
Loss of E-Cadherin Leads to Druggable Vulnerabilities in Sphingolipid Metabolism and Vesicle Trafficking

 , ,
, ,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Drug Screening
2.3. Autophagy Assay
2.4. Organoid Culture
2.5. Fluorescence-Activated Cell Sorting
2.6. Immunofluorescence
2.7. Organoid Drug Screening
3. Results
3.1. Identification of Novel Synthetic Lethal Pathways for CDH1
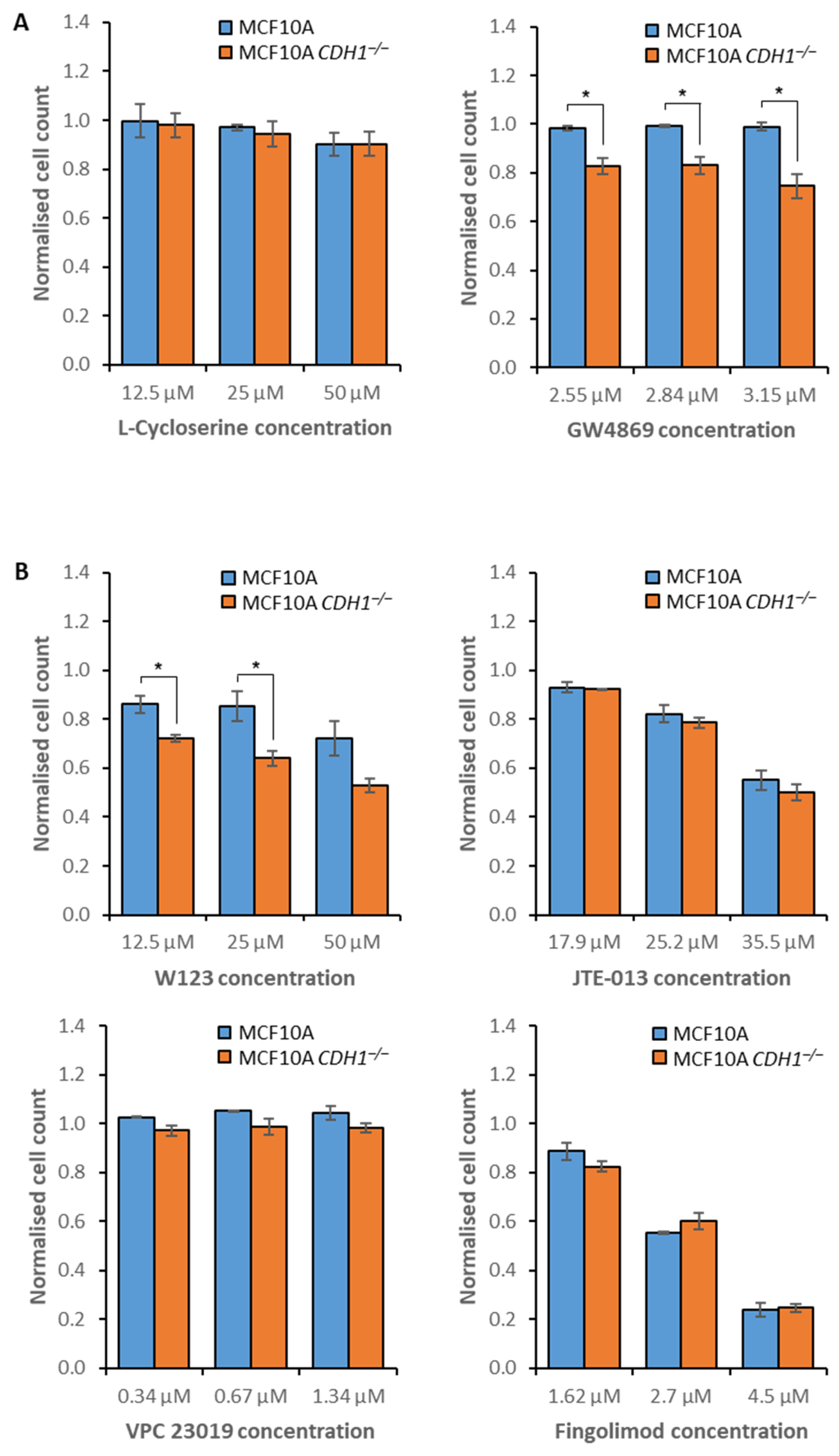
3.2. MCF10A CDH1−/− Cells Are Vulnerable to the Inhibition of Sphingolipid Metabolism and Signaling
3.3. E-Cadherin-Null Cells Exhibit Vulnerabilities in Clathrin- and Flotillin-Mediated Endocytosis
3.4. Disruption of Autophagy Preferentially Inhibits the Growth of Non-Tumorigenic CDH1−/− Cells
3.5. Combination Drug Treatment Enhances Efficacy against MCF10A CDH1−/− Cells
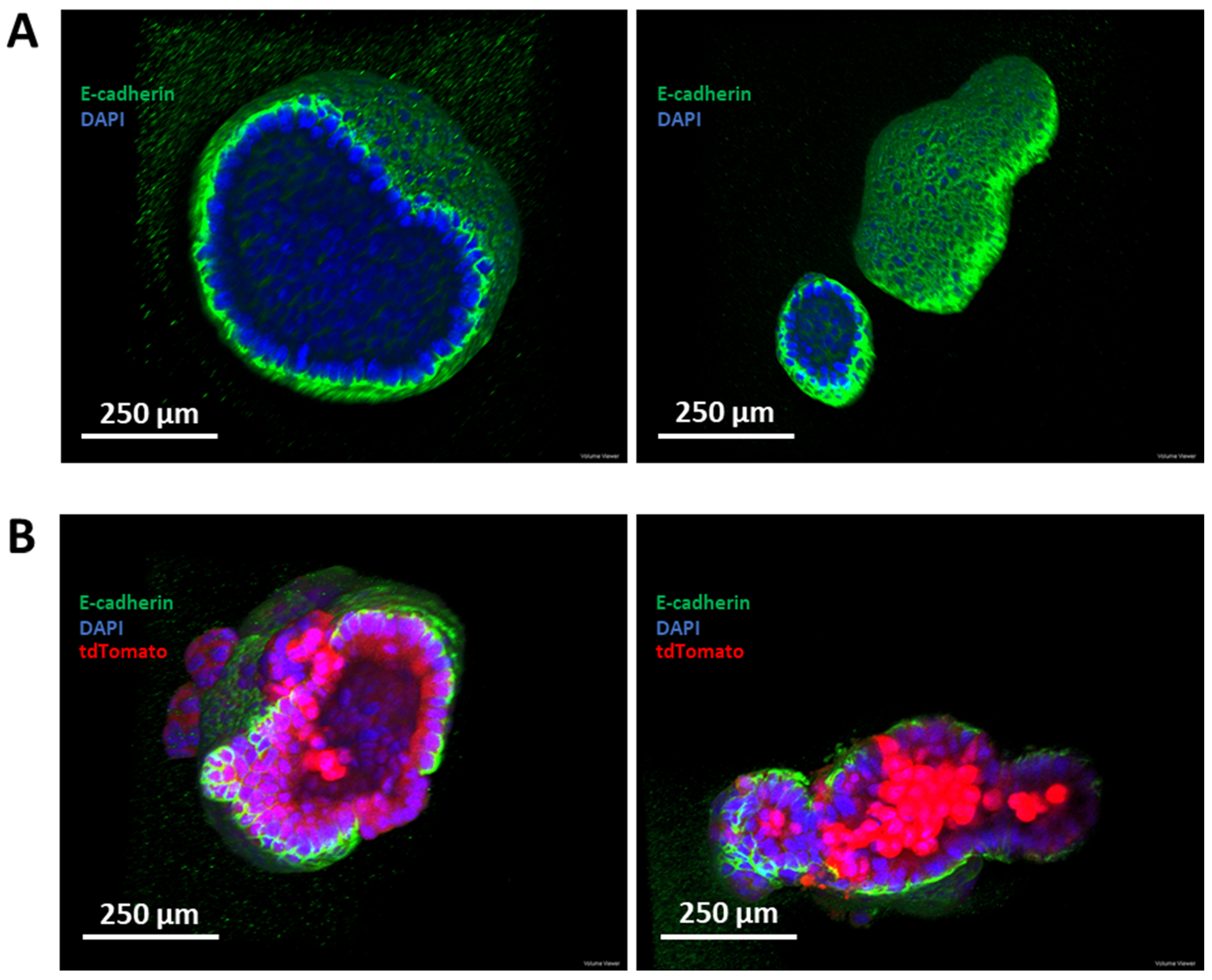
3.6. Establishment of a Murine-Derived Gastric Organoid Model of HDGC
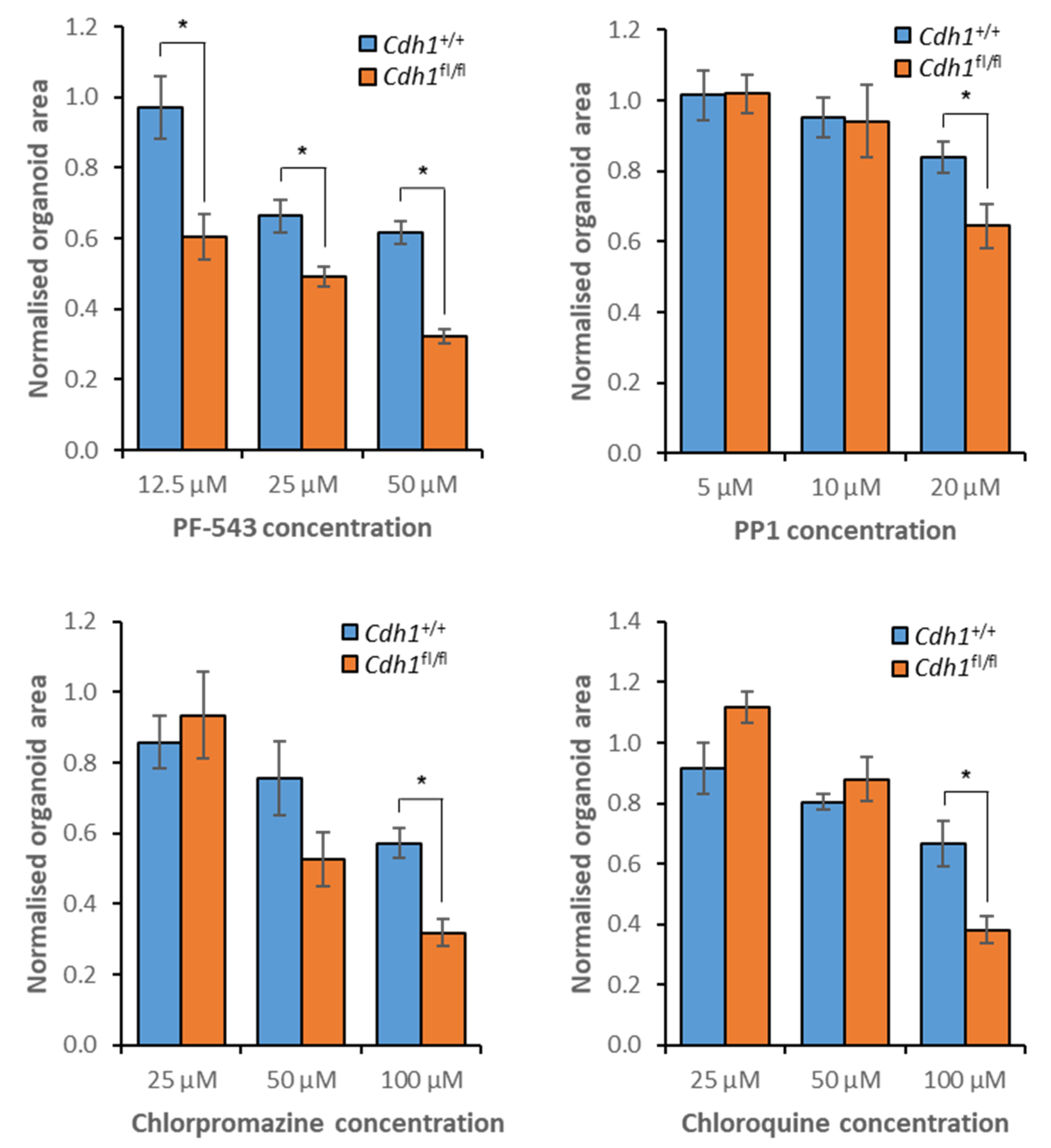
3.7. Validation of Candidate Synthetic Lethal Compounds in Organoid Models of HDGC
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Blair, V.R.; McLeod, M.; Carneiro, F.; Coit, D.G.; D’Addario, J.L.; van Dieren, J.M.; Harris, K.L.; Hoogerbrugge, N.; Oliveira, C.; van der Post, R.S.; et al. Hereditary diffuse gastric cancer: Updated clinical practice guidelines. Lancet Oncol. 2020, 21, e386–e397. [Google Scholar] [CrossRef]
- Guilford, P.; Hopkins, J.; Harraway, J.; McLeod, M.; McLeod, N.; Harawira, P.; Taite, H.; Scoular, R.; Miller, A.; Reeve, A.E. E-cadherin germline mutations in familial gastric cancer. Nature 1998, 392, 402–405. [Google Scholar] [CrossRef] [PubMed]
- Hallowell, N.; Lawton, J.; Badger, S.; Richardson, S.; Hardwick, R.H.; Caldas, C.; Fitzgerald, R.C. The psychosocial impact of undergoing prophylactic total gastrectomy (ptg) to manage the risk of hereditary diffuse gastric cancer (hdgc). J. Genet. Couns. 2017, 26, 752–762. [Google Scholar] [CrossRef] [PubMed]
- Yonemura, S. Cadherin-actin interactions at adherens junctions. Curr. Opin. Cell Biol. 2011, 23, 515–522. [Google Scholar] [CrossRef]
- Lecuit, T.; Yap, A.S. E-cadherin junctions as active mechanical integrators in tissue dynamics. Nat. Cell Biol. 2015, 17, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, M.C.; Halder, G. Regulation of the hippo pathway by cell architecture and mechanical signals. Semin. Cell Dev. Biol. 2012, 23, 803–811. [Google Scholar] [CrossRef]
- Becker, K.F.; Atkinson, M.J.; Reich, U.; Becker, I.; Nekarda, H.; Siewert, J.R.; Hofler, H. E-cadherin gene mutations provide clues to diffuse type gastric carcinomas. Cancer Res. 1994, 54, 3845–3852. [Google Scholar]
- Berx, G.; Cleton-Jansen, A.M.; Strumane, K.; de Leeuw, W.J.; Nollet, F.; van Roy, F.; Cornelisse, C. E-cadherin is inactivated in a majority of invasive human lobular breast cancers by truncation mutations throughout its extracellular domain. Oncogene 1996, 13, 1919–1925. [Google Scholar]
- Carvalho, J.; van Grieken, N.C.; Pereira, P.M.; Sousa, S.; Tijssen, M.; Buffart, T.E.; Diosdado, B.; Grabsch, H.; Santos, M.A.; Meijer, G.; et al. Lack of microrna-101 causes e-cadherin functional deregulation through ezh2 up-regulation in intestinal gastric cancer. J. Pathol. 2012, 228, 31–44. [Google Scholar] [CrossRef]
- Rossi, T.; Tedaldi, G.; Petracci, E.; Abou Khouzam, R.; Ranzani, G.N.; Morgagni, P.; Saragoni, L.; Monti, M.; Calistri, D.; Ulivi, P.; et al. E-cadherin downregulation and micrornas in sporadic intestinal-type gastric cancer. Int. J. Mol. Sci. 2019, 20, 4452. [Google Scholar] [CrossRef]
- Kaelin, W.G., Jr. The concept of synthetic lethality in the context of anticancer therapy. Nat. Rev. Cancer 2005, 5, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Beetham, H.; Chen, A.; Telford, B.J.; Single, A.; Jarman, K.E.; Lackovic, K.; Luxenburger, A.; Guilford, P. A high-throughput screen to identify novel synthetic lethal compounds for the treatment of e-cadherin-deficient cells. Sci. Rep. 2019, 9, 12511. [Google Scholar] [CrossRef] [PubMed]
- Bougen-Zhukov, N.; Nouri, Y.; Godwin, T.; Taylor, M.; Hakkaart, C.; Single, A.; Brew, T.; Permina, E.; Chen, A.; Black, M.A.; et al. Allosteric akt inhibitors target synthetic lethal vulnerabilities in e-cadherin-deficient cells. Cancers 2019, 11, 1359. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Beetham, H.; Black, M.A.; Priya, R.; Telford, B.J.; Guest, J.; Wiggins, G.A.; Godwin, T.D.; Yap, A.S.; Guilford, P.J. E-cadherin loss alters cytoskeletal organization and adhesion in non-malignant breast cells but is insufficient to induce an epithelial-mesenchymal transition. BMC Cancer 2014, 14, 552. [Google Scholar] [CrossRef] [PubMed]
- Godwin, T.D.; Kelly, S.T.; Brew, T.P.; Bougen-Zhukov, N.M.; Single, A.B.; Chen, A.; Stylianou, C.E.; Harris, L.D.; Currie, S.K.; Telford, B.J.; et al. E-cadherin-deficient cells have synthetic lethal vulnerabilities in plasma membrane organisation, dynamics and function. Gastric Cancer 2019, 22, 273–286. [Google Scholar] [CrossRef]
- Telford, B.J.; Chen, A.; Beetham, H.; Frick, J.; Brew, T.P.; Gould, C.M.; Single, A.; Godwin, T.; Simpson, K.J.; Guilford, P. Synthetic lethal screens identify vulnerabilities in gpcr signaling and cytoskeletal organization in e-cadherin-deficient cells. Mol. Cancer Ther. 2015, 14, 1213–1223. [Google Scholar] [CrossRef]
- Debnath, J.; Muthuswamy, S.K.; Brugge, J.S. Morphogenesis and oncogenesis of mcf-10a mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods 2003, 30, 256–268. [Google Scholar] [CrossRef]
- Soule, H.D.; Maloney, T.M.; Wolman, S.R.; Peterson, W.D., Jr.; Brenz, R.; McGrath, C.M.; Russo, J.; Pauley, R.J.; Jones, R.F.; Brooks, S.C. Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, mcf-10. Cancer Res. 1990, 50, 6075–6086. [Google Scholar]
- Single, A.; Beetham, H.; Telford, B.J.; Guilford, P.; Chen, A. A comparison of real-time and endpoint cell viability assays for improved synthetic lethal drug validation. J. Biomol. Screen. 2015, 20, 1286–1293. [Google Scholar] [CrossRef]
- Bartfeld, S.; Clevers, H. Organoids as model for infectious diseases: Culture of human and murine stomach organoids and microinjection of helicobacter pylori. J. Vis. Exp. 2015, 105, 53359. [Google Scholar] [CrossRef]
- Ewald, A.J. Isolation of mouse mammary organoids for long-term time-lapse imaging. Cold Spring Harb. Protoc. 2013, 2013, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Broutier, L.; Andersson-Rolf, A.; Hindley, C.J.; Boj, S.F.; Clevers, H.; Koo, B.K.; Huch, M. Culture and establishment of self-renewing human and mouse adult liver and pancreas 3d organoids and their genetic manipulation. Nat. Protoc. 2016, 11, 1724–1743. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. Nih image to imagej: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Guo, S.; Zhang, X.; Zheng, M.; Zhang, X.; Min, C.; Wang, Z.; Cheon, S.H.; Oak, M.-H.; Nah, S.-Y.; Kim, K.-M. Selectivity of commonly used inhibitors of clathrin-mediated and caveolae-dependent endocytosis of g protein–coupled receptors. Biochim. Biophys. Acta Biomembr. 2015, 1848, 2101–2110. [Google Scholar] [CrossRef]
- Dutta, D.; Donaldson, J.G. Search for inhibitors of endocytosis: Intended specificity and unintended consequences. Cell Logist. 2012, 2, 203–208. [Google Scholar] [CrossRef]
- Neumann-Giesen, C.; Fernow, I.; Amaddii, M.; Tikkanen, R. Role of egf-induced tyrosine phosphorylation of reggie-1/flotillin-2 in cell spreading and signaling to the actin cytoskeleton. J. Cell Sci. 2007, 120, 395–406. [Google Scholar] [CrossRef]
- Carcea, I.; Ma’ayan, A.; Mesias, R.; Sepulveda, B.; Salton, S.R.; Benson, D.L. Flotillin-mediated endocytic events dictate cell type-specific responses to semaphorin 3a. J. Neurosci. 2010, 30, 15317–15329. [Google Scholar] [CrossRef]
- Chou, T.F.; Brown, S.J.; Minond, D.; Nordin, B.E.; Li, K.; Jones, A.C.; Chase, P.; Porubsky, P.R.; Stoltz, B.M.; Schoenen, F.J.; et al. Reversible inhibitor of p97, dbeq, impairs both ubiquitin-dependent and autophagic protein clearance pathways. Proc. Natl. Acad. Sci. USA 2011, 108, 4834–4839. [Google Scholar] [CrossRef]
- Ramanathan, H.N.; Ye, Y. The p97 atpase associates with eea1 to regulate the size of early endosomes. Cell Res. 2012, 22, 346–359. [Google Scholar] [CrossRef] [PubMed]
- Magnaghi, P.; D’Alessio, R.; Valsasina, B.; Avanzi, N.; Rizzi, S.; Asa, D.; Gasparri, F.; Cozzi, L.; Cucchi, U.; Orrenius, C.; et al. Covalent and allosteric inhibitors of the atpase vcp/p97 induce cancer cell death. Nat. Chem. Biol. 2013, 9, 548–556. [Google Scholar] [CrossRef] [PubMed]
- Glanz, V.Y.; Myasoedova, V.A.; Grechko, A.V.; Orekhov, A.N. Inhibition of sialidase activity as a therapeutic approach. Drug Des. Dev. Ther. 2018, 12, 3431–3437. [Google Scholar] [CrossRef] [PubMed]
- Rejman, J.; Bragonzi, A.; Conese, M. Role of clathrin- and caveolae-mediated endocytosis in gene transfer mediated by lipo- and polyplexes. Mol. Ther. 2005, 12, 468–474. [Google Scholar] [CrossRef]
- Solomon, V.R.; Lee, H. Chloroquine and its analogs: A new promise of an old drug for effective and safe cancer therapies. Eur. J. Pharmacol. 2009, 625, 220–233. [Google Scholar] [CrossRef]
- Cook, K.L.; Wärri, A.; Soto-Pantoja, D.R.; Clarke, P.A.; Cruz, M.I.; Zwart, A.; Clarke, R. Hydroxychloroquine inhibits autophagy to potentiate antiestrogen responsiveness in er+ breast cancer. Clin. Cancer Res. 2014, 20, 3222–3232. [Google Scholar] [CrossRef]
- Sáenz, J.B.; Sun, W.J.; Chang, J.W.; Li, J.; Bursulaya, B.; Gray, N.S.; Haslam, D.B. Golgicide a reveals essential roles for gbf1 in golgi assembly and function. Nat. Chem. Biol. 2009, 5, 157–165. [Google Scholar] [CrossRef]
- Miller, S.G.; Carnell, L.; Moore, H.H. Post-golgi membrane traffic: Brefeldin a inhibits export from distal golgi compartments to the cell surface but not recycling. J. Cell Biol. 1992, 118, 267–283. [Google Scholar] [CrossRef]
- Ram, B.M.; Ramakrishna, G. Endoplasmic reticulum vacuolation and unfolded protein response leading to paraptosis like cell death in cyclosporine a treated cancer cervix cells is mediated by cyclophilin b inhibition. Biochim. Biophys. Acta Biomembr. 2014, 1843, 2497–2512. [Google Scholar] [CrossRef] [PubMed]
- Wada, A.; Fukuda, M.; Mishima, M.; Nishida, E. Nuclear export of actin: A novel mechanism regulating the subcellular localization of a major cytoskeletal protein. EMBO J. 1998, 17, 1635–1641. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Martinez-Williams, C.; Gilbert, K.A.; Rannels, D.E. Inhibition of gap junction communication in alveolar epithelial cells by 18alpha-glycyrrhetinic acid. Am. J. Physiol. 1999, 276, L1018–L1026. [Google Scholar]
- Manjarrez-Marmolejo, J.; Franco-Pérez, J. Gap junction blockers: An overview of their effects on induced seizures in animal models. Curr. Neuropharmacol. 2016, 14, 759–771. [Google Scholar] [CrossRef]
- Hojjati, M.R.; Li, Z.; Zhou, H.; Tang, S.; Huan, C.; Ooi, E.; Lu, S.; Jiang, X.-C. Effect of myriocin on plasma sphingolipid metabolism and atherosclerosis in apoe-deficient mice. J. Biol. Chem. 2005, 280, 10284–10289. [Google Scholar] [CrossRef]
- Spiegel, S.; Merrill, A.H., Jr. Sphingolipid metabolism and cell growth regulation. FASEB J. 1996, 10, 1388–1397. [Google Scholar] [CrossRef] [PubMed]
- French, K.J.; Zhuang, Y.; Maines, L.W.; Gao, P.; Wang, W.; Beljanski, V.; Upson, J.J.; Green, C.L.; Keller, S.N.; Smith, C.D. Pharmacology and antitumor activity of abc294640, a selective inhibitor of sphingosine kinase-2. J. Pharmacol. Exp. Ther. 2010, 333, 129–139. [Google Scholar] [CrossRef] [PubMed]
- French, K.J.; Schrecengost, R.S.; Lee, B.D.; Zhuang, Y.; Smith, S.N.; Eberly, J.L.; Yun, J.K.; Smith, C.D. Discovery and evaluation of inhibitors of human sphingosine kinase. Cancer Res. 2003, 63, 5962–5969. [Google Scholar] [PubMed]
- Piali, L.; Froidevaux, S.; Hess, P.; Nayler, O.; Bolli, M.H.; Schlosser, E.; Kohl, C.; Steiner, B.; Clozel, M. The selective sphingosine 1-phosphate receptor 1 agonist ponesimod protects against lymphocyte-mediated tissue inflammation. J. Pharmacol. Exp. Ther. 2011, 337, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Schnute, M.E.; McReynolds, M.D.; Kasten, T.; Yates, M.; Jerome, G.; Rains, J.W.; Hall, T.; Chrencik, J.; Kraus, M.; Cronin, C.N.; et al. Modulation of cellular s1p levels with a novel, potent and specific inhibitor of sphingosine kinase-1. Biochem. J. 2012, 444, 79–88. [Google Scholar] [CrossRef]
- Pyne, N.J.; Adams, D.R.; Pyne, S. Sphingosine kinase 2 in autoimmune/inflammatory disease and the development of sphingosine kinase 2 inhibitors. Trends Pharm. Sci. 2017, 38, 581–591. [Google Scholar] [CrossRef]
- Pitman, M.R.; Powell, J.A.; Coolen, C.; Moretti, P.A.B.; Zebol, J.R.; Pham, D.H.; Finnie, J.W.; Don, A.S.; Ebert, L.M.; Bonder, C.S.; et al. A selective atp-competitive sphingosine kinase inhibitor demonstrates anti-cancer properties. Oncotarget 2015, 6, 7065–7083. [Google Scholar] [CrossRef]
- Sundaram, K.S.; Lev, M. Inhibition of sphingolipid synthesis by cycloserine in vitro and in vivo. J. Neurochem. 1984, 42, 577–581. [Google Scholar] [CrossRef]
- Luberto, C.; Hassler, D.F.; Signorelli, P.; Okamoto, Y.; Sawai, H.; Boros, E.; Hazen-Martin, D.J.; Obeid, L.M.; Hannun, Y.A.; Smith, G.K. Inhibition of tumor necrosis factor-induced cell death in mcf7 by a novel inhibitor of neutral sphingomyelinase. J. Biol. Chem. 2002, 277, 41128–41139. [Google Scholar] [CrossRef]
- Kohno, M.; Momoi, M.; Oo, M.L.; Paik, J.H.; Lee, Y.M.; Venkataraman, K.; Ai, Y.; Ristimaki, A.P.; Fyrst, H.; Sano, H.; et al. Intracellular role for sphingosine kinase 1 in intestinal adenoma cell proliferation. Mol. Cell Biol. 2006, 26, 7211–7223. [Google Scholar] [CrossRef]
- Spiegel, S.; Milstien, S. Sphingosine-1-phosphate: An enigmatic signalling lipid. Nat. Rev. Mol. Cell Biol. 2003, 4, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Davis, M.D.; Heise, C.E.; Albert, R.; Cottens, S.; Hof, R.; Bruns, C.; Prieschl, E.; Baumruker, T.; Hiestand, P.; et al. The immune modulator fty720 targets sphingosine 1-phosphate receptors. J. Biol. Chem. 2002, 277, 21453–21457. [Google Scholar] [CrossRef]
- Davis, M.D.; Clemens, J.J.; Macdonald, T.L.; Lynch, K.R. Sphingosine 1-phosphate analogs as receptor antagonists. J. Biol. Chem. 2005, 280, 9833–9841. [Google Scholar] [CrossRef] [PubMed]
- Osada, M.; Yatomi, Y.; Ohmori, T.; Ikeda, H.; Ozaki, Y. Enhancement of sphingosine 1-phosphate-induced migration of vascular endothelial cells and smooth muscle cells by an edg-5 antagonist. Biochem. Biophys. Res. Commun. 2002, 299, 483–487. [Google Scholar] [CrossRef]
- Wei, S.H.; Rosen, H.; Matheu, M.P.; Sanna, M.G.; Wang, S.K.; Jo, E.; Wong, C.H.; Parker, I.; Cahalan, M.D. Sphingosine 1-phosphate type 1 receptor agonism inhibits transendothelial migration of medullary t cells to lymphatic sinuses. Nat. Immunol. 2005, 6, 1228–1235. [Google Scholar] [CrossRef]
- Gräler, M.H.; Bernhardt, G.; Lipp, M. Edg6, a novel g-protein-coupled receptor related to receptors for bioactive lysophospholipids, is specifically expressed in lymphoid tissue. Genomics 1998, 53, 164–169. [Google Scholar] [CrossRef]
- Im, D.S.; Heise, C.E.; Ancellin, N.; O’Dowd, B.F.; Shei, G.J.; Heavens, R.P.; Rigby, M.R.; Hla, T.; Mandala, S.; McAllister, G.; et al. Characterization of a novel sphingosine 1-phosphate receptor, edg-8. J. Biol. Chem. 2000, 275, 14281–14286. [Google Scholar] [CrossRef]
- Doody, A.M.; Antosh, A.L.; Brown, W.J. Cytoplasmic phospholipase a2 antagonists inhibit multiple endocytic membrane trafficking pathways. Biochem. Biophys. Res. Commun. 2009, 388, 695–699. [Google Scholar] [CrossRef]
- Gillespie, E.J.; Ho, C.L.; Balaji, K.; Clemens, D.L.; Deng, G.; Wang, Y.E.; Elsaesser, H.J.; Tamilselvam, B.; Gargi, A.; Dixon, S.D.; et al. Selective inhibitor of endosomal trafficking pathways exploited by multiple toxins and viruses. Proc. Natl. Acad. Sci. USA 2013, 110, E4904–E4912. [Google Scholar] [CrossRef] [PubMed]
- Bouhamdani, N.; Comeau, D.; Cormier, K.; Turcotte, S. Stf-62247 accumulates in lysosomes and blocks late stages of autophagy to selectively target von hippel-lindau-inactivated cells. Am. J. Physiol. Cell Physiol. 2019, 316, C605–C620. [Google Scholar] [CrossRef]
- Chou, T.C. Drug combination studies and their synergy quantification using the chou-talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef]
- Bieberich, E. Sphingolipids and lipid rafts: Novel concepts and methods of analysis. Chem. Phys. Lipids 2018, 216, 114–131. [Google Scholar] [CrossRef] [PubMed]
- Raghupathy, R.; Anilkumar, A.A.; Polley, A.; Singh, P.P.; Yadav, M.; Johnson, C.; Suryawanshi, S.; Saikam, V.; Sawant, S.D.; Panda, A.; et al. Transbilayer lipid interactions mediate nanoclustering of lipid-anchored proteins. Cell 2015, 161, 581–594. [Google Scholar] [CrossRef] [PubMed]
- Pavoine, C.; Pecker, F. Sphingomyelinases: Their regulation and roles in cardiovascular pathophysiology. Cardiovasc. Res. 2009, 82, 175–183. [Google Scholar] [CrossRef]
- Gomà, A.; Mir, R.; Martínez-Soler, F.; Tortosa, A.; Vidal, A.; Condom, E.; Pérez-Tomás, R.; Giménez-Bonafé, P. Multidrug resistance protein 1 localization in lipid raft domains and prostasomes in prostate cancer cell lines. OncoTargets Ther. 2014, 7, 2215–2225. [Google Scholar]
- D’Aprile, C.; Prioni, S.; Mauri, L.; Prinetti, A.; Grassi, S. Lipid rafts as platforms for sphingosine 1-phosphate metabolism and signalling. Cell. Signal. 2021, 80, 109929. [Google Scholar] [CrossRef]
- Frost, S.C.; Lane, M.D.; Gibbs, E.M. Effect of phenylarsine oxide on fluid phase endocytosis: Further evidence for activation of the glucose transporter. J. Cell. Physiol. 1989, 141, 467–474. [Google Scholar] [CrossRef]
- Massol, P.; Montcourrier, P.; Guillemot, J.C.; Chavrier, P. Fc receptor-mediated phagocytosis requires cdc42 and rac1. EMBO J. 1998, 17, 6219–6229. [Google Scholar] [CrossRef]
- Messa, M.; Fernández-Busnadiego, R.; Sun, E.W.; Chen, H.; Czapla, H.; Wrasman, K.; Wu, Y.; Ko, G.; Ross, T.; Wendland, B.; et al. Epsin deficiency impairs endocytosis by stalling the actin-dependent invagination of endocytic clathrin-coated pits. eLife 2014, 3, e03311. [Google Scholar] [CrossRef]
- Idrissi, F.Z.; Grötsch, H.; Fernández-Golbano, I.M.; Presciatto-Baschong, C.; Riezman, H.; Geli, M.I. Distinct acto/myosin-i structures associate with endocytic profiles at the plasma membrane. J. Cell Biol. 2008, 180, 1219–1232. [Google Scholar] [CrossRef] [PubMed]
- Pontes, B.; Monzo, P.; Gauthier, N.C. Membrane tension: A challenging but universal physical parameter in cell biology. Semin. Cell Dev. Biol. 2017, 71, 30–41. [Google Scholar] [CrossRef]
- Boulant, S.; Kural, C.; Zeeh, J.C.; Ubelmann, F.; Kirchhausen, T. Actin dynamics counteract membrane tension during clathrin-mediated endocytosis. Nat. Cell Biol. 2011, 13, 1124–1131. [Google Scholar] [CrossRef]
- Kaksonen, M.; Roux, A. Mechanisms of clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol. 2018, 19, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Elkin, S.R.; Lakoduk, A.M.; Schmid, S.L. Endocytic pathways and endosomal trafficking: A primer. Wien. Med. Wochenschr. 2016, 166, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Frick, M.; Bright, N.A.; Riento, K.; Bray, A.; Merrified, C.; Nichols, B.J. Coassembly of flotillins induces formation of membrane microdomains, membrane curvature, and vesicle budding. Curr. Biol. 2007, 17, 1151–1156. [Google Scholar] [CrossRef]
- Goswami, D.; Gowrishankar, K.; Bilgrami, S.; Ghosh, S.; Raghupathy, R.; Chadda, R.; Vishwakarma, R.; Rao, M.; Mayor, S. Nanoclusters of gpi-anchored proteins are formed by cortical actin-driven activity. Cell 2008, 135, 1085–1097. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Varma, R.; Sarasij, R.C.; Ira Gousset, K.; Krishnamoorthy, G.; Rao, M.; Mayor, S. Nanoscale organization of multiple gpi-anchored proteins in living cell membranes. Cell 2004, 116, 577–589. [Google Scholar] [CrossRef]
- Mayor, S.; Parton, R.G.; Donaldson, J.G. Clathrin-independent pathways of endocytosis. Cold Spring Harb. Perspect. Biol. 2014, 6, a016758. [Google Scholar] [CrossRef]
- Römer, W.; Pontani, L.-L.; Sorre, B.; Rentero, C.; Berland, L.; Chambon, V.; Lamaze, C.; Bassereau, P.; Sykes, C.; Gaus, K.; et al. Actin dynamics drive membrane reorganization and scission in clathrin-independent endocytosis. Cell 2010, 140, 540–553. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.D.; Jensen, A.R.; Salgia, R.; Posadas, E.M. Fyn: A novel molecular target in cancer. Cancer 2010, 116, 1629–1637. [Google Scholar] [CrossRef]
- Semba, K.; Nishizawa, M.; Miyajima, N.; Yoshida, M.C.; Sukegawa, J.; Yamanashi, Y.; Sasaki, M.; Yamamoto, T.; Toyoshima, K. Yes-related protooncogene, syn, belongs to the protein-tyrosine kinase family. Proc. Natl. Acad. Sci. USA 1986, 83, 5459–5463. [Google Scholar] [CrossRef]
- Yu, J.; Zhou, Z.; Wei, Z.; Wu, J.; OuYang, J.; Huang, W.; He, Y.; Zhang, C. Fyn promotes gastric cancer metastasis by activating stat3-mediated epithelial-mesenchymal transition. Transl. Oncol. 2020, 13, 100841. [Google Scholar] [CrossRef]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef]
- Kast, D.J.; Zajac, A.L.; Holzbaur, E.L.; Ostap, E.M.; Dominguez, R. Whamm directs the arp2/3 complex to the er for autophagosome biogenesis through an actin comet tail mechanism. Curr. Biol. 2015, 25, 1791–1797. [Google Scholar] [CrossRef]
- Mi, N.; Chen, Y.; Wang, S.; Chen, M.; Zhao, M.; Yang, G.; Ma, M.; Su, Q.; Luo, S.; Shi, J.; et al. Capz regulates autophagosomal membrane shaping by promoting actin assembly inside the isolation membrane. Nat. Cell Biol. 2015, 17, 1112–1123. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; McPhee, C.K.; Zheng, L.; Mardones, G.A.; Rong, Y.; Peng, J.; Mi, N.; Zhao, Y.; Liu, Z.; Wan, F.; et al. Termination of autophagy and reformation of lysosomes regulated by mtor. Nature 2010, 465, 942–946. [Google Scholar] [CrossRef]
- Bouhamdani, N.; Joy, A.; Barnett, D.; Cormier, K.; Léger, D.; Chute, I.C.; Lamarre, S.; Ouellette, R.; Turcotte, S. Quantitative proteomics to study a small molecule targeting the loss of von hippel-lindau in renal cell carcinomas. Int. J. Cancer 2017, 141, 778–790. [Google Scholar] [CrossRef] [PubMed]
- Moruno Manchon, J.F.; Uzor, N.E.; Finkbeiner, S.; Tsvetkov, A.S. Sphk1/sphingosine kinase 1-mediated autophagy differs between neurons and sh-sy5y neuroblastoma cells. Autophagy 2016, 12, 1418–1424. [Google Scholar] [CrossRef][Green Version]
- Johannes, L.; Billet, A. Glycosylation and raft endocytosis in cancer. Cancer Metastasis Rev. 2020, 39, 375–396. [Google Scholar] [CrossRef] [PubMed]
- Riento, K.; Zhang, Q.; Clark, J.; Begum, F.; Stephens, E.; Wakelam, M.J.; Nichols, B.J. Flotillin proteins recruit sphingosine to membranes and maintain cellular sphingosine-1-phosphate levels. PLoS ONE 2018, 13, e0197401. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowska, K.; Matveichuk, O.V.; Fronk, J.; Ciesielska, A. Flotillins: At the intersection of protein s-palmitoylation and lipid-mediated signaling. Int. J. Mol. Sci. 2020, 21, 2283. [Google Scholar] [CrossRef]
- Hamada, M.; Kameyama, H.; Iwai, S.; Yura, Y. Induction of autophagy by sphingosine kinase 1 inhibitor pf-543 in head and neck squamous cell carcinoma cells. Cell Death Discov. 2017, 3, 17047. [Google Scholar] [CrossRef]
- Hieronymus, T.; Grötsch, P.; Blank, N.; Grünke, M.; Capraru, D.; Geiler, T.; Winkler, S.; Kalden, J.R.; Lorenz, H.M. Chlorpromazine induces apoptosis in activated human lymphoblasts: A mechanism supporting the induction of drug-induced lupus erythematosus? Arthritis Rheum. 2000, 43, 1994–2004. [Google Scholar] [CrossRef]
- Barker, N.; Huch, M.; Kujala, P.; van de Wetering, M.; Snippert, H.J.; van Es, J.H.; Sato, T.; Stange, D.E.; Begthel, H.; van den Born, M.; et al. Lgr5(+ve) stem cells drive self-renewal in the stomach and build long-lived gastric units in vitro. Cell Stem Cell 2010, 6, 25–36. [Google Scholar] [CrossRef]
- Martin, A.C.; Gelbart, M.; Fernandez-Gonzalez, R.; Kaschube, M.; Wieschaus, E.F. Integration of contractile forces during tissue invagination. J. Cell Biol. 2010, 188, 735–749. [Google Scholar] [CrossRef] [PubMed]
- Miyaguchi, K. Ultrastructure of the zonula adherens revealed by rapid-freeze deep-etching. J. Struct. Biol. 2000, 132, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, T.P.; Harris, A.R.; Lam, M.; Cheng, Q.; Bellis, J.; Dimitracopoulos, A.; Kabla, A.J.; Charras, G.T.; Baum, B. Emergence of homeostatic epithelial packing and stress dissipation through divisions oriented along the long cell axis. Proc. Natl. Acad. Sci. USA 2015, 112, 5726–5731. [Google Scholar] [CrossRef] [PubMed]
- Rocha, J.P.; Gullo, I.; Wen, X.; Devezas, V.; Baptista, M.; Oliveira, C.; Carneiro, F. Pathological features of total gastrectomy specimens from asymptomatic hereditary diffuse gastric cancer patients and implications for clinical management. Histopathology 2018, 73, 878–886. [Google Scholar] [CrossRef]
- Humar, B.; Guilford, P. Hereditary diffuse gastric cancer: A manifestation of lost cell polarity. Cancer Sci. 2009, 100, 1151–1157. [Google Scholar] [CrossRef]
- Abd-Rahman, A.N.; Marquart, L.; Gobeau, N.; Kümmel, A.; Simpson, J.A.; Chalon, S.; Möhrle, J.J.; McCarthy, J.S. Population pharmacokinetics and pharmacodynamics of chloroquine in a plasmodium vivax volunteer infection study. Clin. Pharmacol. Ther. 2020, 108, 1055–1066. [Google Scholar] [CrossRef] [PubMed]
- Adelusi, S.A.; Salako, L.A. Tissue and blood concentrations of chloroquine following chronic administration in the rat. J. Pharm. Pharmacol. 1982, 34, 733–735. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Process | Pathway of Interest | Compound |
|---|---|---|
| Endocytosis | Clathrin-mediated endocytosis | Concanavilin A [24] |
| Phenylarsine oxide [25] | ||
| Chlorpromazine [25] | ||
| Flotillin-mediated endocytosis | PP1 [26] | |
| PP2 [26] | ||
| SU6656 [27] | ||
| Vesicle formation | DBeQ [28,29] | |
| NMS-873 [29,30] | ||
| 3,4-methylenedioxy-β-nitrostyrene (MNS) [28,29] | ||
| Sialic acid-mediated endocytosis | N-Acetyl-2,3-dehydro-2-deoxyneuraminic acid [31] | |
| Oseltamivir [31] | ||
| Caveolae-mediated endocytosis | Genistein [32] | |
| Autophagy | Endosome acidification | Chloroquine [33] |
| Hydroxy-chloroquine [34] | ||
| Intracellular vesicle trafficking | Golgi apparatus vesicle transport | Golgicide A [35] |
| Brefeldin A [36] | ||
| Endoplasmic reticulum vesicle transport | Cyclosporin [37] | |
| Nuclear export | Leptomycin B [38] | |
| Gap junction vesicle transport | 18α-glycyrrhetinic acid [39] | |
| Carbenoxolone [40] | ||
| Plasma membrane organisation | Sphingolipid metabolism | Myriocin [41] |
| Fumonisin B1 [42] | ||
| ABC294640 [43] | ||
| SKI-11 [44] | ||
| Ponesimod [45] | ||
| PF-543 [46] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brew, T.; Bougen-Zhukov, N.; Mitchell, W.; Decourtye, L.; Schulpen, E.; Nouri, Y.; Godwin, T.; Guilford, P. Loss of E-Cadherin Leads to Druggable Vulnerabilities in Sphingolipid Metabolism and Vesicle Trafficking. Cancers 2022, 14, 102. https://doi.org/10.3390/cancers14010102
Brew T, Bougen-Zhukov N, Mitchell W, Decourtye L, Schulpen E, Nouri Y, Godwin T, Guilford P. Loss of E-Cadherin Leads to Druggable Vulnerabilities in Sphingolipid Metabolism and Vesicle Trafficking. Cancers. 2022; 14(1):102. https://doi.org/10.3390/cancers14010102
Chicago/Turabian StyleBrew, Tom, Nicola Bougen-Zhukov, Wilson Mitchell, Lyvianne Decourtye, Emily Schulpen, Yasmin Nouri, Tanis Godwin, and Parry Guilford. 2022. "Loss of E-Cadherin Leads to Druggable Vulnerabilities in Sphingolipid Metabolism and Vesicle Trafficking" Cancers 14, no. 1: 102. https://doi.org/10.3390/cancers14010102
APA StyleBrew, T., Bougen-Zhukov, N., Mitchell, W., Decourtye, L., Schulpen, E., Nouri, Y., Godwin, T., & Guilford, P. (2022). Loss of E-Cadherin Leads to Druggable Vulnerabilities in Sphingolipid Metabolism and Vesicle Trafficking. Cancers, 14(1), 102. https://doi.org/10.3390/cancers14010102