
RAB7A Regulates Vimentin Phosphorylation through AKT and PAK

 ,
,  , , ,
, , ,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Chemicals
2.2. Mutagenesis and Plasmids Construction
2.3. Antibodies
2.4. Transfection and RNA Interference
2.5. Co-Immunoprecipitation
2.6. Pull-Down Experiments
2.7. Western Blotting
2.8. Gelatin Zymography
2.9. Immunofluorescence and Confocal Microscopy
2.10. In-Silico Modeling
2.11. Luciferase Assays
2.12. Quantification and Statistical Analysis
3. Results
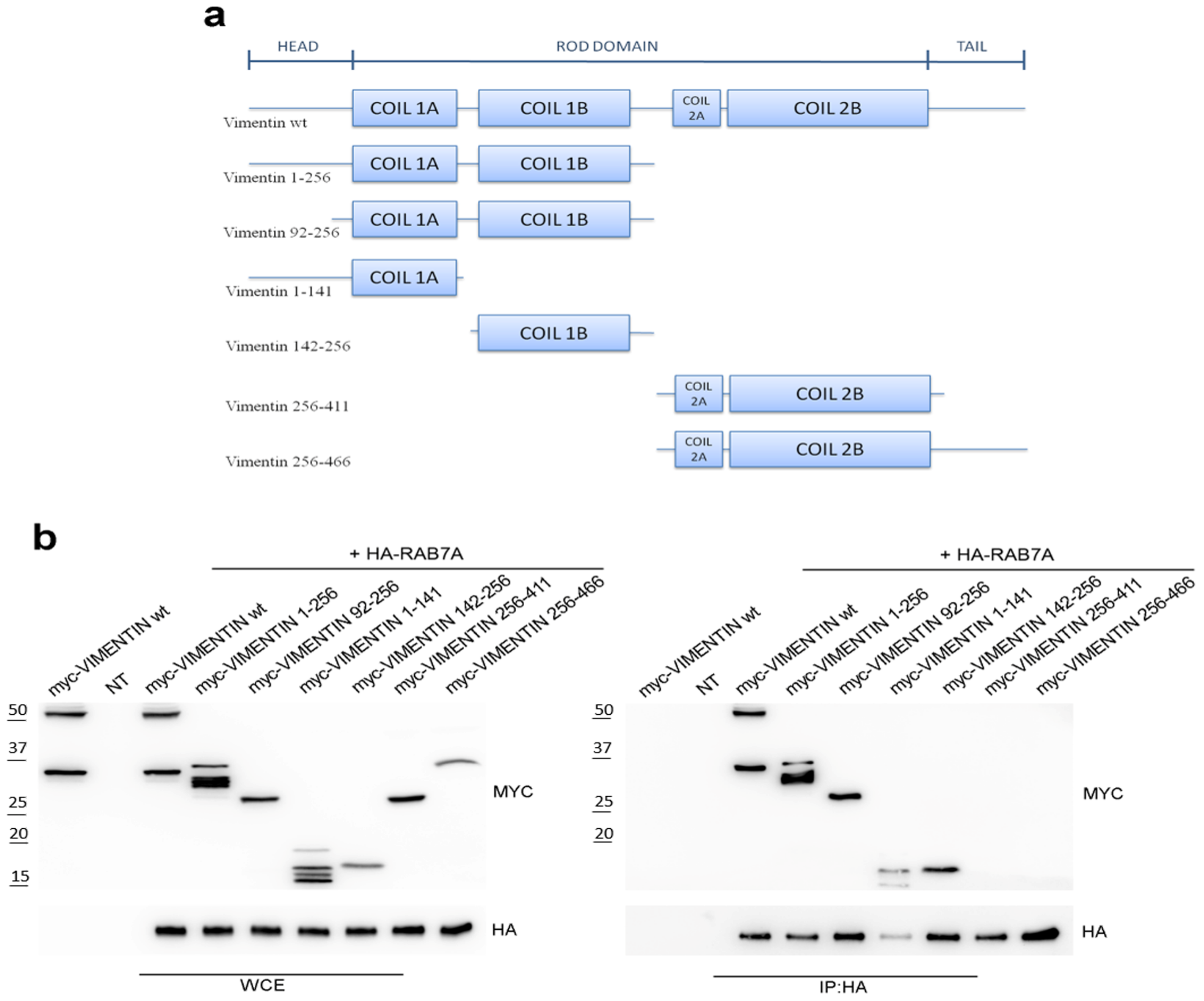
3.1. RAB7A Interacts with the Coil 1 Domain of Vimentin
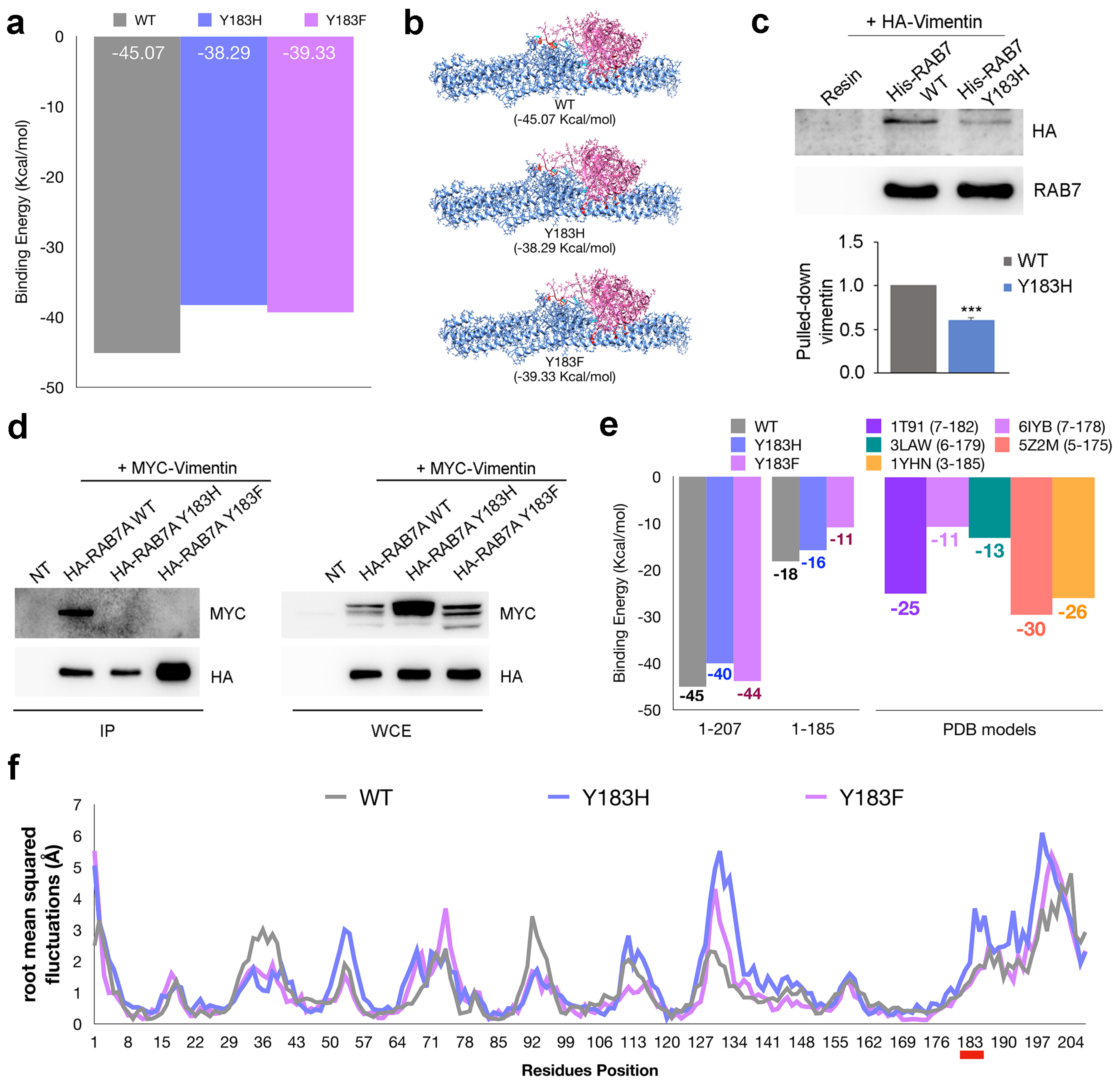
3.2. Mutation of Amino Acid 183 of RAB7A Affects the Interaction with Vimentin
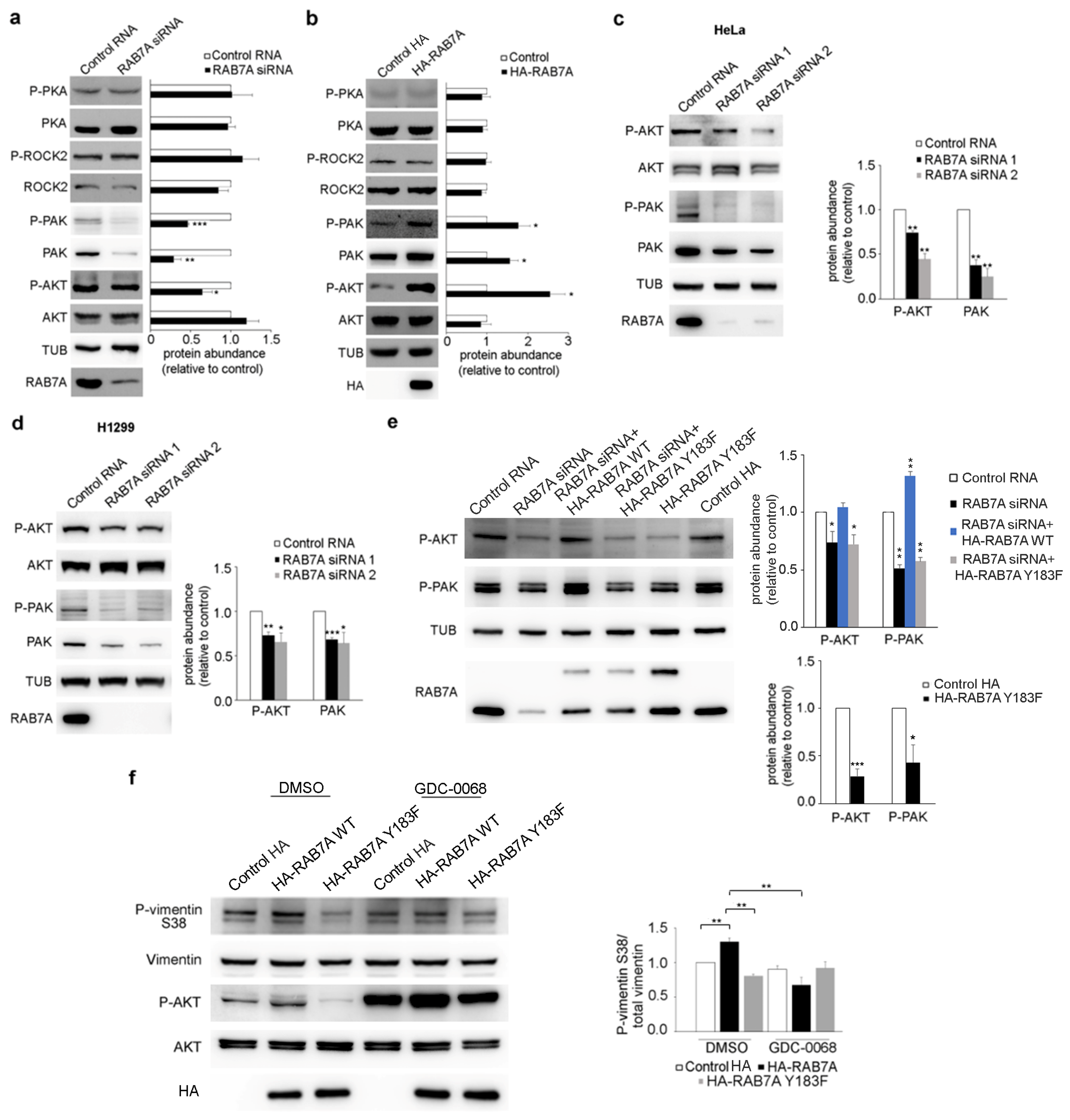
3.3. Modulation of RAB7A Expression Affects AKT and PAK1 but Not ROCK2 and PKA Kinases
3.4. RAB7A Regulates Vimentin Phosphorylation at Ser38 Modulating AKT Activity
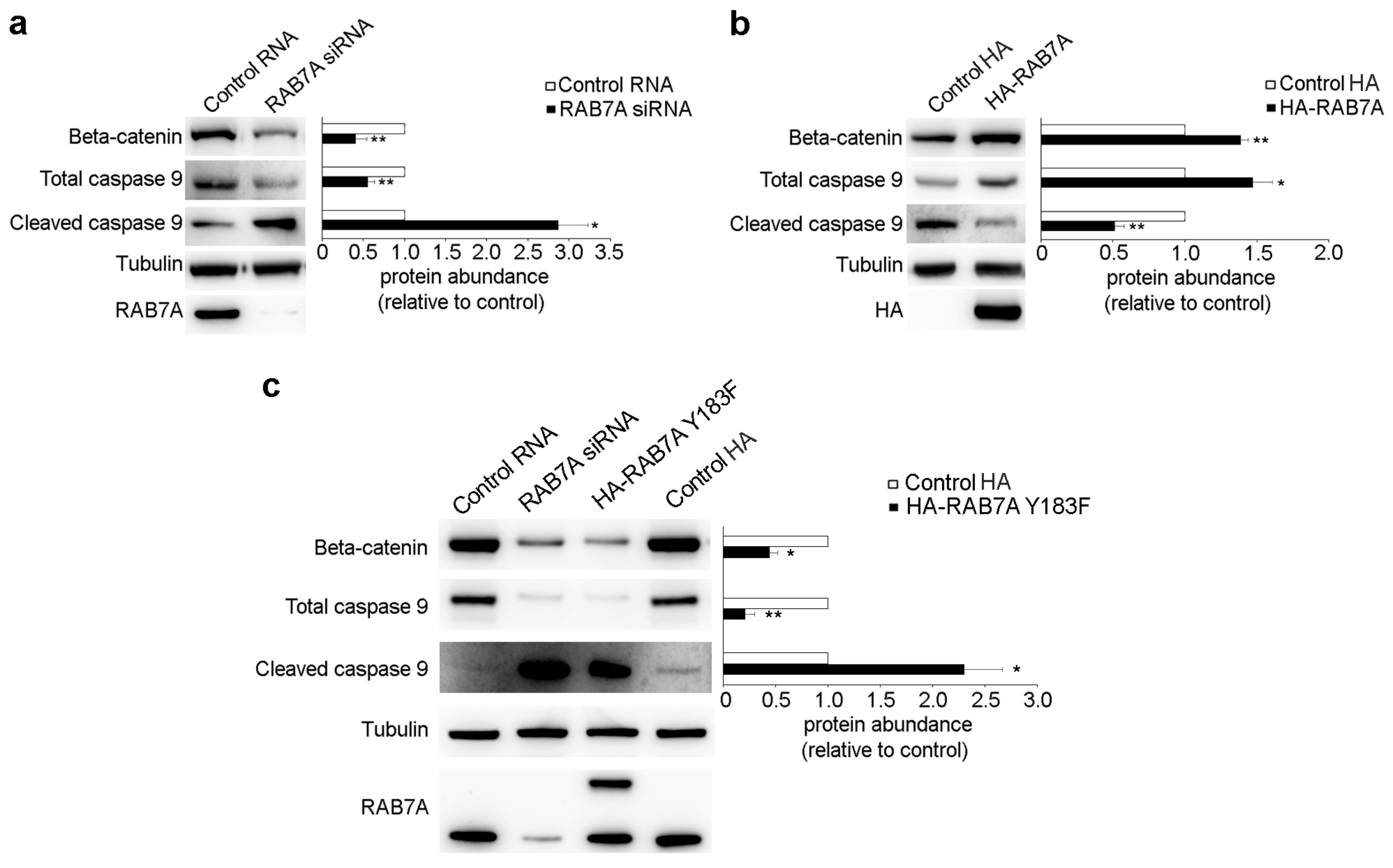
3.5. RAB7A Regulates Beta-Catenin and Caspase 9 Expression
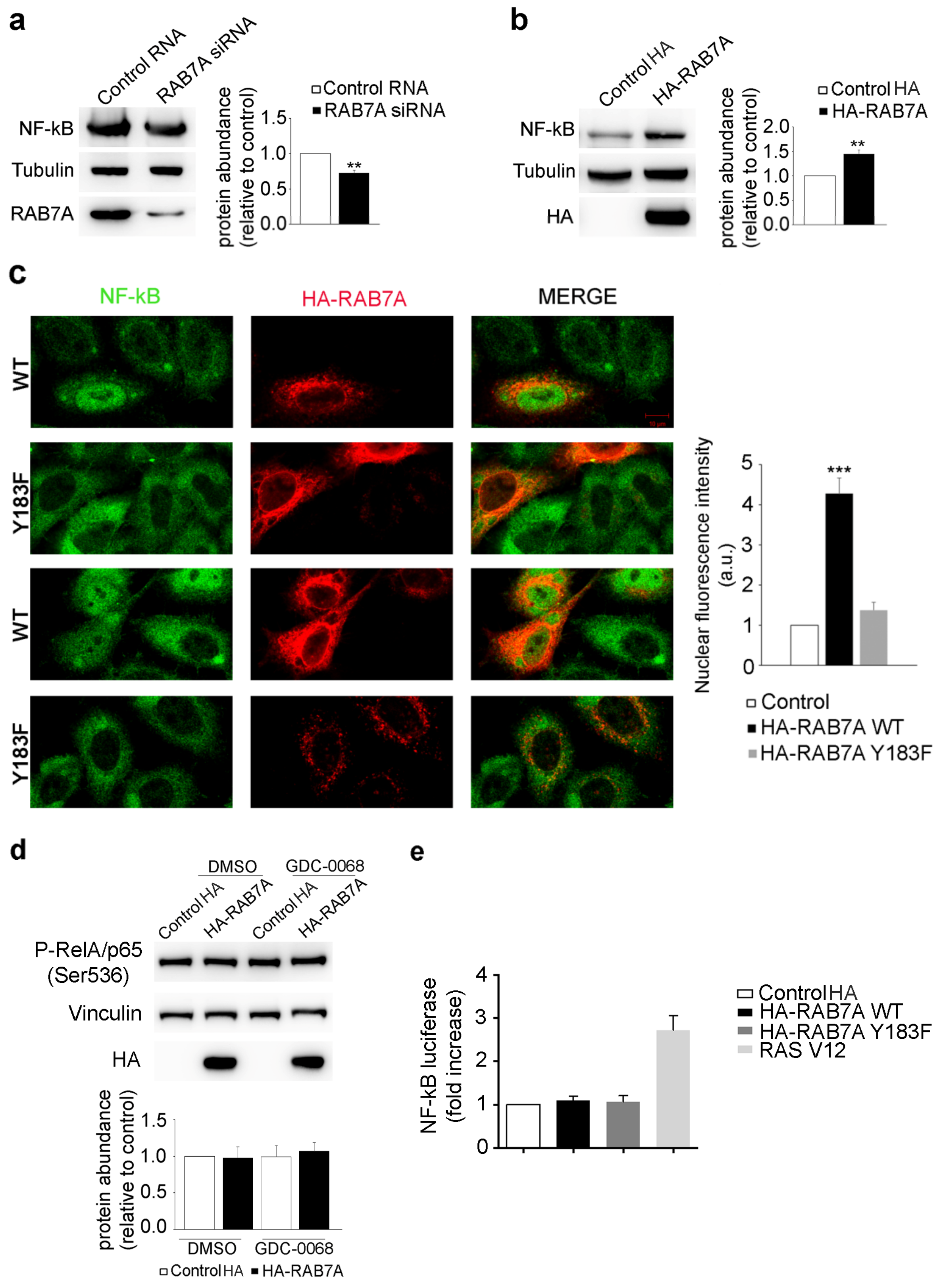
3.6. RAB7A Affects NF-kB Expression and Localization
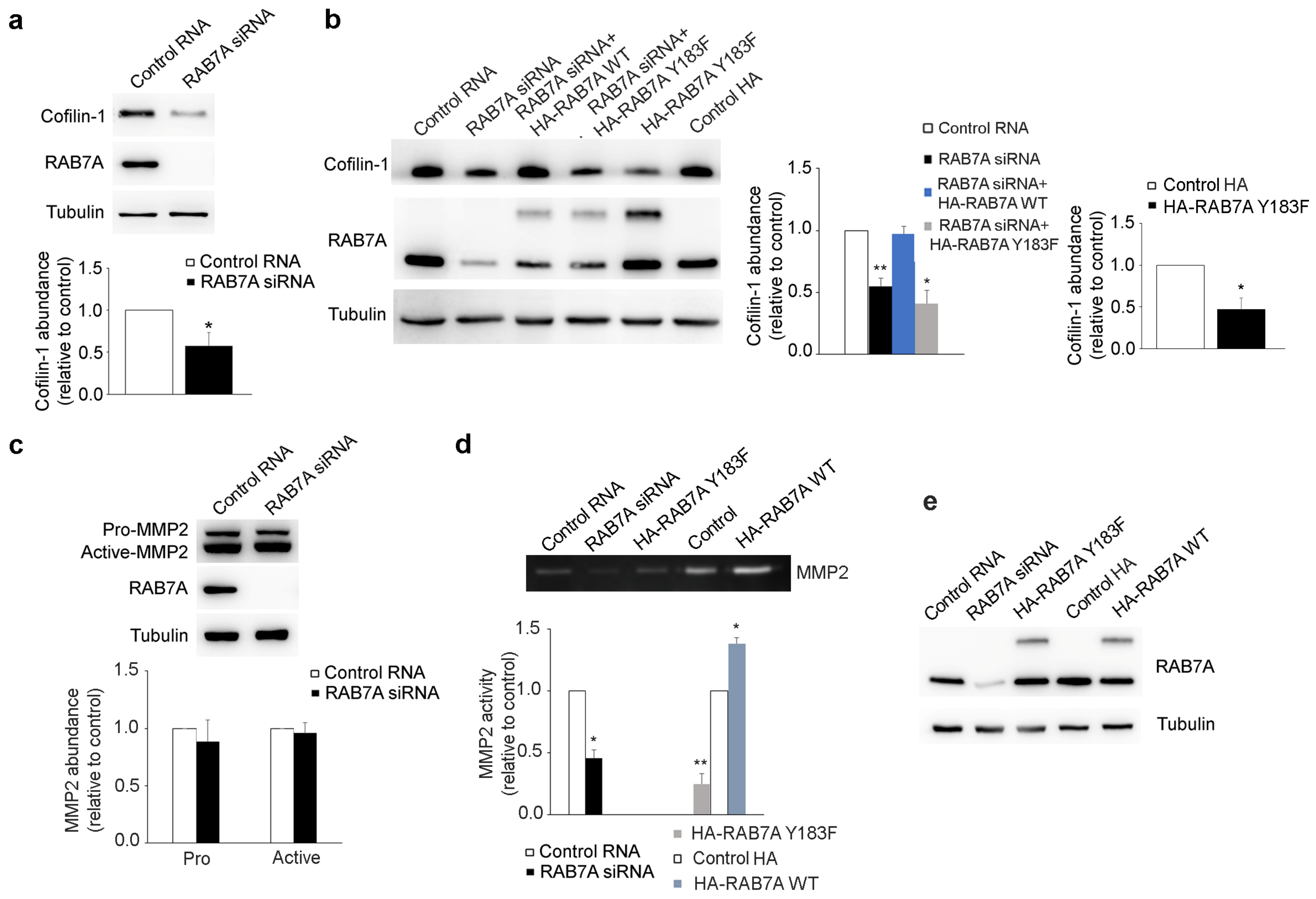
3.7. RAB7A Regulates Cofilin-1 Abundance and MMP2 Activity
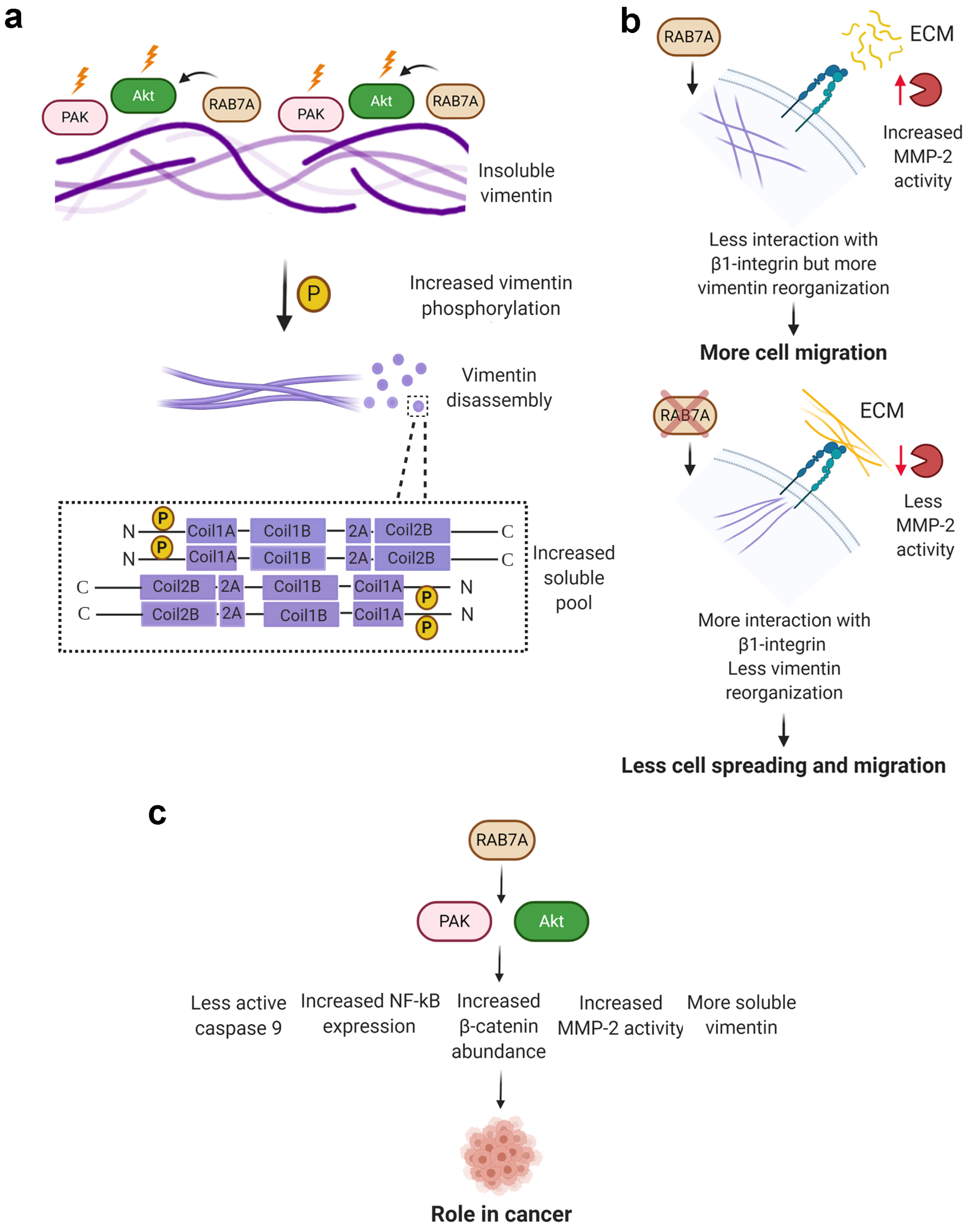
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pfeffer, S.R. Rab gtpases: Master regulators that establish the secretory and endocytic pathways. Mol. Biol. Cell 2017, 28, 712–715. [Google Scholar] [CrossRef] [PubMed]
- Guerra, F.; Bucci, C. Multiple roles of the small gtpase rab7. Cells 2016, 5, 34. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Chen, L.; Wang, S.; Wang, T. Rab7: Roles in membrane trafficking and disease. Biosci. Rep. 2009, 29, 193–209. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Ming, Z.; Xiaochun, W.; Hong, W. Rab7: Role of its protein interaction cascades in endo-lysosomal traffic. Cell Signal. 2011, 23, 516–521. [Google Scholar] [CrossRef]
- Mateus, D.; Marini, E.S.; Progida, C.; Bakke, O. Rab7a modulates er stress and er morphology. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 781–793. [Google Scholar] [CrossRef]
- Raiborg, C.; Wenzel, E.M.; Pedersen, N.M.; Olsvik, H.; Schink, K.O.; Schultz, S.W.; Vietri, M.; Nisi, V.; Bucci, C.; Brech, A.; et al. Repeated er-endosome contacts promote endosome translocation and neurite outgrowth. Nature 2015, 520, 234–238. [Google Scholar] [CrossRef]
- Luo, S.; Valencia, C.A.; Zhang, J.; Lee, N.C.; Slone, J.; Gui, B.; Wang, X.; Li, Z.; Dell, S.; Brown, J.; et al. Biparental inheritance of mitochondrial DNA in humans. Proc. Natl. Acad. Sci. USA 2018, 115, 13039–13044. [Google Scholar] [CrossRef]
- Schroeder, B.; Schulze, R.J.; Weller, S.G.; Sletten, A.C.; Casey, C.A.; McNiven, M.A. The small gtpase rab7 as a central regulator of hepatocellular lipophagy. Hepatology 2015, 61, 1896–1907. [Google Scholar] [CrossRef]
- Cogli, L.; Progida, C.; Bramato, R.; Bucci, C. Vimentin phosphorylation and assembly are regulated by the small gtpase rab7a. Biochim. Biophys. Acta 2013, 1833, 1283–1293. [Google Scholar] [CrossRef]
- Cogli, L.; Progida, C.; Thomas, C.L.; Spencer-Dene, B.; Donno, C.; Schiavo, G.; Bucci, C. Charcot-marie-tooth type 2b disease-causing rab7a mutant proteins show altered interaction with the neuronal intermediate filament peripherin. Acta Neuropathol. 2013, 125, 257–272. [Google Scholar] [CrossRef]
- Zhu, Q.S.; Rosenblatt, K.; Huang, K.L.; Lahat, G.; Brobey, R.; Bolshakov, S.; Nguyen, T.; Ding, Z.; Belousov, R.; Bill, K.; et al. Vimentin is a novel akt1 target mediating motility and invasion. Oncogene 2011, 30, 457–470. [Google Scholar] [CrossRef]
- Goto, H.; Tanabe, K.; Manser, E.; Lim, L.; Yasui, Y.; Inagaki, M. Phosphorylation and reorganization of vimentin by p21-activated kinase (pak). Genes Cells 2002, 7, 91–97. [Google Scholar] [CrossRef]
- Tang, D.D.; Bai, Y.; Gunst, S.J. Silencing of p21-activated kinase attenuates vimentin phosphorylation on ser-56 and reorientation of the vimentin network during stimulation of smooth muscle cells by 5-hydroxytryptamine. Biochem. J. 2005, 388, 773–783. [Google Scholar] [CrossRef]
- Ando, S.; Tanabe, K.; Gonda, Y.; Sato, C.; Inagaki, M. Domain- and sequence-specific phosphorylation of vimentin induces disassembly of the filament structure. Biochemistry 1989, 28, 2974–2979. [Google Scholar] [CrossRef]
- Geisler, N.; Hatzfeld, M.; Weber, K. Phosphorylation in vitro of vimentin by protein kinases a and c is restricted to the head domain. Identification of the phosphoserine sites and their influence on filament formation. Eur. J. Biochem. 1989, 183, 441–447. [Google Scholar] [CrossRef]
- Sin, W.C.; Chen, X.Q.; Leung, T.; Lim, L. Rhoa-binding kinase alpha translocation is facilitated by the collapse of the vimentin intermediate filament network. Mol. Cell Biol. 1998, 18, 6325–6339. [Google Scholar] [CrossRef]
- Bauer, P.O.; Hudec, R.; Goswami, A.; Kurosawa, M.; Matsumoto, G.; Mikoshiba, K.; Nukina, N. Rock-phosphorylated vimentin modifies mutant huntingtin aggregation via sequestration of irbit. Mol. Neurodegener. 2012, 7, 43. [Google Scholar] [CrossRef]
- Goto, H.; Kosako, H.; Tanabe, K.; Yanagida, M.; Sakurai, M.; Amano, M.; Kaibuchi, K.; Inagaki, M. Phosphorylation of vimentin by rho-associated kinase at a unique amino-terminal site that is specifically phosphorylated during cytokinesis. J. Biol. Chem. 1998, 273, 11728–11736. [Google Scholar] [CrossRef]
- Bayascas, J.R.; Alessi, D.R. Regulation of akt/pkb ser473 phosphorylation. Mol. Cell 2005, 18, 143–145. [Google Scholar] [CrossRef]
- Chuang, H.H.; Yang, C.H.; Tsay, Y.G.; Hsu, C.Y.; Tseng, L.M.; Chang, Z.F.; Lee, H.H. Rockii ser1366 phosphorylation reflects the activation status. Biochem. J. 2012, 443, 145–151. [Google Scholar] [CrossRef]
- Higuchi, M.; Onishi, K.; Kikuchi, C.; Gotoh, Y. Scaffolding function of pak in the pdk1-akt pathway. Nat. Cell Biol. 2008, 10, 1356–1364. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Jang, J.; Yang, C.; Kim, E.J.; Jung, H.; Kim, C. Vimentin filament controls integrin α5β1-mediated cell adhesion by binding to integrin through its ser38 residue. FEBS Lett. 2016, 590, 3517–3525. [Google Scholar] [CrossRef] [PubMed]
- Margiotta, A.; Progida, C.; Bakke, O.; Bucci, C. Rab7a regulates cell migration through rac1 and vimentin. Biochim Biophys Acta Mol. Cell Res. 2017, 1864, 367–381. [Google Scholar] [CrossRef] [PubMed]
- Spinosa, M.R.; Progida, C.; De Luca, A.; Colucci, A.M.R.; Alifano, P.; Bucci, C. Functional characterization of rab7 mutant proteins associated with charcot-marie-tooth type 2b disease. J. Neurosci. 2008, 28, 1640–1648. [Google Scholar] [CrossRef]
- Murga, C.; Zohar, M.; Teramoto, H.; Gutkind, J.S. Rac1 and rhog promote cell survival by the activation of pi3k and akt, independently of their ability to stimulate jnk and nf-kappab. Oncogene 2002, 21, 207–216. [Google Scholar] [CrossRef]
- Guerra, F.; Paiano, A.; Migoni, D.; Girolimetti, G.; Perrone, A.M.; De Iaco, P.; Fanizzi, F.P.; Gasparre, G.; Bucci, C. Modulation of rab7a protein expression determines resistance to cisplatin through late endocytic pathway impairment and extracellular vesicular secretion. Cancers 2019, 11, 52. [Google Scholar] [CrossRef]
- Saxena, S.; Bucci, C.; Weis, J.; Kruttgen, A. The small gtpase rab7 controls the endosomal trafficking and neuritogenic signaling of the nerve growth factor receptor trka. J. Neurosci. 2005, 25, 10930–10940. [Google Scholar] [CrossRef]
- Romano, R.; Rivellini, C.; De Luca, M.; Tonlorenzi, R.; Beli, R.; Manganelli, F.; Nolano, M.; Santoro, L.; Eskelinen, E.L.; Previtali, S.C.; et al. Alteration of the late endocytic pathway in charcot-marie-tooth type 2b disease. Cell Mol. Life Sci. 2020, 78, 351–372. [Google Scholar] [CrossRef]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. Ucsf chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The i-tasser suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. Patchdock and symmdock: Servers for rigid and symmetric docking. Nucleic Acids Res. 2005, 33, W363–W367. [Google Scholar] [CrossRef]
- Tovchigrechko, A.; Vakser, I.A. Gramm-x public web server for protein-protein docking. Nucleic Acids Res. 2006, 34, W310–W314. [Google Scholar] [CrossRef]
- Andrusier, N.; Nussinov, R.; Wolfson, H.J. Firedock: Fast interaction refinement in molecular docking. Proteins 2007, 69, 139–159. [Google Scholar] [CrossRef]
- Mashiach, E.; Schneidman-Duhovny, D.; Andrusier, N.; Nussinov, R.; Wolfson, H.J. Firedock: A web server for fast interaction refinement in molecular docking. Nucleic Acids Res. 2008, 36, W229–W232. [Google Scholar] [CrossRef]
- Rodrigues, C.H.; Pires, D.E.; Ascher, D.B. Dynamut: Predicting the impact of mutations on protein conformation, flexibility and stability. Nucleic Acids Res. 2018, 46, W350–W355. [Google Scholar] [CrossRef]
- Huang, X.; Miller, W. A time-efficient, linear-space local similarity algorithm. Adv. Appl. Math. 1991, 12, 337–357. [Google Scholar] [CrossRef]
- Wu, M.; Wang, T.; Loh, E.; Hong, W.; Song, H. Structural basis for recruitment of rilp by small gtpase rab7. EMBO J. 2005, 24, 1491–1501. [Google Scholar] [CrossRef]
- Wang, T.; Zhang, M.; Ma, Z.; Guo, K.; Tergaonkar, V.; Zeng, Q.; Hong, W. A role of rab7 in stabilizing egfr-her2 and in sustaining akt survival signal. J. Cell Physiol. 2012, 227, 2788–2797. [Google Scholar] [CrossRef]
- Lin, J.; Sampath, D.; Nannini, M.A.; Lee, B.B.; Degtyarev, M.; Oeh, J.; Savage, H.; Guan, Z.; Hong, R.; Kassees, R.; et al. Targeting activated akt with gdc-0068, a novel selective akt inhibitor that is efficacious in multiple tumor models. Clin. Cancer Res. 2013, 19, 1760–1772. [Google Scholar] [CrossRef]
- Manning, B.D.; Cantley, L.C. Akt/pkb signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [PubMed]
- Molli, P.R.; Li, D.Q.; Murray, B.W.; Rayala, S.K.; Kumar, R. Pak signaling in oncogenesis. Oncogene 2009, 28, 2545–2555. [Google Scholar] [CrossRef] [PubMed]
- Menard, R.E.; Jovanovski, A.P.; Mattingly, R.R. Active p21-activated kinase 1 rescues mcf10a breast epithelial cells from undergoing anoikis. Neoplasia 2005, 7, 638–645. [Google Scholar] [CrossRef] [PubMed]
- Cardone, M.H.; Roy, N.; Stennicke, H.R.; Salvesen, G.S.; Franke, T.F.; Stanbridge, E.; Frisch, S.; Reed, J.C. Regulation of cell death protease caspase-9 by phosphorylation. Science 1998, 282, 1318–1321. [Google Scholar] [CrossRef]
- Chaturvedi, M.M.; Sung, B.; Yadav, V.R.; Kannappan, R.; Aggarwal, B.B. Nf-kappab addiction and its role in cancer: ‘One size does not fit all’. Oncogene 2011, 30, 1615–1630. [Google Scholar] [CrossRef]
- Bai, D.; Ueno, L.; Vogt, P.K. Akt-mediated regulation of nfkappab and the essentialness of nfkappab for the oncogenicity of pi3k and akt. Int. J. Cancer 2009, 125, 2863–2870. [Google Scholar] [CrossRef]
- Dammann, K.; Khare, V.; Lang, M.; Claudel, T.; Harpain, F.; Granofszky, N.; Evstatiev, R.; Williams, J.M.; Pritchard, D.M.; Watson, A.; et al. Pak1 modulates a ppargamma/nf-kappab cascade in intestinal inflammation. Biochim. Biophys. Acta 2015, 1853, 2349–2360. [Google Scholar] [CrossRef]
- Chen, L.F.; Williams, S.A.; Mu, Y.; Nakano, H.; Duerr, J.M.; Buckbinder, L.; Greene, W.C. Nf-kappab rela phosphorylation regulates rela acetylation. Mol. Cell Biol. 2005, 25, 7966–7975. [Google Scholar] [CrossRef]
- Mizuno, K. Signaling mechanisms and functional roles of cofilin phosphorylation and dephosphorylation. Cell Signal. 2013, 25, 457–469. [Google Scholar] [CrossRef]
- Nishita, M.; Wang, Y.; Tomizawa, C.; Suzuki, A.; Niwa, R.; Uemura, T.; Mizuno, K. Phosphoinositide 3-kinase-mediated activation of cofilin phosphatase slingshot and its role for insulin-induced membrane protrusion. J. Biol. Chem. 2004, 279, 7193–7198. [Google Scholar] [CrossRef]
- Sousa-Squiavinato, A.C.M.; Rocha, M.R.; Barcellos-de-Souza, P.; de Souza, W.F.; Morgado-Diaz, J.A. Cofilin-1 signaling mediates epithelial-mesenchymal transition by promoting actin cytoskeleton reorganization and cell-cell adhesion regulation in colorectal cancer cells. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 418–429. [Google Scholar] [CrossRef]
- Margiotta, A.; Bucci, C. Role of intermediate filaments in vesicular traffic. Cells 2016, 5, 20. [Google Scholar] [CrossRef]
- Aziz, A.; Hess, J.F.; Budamagunta, M.S.; FitzGerald, P.G.; Voss, J.C. Head and rod 1 interactions in vimentin: Identification of contact sites, structure, and changes with phosphorylation using site-directed spin labeling and electron paramagnetic resonance. J. Biol. Chem. 2009, 284, 7330–7338. [Google Scholar] [CrossRef]
- Chernyatina, A.A.; Nicolet, S.; Aebi, U.; Herrmann, H.; Strelkov, S.V. Atomic structure of the vimentin central alpha-helical domain and its implications for intermediate filament assembly. Proc. Natl. Acad. Sci. USA 2012, 109, 13620–13625. [Google Scholar] [CrossRef]
- Shinde, S.R.; Maddika, S. Pten modulates egfr late endocytic trafficking and degradation by dephosphorylating rab7. Nat. Commun. 2016, 7, 10689. [Google Scholar] [CrossRef]
- Manser, E.; Leung, T.; Salihuddin, H.; Zhao, Z.S.; Lim, L. A brain serine/threonine protein kinase activated by cdc42 and rac1. Nature 1994, 367, 40–46. [Google Scholar] [CrossRef]
- Bargagna-Mohan, P.; Lei, L.; Thompson, A.; Shaw, C.; Kasahara, K.; Inagaki, M.; Mohan, R. Vimentin phosphorylation underlies myofibroblast sensitivity to withaferin a in vitro and during corneal fibrosis. PLoS ONE 2015, 10, e0133399. [Google Scholar] [CrossRef]
- Moon, R.T.; Kohn, A.D.; De Ferrari, G.V.; Kaykas, A. Wnt and beta-catenin signalling: Diseases and therapies. Nat. Rev. Genet. 2004, 5, 691–701. [Google Scholar] [CrossRef]
- Mosimann, C.; Hausmann, G.; Basler, K. Beta-catenin hits chromatin: Regulation of wnt target gene activation. Nat. Rev. Mol. Cell Biol. 2009, 10, 276–286. [Google Scholar] [CrossRef]
- Zhu, G.; Wang, Y.; Huang, B.; Liang, J.; Ding, Y.; Xu, A.; Wu, W. A rac1/pak1 cascade controls beta-catenin activation in colon cancer cells. Oncogene 2012, 31, 1001–1012. [Google Scholar] [CrossRef]
- Fang, D.; Hawke, D.; Zheng, Y.; Xia, Y.; Meisenhelder, J.; Nika, H.; Mills, G.B.; Kobayashi, R.; Hunter, T.; Lu, Z. Phosphorylation of beta-catenin by akt promotes beta-catenin transcriptional activity. J. Biol. Chem. 2007, 282, 11221–11229. [Google Scholar] [CrossRef]
- Li, P.; Nijhawan, D.; Budihardjo, I.; Srinivasula, S.M.; Ahmad, M.; Alnemri, E.S.; Wang, X. Cytochrome c and datp-dependent formation of apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 1997, 91, 479–489. [Google Scholar] [CrossRef]
- Ganchi, P.A.; Sun, S.C.; Greene, W.C.; Ballard, D.W. I kappa b/mad-3 masks the nuclear localization signal of nf-kappa b p65 and requires the transactivation domain to inhibit nf-kappa b p65 DNA binding. Mol. Biol. Cell 1992, 3, 1339–1352. [Google Scholar] [CrossRef]
- Ozes, O.N.; Mayo, L.D.; Gustin, J.A.; Pfeffer, S.R.; Pfeffer, L.M.; Donner, D.B. Nf-kappab activation by tumour necrosis factor requires the akt serine-threonine kinase. Nature 1999, 401, 82–85. [Google Scholar] [CrossRef]
- Frost, J.A.; Swantek, J.L.; Stippec, S.; Yin, M.J.; Gaynor, R.; Cobb, M.H. Stimulation of nfkappa b activity by multiple signaling pathways requires pak1. J. Biol. Chem. 2000, 275, 19693–19699. [Google Scholar] [CrossRef]
- Nelson, G.; Paraoan, L.; Spiller, D.G.; Wilde, G.J.; Browne, M.A.; Djali, P.K.; Unitt, J.F.; Sullivan, E.; Floettmann, E.; White, M.R. Multi-parameter analysis of the kinetics of nf-kappab signalling and transcription in single living cells. J. Cell Sci. 2002, 115, 1137–1148. [Google Scholar] [CrossRef]
- Hoeflich, K.P.; Luo, J.; Rubie, E.A.; Tsao, M.S.; Jin, O.; Woodgett, J.R. Requirement for glycogen synthase kinase-3beta in cell survival and nf-kappab activation. Nature 2000, 406, 86–90. [Google Scholar] [CrossRef]
- Kanellos, G.; Frame, M.C. Cellular functions of the adf/cofilin family at a glance. J. Cell Sci. 2016, 129, 3211–3218. [Google Scholar] [CrossRef]
- Li, L.H.; Wu, G.Y.; Lu, Y.Z.; Chen, X.H.; Liu, B.Y.; Zheng, M.H.; Cai, J.C. P21-activated protein kinase 1 induces the invasion of gastric cancer cells through c-jun nh2-terminal kinase-mediated activation of matrix metalloproteinase-2. Oncol. Rep. 2017, 38, 193–200. [Google Scholar] [CrossRef]
- Kwiatkowska, A.; Kijewska, M.; Lipko, M.; Hibner, U.; Kaminska, B. Downregulation of akt and fak phosphorylation reduces invasion of glioblastoma cells by impairment of mt1-mmp shuttling to lamellipodia and downregulates mmps expression. Biochim. Biophys. Acta 2011, 1813, 655–667. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.C.; Coppolino, M.G. Phosphorylation of membrane type 1-matrix metalloproteinase (mt1-mmp) and its vesicle-associated membrane protein 7 (vamp7)-dependent trafficking facilitate cell invasion and migration. J. Biol. Chem. 2011, 286, 43405–43416. [Google Scholar] [CrossRef] [PubMed]
- Clark, E.S.; Whigham, A.S.; Yarbrough, W.G.; Weaver, A.M. Cortactin is an essential regulator of matrix metalloproteinase secretion and extracellular matrix degradation in invadopodia. Cancer Res. 2007, 67, 4227–4235. [Google Scholar] [CrossRef] [PubMed]
- Mascia, A.; Gentile, F.; Izzo, A.; Mollo, N.; De Luca, M.; Bucci, C.; Nitsch, L.; Calì, G. Rab7 regulates cdh1 endocytosis, circular dorsal ruffles genesis and thyroglobulin internalization in a thyroid cell line. J. Cell Physiol. 2016, 231, 1695–1708. [Google Scholar] [CrossRef] [PubMed]
- Bauvois, B. New facets of matrix metalloproteinases mmp-2 and mmp-9 as cell surface transducers: Outside-in signaling and relationship to tumor progression. Biochim. Biophys. Acta 2012, 1825, 29–36. [Google Scholar] [CrossRef]
- Winer, A.; Adams, S.; Mignatti, P. Matrix metalloproteinase inhibitors in cancer therapy: Turning past failures into future successes. Mol. Cancer Ther. 2018, 17, 1147–1155. [Google Scholar] [CrossRef]
- Chetty, C.; Lakka, S.S.; Bhoopathi, P.; Rao, J.S. Mmp-2 alters vegf expression via alphavbeta3 integrin-mediated pi3k/akt signaling in a549 lung cancer cells. Int. J. Cancer 2010, 127, 1081–1095. [Google Scholar] [CrossRef]
- Dufour, A.; Zucker, S.; Sampson, N.S.; Kuscu, C.; Cao, J. Role of matrix metalloproteinase-9 dimers in cell migration: Design of inhibitory peptides. J. Biol. Chem. 2010, 285, 35944–35956. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romano, R.; Calcagnile, M.; Margiotta, A.; Franci, L.; Chiariello, M.; Alifano, P.; Bucci, C. RAB7A Regulates Vimentin Phosphorylation through AKT and PAK. Cancers 2021, 13, 2220. https://doi.org/10.3390/cancers13092220
Romano R, Calcagnile M, Margiotta A, Franci L, Chiariello M, Alifano P, Bucci C. RAB7A Regulates Vimentin Phosphorylation through AKT and PAK. Cancers. 2021; 13(9):2220. https://doi.org/10.3390/cancers13092220
Chicago/Turabian StyleRomano, Roberta, Matteo Calcagnile, Azzurra Margiotta, Lorenzo Franci, Mario Chiariello, Pietro Alifano, and Cecilia Bucci. 2021. "RAB7A Regulates Vimentin Phosphorylation through AKT and PAK" Cancers 13, no. 9: 2220. https://doi.org/10.3390/cancers13092220
APA StyleRomano, R., Calcagnile, M., Margiotta, A., Franci, L., Chiariello, M., Alifano, P., & Bucci, C. (2021). RAB7A Regulates Vimentin Phosphorylation through AKT and PAK. Cancers, 13(9), 2220. https://doi.org/10.3390/cancers13092220