
Frontiers in Anti-Cancer Drug Discovery: Challenges and Perspectives of Metformin as Anti-Angiogenic Add-On Therapy in Glioblastoma

 , , ,
, , ,  , , ,
, , ,  and
and
Abstract
Simple Summary
Abstract
1. Introduction
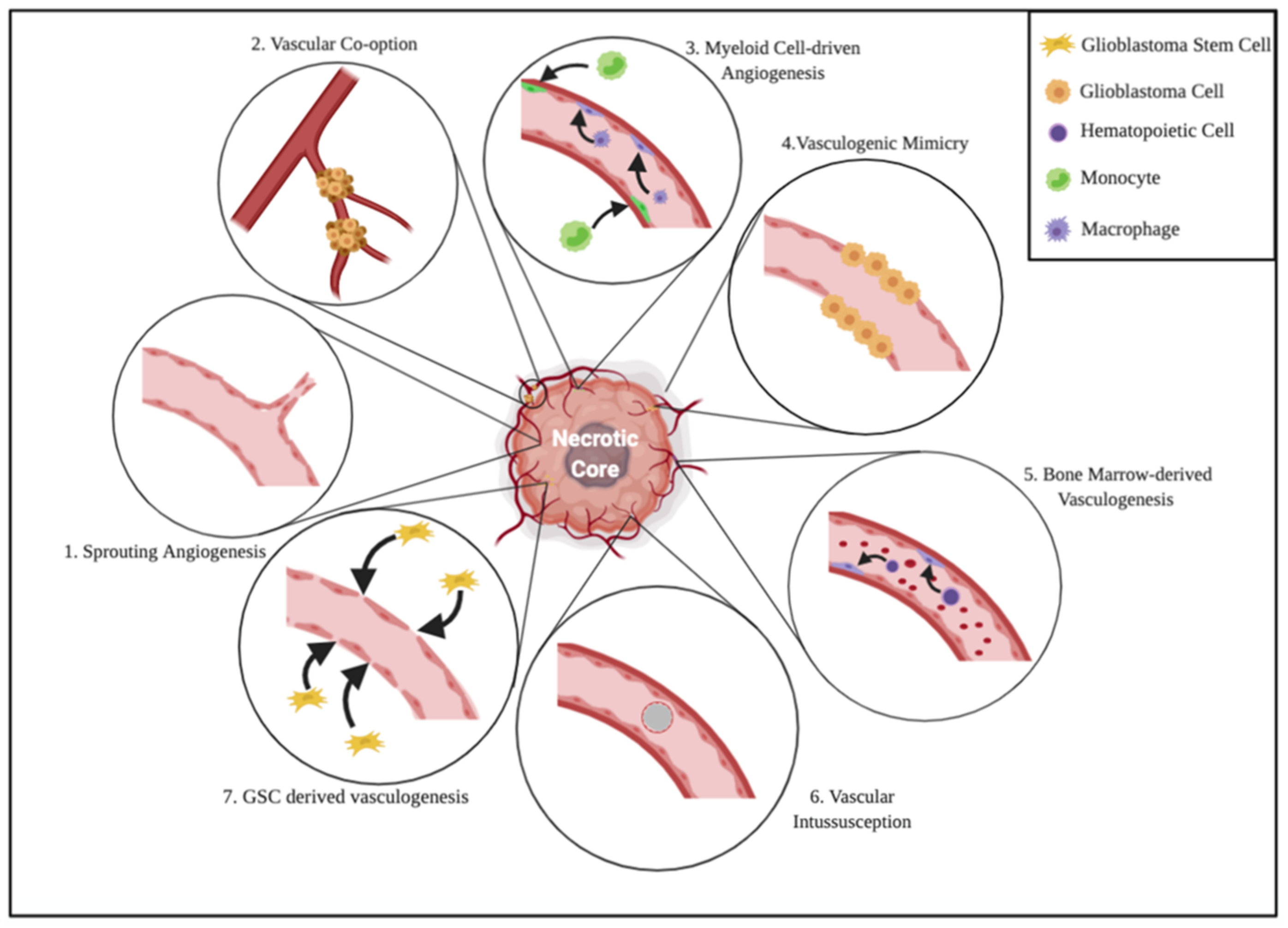
2. Neo-Angiogenesis in Glioblastoma
2.1. Cell Biology of Glioblastoma Angiogenesis
2.2. Angiogenic Signaling Pathways in Glioblastoma
2.3. Angiogenesis as a Plausible Target in Glioblastoma Therapy
3. Metformin
3.1. Molecular Mechanism of Metformin Effect
3.2. Evidence of Metformin Potential on Gliomas
3.3. Pre-Clinical Studies on the Efficacy of Metformin on Glioblastoma
3.4. Metformin Effects on GSCs
3.5. Could Angiogenesis Be a New Target for Metformin in Glioblastoma Therapy?
3.6. Clinical Trial with Metformin in Glioblastoma
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Crocetti, E.; Trama, A.; Stiller, C.; Caldarella, A.; Soffietti, R.; Jaal, J.; Weber, D.C.; Ricardi, U.; Slowinski, J.; Brandes, A.; et al. Epidemiology of glial and non-glial brain tumours in Europe. Eur. J. Cancer 2012, 48, 1532–1542. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; Wick, W.; Aldape, K.; Brada, M.; Berger, M.; Pfister, S.M.; Nishikawa, R.; Rosenthal, M.; Wen, P.Y.; Stupp, R.; et al. Glioma. Nat. Rev. Dis. Prim. 2015, 1, 15017. [Google Scholar] [CrossRef] [PubMed]
- Gavrilovic, I.T.; Posner, J.B. Brain metastases: Epidemiology and pathophysiology. J. Neurooncol. 2005, 75, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Parkin, D.M.; Steliarova-Foucher, E. Estimates of cancer incidence and mortality in Europe in 2008. Eur. J. Cancer 2010, 46, 765–781. [Google Scholar] [CrossRef]
- Modrek, A.S. Brain stem cells as the cell of origin in glioma. World J. Stem Cells 2014, 6, 43. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Gittleman, H.; Xu, J.; Kromer, C.; Wolinsky, Y.; Kruchko, C.B.-S.J. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2009–2013. Neuro Oncol. 2016, 18, v1–v75. [Google Scholar] [CrossRef]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.B.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Lee, E.; Yong, R.L.; Paddison, P.; Zhu, J. Comparison of glioblastoma (GBM) molecular classification methods. Semin. Cancer Biol. 2018, 53, 201–211. [Google Scholar] [CrossRef]
- Theeler, B.J.; Gilbert, M.R. Advances in the treatment of newly diagnosed glioblastoma. BMC Med. 2015, 13, 293. [Google Scholar] [CrossRef] [PubMed]
- Brat, D.J.; Aldape, K.; Colman, H.; Holland, E.C.; Louis, D.N.; Jenkins, R.B.; Kleinschmidt-DeMasters, B.K.; Perry, A.; Reifenberger, G.; Stupp, R.; et al. cIMPACT-NOW update 3: Recommended diagnostic criteria for “Diffuse astrocytic glioma, IDH-wildtype, with molecular features of glioblastoma, WHO grade IV”. Acta Neuropathol. 2018, 136, 805–810. [Google Scholar] [CrossRef] [PubMed]
- Kreisl, T.N.; Kim, L.; Moore, K.; Duic, P.; Royce, C.; Stroud, I.; Garren, N.; Mackey, M.; Butman, J.A.; Camphausen, K.; et al. Phase II trial of single-agent bevacizumab followed by bevacizumab plus irinotecan at tumor progression in recurrent glioblastoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2009, 27, 740–745. [Google Scholar] [CrossRef] [PubMed]
- Norden, A.D.; Drappatz, J.; Wen, P.Y. Novel anti-angiogenic therapies for malignant gliomas. Lancet Neurol. 2008, 7, 1152–1160. [Google Scholar] [CrossRef]
- Saraei, P.; Asadi, I.; Kakar, M.A.; Moradi-Kor, N. The beneficial effects of metformin on cancer prevention and therapy: A comprehensive review of recent advances. Cancer Manag. Res. 2019, 11, 3295–3313. [Google Scholar] [CrossRef] [PubMed]
- Welch, M.R.; Grommes, C. Retrospective analysis of the effects of steroid therapy and antidiabetic medication on survival in diabetic glioblastoma patients. CNS Oncol. 2013, 2, 237–246. [Google Scholar] [CrossRef]
- American Diabetes Association. Standards of medical care in diabetes-2014. Diabetes Care 2014, 37, S14–S80. [Google Scholar] [CrossRef]
- Pryor, R.; Cabreiro, F. Repurposing metformin: An old drug with new tricks in its binding pockets. Biochem. J. 2015, 471, 307–322. [Google Scholar] [CrossRef]
- Bailey, C.J. Metformin: Historical overview. Diabetologia 2017, 60, 1566–1576. [Google Scholar] [CrossRef]
- Thomas, I.; Gregg, B. Metformin; a review of its history and future: From lilac to longevity. Pediatr. Diabetes 2017, 18, 10–16. [Google Scholar] [CrossRef]
- Inzucchi, S.E. Oral antihyperglycemic therapy for type 2 diabetes: Scientific review. J. Am. Med. Assoc. 2002, 287, 360–372. [Google Scholar] [CrossRef]
- Nathan, D.M.; Buse, J.B.; Davidson, M.B.; Ferrannini, E.; Holman, R.R.; Sherwin, R.; Zinman, B. Medical management of hyperglycemia in type 2 diabetes: A consensus algorithm for the initiation and adjustment of therapy. Diabetes Care 2009, 32, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Mallik, R.; Chowdhury, T.A. Metformin in cancer. Diabetes Res. Clin. Pract. 2018, 143, 409–419. [Google Scholar] [CrossRef]
- Zhang, H.H.; Guo, X.L. Combinational strategies of metformin and chemotherapy in cancers. Cancer Chemother. Pharmacol. 2016, 78, 13–26. [Google Scholar] [CrossRef]
- Josko, J.; Gwozdz, B.; Jedrzejowska-Szypulka, H.; Hendryk, S. Vascular endothelial growth factor (VEGF) and its effect on angiogenesis. Med. Sci. Monit. 2000, 6, 1047–1052. [Google Scholar]
- Gupta, M.K.; Qin, R.Y. Mechanism and its regulation of tumor-induced angiogenesis. World J. Gastroenterol. 2003, 9, 1144–1155. [Google Scholar] [CrossRef]
- Sherwood, L.M.; Parris, E.E.; Folkman, J. Tumor Angiogenesis: Therapeutic Implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [CrossRef]
- Carmeliet, P.; Jain, R.K. Angiogenesis in cancer and other diseases. Nature 2000, 407, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Plate, K.H.; Scholz, A.; Dumont, D.J. Tumor angiogenesis and anti-angiogenic therapy in malignant gliomas revisited. Acta Neuropathol. 2012, 124, 763–775. [Google Scholar] [CrossRef]
- Simon, M.P.; Tournaire, R.; Pouyssegur, J. The angiopoietin-2 gene of endothelial cells is up-regulated in hypoxia by a HIF binding site located in its first intron and by the central factors GATA-2 and Ets-1. J. Cell Physiol. 2008, 217, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-inducible factors in physiology and medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Wu, Q.; Sathornsumetee, S.; Hao, Y.; Li, Z.; Hjelmeland, A.B.; Shi, Q.; McLendon, R.E.; Bigner, D.D.; Rich, J.N. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res. 2006, 66, 7843–7848. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.S.; Costello, M.A.; Talsma, C.E.; Flack, C.G.; Crowley, J.G.; Hamm, L.L.; He, X.; Hervey-Jumper, S.L.; Heth, J.A.; Muraszko, K.M.; et al. Endothelial cells create a stem cell niche in glioblastoma by providing NOTCH ligands that nurture self-renewal of cancer stem-like cells. Cancer Res. 2011, 71, 6061–6072. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.-J.; Shirakawa, T.; Li, Y.; Soma, A.; Oka, M.; Dotto, G.P.; Fairman, R.M.; Velazquez, O.C.; Herlyn, M. Regulation of Notch1 and Dll4 by Vascular Endothelial Growth Factor in Arterial Endothelial Cells: Implications for Modulating Arteriogenesis and Angiogenesis. Mol. Cell Biol. 2003, 23, 14–25. [Google Scholar] [CrossRef]
- Lobov, I.B.; Renard, R.A.; Papadopoulos, N.; Gale, N.W.; Thurston, G.; Yancopoulos, G.D.; Wiegand, S.J. Delta-like ligand 4 (DII4) is induced by VEGF as a negative regulator of angiogenic sprouting. Proc. Natl. Acad. Sci. USA 2007, 104, 3219–3224. [Google Scholar] [CrossRef]
- Charles, N.; Ozawa, T.; Squatrito, M.; Bleau, A.M.; Brennan, C.W.; Hambardzumyan, D.; Holland, E.C. Perivascular Nitric Oxide Activates Notch Signaling and Promotes Stem-like Character in PDGF-Induced Glioma Cells. Cell Stem Cell 2010, 6, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Eyler, C.E.; Wu, Q.; Yan, K.; MacSwords, J.M.; Chandler-Militello, D.; Misuraca, K.L.; Lathia, J.D.; Forrester, M.T.; Lee, J.; Stamler, J.S.; et al. Glioma stem cell proliferation and tumor growth are promoted by nitric oxide synthase-2. Cell 2011, 146, 53–66. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Ferreira, V.; Breier, G.; Pollefeyt, S.; Kieckens, L.; Gertsenstein, M.; Fahrig, M.; Vandenhoeck, A.; Harpal, K.; Eberhardt, C.; et al. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature 1996, 380, 435–439. [Google Scholar] [CrossRef]
- Fong, G.; Zhang, L.; Bryce, D.M.; Peng, J. Increased Hemangioblasts in flt-1–/–embryos. Development 1999, 126, 3015–3025. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Lampugnani, M.G.; Moons, L.; Breviario, F.; Compernolle, V.; Bono, F.; Balconi, G.; Spagnuolo, R.; Oosthuyse, B.; Dewerchin, M.; et al. Targeted deficiency or cytosolic truncation of the VE-cadherin gene in mice impairs VEGF-mediated endothelial survival and angiogenesis. Cell 1999, 98, 147–157. [Google Scholar] [CrossRef]
- Barbato, L.; Bocchetti, M.; Di Biase, A.; Regad, T. Cancer Stem Cells and Targeting Strategies. Cells 2019, 8, 926. [Google Scholar] [CrossRef]
- Navone, S.E.; Guarnaccia, L.; Cordiglieri, C.; Crisà, F.M.; Caroli, M.; Locatelli, M.; Schisano, L.; Rampini, P.; Miozzo, M.; La Verde, N.; et al. Aspirin Affects Tumor Angiogenesis and Sensitizes Human Glioblastoma Endothelial Cells to Temozolomide, Bevacizumab, and Sunitinib, Impairing Vascular Endothelial Growth Factor-Related Signaling. World Neurosurg. 2018, 120, e380–e391. [Google Scholar] [CrossRef] [PubMed]
- Guarnaccia, L.; Navone, S.E.; Trombetta, E.; Cordiglieri, C.; Cherubini, A.; Crisà, F.M.; Rampini, P.; Miozzo, M.; Fontana, L.; Caroli, M.; et al. Angiogenesis in human brain tumors: Screening of drug response through a patient-specific cell platform for personalized therapy. Sci. Rep. 2018, 8, 8748. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K.; di Tomaso, E.; Duda, D.G.; Loeffler, J.S.; Sorensen, A.G.; Batchelor, T.T. Angiogenesis in brain tumours. Nat. Rev. Neurosci. 2007, 8, 610–622. [Google Scholar] [CrossRef] [PubMed]
- Benedito, R.; Rocha, S.F.; Woeste, M.; Zamykal, M.; Radtke, F.; Casanovas, O.; Duarte, A.; Pytowski, B.; Adams, R.H. Notch-dependent VEGFR3 upregulation allows angiogenesis without VEGF-VEGFR2 signalling. Nature 2012, 484, 110–114. [Google Scholar] [CrossRef]
- Jubb, A.M.; Browning, L.; Campo, L.; Turley, H.; Steers, G.; Thurston, G.; Harris, A.L.; Ansorge, O. Expression of vascular Notch ligands Delta-like 4 and Jagged-1 in glioblastoma. Histopathology 2012, 60, 740–747. [Google Scholar] [CrossRef]
- Kim, H.; Koh, G.Y. Ang2, the instigator of inflammation. Blood 2011, 118, 4767–4768. [Google Scholar] [CrossRef]
- Scholz, A.; Lang, V.; Henschler, R.; Czabanka, M.; Vajkoczy, P.; Chavakis, E.; Drynski, J.; Harter, P.N.; Mittelbronn, M.; Dumont, D.J.; et al. Angiopoietin-2 promotes myeloid cell infiltration in a β2-integrin-dependent manner. Blood 2011, 118, 5050–5059. [Google Scholar] [CrossRef]
- Calabrese, C.; Poppleton, H.; Kocak, M.; Hogg, T.L.; Fuller, C.; Hamner, B.; Oh, E.Y.; Gaber, M.W.; Finklestein, D.; Allen, M.; et al. A perivascular niche for brain tumor stem cells. Cancer Cell 2007, 11, 69–82. [Google Scholar] [CrossRef]
- Norden, A.D.; Drappatz, J.W.P. Antiangiogenic therapies for high-grade glioma. Nat. Rev. Neurol. 2009, 5, 610–620. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K.; Duda, D.G.; Clark, J.W.; Loeffler, J.S. Lessons from phase III clinical trials on anti-VEGF therapy for cancer. Nat. Clin. Pract. Oncol. 2006, 3, 24–40. [Google Scholar] [CrossRef]
- Benjamin, L.E.; Hemo, I.; Keshet, E. A plasticity window for blood vessel remodelling is defined by pericyte coverage of the preformed endothelial network and is regulated by PDGF-B and VEGF. Development 1998, 125, 1591–1598. [Google Scholar] [CrossRef]
- Batchelor, T.T.; Sorensen, A.G.; di Tomaso, E.; Zhang, W.T.; Duda, D.G.; Cohen, K.S.; Kozak, K.R.; Cahill, D.P.; Chen, P.J.; Zhu, M.; et al. AZD2171, a pan-VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer Cell 2007, 11, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Normalization of tumor vasculature: An emerging concept in antiangiogenic therapy. Science 2005, 307, 58–62. [Google Scholar] [CrossRef]
- Kerbel, R.S. Antiangiogenic therapy: A universal chemosensitization strategy for cancer? Science 2006, 312, 1171–1175. [Google Scholar] [CrossRef] [PubMed]
- Winkler, F.; Kozin, S.V.; Tong, R.T.; Chae, S.S.; Booth, M.F.; Garkavtsev, I.; Xu, L.; Hicklin, D.J.; Fukumura, D.; di Tomaso, E.; et al. Kinetics of vascular normalization by VEGFR2 blockade governs brain tumor response to radiation: Role of oxygenation, angiopoietin-1, and matrix metalloproteinases. Cancer Cell 2004, 6, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Ameratunga, M.; Pavlakis, N.; Wheeler, H.; Grant, R.; Simes, J.; Khasraw, M. Anti-angiogenic therapy for high-grade glioma. Cochrane Database Syst. Rev. 2018, 11, CD008218. [Google Scholar] [CrossRef] [PubMed]
- De Groot, J.F.; Fuller, G.; Kumar, A.J.; Piao, Y.; Eterovic, K.; Ji, Y.; Conrad, C. Tumor invasion after treatment of glioblastoma with bevacizumab: Radiographic and pathologic correlation in humans and mice. Neuro Oncol. 2010, 12, 233–242. [Google Scholar] [CrossRef]
- Narayana, A.; Gruber, D.; Kunnakkat, S.; Golfinos, J.G.; Parker, E.; Raza, S.; Zagzag, D.; Eagan, P.; Gruber, M. A clinical trial of bevacizumab, temozolomide, and radiation for newly diagnosed glioblastoma. J. Neurosurg. 2012, 116, 341–345. [Google Scholar] [CrossRef]
- Suissa, S.; Azoulay, L. Metformin and cancer: Mounting evidence against an association. Diabetes Care 2014, 37, 1786–1788. [Google Scholar] [CrossRef]
- Samsuri, N.A.B.; Leech, M.; Marignol, L. Metformin and improved treatment outcomes in radiation therapy- A review. Cancer Treat. Rev. 2017, 55, 150–162. [Google Scholar] [CrossRef] [PubMed]
- Viollet, B.; Guigas, B.; Sanz Garcia, N.; Leclerc, J.; Foretz, M.; Andreelli, F. Cellular and molecular mechanisms of metformin: An overview. Clin. Sci. 2012, 122, 253–270. [Google Scholar] [CrossRef]
- Mazza, A.; Fruci, B.; Garinis, G.A.; Giuliano, S.; Malaguarnera, R.; Belfiore, A. The role of metformin in the management of NAFLD. Exp. Diabetes Res. 2012, 2012, 716404. [Google Scholar] [CrossRef] [PubMed]
- Ghatak, S.B.; Dhamecha, P.S.; Bhadada, S.V.; Panchal, S.J. Investigation of the potential effects of metformin on atherothrombotic risk factors in hyperlipidemic rats. Eur. J. Pharmacol. 2011, 659, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Diamanti-Kandarakis, E.; Economou, F.; Palimeri, S.; Christakou, C. Metformin in polycystic ovary syndrome. Ann. N. Y. Acad. Sci. 2010, 1205, 192–198. [Google Scholar] [CrossRef]
- Foretz, M.; Guigas, B.; Bertrand, L.; Pollak, M.; Viollet, B. Metformin: From mechanisms of action to therapies. Cell Metab. 2014, 20, 953–966. [Google Scholar] [CrossRef]
- Malek, M.; Aghili, R.; Emami, Z.; Khamseh, M.E. Risk of cancer in diabetes: The effect of metformin. ISRN Endocrinol. 2013, 2013, 636927. [Google Scholar] [CrossRef]
- Hirsch, H.A.; Iliopoulos, D.; Tsichlis, P.N.; Struhl, K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. 2009, 69, 7507–7511. [Google Scholar] [CrossRef]
- Ugwueze, C.V.; Ogamba, O.J.; Young, E.E.; Onyenekwe, B.M.; Ezeokpo, B.C. Metformin: A Possible Option in Cancer Chemotherapy. Anal. Cell Pathol. 2020, 2020, 7180923. [Google Scholar] [CrossRef]
- Pakkir Maideen, N.M.; Jumale, A.; Balasubramaniam, R. Drug Interactions of Metformin Involving Drug Transporter Proteins. Adv. Pharm. Bull. 2017, 7, 501–505. [Google Scholar] [CrossRef]
- He, L.; Wondisford, F.E. Metformin action: Concentrations matter. Cell Metab. 2015, 21, 159–162. [Google Scholar] [CrossRef]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S.; et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. eLife 2014, 3, e02242. [Google Scholar] [CrossRef]
- Madiraju, A.K.; Erion, D.M.; Rahimi, Y.; Zhang, X.M.; Braddock, D.T.; Albright, R.A.; Prigaro, B.J.; Wood, J.L.; Bhanot, S.; MacDonald, M.J.; et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature 2014, 510, 542–546. [Google Scholar] [CrossRef]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef]
- Todd, J.N.; Florez, J. An update on the pharmacogenomics of metformin: Progress, problems and potential. Pharmacogenomics 2014, 15, 529–539. [Google Scholar] [CrossRef]
- Whitaker-Menezes, D.; Martinez-Outschoorn, U.E.; Flomenberg, N.; Birbe, R.C.; Witkiewicz, A.K.; Howell, A.; Pavlides, S.; Tsirigos, A.; Ertel, A.; Pestell, R.G.; et al. Hyperactivation of oxidative mitochondrial metabolism in epithelial cancer cells in situ: Visualizing the therapeutic effects of metformin in tumor tissue. Cell Cycle 2011, 10, 4047–4064. [Google Scholar] [CrossRef]
- Kourelis, T.V.; Siegel, R. Metformin and cancer: New applications for an old drug. Med. Oncol. 2012, 29, 1314–1327. [Google Scholar] [CrossRef] [PubMed]
- Jin, D.H.; Kim, Y.; Lee, B.B.; Han, J.; Kim, H.K.; Shim, Y.M.; Kim, D. Metformin induces cell cycle arrest at the G1 phase through E2F8 suppression in lung cancer cells. Oncotarget 2017, 8, 101509–101519. [Google Scholar] [CrossRef] [PubMed]
- Whitburn, J.; Edwards, C.M.; Sooriakumaran, P. Metformin and prostate cancer: A new role for an old drug. Curr. Urol. Rep. 2017, 18, 46. [Google Scholar] [CrossRef] [PubMed]
- Muaddi, H.; Chowdhury, S.; Vellanki, R.; Zamiara, P.; Koritzinsky, M. Contributions of AMPK and p53 dependent signaling to radiation response in the presence of metformin. Radiother. Oncol. 2013, 108, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Orgel, E.M.S. The links between insulin resistance, diabetes, and cancer. Curr. Diab. Rep. 2013, 13, 213–222. [Google Scholar] [CrossRef]
- Abdelgadir, E.; Ali, R.; Rashid, F.B.A. Effect of Metformin on Different Non-Diabetes Related Conditions, a Special Focus on Malignant Conditions: Review of Literature. J. Clin. Med. Res. 2017, 9, 388–395. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Tang, G.; Li, Y.; Wang, Y.; Chen, X.; Gu, X.; Zhang, Z.; Wang, Y.; Yang, G.-Y. Metformin attenuates blood-brain barrier disruption in mice following middle cerebral artery occlusion. J. Neuroinflamm. 2014, 11, 177. [Google Scholar] [CrossRef]
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res. Clin. Pract. 2019, 157, 107843. [Google Scholar] [CrossRef]
- Chaichana, K.L.; McGirt, M.J.; Woodworth, G.F.; Datoo, G.; Tamargo, R.J.; Weingart, J.; Olivi, A.; Brem, H.; Quinones-Hinojosa, A. Persistent outpatient hyperglycemia is independently associated with survival, recurrence and malignant degeneration following surgery for hemispheric low grade gliomas. Neurol. Res. 2010, 32, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Montemurro, N.; Perrini, P.; Rapone, B. Clinical Risk and Overall Survival in Patients with Diabetes Mellitus, Hyperglycemia and Glioblastoma Multiforme. A Review of the Current Literature. Int. J. Environ. Res. Public Health 2020, 17, 8501. [Google Scholar] [CrossRef]
- Derr, R.L.; Ye, X.; Islas, M.U.; Desideri, S.; Saudek, C.D.; Grossman, S. Association between hyperglycemia and survival in patients with newly diagnosed glioblastoma. J. Clin. Oncol. 2009, 27, 1082–1086. [Google Scholar] [CrossRef]
- Carr, M.T.; Hochheimer, C.J.; Rock, A.K.; Dincer, A.; Ravindra, L.; Zhang, F.L.; Opalak, C.F.; Poulos, N.; Sima, A.P.; Broaddus, W.C. Comorbid Medical Conditions as Predictors of Overall Survival in Glioblastoma Patients. Sci. Rep. 2019, 9, 20018. [Google Scholar] [CrossRef]
- Pyaskovskaya, O.N.; Kolesnik, D.L.; Fedorchuk, A.G.; Gorbik, G.V.; Solyanik, G.I. Cytotoxic activity of metformin in vitro does not correlate with its antitumor action in vivo. Exp. Oncol. 2017, 39, 264–268. [Google Scholar] [CrossRef]
- Adeberg, S.; Bernhardt, D.; Foerster, R.; Bostel, T.; Koerber, S.A.; Mohr, A.; Koelsche, C.; Rieken, S.; Debus, J. The influence of hyperglycemia during radiotherapy on survival in patients with primary glioblastoma. Acta Oncol. 2016, 55, 201–207. [Google Scholar] [CrossRef]
- Seliger, C.; Luber, C.; Gerken, M.; Schaertl, J.; Proescholdt, M.; Riemenschneider, M.J.; Meier, C.R.; Bogdahn, U.; Leitzmann, M.F.; Klinkhammer-Schalke, M.; et al. Use of metformin and survival of patients with high-grade glioma. Int. J. Cancer 2019, 144, 273–280. [Google Scholar] [CrossRef]
- Seliger, C.; Genbrugge, E.; Gorlia, T.; Chinot, O.; Stupp, R.; Nabors, B.; Weller, M.; Hau, P. Use of metformin and outcome of patients with newly diagnosed glioblastoma: Pooled analysis. Int. J. Cancer 2020, 146, 803–809. [Google Scholar] [CrossRef]
- Mazurek, M.; Litak, J.; Kamieniak, P.; Kulesza, B.; Jonak, K.; Baj, J.; Grochowski, C. Metformin as Potential Therapy for High-Grade Glioma. Cancers 2020, 12, 210. [Google Scholar] [CrossRef]
- Zakikhani, M.; Dowling, R.; Fantus, I.G.; Sonenberg, N.; Pollak, M. Metformin is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer Res. 2006, 66, 10269–10273. [Google Scholar] [CrossRef]
- Pollak, M.N. Investigating metformin for cancer prevention and treatment: The end of the beginning. Cancer Discov. 2012, 2, 778–790. [Google Scholar] [CrossRef]
- Rattan, R.; Giri, S.; Hartmann, L.C.; Shridhar, V. Metformin attenuates ovarian cancer cell growth in an AMP-kinase dispensable manner. J. Cell Mol. Med. 2011, 15, 166–178. [Google Scholar] [CrossRef] [PubMed]
- Lutz, T.A.; Estermann, A.; Haag, S.; Scharrer, E. Depolarization of the liver cell membrane by metformin. Biochim. Biophys. Acta 2001, 1513, 176–184. [Google Scholar] [CrossRef][Green Version]
- Łabuzek, K.; Suchy, D.; Gabryel, B.; Bielecka, A.; Liber, S.; Okopień, B. Quantification of metformin by the HPLC method in brain regions, cerebrospinal fluid and plasma of rats treated with lipopolysaccharide. Pharmacol. Rep. 2010, 62, 956–965. [Google Scholar] [CrossRef]
- Sesen, J.; Dahan, P.; Scotland, S.J.; Saland, E.; Dang, V.T.; Lemarié, A.; Tyler, B.M.; Brem, H.; Toulas, C.; Moyal, E.C.J.; et al. Metformin inhibits growth of human glioblastoma cells and enhances therapeutic response. PLoS ONE 2015, 10, e0123721. [Google Scholar] [CrossRef]
- Yu, Z.; Zhao, G.; Li, P.; Li, Y.; Zhou, G.; Chen, Y.; Xie, G. Temozolomide in combination with metformin act synergistically to inhibit proliferation and expansion of glioma stem-like cells. Oncol. Lett. 2016, 11, 2792–2800. [Google Scholar] [CrossRef] [PubMed]
- Lo Dico, A.; Valtorta, S.; Ottobrini, L.; Moresco, R.M. Role of Metformin and AKT Axis Modulation in the Reversion of Hypoxia Induced TMZ-Resistance in Glioma Cells. Front. Oncol. 2019, 9, 463. [Google Scholar] [CrossRef]
- Gritti, M.; Würth, R.; Angelini, M.; Barbieri, F.; Peretti, M.; Pizzi, E.; Pattarozzi, A.; Carra, E.; Sirito, R.; Daga, A.; et al. Metformin repositioning as antitumoral agent: Selective antiproliferative effects in human glioblastoma stem cells, via inhibition of CLIC1-mediated ion current. Oncotarget 2014, 5, 11252–11268. [Google Scholar] [CrossRef]
- Jeon, S.-M.; Hay, N. The dark face of AMPK as an essential tumor promoter. Cell Logist. 2012, 2, 197–202. [Google Scholar] [CrossRef]
- Xiong, Z.S.; Gong, S.F.; Si, W.; Jiang, T.; Li, Q.L.; Wang, T.J.; Wang, W.J.; Wu, R.Y.; Jiang, K. Effect of metformin on cell proliferation, apoptosis, migration and invasion in A172 glioma cells and its mechanisms. Mol. Med. Rep. 2019, 20, 887–894. [Google Scholar] [CrossRef]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef]
- Al Hassan, M.; Fakhoury, I.; El Masri, Z.; Ghazale, N.; Dennaoui, R.; El Atat, O.; Kanaan, A.; El-Sibai, M. Metformin Treatment Inhibits Motility and Invasion of Glioblastoma Cancer Cells. Anal. Cell Pathol. 2018, 2018, 5917470. [Google Scholar] [CrossRef] [PubMed]
- Zarnescu, O.; Brehar, F.M.; Chivu, M.; Ciurea, A.V. Immunohistochemical localization of caspase-3, caspase-9 and Bax in U87 glioblastoma xenografts. J. Mol. Histol. 2008, 39, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Sato, A.; Sunayama, J.; Okada, M.; Watanabe, E.; Seino, S.; Shibuya, K.; Suzuki, K.; Narita, Y.; Shibui, S.; Kayama, T.; et al. Glioma-Initiating Cell Elimination by Metformin Activation of FOXO3 via AMPK. Stem Cells Transl. Med. 2012, 1, 811–824. [Google Scholar] [CrossRef]
- Leidgens, V.; Proske, J.; Rauer, L.; Moeckel, S.; Renner, K.; Bogdahn, U.; Riemenschneider, M.J.; Proescholdt, M.; Vollmann-Zwerenz, A.; Hau, P.; et al. Stattic and metformin inhibit brain tumor initiating cells by reducing STAT3-phosphorylation. Oncotarget 2017, 8, 8250–8263. [Google Scholar] [CrossRef]
- Yuan, X.; Wei, W.; Bao, Q.; Chen, H.; Jin, P.; Jiang, W. Metformin inhibits glioma cells stemness and epithelial-mesenchymal transition via regulating YAP activity. Biomed. Pharmacother. 2018, 102, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Dowling, R.J.O.; Niraula, S.; Stambolic, V.; Goodwin, P.J. Metformin in cancer: Translational challenges. J. Mol. Endocrinol. 2012, 48, R31–R43. [Google Scholar] [CrossRef]
- Chandel, N.S.; Avizonis, D.; Reczek, C.R.; Weinberg, S.E.; Menz, S.; Neuhaus, R.; Christian, S.; Haegebarth, A.; Algire, C.; Pollak, M. Are Metformin Doses Used in Murine Cancer Models Clinically Relevant? Cell Metab. 2016, 23, 569–570. [Google Scholar] [CrossRef]
- Facchino, S.; Abdouh, M.; Bernier, G. Brain cancer stem cells: Current status on glioblastoma multiforme. Cancers 2011, 3, 1777–1797. [Google Scholar] [CrossRef] [PubMed]
- Son, M.J.; Woolard, K.; Nam, D.H.; Lee, J.; Fine, H.A. SSEA-1 Is an Enrichment Marker for Tumor-Initiating Cells in Human Glioblastoma. Cell Stem Cell 2009, 4, 440–452. [Google Scholar] [CrossRef] [PubMed]
- Gilbertson, R.J.; Rich, J.N. Making a tumour’s bed: Glioblastoma stem cells and the vascular niche. Nat. Rev. Cancer 2007, 7, 733–736. [Google Scholar] [CrossRef]
- Yu, Z.; Zhao, G.; Xie, G.; Zhao, L.; Chen, Y.; Yu, H.; Zhang, Z.; Li, C.; Li, Y. Metformin and temozolomide act synergistically to inhibit growth of glioma cells and glioma stem cells in vitro and in vivo. Oncotarget 2015, 6, 32930–32943. [Google Scholar] [CrossRef] [PubMed]
- Würth, R.; Pattarozzi, A.; Gatti, M.; Bajetto, A.; Corsaro, A.; Parodi, A.; Sirito, R.; Massollo, M.; Marini, C.; Zona, G.; et al. Metformin selectively affects human glioblastoma tumor-initiating cell viability: A role for metformin-induced inhibition of Akt. Cell Cycle 2013, 12, 145–156. [Google Scholar] [CrossRef]
- Würth, R.; Barbieri, F.; Florio, T. New molecules and old drugs as emerging approaches to selectively target human glioblastoma cancer stem cells. Biomed. Res. Int. 2014, 2014, 126586. [Google Scholar] [CrossRef]
- Najbauer, J.; Kraljik, N.; Németh, P. Glioma stem cells: Markers, hallmarks and therapeutic targeting by metformin. Pathol. Oncol. Res. 2014, 20, 789–797. [Google Scholar] [CrossRef]
- Kannarkatt, J.; Alkharabsheh, O.; Tokala, H.; Dimitrov, N.V. Metformin and Angiogenesis in Cancer-Revisited. Oncology 2016, 91, 179–184. [Google Scholar] [CrossRef]
- Hirsch, H.A.; Iliopoulos, D.; Struhl, K. Metformin inhibits the inflammatory response associated with cellular transformation and cancer stem cell growth. Proc. Natl. Acad. Sci. USA 2013, 110, 972–977. [Google Scholar] [CrossRef] [PubMed]
- Xavier, D.O.; Amaral, L.S.; Gomes, M.A.; Rocha, M.A.; Campos, P.R.; Cota, B.D.C.V.; Tafuri, L.S.A.; Paiva, A.M.R.; Silva, J.H.; Andrade, S.P.; et al. Metformin inhibits inflammatory angiogenesis in a murine sponge model. Biomed. Pharmacother. 2010, 64, 220–225. [Google Scholar] [CrossRef]
- Dallaglio, K.; Bruno, A.; Cantelmo, A.R.; Esposito, A.I.; Ruggiero, L.; Orecchioni, S.; Calleri, A.; Bertolini, F.; Pfeffer, U.; Noonan, D.M.; et al. Paradoxic effects of metformin on endothelial cells and angiogenesis. Carcinogenesis 2014, 35, 1055–1066. [Google Scholar] [CrossRef]
- Orecchioni, S.; Reggiani, F.; Talarico, G.; Mancuso, P.; Calleri, A.; Gregato, G.; Labanca, V.; Noonan, D.M.; Dallaglio, K.; Albini, A.; et al. The biguanides metformin and phenformin inhibit angiogenesis, local and metastatic growth of breast cancer by targeting both neoplastic and microenvironment cells. Int. J. Cancer 2015, 136, E534–E544. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Li, Y.; Guo, Z.; Zeng, Y.; Zhang, W.; Wang, H. Metformin: Current clinical applications in nondiabetic patients with cancer. Aging 2020, 12, 3993–4009. [Google Scholar] [CrossRef] [PubMed]
- Maraka, S.; Groves, M.D.; Mammoser, A.G.; Melguizo-Gavilanes, I.; Conrad, C.A.; Tremont-Lukats, I.W.; Loghin, M.E.; O’Brien, B.J.; Puduvalli, V.K.; Sulman, E.P.; et al. Phase 1 lead-in to a phase 2 factorial study of temozolomide plus memantine, mefloquine, and metformin as postradiation adjuvant therapy for newly diagnosed glioblastoma. Cancer 2019, 125, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Shenouda, G.; Souhami, L.; Petrecca, K.; Panet-Raymond, V.; Owen, S.; Diaz, R.J.; Hall, J.; Guiot, M.C.; Carvalho, T.; Abdulkarim, B.S. A Phase 2 Study of Neo-adjuvant Metformin and Temozolomide followed by Hypofractionated Accelerated RadioTherapy (HART) with Concomitant and Adjuvant Metformin and Temozolomide (TMZ) in Patients with Glioblastoma. Int. J. Radiat. Oncol. 2020, 108, S21. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Metformin Properties | |
|---|---|
| Structure |  |
| Chemical formula | C4H11N5 |
| Weight | 129.16 KDa |
| Indication | Tablet, oral administration |
| Associated conditions | T2DM; PCOS |
| Route of elimination | Kidney |
| Half-life | Plasma and blood: ~6.2 h Elimination half-life: ~17.6 h |
| Clearance | 510 ± 120 mL/min |
| Pharmacokinetics | Trough steady-state metformin plasma concentration: 54–4133 ng/ml |
| Intestinal/hepatic uptake | PMAT, Oct1/3 |
| Carrier | Oct1/3 for absorption; Oct2 for excretion |
| BBB permeability | Yes |
| Metformin Effects | Molecular Pathways | Reference |
|---|---|---|
| Metformin specifically acts on neoplastic or glioma stem cells, while not affecting normal cells | Metformin acts by blocking the chloride channel CLIC1. The downstream cascade is yet to be studied | [102] |
| Metformin alters cells metabolism by acting on ETC I and, consequently, by impairing the ATP/AMP ratio and activating AMPK | Metformin decreases oxidative phosphorylation while increasing the amount of ATP produced through anaerobic glycolysis | [99] |
| Metformin decreases the protein synthesis through the inhibition of mTOR while inducing the predominance of catabolic processes | [103] | |
| Metformin increases oxidative stress in glioblastoma cells | Metformin blocks ETC I, generating an impaired mitochondria action and leading to an increase in ROS production | [99] |
| Metformin inhibits mitochondrial superoxide dismutase, increasing ROS production | [104] | |
| Metformin inhibits cell proliferation | By activating AMPK, through the phosphorylation of PIKE-A, Metformin inhibits the Akt/mTOR axis | [104] |
| By activating TSC2 and RAPTOR, Metformin inhibits mTOR | [105] | |
| Metformin inhibits cell motility and invasiveness | By activating AMPK, through the phosphorylation of PIKE-A, Metformin inhibits the Akt/mTOR axis | [106] |
| Metformin moderately increases apoptosis | Metformin increases the levels of caspase 3 | [86,106] |
| Metformin increases the levels of caspase 9 | [107] | |
| Metformin increases the levels of Bax, while reducing the levels of Bcl-2 | [99,106] | |
| Metformin increases sensitivity to chemo- and radiotherapy | Metformin inhibits HIF and its downstream effects | [60,101] |
| Metformin acts on GSCs | Together with TMZ, Metformin inhibits proliferation and promotes apoptosis | [101,107] |
| Metformin induces GSCs differentiation by activating FOXO3 | [108] | |
| Metformin induces GSCs differentiation by inhibiting STAT3, through AMPK (phosphorylation site Ser727) or directly (phosphorylation site Y705) | [109] | |
| Metformin inhibits GSCs EMT through the inhibition of the axis YAP/Hippo | [110] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guarnaccia, L.; Marfia, G.; Masseroli, M.M.; Navone, S.E.; Balsamo, M.; Caroli, M.; Valtorta, S.; Moresco, R.M.; Campanella, R.; Garzia, E.; et al. Frontiers in Anti-Cancer Drug Discovery: Challenges and Perspectives of Metformin as Anti-Angiogenic Add-On Therapy in Glioblastoma. Cancers 2022, 14, 112. https://doi.org/10.3390/cancers14010112
Guarnaccia L, Marfia G, Masseroli MM, Navone SE, Balsamo M, Caroli M, Valtorta S, Moresco RM, Campanella R, Garzia E, et al. Frontiers in Anti-Cancer Drug Discovery: Challenges and Perspectives of Metformin as Anti-Angiogenic Add-On Therapy in Glioblastoma. Cancers. 2022; 14(1):112. https://doi.org/10.3390/cancers14010112
Chicago/Turabian StyleGuarnaccia, Laura, Giovanni Marfia, Matteo Maria Masseroli, Stefania Elena Navone, Melissa Balsamo, Manuela Caroli, Silvia Valtorta, Rosa Maria Moresco, Rolando Campanella, Emanuele Garzia, and et al. 2022. "Frontiers in Anti-Cancer Drug Discovery: Challenges and Perspectives of Metformin as Anti-Angiogenic Add-On Therapy in Glioblastoma" Cancers 14, no. 1: 112. https://doi.org/10.3390/cancers14010112
APA StyleGuarnaccia, L., Marfia, G., Masseroli, M. M., Navone, S. E., Balsamo, M., Caroli, M., Valtorta, S., Moresco, R. M., Campanella, R., Garzia, E., Riboni, L., & Locatelli, M. (2022). Frontiers in Anti-Cancer Drug Discovery: Challenges and Perspectives of Metformin as Anti-Angiogenic Add-On Therapy in Glioblastoma. Cancers, 14(1), 112. https://doi.org/10.3390/cancers14010112