.; Bandlamudi, C.; Srinivasan, P.; Tap, W.D.; et al. Genomic Landscape of Uterine Sarcomas Defined Through Prospective Clinical Sequencing. Clin. Cancer Res. 2020, 26, 3881–3888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.-E.; Huang, C.-Y.; Chen, C.-P.; Pan, Y.-R.; Chang, J.; Chen, J.-S.; Yeh, C.-N.; Lunec, J. WIP1 Inhibition by GSK2830371 Potentiates HDM201 through Enhanced p53 Phosphorylation and Activation in Liver Adenocarcinoma Cells. Cancers 2021, 13, 3876. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, N.; Amato, C.M.; Skees, J.; Todd, K.J.; Lambert, K.A.; Robinson, W.A.; Van Gulick, R.; Weight, R.M.; Dart, C.R.; Tobin, R.P.; et al. Simultaneously Inhibiting BCL2 and MCL1 Is a Therapeutic Option for Patients with Advanced Melanoma. Cancers 2020, 12, 2182. [Google Scholar] [CrossRef]
- Seiller, C.; Maiga, S.; Touzeau, C.; Bellanger, C.; Kervoëlen, C.; Descamps, G.; Maillet, L.; Moreau, P.; Deceunynck, C.; Bougie, P.G.; et al. Dual targeting of BCL2 and MCL1 rescues myeloma cells resistant to BCL2 and MCL1 inhibitors associated with the formation of BAX/BAK hetero-complexes. Cell Death Dis. 2020, 11, 1–14. [Google Scholar] [CrossRef]
- Jeay, S.; Gaulis, S.; Ferretti, S.; Bitter, H.; Ito, M.; Valat, T.; Murakami, M.; Ruetz, S.; Guthy, D.A.; Rynn, C. A distinct p53 target gene set predicts for response to the selective p53-HDM2 inhibitor NVP-CGM097. Elife 2015, 4, e06498. [Google Scholar] [CrossRef] [PubMed]
- Sonkin, D. Expression signature based on TP53 target genes doesn’t predict response to TP53-MDM2 inhibitor in wild type TP53 tumors. ELife 2015, 4, e10279. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chamberlain, V.; Drew, Y.; Lunec, J. Tipping Growth Inhibition into Apoptosis by Combining Treatment with MDM2 and WIP1 Inhibitors in p53WT Uterine Leiomyosarcoma. Cancers 2022, 14, 14. https://doi.org/10.3390/cancers14010014
Chamberlain V, Drew Y, Lunec J. Tipping Growth Inhibition into Apoptosis by Combining Treatment with MDM2 and WIP1 Inhibitors in p53WT Uterine Leiomyosarcoma. Cancers. 2022; 14(1):14. https://doi.org/10.3390/cancers14010014
Chicago/Turabian StyleChamberlain, Victoria, Yvette Drew, and John Lunec. 2022. "Tipping Growth Inhibition into Apoptosis by Combining Treatment with MDM2 and WIP1 Inhibitors in p53WT Uterine Leiomyosarcoma" Cancers 14, no. 1: 14. https://doi.org/10.3390/cancers14010014
APA StyleChamberlain, V., Drew, Y., & Lunec, J. (2022). Tipping Growth Inhibition into Apoptosis by Combining Treatment with MDM2 and WIP1 Inhibitors in p53WT Uterine Leiomyosarcoma. Cancers, 14(1), 14. https://doi.org/10.3390/cancers14010014