
Intra-Tumoral Genomic Heterogeneity in Rectal Cancer: Mutational Status Is Dependent on Preoperative Biopsy Depth and Location

 ,
,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Tumor Identification and DNA Isolation
2.3. SmMIP Sequencing
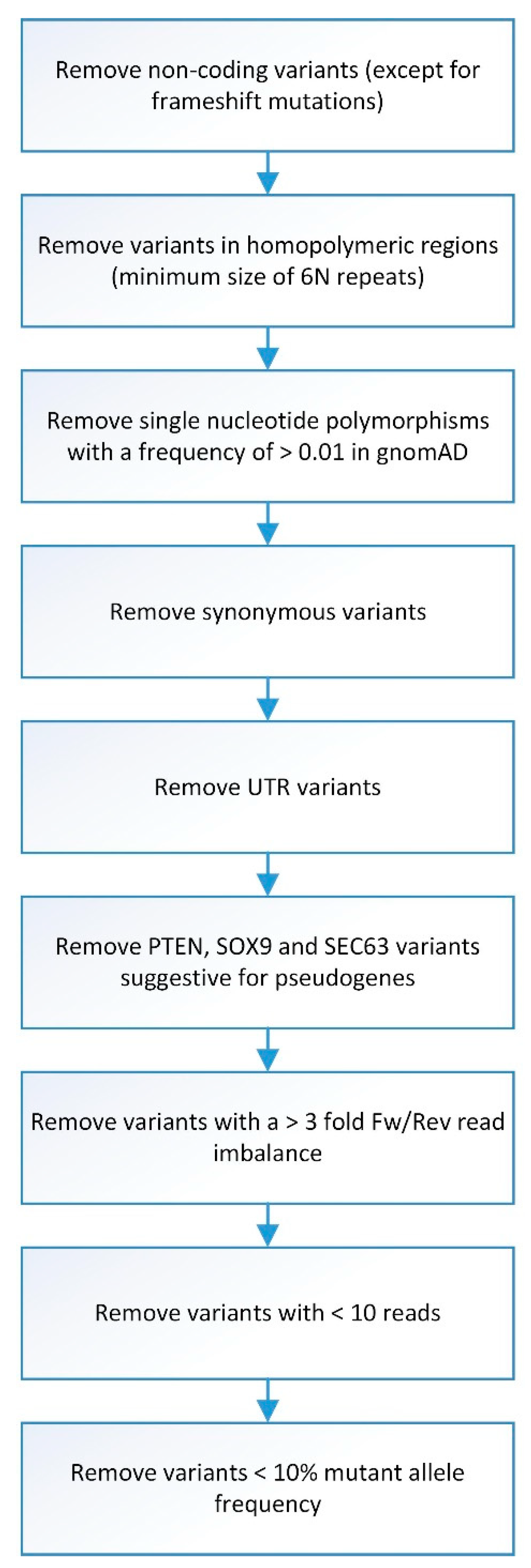
2.4. Sequence Data Analysis
2.5. Statistical Analysis
3. Results
3.1. Patients
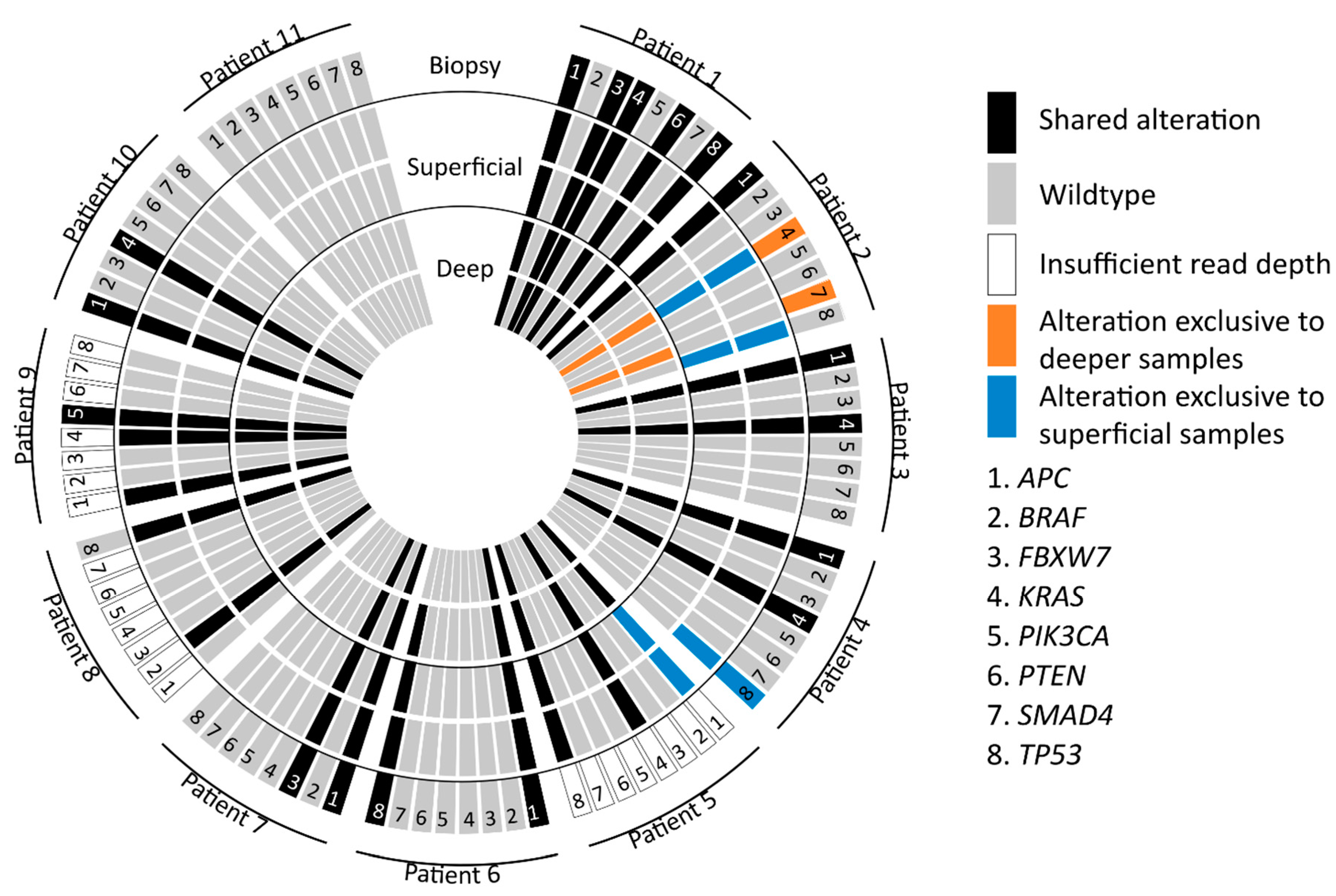
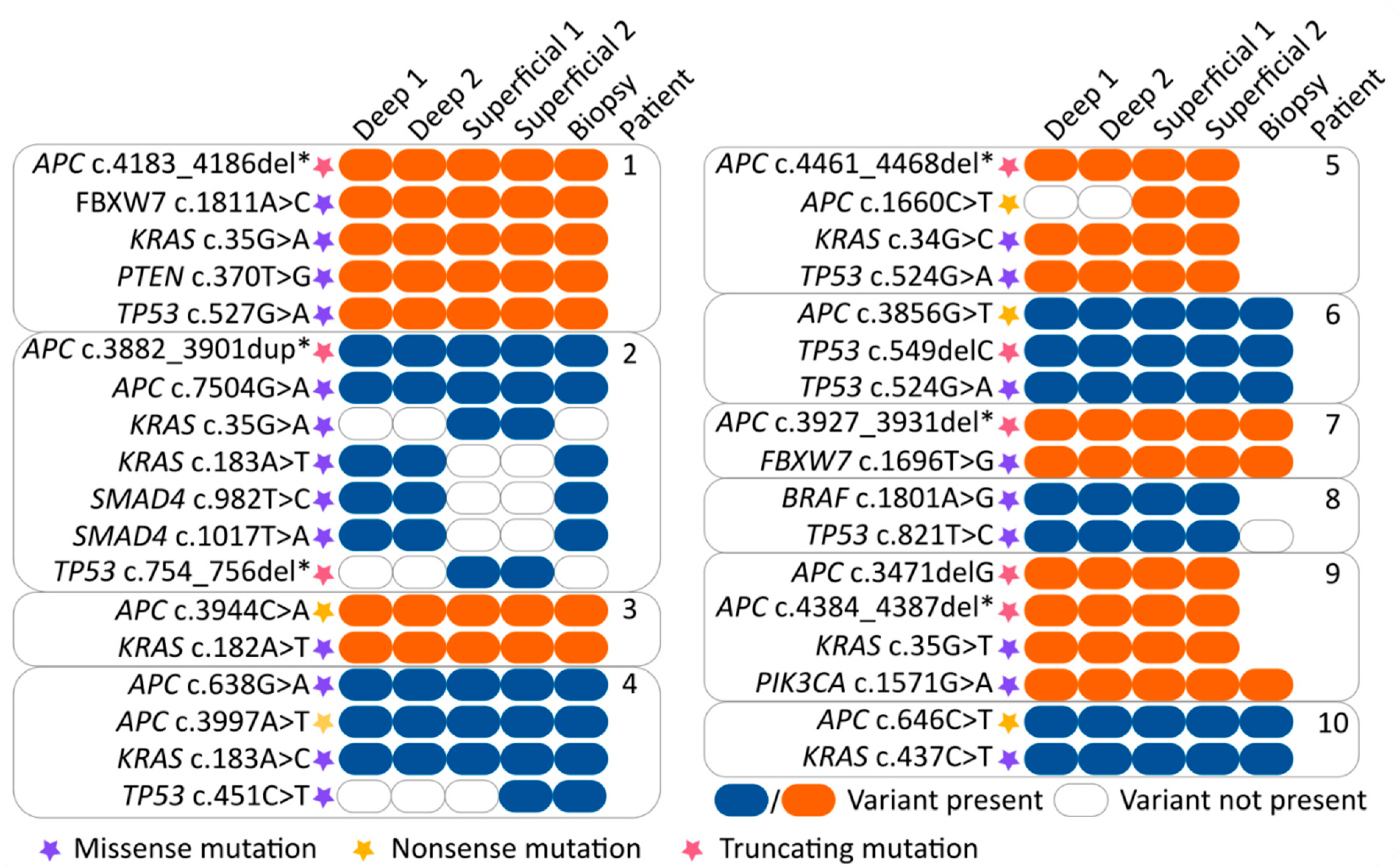
3.2. Mutation Concordance
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Beets-Tan, R.G.H.; Lambregts, D.M.J.; Maas, M.; Bipat, S.; Barbaro, B.; Curvo-Semedo, L.; Fenlon, H.M.; Gollub, M.J.; Gourtsoyianni, S.; Halligan, S.; et al. Magnetic resonance imaging for clinical management of rectal cancer: Updated recommendations from the 2016 European Society of Gastrointestinal and Abdominal Radiology (ESGAR) consensus meeting. Eur. Radiol. 2018, 28, 1465–1475. [Google Scholar] [CrossRef]
- Maas, M.; Nelemans, P.J.; Valentini, V.; Das, P.; Rödel, C.; Kuo, L.-J.; Calvo, F.A.; García-Aguilar, J.; Glynne-Jones, R.; Haustermans, K.; et al. Long-term outcome in patients with a pathological complete response after chemoradiation for rectal cancer: A pooled analysis of individual patient data. Lancet Oncol. 2010, 11, 835–844. [Google Scholar] [CrossRef]
- Appelt, A.L.; Pløen, J.; Harling, H.; Jensen, F.S.; Jensen, L.H.; Jørgensen, J.C.R.; Lindebjerg, J.; Rafaelsen, S.R.; Jakobsen, A. High-dose chemoradiotherapy and watchful waiting for distal rectal cancer: A prospective observational study. Lancet Oncol. 2015, 16, 919–927. [Google Scholar] [CrossRef]
- Park, J.-S.; Baek, J.-H.; Lee, W.-S.; Yang, J.-Y.; Lee, W.-K.; Kim, K.-K.; Park, Y.-H. Long-term oncologic outcomes in pathologic tumor response after neoadjuvant chemoradiation for locally advanced rectal cancer. Korean J. Clin. Oncol. 2018, 14, 37–42. [Google Scholar] [CrossRef]
- Landelijke Werkgroep Gastro Intestinale Tumoren Oncoline. Available online: https://www.oncoline.nl/colorectaalcarcinoom (accessed on 6 November 2019).
- Frydrych, L.M.; Ulintz, P.; Bankhead, A.; Sifuentes, C.; Greenson, J.; Maguire, L.; Irwin, R.; Fearon, E.R.; Hardiman, K.M. Rectal cancer sub-clones respond differentially to neoadjuvant therapy. Neoplasia 2019, 21, 1051–1062. [Google Scholar] [CrossRef]
- Hinohara, K.; Polyak, K. Intratumoral Heterogeneity: More Than Just Mutations. Trends Cell Biol. 2019, 29, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Parsons, B.L. Many different tumor types have polyclonal tumor origin: Evidence and implications. Mutat. Res. 2008, 659, 232–247. [Google Scholar] [CrossRef] [PubMed]
- Molinari, C.; Marisi, G.; Passardi, A.; Matteucci, L.; De Maio, G.; Ulivi, P. Heterogeneity in Colorectal Cancer: A Challenge for Personalized Medicine? Int. J. Mol. Sci. 2018, 19, 3733. [Google Scholar] [CrossRef]
- Grady, W.M.; Carethers, J.M. Genomic and epigenetic instability in colorectal cancer pathogenesis. Gastroenterology 2008, 135, 1079–1099. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Habr-Gama, A.; São Julião, G.P.; Gama-Rodrigues, J.; Vailati, B.B.; Ortega, C.; Fernandez, L.M.; Araújo, S.E.A.; Perez, R.O.; Baseline, T. Classification Predicts Early Tumor Regrowth After Nonoperative Management in Distal Rectal Cancer After Extended Neoadjuvant Chemoradiation and Initial Complete Clinical Response. Dis. Colon Rectum 2017, 60, 586–594. [Google Scholar] [CrossRef] [PubMed]
- Joye, I.; Deroose, C.M.; Vandecaveye, V.; Haustermans, K. The role of diffusion-weighted MRI and (18)F-FDG PET/CT in the prediction of pathologic complete response after radiochemotherapy for rectal cancer: A systematic review. Radiother. Oncol. J. Eur. Soc. Ther. Radiol. Oncol. 2014, 113, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Giralt, J.; Eraso, A.; Armengol, M.; Rosselló, J.; Majó, J.; Ares, C.; Espin, E.; Benavente, S.; de Torres, I. Epidermal growth factor receptor is a predictor of tumor response in locally advanced rectal cancer patients treated with preoperative radiotherapy. Int. J. Radiat. Oncol. 2002, 54, 1460–1465. [Google Scholar] [CrossRef]
- Reerink, O.; Karrenbeld, A.; Plukker, J.T.M.; Verschueren, R.C.J.; Szabo M, B.G.; Sluiter, W.J.; Hospers, G.A.P.; Mulder, N.H. Molecular Prognostic Factors in Locally Irresectable Rectal Cancer Treated Preoperatively by Chemo-radiotherapy. Anticancer Res. 2004, 24, 1217–1222. [Google Scholar] [PubMed]
- Tsang, J.S.; Vencken, S.; Sharaf, O.; Leen, E.; Kay, E.W.; McNamara, D.A.; Deasy, J.; Mulligan, E.D. Global DNA methylation is altered by neoadjuvant chemoradiotherapy in rectal cancer and may predict response to treatment—A pilot study. Eur. J. Surg. Oncol. EJSO 2014, 40, 1459–1466. [Google Scholar] [CrossRef]
- Lièvre, A.; Bachet, J.-B.; Le Corre, D.; Boige, V.; Landi, B.; Emile, J.-F.; Côté, J.-F.; Tomasic, G.; Penna, C.; Ducreux, M.; et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006, 66, 3992–3995. [Google Scholar] [CrossRef]
- Linardou, H.; Dahabreh, I.J.; Kanaloupiti, D.; Siannis, F.; Bafaloukos, D.; Kosmidis, P.; Papadimitriou, C.A.; Murray, S. Assessment of somatic k-RAS mutations as a mechanism associated with resistance to EGFR-targeted agents: A systematic review and meta-analysis of studies in advanced non-small-cell lung cancer and metastatic colorectal cancer. Lancet Oncol. 2008, 9, 962–972. [Google Scholar] [CrossRef]
- Greenbaum, A.; Martin, D.R.; Bocklage, T.; Lee, J.-H.; Ness, S.A.; Rajput, A. Tumor Heterogeneity as a Predictor of Response to Neoadjuvant Chemotherapy in Locally Advanced Rectal Cancer. Clin. Colorectal Cancer 2019, 18, 102–109. [Google Scholar] [CrossRef]
- Oh, B.Y.; Shin, H.-T.; Yun, J.W.; Kim, K.-T.; Kim, J.; Bae, J.S.; Cho, Y.B.; Lee, W.Y.; Yun, S.H.; Park, Y.A.; et al. Intratumor heterogeneity inferred from targeted deep sequencing as a prognostic indicator. Sci. Rep. 2019, 9, 1–8. [Google Scholar] [CrossRef]
- De Roock, W.; Claes, B.; Bernasconi, D.; De Schutter, J.; Biesmans, B.; Fountzilas, G.; Kalogeras, K.T.; Kotoula, V.; Papamichael, D.; Laurent-Puig, P.; et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: A retrospective consortium analysis. Lancet Oncol. 2010, 11, 753–762. [Google Scholar] [CrossRef]
- Jhawer, M.; Goel, S.; Wilson, A.J.; Montagna, C.; Ling, Y.-H.; Byun, D.-S.; Nasser, S.; Arango, D.; Shin, J.; Klampfer, L.; et al. PIK3CA mutation/PTEN expression status predicts response of colon cancer cells to the epidermal growth factor receptor inhibitor cetuximab. Cancer Res. 2008, 68, 1953–1961. [Google Scholar] [CrossRef]
- Tian, S.; Simon, I.; Moreno, V.; Roepman, P.; Tabernero, J.; Snel, M.; van’t Veer, L.; Salazar, R.; Bernards, R.; Capella, G. A combined oncogenic pathway signature of BRAF, KRAS and PI3KCA mutation improves colorectal cancer classification and cetuximab treatment prediction. Gut 2013, 62, 540–549. [Google Scholar] [CrossRef]
- Sartore-Bianchi, A.; Martini, M.; Molinari, F.; Veronese, S.; Nichelatti, M.; Artale, S.; Di Nicolantonio, F.; Saletti, P.; De Dosso, S.; Mazzucchelli, L.; et al. PIK3CA mutations in colorectal cancer are associated with clinical resistance to EGFR-targeted monoclonal antibodies. Cancer Res. 2009, 69, 1851–1857. [Google Scholar] [CrossRef]
- Douillard, J.-Y.; Oliner, K.S.; Siena, S.; Tabernero, J.; Burkes, R.; Barugel, M.; Humblet, Y.; Bodoky, G.; Cunningham, D.; Jassem, J.; et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N. Engl. J. Med. 2013, 369, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- Joung, J.-G.; Oh, B.Y.; Hong, H.K.; Al-Khalidi, H.; Al-Alem, F.; Lee, H.-O.; Bae, J.S.; Kim, J.; Cha, H.-U.; Alotaibi, M.; et al. Tumor Heterogeneity Predicts Metastatic Potential in Colorectal Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 7209–7216. [Google Scholar] [CrossRef] [PubMed]
- Baldus, S.E.; Schaefer, K.-L.; Engers, R.; Hartleb, D.; Stoecklein, N.H.; Gabbert, H.E. Prevalence and heterogeneity of KRAS, BRAF, and PIK3CA mutations in primary colorectal adenocarcinomas and their corresponding metastases. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2010, 16, 790–799. [Google Scholar] [CrossRef]
- Losi, L.; Baisse, B.; Bouzourene, H.; Benhattar, J. Evolution of intratumoral genetic heterogeneity during colorectal cancer progression. Carcinogenesis 2005, 26, 916–922. [Google Scholar] [CrossRef]
- Eijkelenboom, A.; Kamping, E.J.; Kastner-van Raaij, A.W.; Hendriks-Cornelissen, S.J.; Neveling, K.; Kuiper, R.P.; Hoischen, A.; Nelen, M.R.; Ligtenberg, M.J.L.; Tops, B.B.J. Reliable Next-Generation Sequencing of Formalin-Fixed, Paraffin-Embedded Tissue Using Single Molecule Tags. J. Mol. Diagn. 2016, 18, 851–863. [Google Scholar] [CrossRef] [PubMed]
- Bouaoun, L.; Sonkin, D.; Ardin, M.; Hollstein, M.; Byrnes, G.; Zavadil, J.; Olivier, M. TP53 Variations in Human Cancers: New Lessons from the IARC TP53 Database and Genomics Data. Hum. Mutat. 2016, 37, 865–876. [Google Scholar] [CrossRef]
- Dahlman, K.B.; Xia, J.; Hutchinson, K.; Ng, C.; Hucks, D.; Jia, P.; Atefi, M.; Su, Z.; Branch, S.; Lyle, P.L.; et al. BRAF(L597) mutations in melanoma are associated with sensitivity to MEK inhibitors. Cancer Discov. 2012, 2, 791–797. [Google Scholar] [CrossRef]
- Yao, Z.; Yaeger, R.; Rodrik-Outmezguine, V.S.; Tao, A.; Torres, N.M.; Chang, M.T.; Drosten, M.; Zhao, H.; Cecchi, F.; Hembrough, T.; et al. Tumours with class 3 BRAF mutants are sensitive to the inhibition of activated RAS. Nature 2017, 548, 234–238. [Google Scholar] [CrossRef]
- Acquaviva, G.; de Biase, D.; Diquigiovanni, C.; Argento, C.M.; De Leo, A.; Bonora, E.; Rhoden, K.J.; Pession, A.; Tallini, G. BRAF Exon 15 Mutations in Papillary Carcinoma and Adjacent Thyroid Parenchyma: A Search for the Early Molecular Events Associated with Tumor Development. Cancers 2020, 12, 430. [Google Scholar] [CrossRef]
- Willett, C.G.; Chang, D.T.; Czito, B.G.; Meyer, J.; Wo, J. Cancer Genome Atlas Network Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar]
- Hardiman, K.M.; Ulintz, P.J.; Kuick, R.D.; Hovelson, D.H.; Gates, C.M.; Bhasi, A.; Rodrigues Grant, A.; Liu, J.; Cani, A.K.; Greenson, J.K.; et al. Intra-tumor genetic heterogeneity in rectal cancer. Lab. Investig. 2016, 96, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Bettoni, F.; Masotti, C.; Habr-Gama, A.; Correa, B.R.; Gama-Rodrigues, J.; Vianna, M.R.; Vailati, B.B.; São Julião, G.P.; Fernandez, L.M.; Galante, P.A.; et al. Intratumoral Genetic Heterogeneity in Rectal Cancer: Are Single Biopsies representative of the entirety of the tumor? Ann. Surg. 2017, 265, e4–e6. [Google Scholar] [CrossRef] [PubMed]
- Dijkstra, J.R.; Tops, B.B.J.; Nagtegaal, I.D.; van Krieken, J.H.J.M.; Ligtenberg, M.J.L. The homogeneous mutation status of a 22 gene panel justifies the use of serial sections of colorectal cancer tissue for external quality assessment. Virchows Arch. Int. J. Pathol. 2015, 467, 273–278. [Google Scholar] [CrossRef]
- Mroz, E.A.; Rocco, J.W. MATH, a novel measure of intratumor genetic heterogeneity, is high in poor-outcome classes of head and neck squamous cell carcinoma. Oral Oncol. 2013, 49, 211–215. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Gene | Transcript ID (RefSeq) | Transcript ID (Ensembl) | Exon Number | Targeted Regions | Positions Analyzed for Variants |
|---|---|---|---|---|---|
| ACVR1B | ENST00000257963 | NM_004302 | 02 | Activin types I and II receptor domain | c.92−5 to c.331+5 |
| 03–09 | Transforming growth factor beta type I GS-motif | c.556 to c.1518+5 | |||
| ACVR2A | ENST00000241416 | NM_001616 | 06–11 | Protein kinase domain | c.673−5 to c.1542+5 |
| AMER1 | ENST00000330258 | NM_152424 | 02 | WTX Protein | c.639 to c.1629 |
| APC | ENST00000257430 | NM_000038 | 01–16 | Whole gene | c1−5 to c.8532+5 |
| ARID1A | ENST00000324856 | NM_006015 | 11–12 | ARID DNA-binding domain | c.2989 to c.3397 |
| 20 | SWI/SNF-like complex subunit BAF250/Osa | c.5820 to c.6777 | |||
| B2M | ENST00000558401 | NM_004048 | 02 | Immunoglobulin C1-set domain | c.68−5 to c.346+5 |
| BRAF | ENST00000288602 | NM_004333 | 15 | Codon D594-K601 | c.1742−5 to c.1860+5 |
| CASP5 | ENST00000393141 | NM_004347 | 02–03 | CARD domain | c.8 to c.433+5 |
| CASP8 | ENST00000358485 | NM_001080125 | 07–09 | Caspase domain | c.838 to c.1617+5 |
| CTNNB1 | ENST00000349496 | NM_001904 | 03 | Codon D32-S45 | c.36 to c.163 |
| 08 | Codon W383-N387 | c.1082−5 to c.1185+5 | |||
| EGFR | ENST00000275493 | NM_005228 | 12 | Receptor L domain | c.1391 to c.1498+5 |
| 18–21 | Protein tyrosine kinase | c.2062−5 to c.2625+5 | |||
| ERBB2 | ENST00000269571 | NM_004448 | 18–24 | Protein tyrosine kinase | c.2101 to c.2970+5 |
| FBXW7 | ENST00000281708 | NM_033632 | 07–12 | WD domain, G-beta repeat | c.1035 to c.2124+5 |
| GNAS | ENST00000371085 | NM_000516 | 08–09 | Codon R201 and Q227 | c.586−5 to c.718+5 |
| IDH2 | ENST00000330062 | NM_002168 | 04 | Codon R140 and R172 | c.374−5 to c.534+5 |
| KRAS | ENST00000311936 | NM_004985 | 02 | Codon G12, and G13 | c.1−5 to c.111+5 |
| 03 | Codon A59 and Q61 | c.112−5 to c.232 | |||
| 04 | Codon K117 and A146 | c.291−5 to c.385 and c.402 to c.450+5 | |||
| MET | ENST00000318493 | NM_001127500 | 15–21 | Protein tyrosine kinase | c.3140 to c.4227+5 |
| NRAS | ENST00000369535 | NM_002524 | 02 | Codon G12 and G13 | c.1−5 to c.99 |
| 03 | Codon A59 and Q61 | c.135 to c.272 | |||
| PIK3CA | ENST00000263967 | NM_006218 | 10 | Codon E542 to Q546 | c.1557 to c.1664+5 |
| 21 | Codon M1043 to G1049 | c.3041 to c.3207+5 | |||
| POLE | ENST00000320574 | NM_006231 | 03–13 | DNA-directed DNA polymerase, family B, exonuclease domain | c.205−5 to c.1301 |
| PTEN | ENST00000371953 | NM_000314 | 05–08 | Dual specificity phosphatase, catalytic domain, C2 domain of PTEN tumor-suppressor protein | c.310 to c.1026+5 |
| RNF43 | ENST00000407977 | NM_017763 | 02–10 | Whole CDS | c.1−5 to c.2352+5 |
| SMAD2 | ENST00000262160 | NM_005901 | 02–11 | Whole CDS | c.1−5 to c.1404+5 |
| SMAD4 | ENST00000342988 | NM_005359 | 03–04 | MH1 domain | c.250−5 to c.454+5 |
| 09–12 | MH2 domain | c.956−5 to c.1659+5 | |||
| SMARCA2 | ENST00000349721 | NM_003070 | 15–21 | SNF2-related, N-terminal domain | c.2185−5 to c.3078+5 |
| 23–25 | Helicase, C-terminal | c.3136 to c.3684+5 | |||
| SMARCA4 | ENST00000450717 | NM_001128846 | 15–21 | SNF2-related, N-terminal domain | c.2275−5 to c.3168+5 |
| 23–25 | Helicase, C-terminal | c.3324 to c.3374+5 | |||
| SMARCB1 | ENST00000263121 | NM_003073 | 05–09 | SNF5/SMARCB1/INI1 | c.501−5 to c.1158+5 |
| SOX9 | ENST00000245479 | NM_000346 | 01–03 | Whole CDS | c.1−5 to c.1530+5 |
| TCF7L2 | ENST00000369397 | NM_030756 | 01–06 | CTNNB1 binding, N-terminal | c.1−5 to c.719+5 |
| 09–10 | High mobility group box domain | c.933−5 to c.1200+5 | |||
| TGFBR2 | ENST00000359013 | NM_001024847 | 04 | Codon E125 | c.339−5 to c.529+5 |
| TP53 | ENST00000269305 | NM_000546 | 03–08 | P53 DNA-binding domain | c.83 to c.919+5 |
| Variables | N = 11 | |
|---|---|---|
| Age (years) | Mean (SD) | 72.2 (27.4) |
| Gender | Male | 6 (55%) |
| Female | 5 (45%) | |
| pT | 3 | 9 (82%) |
| 4 | 2 (18%) | |
| pN | 0 | 6 (55%) |
| 1 | 3 (27%) | |
| UICC stage | 2 2A 3A 3C | 2 (18%) 6 4 1 |
| EMVI Differentiation (UICC grade) | Yes | 4 (36%) |
| No | 6 (55%) | |
| Missing | 1 (9%) | |
| Well/moderate (UICC grade 1–2) | 9 (82%) | |
| Poor (UICC grade 3) | 1 (9%) | |
| Missing | 1 (9%) | |
| Distance to CRM (mm) | Mean (SD) | 14.1 (7.7) |
| Diameter tumor (mm) | Mean (SD) | 53.5 (21.6) |
| Total number of lymph nodes | Median (IQR) | 15 (12–19) |
| Number of tumor positive lymph nodes | Median (IQR) | 0 (0–3) |
| Distance from anal verge (mm) | Mean (SD) | 57.8 (46.3) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vuijk, F.A.; van de Water, C.; Lent-van Vliet, S.; van der Valk, M.J.M.; Simmer, F.; van de Velde, C.J.H.; Vahrmeijer, A.L.; Nagtegaal, I.D.; Hilling, D.E. Intra-Tumoral Genomic Heterogeneity in Rectal Cancer: Mutational Status Is Dependent on Preoperative Biopsy Depth and Location. Cancers 2021, 13, 2271. https://doi.org/10.3390/cancers13092271
Vuijk FA, van de Water C, Lent-van Vliet S, van der Valk MJM, Simmer F, van de Velde CJH, Vahrmeijer AL, Nagtegaal ID, Hilling DE. Intra-Tumoral Genomic Heterogeneity in Rectal Cancer: Mutational Status Is Dependent on Preoperative Biopsy Depth and Location. Cancers. 2021; 13(9):2271. https://doi.org/10.3390/cancers13092271
Chicago/Turabian StyleVuijk, Floris A., Carlijn van de Water, Shannon Lent-van Vliet, Maxime J. M. van der Valk, Femke Simmer, Cornelis J. H. van de Velde, Alexander L. Vahrmeijer, Iris D. Nagtegaal, and Denise E. Hilling. 2021. "Intra-Tumoral Genomic Heterogeneity in Rectal Cancer: Mutational Status Is Dependent on Preoperative Biopsy Depth and Location" Cancers 13, no. 9: 2271. https://doi.org/10.3390/cancers13092271
APA StyleVuijk, F. A., van de Water, C., Lent-van Vliet, S., van der Valk, M. J. M., Simmer, F., van de Velde, C. J. H., Vahrmeijer, A. L., Nagtegaal, I. D., & Hilling, D. E. (2021). Intra-Tumoral Genomic Heterogeneity in Rectal Cancer: Mutational Status Is Dependent on Preoperative Biopsy Depth and Location. Cancers, 13(9), 2271. https://doi.org/10.3390/cancers13092271