
Identification of Neoantigens in Two Murine Gastric Cancer Cell Lines Leading to the Neoantigen-Based Immunotherapy

 , , , , ,
, , , , ,  and
and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Mice, Tumor Cells and Reagents
2.2. Transplantation of YTN2 and YTN16
2.3. Whole-Exome Sequencing (WES)
2.4. RNA Sequencing (RNA-Seq)
2.5. Epitope Prediction
2.6. MHC Class I Stabilization Assay
2.7. MHC Class I Ligandome Analysis for Neoantigen Identification
2.8. Mild Acid Elution of MHC Class I-Binding Peptides
2.9. Screening of Candidate Neoepitope Peptides
2.10. Establishment of Neoantigen-Specific CD8+ T Cell Lines
2.11. Cell Preparation and Flow Cytometry
2.12. Dendritic Cell (DC) Vaccine
2.13. Adoptive Transfer of Neoantigen-Reactive CD8+ T Cells
2.14. Statistical Analysis
3. Results
3.1. Comparison of Intratumoral Immune Responses to Gastric Cancer Cell Lines YTN2 and YTN16
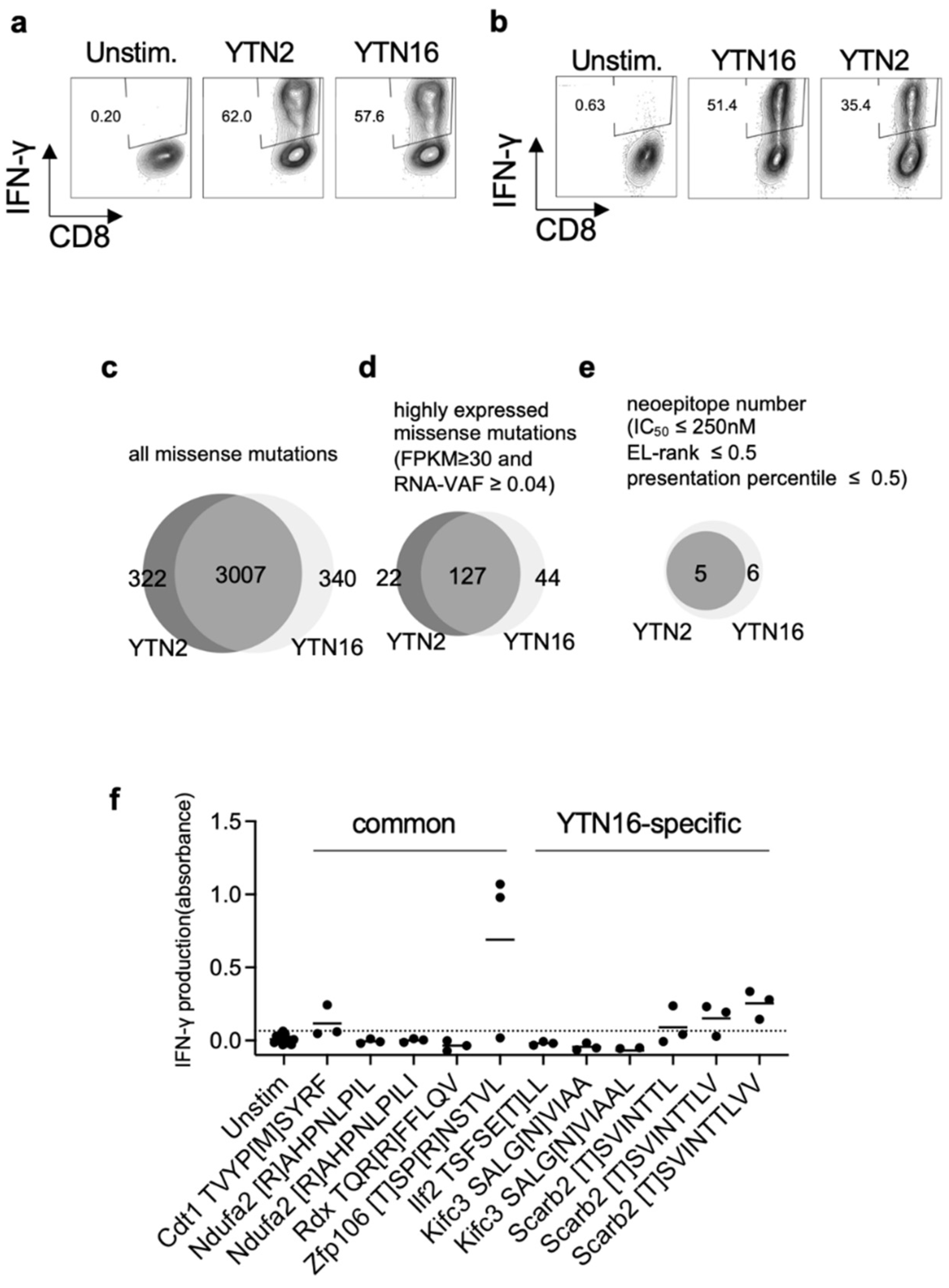
3.2. Identification of Neoantigens in YTN2 and YTN16 Tumors
3.3. Neoepitope-Specific CD8+ T Cell Lines
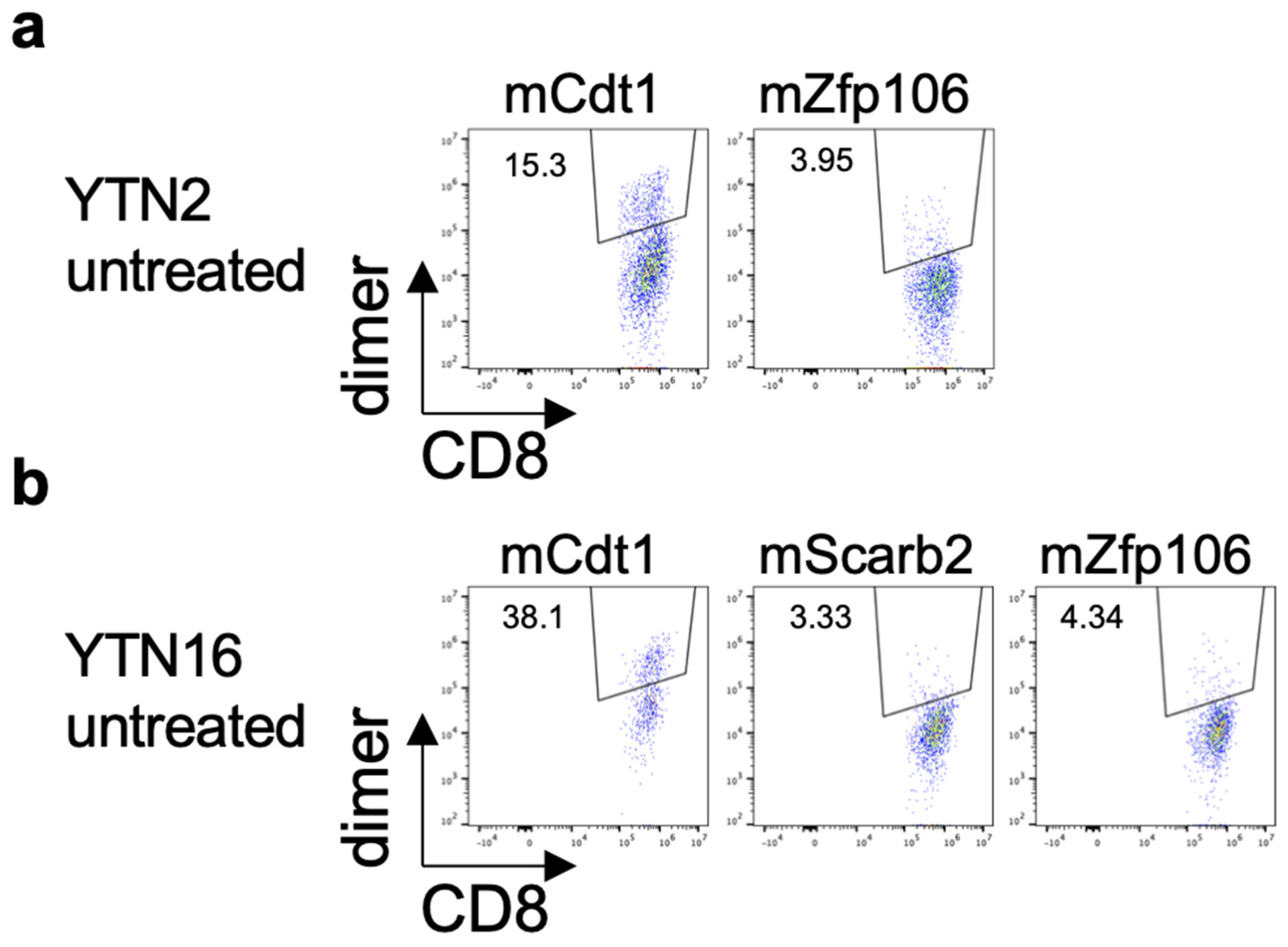
3.4. Existence of Neoepitope-Reactive CD8+ T Cells in YTN2 and YTN16 Tumors
3.5. MHC Class I Ligandome Analysis with Mass Spectrometry
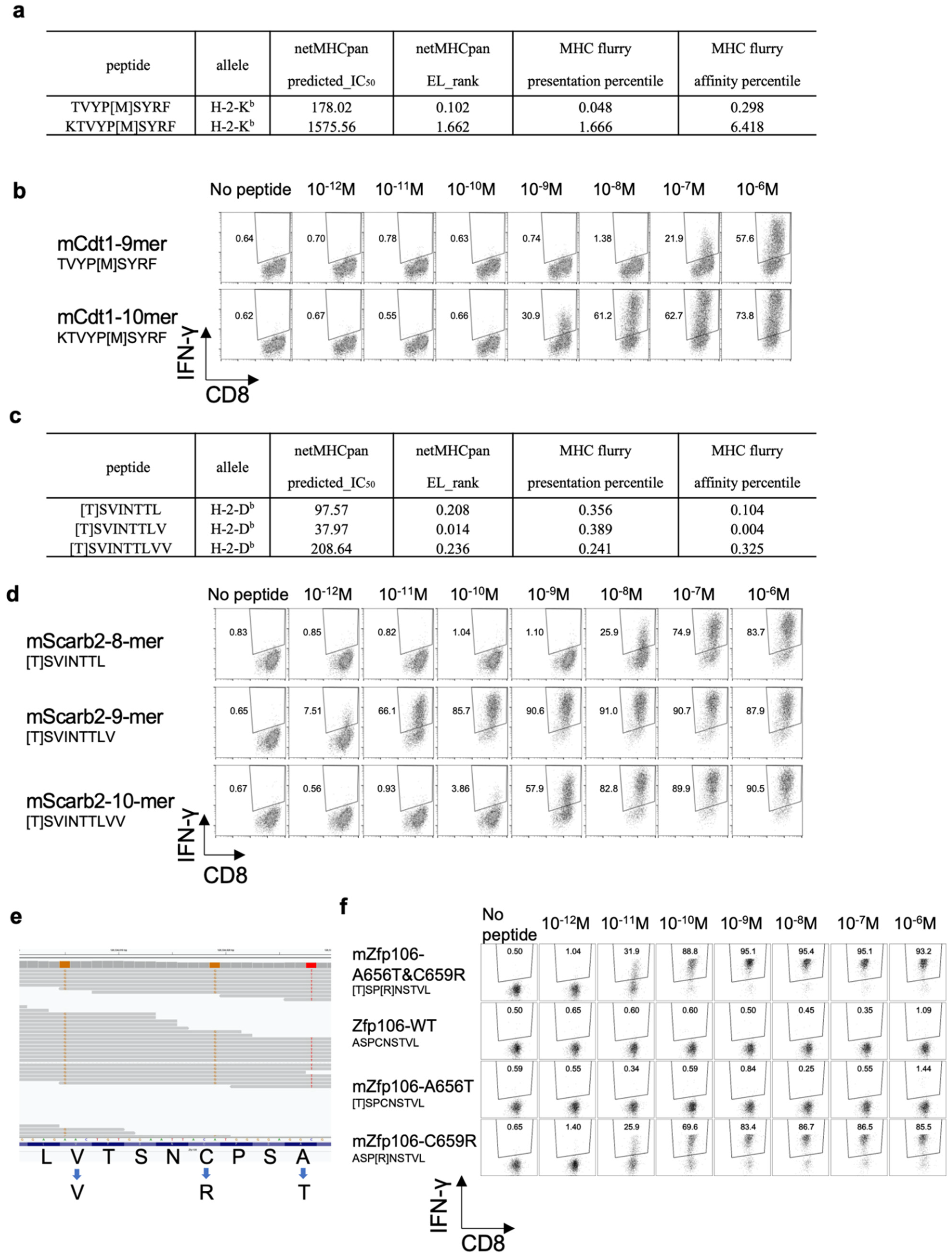
3.6. Determining the Optimal Epitope for mCdt1-Reactive CD8+ T Cells
3.7. Optimal Epitope Peptide for mScarb2-Reactive CD8+ T Cells
3.8. Proximal Variants That Alter the Peptide Sequence
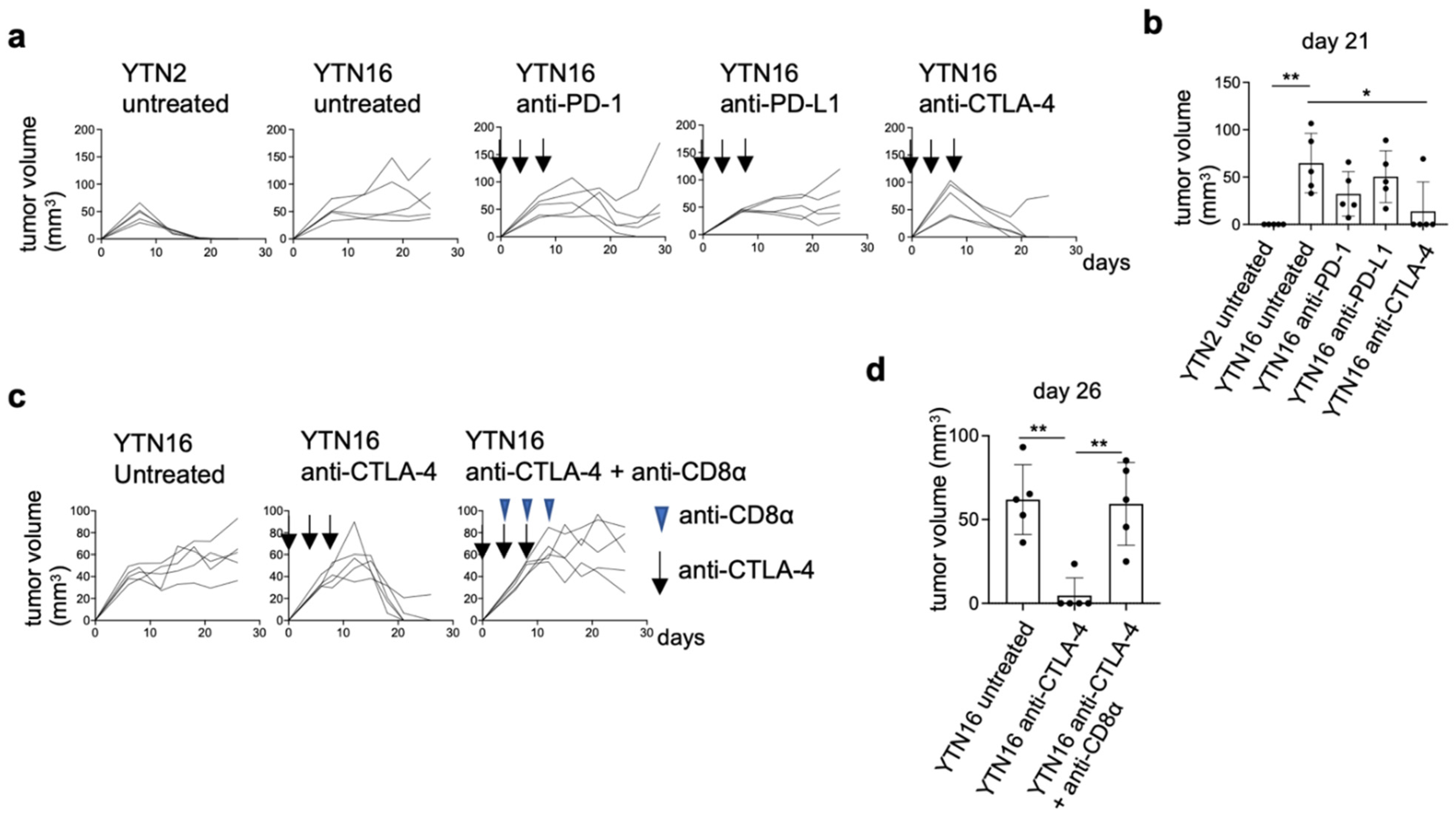
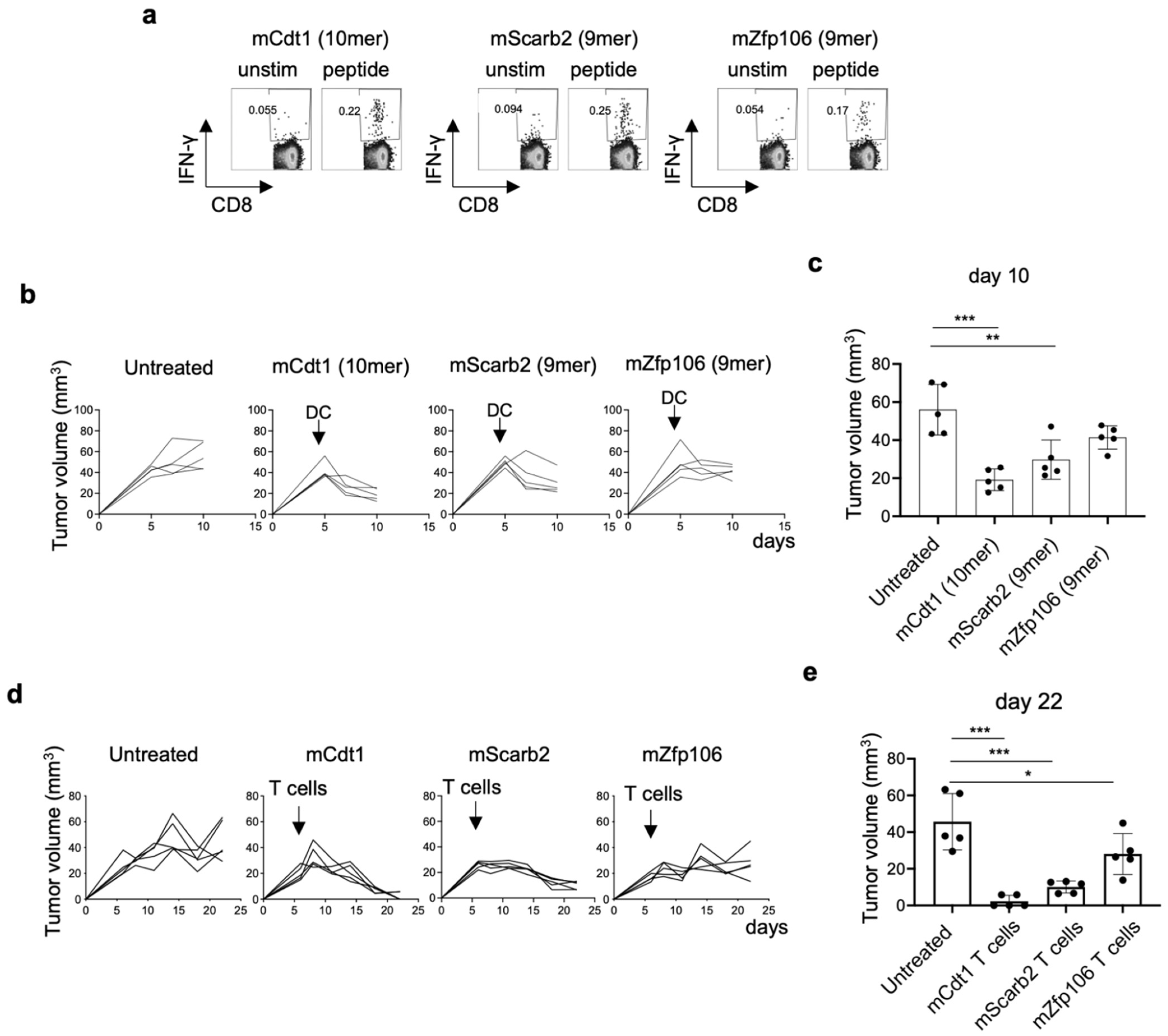
3.9. Neoantigen-Based Immunotherapy for YTN16 Tumors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, C.S.; Doi, T.; Jang, R.W.; Muro, K.; Satoh, T.; Machado, M.; Sun, W.; Jalal, S.I.; Shah, M.A.; Metges, J.P.; et al. Safety and Efficacy of Pembrolizumab Monotherapy in Patients With Previously Treated Advanced Gastric and Gastroesophageal Junction Cancer: Phase 2 Clinical KEYNOTE-059 Trial. JAMA Oncol. 2018, 4, e180013. [Google Scholar] [CrossRef]
- Kang, Y.K.; Boku, N.; Satoh, T.; Ryu, M.H.; Chao, Y.; Kato, K.; Chung, H.C.; Chen, J.S.; Muro, K.; Kang, W.K.; et al. Nivolumab in patients with advanced gastric or gastro-oesophageal junction cancer refractory to, or intolerant of, at least two previous chemotherapy regimens (ONO-4538-12, ATTRACTION-2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 2461–2471. [Google Scholar] [CrossRef]
- Kim, S.T.; Cristescu, R.; Bass, A.J.; Kim, K.M.; Odegaard, J.I.; Kim, K.; Liu, X.Q.; Sher, X.; Jung, H.; Lee, M.; et al. Comprehensive molecular characterization of clinical responses to PD-1 inhibition in metastatic gastric cancer. Nat. Med. 2018, 24, 1449–1458. [Google Scholar] [CrossRef] [PubMed]
- Janjigian, Y.Y.; Shitara, K.; Moehler, M.; Garrido, M.; Salman, P.; Shen, L.; Wyrwicz, L.; Yamaguchi, K.; Skoczylas, T.; Campos Bragagnoli, A.; et al. First-line nivolumab plus chemotherapy versus chemotherapy alone for advanced gastric, gastro-oesophageal junction, and oesophageal adenocarcinoma (CheckMate 649): A randomised, open-label, phase 3 trial. Lancet 2021, 398, 27–40. [Google Scholar] [CrossRef]
- Yamamoto, M.; Nomura, S.; Hosoi, A.; Nagaoka, K.; Iino, T.; Yasuda, T.; Saito, T.; Matsushita, H.; Uchida, E.; Seto, Y.; et al. Established gastric cancer cell lines transplantable into C57BL/6 mice show fibroblast growth factor receptor 4 promotion of tumor growth. Cancer Sci. 2018, 109, 1480–1492. [Google Scholar] [CrossRef]
- Nagaoka, K.; Shirai, M.; Taniguchi, K.; Hosoi, A.; Sun, C.; Kobayashi, Y.; Maejima, K.; Fujita, M.; Nakagawa, H.; Nomura, S.; et al. Deep immunophenotyping at the single-cell level identifies a combination of anti-IL-17 and checkpoint blockade as an effective treatment in a preclinical model of data-guided personalized immunotherapy. J. Immunother. Cancer 2020, 8, e001358. [Google Scholar] [CrossRef]
- Hoof, I.; Peters, B.; Sidney, J.; Pedersen, L.E.; Sette, A.; Lund, O.; Buus, S.; Nielsen, M. NetMHCpan, a method for MHC class I binding prediction beyond humans. Immunogenetics 2009, 61, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reynisson, B.; Alvarez, B.; Paul, S.; Peters, B.; Nielsen, M. NetMHCpan-4.1 and NetMHCIIpan-4.0: Improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 2020, 48, W449–W454. [Google Scholar] [CrossRef]
- O’Donnell, T.J.; Rubinsteyn, A.; Laserson, U. MHCflurry 2.0: Improved Pan-Allele Prediction of MHC Class I-Presented Peptides by Incorporating Antigen Processing. Cell Syst. 2020, 11, 42–48.e47. [Google Scholar] [CrossRef]
- Udaka, K.; Wiesmuller, K.H.; Kienle, S.; Jung, G.; Walden, P. Decrypting the structure of major histocompatibility complex class I-restricted cytotoxic T lymphocyte epitopes with complex peptide libraries. J. Exp. Med. 1995, 181, 2097–2108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Udaka, K.; Mamitsuka, H.; Nakaseko, Y.; Abe, N. Empirical evaluation of a dynamic experiment design method for prediction of MHC class I-binding peptides. J. Immunol. 2002, 169, 5744–5753. [Google Scholar] [CrossRef] [Green Version]
- Hirama, T.; Tokita, S.; Nakatsugawa, M.; Murata, K.; Nannya, Y.; Matsuo, K.; Inoko, H.; Hirohashi, Y.; Hashimoto, S.; Ogawa, S.; et al. Proteogenomic identification of an immunogenic HLA class I neoantigen in mismatch repair-deficient colorectal cancer tissue. JCI Insight 2021, 6, e146356. [Google Scholar] [CrossRef] [PubMed]
- Storkus, W.J.; Zeh, H.J., 3rd; Salter, R.D.; Lotze, M.T. Identification of T-cell epitopes: Rapid isolation of class I-presented peptides from viable cells by mild acid elution. J. Immunother. Emphasis Tumor Immunol. 1993, 14, 94–103. [Google Scholar] [CrossRef]
- Nagaoka, K.; Hosoi, A.; Iino, T.; Morishita, Y.; Matsushita, H.; Kakimi, K. Dendritic cell vaccine induces antigen-specific CD8+ T cells that are metabolically distinct from those of peptide vaccine and is well-combined with PD-1 checkpoint blockade. Oncoimmunology 2018, 7, e1395124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, E.; Robbins, P.F.; Rosenberg, S.A. ‘Final common pathway’ of human cancer immunotherapy: Targeting random somatic mutations. Nat. Immunol. 2017, 18, 255–262. [Google Scholar] [CrossRef]
- Leko, V.; Rosenberg, S.A. Identifying and Targeting Human Tumor Antigens for T Cell-Based Immunotherapy of Solid Tumors. Cancer Cell 2020, 38, 454–472. [Google Scholar] [CrossRef]
- Kanaseki, T.; Tokita, S.; Torigoe, T. Proteogenomic discovery of cancer antigens: Neoantigens and beyond. Pathol. Int. 2019, 69, 511–518. [Google Scholar] [CrossRef] [Green Version]
- Hundal, J.; Kiwala, S.; Feng, Y.Y.; Liu, C.J.; Govindan, R.; Chapman, W.C.; Uppaluri, R.; Swamidass, S.J.; Griffith, O.L.; Mardis, E.R.; et al. Accounting for proximal variants improves neoantigen prediction. Nat. Genet. 2019, 51, 175–179. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdottir, H.; Wenger, A.M.; Zehir, A.; Mesirov, J.P. Variant Review with the Integrative Genomics Viewer. Cancer Res. 2017, 77, e31–e34. [Google Scholar] [CrossRef] [Green Version]
- Kesmir, C.; Nussbaum, A.K.; Schild, H.; Detours, V.; Brunak, S. Prediction of proteasome cleavage motifs by neural networks. Protein Eng. 2002, 15, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Larsen, M.V.; Lundegaard, C.; Lamberth, K.; Buus, S.; Brunak, S.; Lund, O.; Nielsen, M. An integrative approach to CTL epitope prediction: A combined algorithm integrating MHC class I binding, TAP transport efficiency, and proteasomal cleavage predictions. Eur. J. Immunol. 2005, 35, 2295–2303. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| YTN2 | YTN16 | ||
|---|---|---|---|
| mutation | mCdt1 | T258M | T258M |
| mScarb2 | wild type | K426T | |
| mZfp106 | A656T & C659R | A656T & C659R | |
| gene expression (FPKM) | Cdt1 | 50.67 | 47.91 |
| Scarb2 | 29.73 | 54.31 | |
| Zfp106 | 43.58 | 38.22 | |
| RNA variant allele frequency (VAF) | mCdt1 | 0.52 | 0.41 |
| mScarb2 | 0.00 | 0.09 | |
| mZfp106 | 1.00 | 1.00 | |
| MHC ligandome | mCdt1 | T258M KTVYP[M]SYRF | not detected |
| mScarb2 | not detected | not detected | |
| mZfp106 | not detected | not detected |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagaoka, K.; Sun, C.; Kobayashi, Y.; Kanaseki, T.; Tokita, S.; Komatsu, T.; Maejima, K.; Futami, J.; Nomura, S.; Udaka, K.; et al. Identification of Neoantigens in Two Murine Gastric Cancer Cell Lines Leading to the Neoantigen-Based Immunotherapy. Cancers 2022, 14, 106. https://doi.org/10.3390/cancers14010106
Nagaoka K, Sun C, Kobayashi Y, Kanaseki T, Tokita S, Komatsu T, Maejima K, Futami J, Nomura S, Udaka K, et al. Identification of Neoantigens in Two Murine Gastric Cancer Cell Lines Leading to the Neoantigen-Based Immunotherapy. Cancers. 2022; 14(1):106. https://doi.org/10.3390/cancers14010106
Chicago/Turabian StyleNagaoka, Koji, Changbo Sun, Yukari Kobayashi, Takayuki Kanaseki, Serina Tokita, Toshihiro Komatsu, Kazuhiro Maejima, Junichiro Futami, Sachiyo Nomura, Keiko Udaka, and et al. 2022. "Identification of Neoantigens in Two Murine Gastric Cancer Cell Lines Leading to the Neoantigen-Based Immunotherapy" Cancers 14, no. 1: 106. https://doi.org/10.3390/cancers14010106
APA StyleNagaoka, K., Sun, C., Kobayashi, Y., Kanaseki, T., Tokita, S., Komatsu, T., Maejima, K., Futami, J., Nomura, S., Udaka, K., Nakagawa, H., Torigoe, T., & Kakimi, K. (2022). Identification of Neoantigens in Two Murine Gastric Cancer Cell Lines Leading to the Neoantigen-Based Immunotherapy. Cancers, 14(1), 106. https://doi.org/10.3390/cancers14010106