Simple Summary
In recent years, next-generation sequencing has become a major tool in the management of cancer, advancing the diagnosis and treatment of hematological malignancies. However, the gold standard for cancer diagnosis and monitoring still involves invasive and painful procedures, such as tissue and bone marrow biopsies. These procedures involve physical risks, and a single biopsy cannot account for the spatial heterogeneity of tumors. The validity of circulating tumor DNA-mediated liquid biopsies has been receiving increasing attention. This review provides a brief overview of research on liquid biopsy in hematological malignancies, with special emphasis on circulating tumor DNA technologies, which may, in the near future, guide real-world decision making by hematologists.
Abstract
With the recent advances in noninvasive approaches for cancer diagnosis and surveillance, the term “liquid biopsy” has become more familiar to clinicians, including hematologists. Liquid biopsy provides a variety of clinically useful genetic data. In this era of personalized medicine, genetic information is critical to early diagnosis, aiding risk stratification, directing therapeutic options, and monitoring disease relapse. The validity of circulating tumor DNA (ctDNA)-mediated liquid biopsies has received increasing attention. This review summarizes the current knowledge of liquid biopsy ctDNA in hematological malignancies, focusing on the feasibility, limitations, and key areas of clinical application. We also highlight recent advances in the minimal residual disease monitoring of leukemia using ctDNA. This article will be useful to those involved in the clinical practice of hematopoietic oncology.
1. Introduction
With the aging population on the increase, the global cancer incidence rate has been on the rise. Hematopoietic tumors are no exception, with one person diagnosed with blood cancer every three minutes and one person dying of it every nine minutes in the United States. Interest in the Precision Medicine Initiative announced in President Obama’s State of the Union address in January 2015 has spread worldwide. Precision medicine encompasses the prevention and treatment of diseases based on a detailed analysis of the genomic information and lifestyle of a patient and environmental factors. In cancer genome medicine, the practice of establishing a foundation for individual treatment based on a diagnosis guided by a comprehensive genetic analysis is being developed. However, invasive diagnostic methods are still widely used and remain an issue. This review summarizes the literature on circulating tumor DNA (ctDNA)-mediated liquid biopsy, a noninvasive and useful personal diagnostic approach based on genetic information. The clinical applications of ctDNA, which will be useful to those involved in the clinical management of hematopoietic tumors, are specifically emphasized.
2. What Is a Liquid Biopsy?
The term “biopsy” was coined by Besnier (France) in the late 1870s when conducting dermatological-related experiments. The word is derived from bio (life) and opsis (to see) [1]. Currently, biopsy is defined as “the removal of cells or tissues for examination by a pathologist” (https://www.cancer.gov/publications/dictionaries/cancer-terms/def/biopsy, accessed on 1 April 2021). There are various problems associated with traditional tissue biopsies. First, complications like pain and bleeding at the puncture site after biopsy are frequent [2,3,4]. Second, the detection of an early-stage tumor or residual lesions is unsatisfactory in tissue biopsy, and its application in determining treatment efficacy and prognosis is limited [5]. Further, it is difficult to capture the inherent molecular heterogeneity of metastatic tumors and the ability of cancer genomes to evolve dynamically based on a single local biopsy [6,7].
The term “liquid biopsy” was first used by Pantel et al. in their 2010 review of circulating tumor cells (CTCs) to refer to the clinical utility of peripheral blood sample analysis [8]. Now, in a broad sense, liquid biopsy indicates the isolation and analysis of tumor-derived materials (e.g., DNA, RNA, or even intact cells) from blood or other bodily fluids [9,10]. Samples for liquid biopsy include plasma/serum, urine [11], saliva [12], stool [13], and cerebrospinal fluid [14]. Some argue that cerebrospinal fluid does not qualify as a fluid for liquid biopsy because of the invasive nature of its collection.
3. Contents of a Liquid Biopsy
In a conventional biopsy, tissue from the lesion is collected for the morphological evaluation and quantification of nucleic acids (DNA, RNA), proteins, and metabolites contained in the cells. On the other hand, in liquid biopsy, CTCs, cell-free DNA/RNA, extracellular vesicles, and microRNAs (miRNAs) in the collected liquid are used to evaluate the characteristics of the origin cell. CTCs are thought to be part of cancer stem cells released into the bloodstream via the epithelial–mesenchymal transition and undergo hematogenous metastasis [15]. Therefore, analysis of the CTCs detected in the peripheral blood (PB) of cancer patients is useful for early cancer screening, elucidation of the metastatic process, prediction of prognosis, determination of therapeutic efficacy, and analysis of the mechanisms of therapeutic resistance [16,17,18,19,20,21]. There is a paucity of literature on CTCs in hematopoietic tumors, probably because, unlike solid tumors, the concept of metastasis is rarely used and because the detection of CTCs requires complex enrichment methods [22,23]. In addition, in multiple myeloma (MM), CTCs have been associated with the spread of extramedullary lesions [24,25,26,27] and have been applied in the genetic profiling of tumors [28].
In 1983, Johnstone and Harding discovered vesicles secreted by reticulocytes, and the vesicles were named exosomes in 1987 [29,30,31]. Since then, vesicles of different sizes and origins have, among others, been referred to as ectosomes, microvesicles, and shedding vesicles. These are now collectively referred to as extracellular vesicles (EVs). Their contents include proteins, nucleic acids (miRNAs and messenger RNAs [mRNAs]), lipids, and metabolites, and they have been shown to be a tool for intercellular communication [32,33,34]. In particular, miRNAs encoded by EVs are one of the bioactive molecules involved in tumor growth and drug resistance and have attracted attention in malignant lymphoma [35] and MM studies [36,37].
4. Cell-Free DNA and Circulating Tumor DNA
Cell-free DNA (cfDNA) refers to all non-encapsulated DNA in the bloodstream. In 1948, Mandel and Metais were the first to report the presence of cfDNA in the plasma of patients with systemic lupus erythematosus [38]. In healthy individuals, the concentration of cfDNA ranges from 0 to 100 ng/mL of blood (average ~ 30 ng/mL) [39]; the main sources are apoptotic or necrotic cells [40,41]. Normally, the DNA of apoptotic cells is rapidly degraded by DNase [42]. However, when the uptake of apoptotic bodies is impaired or when a large number of apoptotic cells are generated, such as following acute trauma [43], stroke [44], exercise [45], transplantation [46], infection [47], and cancer [48,49], the cfDNA concentration in plasma increases. In oncology, the tumor-derived fraction of cfDNA is known as ctDNA [50]. ctDNA can be used to analyze not only mutations but also methylation status, size fragment patterns, transcriptomics, and viral load [10,51]. Studies by Dennis Lo et al. regarding clearance time revealed that the circulating fetal DNA, after delivery, has a mean half-life of 16.3 min. The study suggested that tumor-derived DNA may be removed rapidly [52].
Diehl et al. sampled the plasma of colorectal cancer patients who underwent tumor resection and showed that the ctDNA levels determined before surgery varied widely and that the postoperative ctDNA half-life was 114 min. In the study, ctDNA levels reflected the total systemic tumor burden, in that, the levels decreased upon complete surgery and generally increased as new lesions became apparent upon radiological examination [49]. Bettegowda et al. evaluated the use of ctDNA to detect tumors in 640 patients with various cancer types. They found that detectable levels of ctDNA correlated with the stage of cancer; ctDNA was detected in 47% of patients with stage I cancer, 55% with stage II, 69% with stage III, and 82% with stage IV [53]. In a study using 3D volume reconstruction of computed tomography (CT) images, Parkinson et al. clearly demonstrated that the amount of ctDNA in the plasma of high-grade serous ovarian cancer patients reflects the degree of the tumor [54]. From these studies, ctDNA analysis is considered a real-time snapshot of disease burden.
Fan et al. measured the size of fetal cfDNA and found a dominant peak at ~162 bp and a minor peak at ~340 bp [55]. The size of cfDNA in cancer patients has also been reported to peak at ~180 bp, which is considered to be the result of protection from enzymatic degradation by histone binding to nuclear DNA during apoptosis. In contrast, DNA fragments larger than ~10,000 bp could originate from cells dying via necrosis [40].
For cfDNA sampling, plasma centrifuged from whole blood or commercially available blood collection tubes for cell-free DNA sampling is used [56]. In addition, the phenol–chloroform method, sodium iodide method, magnetic bead method, and commercial DNA isolation kits, are commonly used for extraction [57]. Depending on the experimental design, there may be concerns about sampling bias because of the use of samples that have been stored for a long time and the differences in collection methods among facilities.
The detection of ctDNA, which is present in peripheral blood at very low allele frequencies, requires high technology, and with the evolution of PCR methods (ASO-PCR, ddPCR) and the improvement of the data output of sequencers, highly sensitive detection methods have become feasible (Table 1). However, there is still no method that meets all the requirements, such as the cost and the number of facilities where it can be performed, for progress in clinical applicability [56,58].

Table 1.
Methods of circulating tumor DNA (ctDNA) detection in peripheral blood.
Clinical trials are being conducted to use ctDNA to select treatments in solid tumors, and screen for cancer in healthy individuals [64,65,66,67,68,69]. In the case of hematopoietic tumors, there is a need to research cfDNA independently because the tissue collection method (hematopoietic tumor cells are contained in various tissues such as bone marrow, peripheral blood, and lymph nodes) and the genetic abnormalities observed in these tumors are different from those in solid tumors. Currently, ctDNA is being studied in hematopoietic tumors for the detection of tumor-specific mutations, evaluation of therapeutic efficacy, detection of minimal residual disease (MRD), prognosis prediction, and assessment of genomic heterogeneity. The application of cfDNA in hematopoietic tumors is described in this review.
5. Utility of ctDNA Characterization in Hematopoietic Tumors
5.1. Acute Myeloid Leukemia (AML)/Myelodysplastic Syndrome (MDS)
The study of cfDNA started again in 1994 [70] in AML/MDS, about half a century after the discovery of cfDNA in plasma by Mandel et al. In 2004, Rogers et al. used capillary electrophoresis to detect loss of heterozygosity in ctDNA from the plasma of patients with cytogenetically identified chromosomal abnormalities. They found that the plasma may be a potential substitute for bone marrow (BM) as a material for chromosome testing [71]. Although similar studies have not been conducted since then, the detection of chromosomal aberrations by ctDNA is expected to be re-examined using whole-genome sequences.
cfDNA extraction using off-the-shelf kits has become widespread since the 2000s (Table 2). In 2010, Gao et al. focused on the quality of cfDNA extracted using a QIAamp DNA Blood Kit (Qiagen, Hilden, Germany) and examined the plasma ctDNA integrity index of 60 acute leukemia patients using quantitative real-time PCR (qPCR) amplification of the β-actin gene. They concluded that plasma DNA integrity is incremental in acute leukemia and may serve as a biomarker for monitoring MRD [72]. This study became the basis for validating the usefulness of ctDNA in monitoring MRD in leukemia, which was later published in numerous publications. The ultimate goal of many researchers has shifted from conventional diagnostic approaches to using cfDNA-detected genetic mutations for diagnosis, treatment decisions, and prognosis. Iriyama et al. performed a global methylation analysis using bisulfite pyrosequencing based on the specific CpG sites of the LINE-1 promoter and quantified a TET2 mutation in DNA from plasma. The results showed that the methylation rate decreased rapidly in plasma ctDNA after azacitidine administration. Furthermore, in the quantification of the TET2 mutant gene in ctDNA, they observed that the ratio of the mutant gene was almost at the same level as that in the BM CD34+/38− stem cell population. They compared their results with those of BM cells and PBMCs analyzed simultaneously and showed that ctDNA could be used for genetic/epigenetic analysis with higher sensitivity [73]. In 2015, Quan et al. quantified the copies of circulating nucleophosmin (NPM) mutations in plasma DNA of AML patients using qPCR and analyzed the association between ctDNA copies and clinical characterization. They showed that patients with high PB white blood cell and platelet counts and high BM blast rates had significantly high copy numbers of the circulating NPM mutant gene [74].
Table 2.
Description of the studies examining cell-free DNA (cfDNA) for acute myeloid leukemia (AML)/myelodysplastic syndrome (MDS).
Hiseq2000 (Illumina) was introduced in 2010. With the development of next-generation sequencers, the target area for analyzing cfDNA mutations has also expanded rapidly. In 2016, Albitar et al. subjected the cfDNA of MDS patients to a panel genetic analysis targeting 14 genes. All samples were found to have at least one mutated gene, confirming the presence of an abnormal clone consistent with MDS. The authors noted that the diagnosis of MDS requires morphological and cytogenetic diagnoses but that neither of these can sometimes provide clear evidence and that molecular tests to detect abnormal clones have been relied upon in recent years [75]. NGS of cfDNA is expected to be a highly sensitive method to detect abnormal clones in MDS, where cytopenia may prevent the collection of sufficient peripheral blood nucleated cells. Suzuki et al., from the same research group as Iriyama et al., who performed ctDNA methylation analysis, evaluated the validity of targeted sequencing of cfDNA by comparing somatic mutations detected in MDS, BM DNA, and cfDNA. The Sanger method was used for most cases in this study; only two cases underwent NGS on BM DNA/cfDNA pairs; therefore, additional validation is needed [76]. cfDNA mutation analysis is now being validated using a panel targeting more genes; Zhao et al. performed cfDNA genetic analysis of MDS on a panel of 127 genes (Roche NimbleGen liquid phase hybrid capture chip). They concluded that ctDNA reflected genetic variation in BM DNA [77]. With the rise of digital PCR in the late 2010s, MRD monitoring studies targeting mutated genes became mainstream. Yeh et al. performed targeted sequencing using the BM of MDS patients treated with azacitidine and eltrombopag; here, detected driver mutations and karyotypic abnormalities were tracked in cfDNA using digital PCR during treatment. They demonstrated that serial monitoring of ctDNA allowed the concurrent tracking of both mutations and karyotypic abnormalities throughout therapy, and they were able to anticipate treatment failure. In addition, they revealed that ctDNA exhibited a differential response in the malignant subclones during therapy [78].
At the time of diagnosis of acute leukemia, 1012 leukemic cells were present in the patient’s body. Even after successful induction therapy and complete morphological remission with less than 5% leukemic cells in the BM on speculum, 109 leukemic cells remained in the body. The number of leukemic cells below 109 undetectable by light microscopy are termed as MRD and are considered important because they can proliferate and cause relapse after remission [79,80,81]. The MRD evaluation method used in clinical practice, qPCR, which detects genetic mutations in bone marrow DNA, has a high sensitivity of 0.1%. However, only patients with gene mutations detectable by commercial-based tests, such as APL (PML-RALα) and CML (BCR-ABL), can benefit from it. Currently, allogeneic hematopoietic stem cell transplantation (alloSCT) [82] is the only curative option for patients with high-risk or refractory AML and MDS. However, many post-alloSCT AML patients experience relapse and suffer another severe disease course [83,84]. In addition, AML patients with positive MRD pre- or post-transplantation have a higher risk of post-transplant recurrence and a lower survival rate [85]. In 2018 and 2019, we developed a ctDNA-based noninvasive MRD monitoring and prognosis system for patients with AML/MDS undergoing alloSCT [86,87]. We retrospectively performed whole-exome sequencing of tumor samples from AML/MDS patients (using the patient oral mucosa cells as a control) to identify driver mutations in the tumors, construct patient-specific ddPCR assays, and monitor MRD with ctDNA obtained from serum at 1 month and 3 months after transplantation (Figure 1). We found that patients who were MRD-positive at 1 month and 3 months after transplantation had an increased relapse rate 3 years after transplantation. We are currently conducting a prospective study of patients with AML/MDS undergoing alloSCT to verify the usefulness and feasibility of MRD monitoring using ctDNA. We will be able to report on this in the near future. In 2020, Nicholas et al. performed targeted NGS of ctDNA and BM DNA in patients with AML and suggested that they may be complementary to the assessment and monitoring of patients [88]. Zhong et al. detected monoclonal immunoglobulin gene rearrangement in AML ctDNA, which is expected to be applied in MRD monitoring [89]. The detection of IgH and TCR rearrangement is common in lymphoid malignancy but rare in AML. As we have shown, MRD monitoring by ddPCR has been used in many studies, but Christenson et al. did not consider it substantial. If the expression of SNPs in the wild-type gene sequence is not taken into account when creating a ddPCR assay, the correct allele frequency may not be detected [90]. In addition, the presence of clonal hematopoiesis with indeterminate potential (CHIP) should be noted in the evaluation of MRD by ctDNA. Clonal hematopoiesis is a condition in which a single or a very small number of clones maintain hematopoiesis. Mutations in epigenomic regulatory genes such as DNMT3A, TET2, and ASXL1 are the most common causes of CHIP [91,92]. It has been reported that the majority of mutations detected in cfDNA of healthy individuals are CHIP, and if these are detected as mutations in ctDNA and subjected to MRD evaluation, the reliability of the test will be reduced [93]. There is an opinion that it is possible to exclude CHIP mutations by paired sequencing of leukocyte DNA and plasma cfDNA [94], but further validation is needed in terms of cost and time. It is hoped that MRD monitoring by ctDNA will be implemented in the clinical environment after repeated trial and error and sufficient validation.
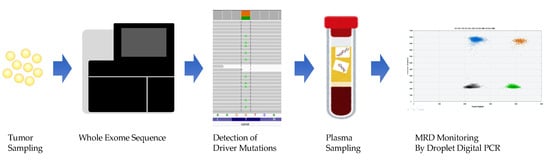
Figure 1.
Whole-exome sequencing of bone marrow fluid before treatment to identify driver mutations and monitoring of minimal residual disease (MRD) by creating a droplet digital PCR assay for each patient.
The monitoring of ctDNA by continuous specimen collection is promising not only as a clinical test as described above but also as an evaluation tool in clinical trials to develop new therapeutic agents. Zeidan et al. performed ctDNA monitoring in a Phase Ib study of PLK1 inhibitor, onvansertib, for AML. They monitored VAFs with ctDNA before and after treatment with a gene mutation that was previously detected in the target sequence of tumor DNA. They concluded that clinical response to onvansertib could be predicted from changes in VAF after treatment [95]. This study did not have a large enough sample size to create a receiver operating characteristic curve, but as more such trials are conducted, the use of ctDNA as a test endpoint will be optimized.
5.2. Lymphoid Malignancy
The most frequently reported studies on ctDNA in hematopoietic tumors are in the field of lymphoid malignancy. In April 2021, we searched PubMed for “circulating tumor DNA, Leukemia, NOT review,” “circulating tumor DNA, Lymphoma, NOT review,” and “circulating tumor DNA, Multiple Myeloma, NOT review,” and found 159, 224, and 38 hits, respectively. This may be because lymphoma incidence is the highest among hematopoietic tumors (Cancer Facts & Figures, 2020; American Cancer Society, 2020).
In the case of myeloid malignancy, most studies have used ctDNA SNVs/Indels as detection targets. On the other hand, in lymphoid malignancy, immunoglobulin-heavy chain (IgH) rearrangement was initially emphasized. In 1997, using PCR, Frickhofen et al. detected clonal DNA from a rearranged IgH locus in the plasma samples of patients with non-Hodgkin lymphoma and acute B-precursor lymphoblastic leukemia [97]. Further, Zhong et al. detected IgH and T cell receptor γ gene rearrangements in plasma cfDNA from patients with non-Hodgkin lymphoma [98]. In 2011, He et al. detected specifically rearranged DNA fragments in patient plasma using IgCap, which captures and sequences the IgH genomic region [99]. Subsequently, the data output of NGS techniques increased, and high-throughput deep sequencing of immunoglobulins (IgHTS), a technique for the comprehensive analysis of CDR3 sequences of BCR genes by multiplex PCR, became available commercially. Armand et al. analyzed plasma ctDNA from 16 DLBCL and MLBCL cases using Sequenta Lympho SIGHT (Sequenta Inc., South San Francisco, CA, USA) and successfully detected IgH and IgK reconstitution [100]; Kurtz et al. compared ctDNA analysis with IgHTS and imaging diagnosis using PET-CT in 75 cases of DLBCL. They compared ctDNA analysis using IgHTS and imaging diagnosis with PET-CT [101]. In addition, Roschewski et al. argued the usefulness of ctDNA surveillance by IgHTS based on a 5-year post-treatment follow-up of 126 DLBCL cases [102]. Sarkozy et al. used IgHTS of follicular lymphoma (FL) cases to compare the chronotype of tumor DNA and plasma ctDNA and showed that the subclonal distribution between tumor and plasma was different in more than half the cases [103]. IgHTS of ctDNA for other diseases including HIV-related B cell lymphoma was performed by Wagner-Johnston et al. [104] and MCL by Kumar et al. [105]. For B cell malignancy, there are many reports, as mentioned above. Regarding T cell lymphoma, Zhang et al. performed T cell receptor HTS in 2021 and detected TCR rearrangement in 78% of the cases; hence, only a few reports exist [106].
Regarding the methods for detecting SNV/Indel, Hosny et al., in 2009, detected the TP53 mutation in ctDNA of NHL patients using direct sequencing, but this method was limited to the target genes [107]. With the advent of CAPP-seq (cancer personalized profiling by deep sequencing), panel sequence, and low-pass WGS, the target gene region has been greatly expanded, and continuous monitoring by ddPCR as in myeloid malignancy has been reported [108,109,110,111,112]. CAPP-seq is an economical and ultrasensitive hybrid capture-based target sequence method developed by Newman et al. in 2014 [62]. WGS and WES have the advantage of measuring a wide range of genetic regions, but they have the disadvantage of high costs to achieve high detection sensitivity. However, CAPP-seq can detect four types of mutations (single-nucleotide polymorphism, insertion/deletion, copy number polymorphism, and fusion region) in cell-free tumor DNA with high efficiency by simultaneously sequencing cancer-specific mutated gene regions. Newman et al. at Stanford University performed CAPP-seq on ctDNA of NHL and showed better sensitivity than IgHTS in genotyping ctDNA [113,114]. This technique was applied to DLBCL by Rossi et al. [115] and classical Hodgkin lymphoma (cHL) by Spina et al. [116]. While CAPP-seq has the advantage of obtaining genotypic information with high sensitivity, the number of facilities that can perform CAPP-seq for hematological malignancy is limited.
A target sequence, which can analyze genetic variation in a specific genomic region, is widely used. Bohers et al. first published the detection of SNV/Indels in lymphoid malignancy [117]. This was followed by reports showing the evaluation of the validity of SNV/Indel detection in ctDNA [118,119,120], the association of genotyping-derived profiling with treatment response and prognosis [121,122,123,124,125,126,127,128], and the study of its applicability as a MRD monitoring material [129,130]. In DLBCL, Rushton et al. performed a 63-gene target sequence for ctDNA in 135 cases [125]. Camus et al. performed the first prospective ctDNA genotyping after cHL chemotherapy and showed that MRD detection by ctDNA was superior to that by PET-CT [129]. The target sequence is highly versatile because it is available commercially, but it is less sensitive in identifying mutations in cfDNA. Technical considerations, such as the expansion of the target gene region, are necessary. For CNS lymphoma, there are scattered reports on ctDNA in cerebrospinal fluid (CSF) [131,132,133,134], and some argue that ctDNA in plasma is not sensitive enough for detection [135,136]. The applicability of ctDNA in plasma should be carefully evaluated considering the characteristics of the disease.
Low-pass WGS can also detect fusion and CNV in addition to SNV/Indels. It has the advantage of providing richer information in samples with high tumor volume; Agarwal et al. used it to elucidate treatment strategies with ibrutinib and venetoclax [137].
Cytosine methylation in the CpG islands of gene promoter regions is another factor that has a significant impact on gene expression. In cancer cells, the expression of tumor suppressor genes is repressed by the aberrant methylation of CpG islands [138]. In the field of leukemia–lymphoma, methylation analysis of tumor suppressor genes has been actively pursued [139,140]. In 2003, Deligezer et al. suggested the presence of methylated tumor suppressor gene 16 in the cfDNA of patients with lymphoproliferative diseases [141]. In 2007, Shi et al. established CpG island DNA microarray methylation profiling in cancer; they detected methylation of the tumor suppressor candidate gene DLC-1 in plasma DNA from non-Hodgkin lymphoma patients. They further reported that DLC-1 methylation disappeared after response to chemotherapy as determined by quantitative methylation-specific PCR analysis [142]. Thus, DNA methylation analysis has been applied to the genetic characterization and monitoring of lymphoid malignancy.
Validation of ctDNA levels and integrity has also been active in lymphoma [143,144,145,146]. Delfau-Larue et al. quantified ctDNA using ddPCR and compared it to tumor volume measured by PET-CT, and showed that in FL, pre-treatment ctDNA levels correlated with total metabolic tumor volume measured by PET-CT and total tumor volume [147]. Kurtz et al. also developed the continuous individualized risk index (CIRI), a risk assessment scale that incorporates pre-treatment ctDNA levels for DLBCL divided into high and low [148]. A challenge for social implementation is to clarify the interpretation of ctDNA analysis results in combination with existing laboratory findings such as imaging, especially in lymphoma.
5.3. Multiple Myeloma
MM is characterized by monoclonal proliferation of plasma cells, the final differentiation stage of B cells. Most cases start as monoclonal gammopathy of undetermined significance (MGUS) and develop into smoldering multiple myeloma (SMM) with no clinical symptoms. Eventually, one of the four symptoms (CRAB) of hypercalcemia, renal failure, anemia, and bone lesions become associated with the diagnosis of MM [149]. At diagnosis, there is clonal diversity with the coexistence of dominant and minor subclones that have evolved from a common ancestral tumor-initiating cell or stem cell. At recurrence, clones are also heterogeneous and may be dominated by the same or different subclones from those at first appearance [150]. Tumors are multifocal in BM and secrete monoclonal immunoglobulins (M-proteins) in blood and urine. They are morphologically heterogeneous and have varying responses to therapy and tumor progression. Therefore, BM puncture at a single site alone is subject to sampling bias and yields only a limited molecular profile, which cannot reflect the diverse pathogenesis of various subclones. The measurement of cfDNA, which comprehensively reflects the entire tumor, is ideal for clarifying the pathogenesis. In particular, MRD monitoring, which has gained clinical significance with the development of new drugs [151], is expected to be applied to ctDNA as well. In this context, Sata et al. [152] and Oberle et al. [153] reported the detection of immunoglobulin rearrangements in ctDNA. Mazzotti et al. analyzed 47 cases of MM using the NGS MRD assay (IgHTS) from Adaptive Biotechnologies, claiming that MRD monitoring is not possible with NGS alone, which targets Ig gene rearrangement [154]. On the other hand, some argue that quantification of IgH rearrangements can be used for prognostic stratification [59,155].
For SNV/Indel, the analysis focuses on targeting KRAS, NRAS, and BRAF [156,157,158]. Mithraprabhu et al. analyzed cfDNA of MM patients for target gene mutations, including KRAS, NRAS, and BRAF, using ddPCR and continuously quantified and monitored ctDNA. They analyzed paired BM cell DNA and ctDNA from 33 relapsed or refractory MM patients and 15 newly diagnosed patients using targeted deep sequencing. ctDNA mutations were detected at a higher frequency in relapsed or refractory patients than in newly diagnosed patients (27.2% vs. 6.6%, respectively), authenticating the existence of spatial and genetic heterogeneity in advanced disease [159,160]. The authors also monitored tumor burden and therapeutic response through ctDNA analysis and reported that a decrease in ctDNA levels at day 5 of treatment cycle 1 correlated with superior PFS (P = 0.017). It was also concluded that ctDNA is useful for predicting disease outcomes in MM patients [161]. As mentioned earlier, MM has a precancerous state, but ctDNA detection in SMM and MGUS is still difficult [162]. In addition, as with other hematopoietic tumors, targeted gene regions have been expanded [163]. Deshpande et al. assessed whether ctDNA levels varied according to risk status defined by the 70-gene expression profile. Patients with high ctDNA levels were associated with worse PFS (hazard ratio 6.4; 95% CI 1.9–22) and overall survival rates (hazard ratio 4.4; 95% CI 1.2–15.7); ctDNA level was also elevated in the high-risk group. These findings showed that cfDNA is a dynamic tool to capture genetic events in MM [56].
Fusion and CNV of MM have been developed using WES and low-pass WGS (Guo, Manier) [164,165]. For ctDNA analysis of MM, many issues need to be resolved in the future, such as improving detection sensitivity by improving sequencing technology and prospective validation together with existing MRD measurement methods.
6. Future Directions and Conclusions
In 2019, Lenaerts et al. made a grand attempt to predict malignancy from copy number abnormalities by applying WGS (Genomewide Imbalance Profiling sequence) to the cfDNA of 1002 individuals with no history of malignancy. As a result, four malignant lymphoma cases and one MDS case with excess blasts were detected [166]. This announcement signifies the development from the era of cfDNA testing to the era of cfDNA-based testing. It is also true that cfDNA has its limitations. With the progress of NGS and other comprehensive genetic analysis technologies, many insights into diagnosis and treatment are moving toward clinical application. However, to expand the application of cfDNA, many issues remain to be addressed, such as the establishment of specimen processing procedures, dissemination of analytical methods that take into account cost and sensitivity, and interpretation of results in conjunction with existing testing methods. If clinicians are unaware of the benefits and limitations of cfDNA, they will not make the right decisions regarding its application to individual patients.
Author Contributions
M.O. and K.Y. wrote the manuscript. S.I. and A.T. reviewed and edited the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
Not applicable.
Conflicts of Interest
K.Y. received research funding from Nippon Shinyaku Co., Ltd. outside the submitted work. A.T. received personal fees from Sysmex, Otsuka Pharmaceutical Co., Ltd., Bristol-Myers Squib, and Takeda Pharmaceutical Co., Ltd., and Daiichi-Sankyo, grants and personal fees from Pfizer and Chugai Pharmaceutical, and research funding from LiquidMine outside the submitted work.
References
- Besnier, E. Études nouvelles de dermatologie: Sur un cas de dégénérescence colloïde du derme, affection non décrite ou improprement appelée colloid milium. Gaz. Hebd. Med. Chir. 1879, 41, 645–650. [Google Scholar]
- Tural-Kara, T.; Özdemir, H.; Fitöz, S.; Çiftçi, E.; Yalçınkaya, F. Bone marrow aspiration complications: Iliopsoas abscess and sacroiliac osteomyelitis. Turk. J. Pediatr. 2016, 58, 562–565. [Google Scholar] [CrossRef]
- Borghesi, M.; Ahmed, H.; Nam, R.; Schaeffer, E.; Schiavina, R.; Taneja, S.; Weidner, W.; Loeb, S. Complications After Systematic, Random, and Image-guided Prostate Biopsy [figure presented]. Eur. Urol. 2017, 71, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Abhishek, K.; Khunger, N. Complications of skin biopsy. J. Cutan. Aesthet. Surg. 2015, 8, 239–241. [Google Scholar] [CrossRef] [PubMed]
- Yong, E. Written in blood: DNA circulating in the bloodstream could guide cancer treatment-if researchers can work out how best to use it. Nature 2014, 511, 524–526. [Google Scholar] [CrossRef] [PubMed]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; Tarpey, P.; et al. Intratumor Heterogeneity and Branched Evolution Revealed by Multiregion Sequencing Marco. NEJM 2012, 366, 883–892. [Google Scholar] [CrossRef]
- Osumi, H.; Shinozaki, E.; Yamaguchi, K.; Zembutsu, H. Clinical utility of circulating tumor DNA for colorectal cancer. Cancer Sci. 2019, 110, 1148–1155. [Google Scholar] [CrossRef] [PubMed]
- Pantel, K.; Alix-Panabières, C. Circulating tumour cells in cancer patients: Challenges and perspectives. Trends Mol. Med. 2010, 16, 398–406. [Google Scholar] [CrossRef]
- Siravegna, G.; Marsoni, S.; Siena, S.; Bardelli, A. Integrating liquid biopsies into the management of cancer. Nat. Rev. Clin. Oncol. 2017, 14, 531–548. [Google Scholar] [CrossRef]
- Corcoran, R.B.; Chabner, B.A. Application of Cell-free DNA Analysis to Cancer Treatment. N. Engl. J. Med. 2018, 379, 1754–1765. [Google Scholar] [CrossRef]
- Togneri, F.S.; Ward, D.G.; Foster, J.M.; Devall, A.J.; Wojtowicz, P.; Alyas, S.; Vasques, F.R.; Oumie, A.; James, N.D.; Cheng, K.K.; et al. Genomic complexity of urothelial bladder cancer revealed in urinary cfDNA. Eur. J. Hum. Genet. 2016, 24, 1167–1174. [Google Scholar] [CrossRef]
- Kaczor-Urbanowicz, K.E.; Wei, F.; Rao, S.L.; Kim, J.; Shin, H.; Cheng, J.; Tu, M.; Wong, D.T.W.; Kim, Y. Clinical validity of saliva and novel technology for cancer detection. Biochim. Biophys. Acta Rev. Cancer 2019, 1872, 49–59. [Google Scholar] [CrossRef]
- Imperiale, T.F.; Ransohoff, D.F.; Itzkowitz, S.H.; Levin, T.R.; Lavin, P.; Lidgard, G.P.; Ahlquist, D.A.; Berger, B.M. Multitarget Stool DNA Testing for Colorectal-Cancer Screening. N. Engl. J. Med. 2014, 370, 1287–1297. [Google Scholar] [CrossRef] [PubMed]
- De Mattos-Arruda, L.; Mayor, R.; Ng, C.K.Y.; Weigelt, B.; Martínez-Ricarte, F.; Torrejon, D.; Oliveira, M.; Arias, A.; Raventos, C.; Tang, J.; et al. Cerebrospinal fluid-derived circulating tumour DNA better represents the genomic alterations of brain tumours than plasma. Nat. Commun. 2015, 6, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Cristofanilli, M. Circulating Tumor Cells, Disease Progression, and Survival in Metastatic Breast Cancer. Semin. Oncol. 2006, 33, 9–14. [Google Scholar] [CrossRef]
- Massagué, J.; Obenauf, A.C. Metastatic colonization by circulating tumour cells. Nature 2016, 529, 298–306. [Google Scholar] [CrossRef]
- Szczerba, B.M.; Castro-Giner, F.; Vetter, M.; Krol, I.; Gkountela, S.; Landin, J.; Scheidmann, M.C.; Donato, C.; Scherrer, R.; Singer, J.; et al. Neutrophils escort circulating tumour cells to enable cell cycle progression. Nature 2019, 566, 553–557. [Google Scholar] [CrossRef]
- Fiorelli, A.; Accardo, M.; Carelli, E.; Angioletti, D.; Santini, M.; Di Domenico, M. Circulating tumor cells in diagnosing lung cancer: Clinical and morphologic analysis. Ann. Thorac. Surg. 2015, 99, 1899–1905. [Google Scholar] [CrossRef]
- Kim, H.; Lim, M.; Kim, J.Y.; Shin, S.J.; Cho, Y.K.; Cho, C.H. Circulating tumor cells enumerated by a centrifugal microfluidic device as a predictive marker for monitoring ovarian cancer treatment: A pilot study. Diagnostics 2020, 10, 249. [Google Scholar] [CrossRef]
- Ma, J.; Yao, S.; Li, X.S.; Kang, H.R.; Yao, F.F.; Du, N. Neoadjuvant therapy of DOF Regimen plus bevacizumab can increase surgical resection rate in locally advanced gastric cancer: A randomized, controlled study. Medicine 2015, 94, e1489. [Google Scholar] [CrossRef]
- Gradilone, A.; Raimondi, C.; Naso, G.; Silvestri, I.; Repetto, L.; Palazzo, A.; Gianni, W.; Frati, L.; Cortesi, E.; Gazzaniga, P. How circulating tumor cells escape from multidrug resistance: Translating molecular mechanisms in metastatic breast cancer treatment. Am. J. Clin. Oncol. Cancer Clin. Trials 2011, 34, 625–627. [Google Scholar] [CrossRef]
- Alix-Panabières, C.; Pantel, K. Challenges in circulating tumour cell research. Nat. Rev. Cancer 2014, 14, 623–631. [Google Scholar] [CrossRef]
- Desitter, I.; Desitter, I.; Guerrouahen, B.S.; Guerrouahen, B.S.; Benali-Furet, N.; Benali-Furet, N.; Wechsler, J.; Wechsler, J.; Jänne, P.A.; Jänne, P.A.; et al. A new device for rapid isolation by size and characterization of rare circulating tumor cells. Anticancer Res. 2011, 31, 427–441. [Google Scholar] [PubMed]
- Bianchi, G.; Kyle, R.A.; Larson, D.R.; Witzig, T.E.; Kumar, S.; Dispenzieri, A.; Morice, W.G.; Rajkumar, S.V. High levels of peripheral blood circulating plasma cells as a specific risk factor for progression of smoldering multiple myeloma. Leukemia 2013, 27, 680–685. [Google Scholar] [CrossRef]
- Gonsalves, W.I.; Rajkumar, S.V.; Gupta, V.; Morice, W.G.; Timm, M.M.; Singh, P.P.; Dispenzieri, A.; Buadi, F.K.; Lacy, M.Q.; Kapoor, P.; et al. Quantification of clonal circulating plasma cells in newly diagnosed multiple myeloma: Implications for redefining high-risk myeloma. Leukemia 2014, 28, 2060–2065. [Google Scholar] [CrossRef]
- Sanoja-Flores, L.; Flores-Montero, J.; Garcés, J.J.; Paiva, B.; Puig, N.; García-Mateo, A.; García-Sánchez, O.; Corral-Mateos, A.; Burgos, L.; Blanco, E.; et al. Next generation flow for minimally-invasive blood characterization of MGUS and multiple myeloma at diagnosis based on circulating tumor plasma cells (CTPC). Blood Cancer J. 2018, 8, 117. [Google Scholar] [CrossRef]
- Kumar, S.; Rajkumar, S.V.; Kyle, R.A.; Lacy, M.Q.; Dispenzieri, A.; Fonseca, R.; Lust, J.A.; Gertz, M.A.; Greipp, P.R.; Witzig, T.E. Prognostic value of circulating plasma cells in monoclonal gammopathy of undetermined significance. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2005, 23, 5668–5674. [Google Scholar] [CrossRef] [PubMed]
- Garcés, J.J.; Bretones, G.; Burgos, L.; Valdes-Mas, R.; Puig, N.; Cedena, M.T.; Alignani, D.; Rodriguez, I.; Puente, D.Á.; Álvarez, M.G.; et al. Circulating tumor cells for comprehensive and multiregional non-invasive genetic characterization of multiple myeloma. Leukemia 2020, 34, 3007–3018. [Google Scholar] [CrossRef] [PubMed]
- Pan, B.T.; Johnstone, R.M. Fate of the transferrin receptor during maturation of sheep reticulocytes in vitro: Selective externalization of the receptor. Cell 1983, 33, 967–978. [Google Scholar] [CrossRef]
- Harding, C.; Heuser, J.; Stahl, P. Receptor-mediated endocytosis of transferrin and recycling of the transferrin receptor in rat reticulocytes. J. Cell Biol. 1983, 97, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, R.M.; Adam, M.; Hammond, J.R.; Orr, L.; Turbide, C. Vesicle formation during reticulocyte maturation. Association of plasma membrane activities with released vesicles (exosomes). J. Biol. Chem. 1987, 262, 9412–9420. [Google Scholar] [CrossRef]
- Kadota, T.; Yoshioka, Y.; Fujita, Y.; Ochiya, T. Exosomes: Toward clinical application. Folia Pharmacol. Jpn. 2017, 149, 119–122. [Google Scholar] [CrossRef]
- Edgar, J.R. Q & A: What are exosomes, exactly? BMC Biol. 2016, 14, 1–7. [Google Scholar] [CrossRef]
- Zomer, A.; Maynard, C.; Verweij, F.J.; Kamermans, A.; Schäfer, R.; Beerling, E.; Schiffelers, R.M.; De Wit, E.; Berenguer, J.; Ellenbroek, S.I.J.; et al. In vivo imaging reveals extracellular vesicle-mediated phenocopying of metastatic behavior. Cell 2015, 161, 1046–1057. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, H.; Yamakawa, N.; Imadome, K.-I.; Yahata, T.; Kotaki, R.; Ogata, J.; Kakizaki, M.; Fujita, K.; Lu, J.; Yokoyama, K.; et al. Role of exosomes as a proinflammatory mediator in the development of EBV-associated lymphoma. Blood 2018, 131, 2552–2567. [Google Scholar] [CrossRef]
- Manier, S.; Liu, C.J.; Avet-Loiseau, H.; Park, J.; Shi, J.; Campigotto, F.; Salem, K.Z.; Huynh, D.; Glavey, S.V.; Rivotto, B.; et al. Prognostic role of circulating exosomal miRNAs in multiple myeloma. Blood 2017, 129, 2429–2436. [Google Scholar] [CrossRef]
- Zhang, J.; Xiao, X.J.; Liu, J. The role of circulating miRNAs in multiple myeloma. Sci. China Life Sci. 2015, 58, 1262–1269. [Google Scholar] [CrossRef]
- Mandel, P.; Metais, P. Les acides nucléiques du plasma sanguin chez l’homme. C. R. Seances Soc. Biol. Fil. 1948, 142, 241–243. [Google Scholar]
- Alborelli, I.; Generali, D.; Jermann, P.; Cappelletti, M.R.; Ferrero, G.; Scaggiante, B.; Bortul, M.; Zanconati, F.; Nicolet, S.; Haegele, J.; et al. Cell-free DNA analysis in healthy individuals by next-generation sequencing: A proof of concept and technical validation study. Cell Death Dis. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Jahr, S.; Hentze, H.; Englisch, S.; Hardt, D.; Fackelmayer, F.O.; Hesch, R.D.; Knippers, R. DNA fragments in the blood plasma of cancer patients: Quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 2001, 61, 1659–1665. [Google Scholar]
- Stroun, M.; Lyautey, J.; Lederrey, C.; Olson-Sand, A.; Anker, P. About the possible origin and mechanism of circulating DNA: Apoptosis and active DNA release. Clin. Chim. Acta 2001, 313, 139–142. [Google Scholar] [CrossRef]
- Nagata, S.; Nagase, H.; Kawane, K.; Mukae, N.; Fukuyama, H. Degradation of chromosomal DNA during apoptosis. Cell Death Differ. 2003, 10, 108–116. [Google Scholar] [CrossRef]
- Rodrigues Filho, E.M.; Simon, D.; Ikuta, N.; Klovan, C.; Dannebrock, F.A.; Oliveira De Oliveira, C.; Regner, A. Elevated cell-free plasma DNA level as an independent predictor of mortality in patients with severe traumatic brain injury. J. Neurotrauma 2014, 31, 1639–1646. [Google Scholar] [CrossRef]
- O’Connell, G.C.; Petrone, A.B.; Tennant, C.S.; Lucke-Wold, N.; Kabbani, Y.; Tarabishy, A.R.; Chantler, P.D.; Barr, T.L. Circulating extracellular DNA levels are acutely elevated in ischaemic stroke and associated with innate immune system activation. Brain Inj. 2017, 31, 1369–1375. [Google Scholar] [CrossRef]
- Tug, S.; Helmig, S.; Deichmann, E.R.; Schmeier-Jürchott, A.; Wagner, E.; Zimmermann, T.; Radsak, M.; Giacca, M.; Simon, P. Exercise-induced increases in cell free DNA in human plasma originate predominantly from cells of the haematopoietic lineage. Exerc. Immunol. Rev. 2015, 21, 164–173. [Google Scholar]
- De Vlaminck, I.; Martin, L.; Kertesz, M.; Patel, K.; Kowarsky, M.; Strehl, C.; Cohen, G.; Luikart, H.; Neff, N.F.; Okamoto, J.; et al. Noninvasive monitoring of infection and rejection after lung transplantation. Proc. Natl. Acad. Sci. USA 2015, 112, 13336–13341. [Google Scholar] [CrossRef] [PubMed]
- Phuong, N.T.N.; Manh, D.H.; Dumre, S.P.; Mizukami, S.; Weiss, L.N.; Van Thuong, N.; Ha, T.T.N.; Phuc, L.H.; Van An, T.; Tieu, T.M.; et al. Plasma cell-free DNA: A potential biomarker for early prediction of severe dengue. Ann. Clin. Microbiol. Antimicrob. 2019, 18, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Leon, S.A.; Shapiro, B.; Sklaroff, D.M.; Yaros, M.J. Free DNA in the Serum of Cancer Patients and the Effect of Therapy. Cancer Res. 1977, 37, 646–650. [Google Scholar] [PubMed]
- Diehl, F.; Schmidt, K.; Choti, M.A.; Romans, K.; Goodman, S.; Li, M.; Thornton, K.; Agrawal, N.; Sokoll, L.; Szabo, S.A.; et al. Circulating mutant DNA to assess tumor dynamics. Nat. Med. 2008, 14, 985–990. [Google Scholar] [CrossRef]
- Stroun, M.; Anker, P.; Maurice, P.; Lyautey, J.; Lederrey, C.; Beljanski, M. Neoplastic characteristics of the DNA found in the plasma of cancer patients. Oncology 1989, 46, 318–322. [Google Scholar] [CrossRef] [PubMed]
- Keller, L.; Belloum, Y.; Wikman, H.; Pantel, K. Clinical relevance of blood-based ctDNA analysis: Mutation detection and beyond. Br. J. Cancer 2020. [Google Scholar] [CrossRef] [PubMed]
- Dennis Lo, Y.M.; Zhang, J.; Leung, T.N.; Lau, T.K.; Chang, A.M.Z.; Magnus Hjelm, N. Rapid clearance of fetal DNA from maternal plasma. Am. J. Hum. Genet. 1999, 64, 218–224. [Google Scholar] [CrossRef]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014, 6. [Google Scholar] [CrossRef]
- Parkinson, C.A.; Gale, D.; Piskorz, A.M.; Biggs, H.; Hodgkin, C.; Addley, H.; Freeman, S.; Moyle, P.; Sala, E.; Sayal, K.; et al. Exploratory Analysis of TP53 Mutations in Circulating Tumour DNA as Biomarkers of Treatment Response for Patients with Relapsed High-Grade Serous Ovarian Carcinoma: A Retrospective Study. PLoS Med. 2016, 13, 1–25. [Google Scholar] [CrossRef]
- Fan, H.C.; Blumenfeld, Y.J.; Chitkara, U.; Hudgins, L.; Quake, S.R. Analysis of the size distributions of fetal and maternal cell-free DNA by paired-end sequencing. Clin. Chem. 2010, 56, 1279–1286. [Google Scholar] [CrossRef]
- Thakral, D.; Das, N.; Basnal, A.; Gupta, R. Cell-free DNA for genomic profiling and minimal residual disease monitoring in Myeloma- are we there yet? Am. J. Blood Res. 2020, 10, 26–45. [Google Scholar]
- Mauger, F.; Dulary, C.; Daviaud, C.; Deleuze, J.-F.; Tost, J. Comprehensive evaluation of methods to isolate, quantify, and characterize circulating cell-free DNA from small volumes of plasma. Anal. Bioanal. Chem. 2015, 407, 6873–6878. [Google Scholar] [CrossRef]
- Kurtz, D.M. Prognostication with circulating tumor DNA: Is it ready for prime time? Hematology 2019, 2019, 47–52. [Google Scholar] [CrossRef]
- Vrabel, D.; Sedlarikova, L.; Besse, L.; Rihova, L.; Bezdekova, R.; Almasi, M.; Kubaczkova, V.; Brožová, L.; Jarkovsky, J.; Plonkova, H.; et al. Dynamics of tumor-specific cfDNA in response to therapy in multiple myeloma patients. Eur. J. Haematol. 2020, 104, 190–197. [Google Scholar] [CrossRef]
- Vogelstein, B.; Kinzler, K.W. Digital PCR. Proc. Natl. Acad. Sci. USA 1999, 96, 9236–9241. [Google Scholar] [CrossRef] [PubMed]
- Forshew, T.; Murtaza, M.; Parkinson, C.; Gale, D.; Tsui, D.W.Y.; Kaper, F.; Dawson, S.J.; Piskorz, A.M.; Jimenez-Linan, M.; Bentley, D.; et al. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci. Transl. Med. 2012, 4. [Google Scholar] [CrossRef]
- Newman, A.M.; Bratman, S.V.; To, J.; Wynne, J.F.; Eclov, N.C.W.; Modlin, L.A.; Liu, C.L.; Neal, J.W.; Wakelee, H.A.; Merritt, R.E.; et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat. Med. 2014, 20, 548–554. [Google Scholar] [CrossRef]
- Adalsteinsson, V.A.; Ha, G.; Freeman, S.S.; Choudhury, A.D.; Stover, D.G.; Parsons, H.A.; Gydush, G.; Reed, S.C.; Rotem, D.; Rhoades, J.; et al. Scalable whole-exome sequencing of cell-free DNA reveals high concordance with metastatic tumors. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Abbosh, C.; Birkbak, N.J.; Wilson, G.A.; Jamal-Hanjani, M.; Constantin, T.; Salari, R.; Le Quesne, J.; Moore, D.A.; Veeriah, S.; Rosenthal, R.; et al. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature 2017, 545, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Jamal-Hanjani, M.; Wilson, G.A.; McGranahan, N.; Birkbak, N.J.; Watkins, T.B.K.; Veeriah, S.; Shafi, S.; Johnson, D.H.; Mitter, R.; Rosenthal, R.; et al. Tracking the Evolution of Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 376, 2109–2121. [Google Scholar] [CrossRef]
- Cohen, J.D.; Li, L.; Wang, Y.; Thoburn, C.; Afsari, B.; Danilova, L.; Douville, C.; Javed, A.A.; Wong, F.; Mattox, A.; et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 2018, 359, 926–930. [Google Scholar] [CrossRef] [PubMed]
- Lennon, A.M.; Buchanan, A.H.; Kinde, I.; Warren, A.; Honushefsky, A.; Cohain, A.T.; Ledbetter, D.H.; Sanfilippo, F.; Sheridan, K.; Rosica, D.; et al. Feasibility of blood testing combined with PET-CT to screen for cancer and guide intervention. Science 2020, 369, eabb9601. [Google Scholar] [CrossRef] [PubMed]
- Kuwata, T.; Wakabayashi, M.; Hatanaka, Y.; Morii, E.; Oda, Y.; Taguchi, K.; Noguchi, M.; Ishikawa, Y.; Nakajima, T.; Sekine, S.; et al. Impact of DNA integrity on the success rate of tissue-based next-generation sequencing: Lessons from nationwide cancer genome screening project SCRUM-Japan GI-SCREEN. Pathol. Int. 2020, 70, 932–942. [Google Scholar] [CrossRef]
- Nakamura, Y.; Taniguchi, H.; Ikeda, M.; Bando, H.; Kato, K.; Morizane, C.; Esaki, T.; Komatsu, Y.; Kawamoto, Y.; Takahashi, N.; et al. Clinical utility of circulating tumor DNA sequencing in advanced gastrointestinal cancer: SCRUM-Japan GI-SCREEN and GOZILA studies. Nat. Med. 2020. [Google Scholar] [CrossRef]
- Vasioukhin, V.; Anker, P.; Maurice, P.; Lyautey, J.; Lederrey, C.; Stroun, M. Point mutations of the N-ras gene in the blood plasma DNA of patients with myelodysplastic syndrome or acute myelogenous leukaemia. Br. J. Haematol. 1994, 86, 774–779. [Google Scholar] [CrossRef] [PubMed]
- Rogers, A.; Joe, Y.; Manshouri, T.; Dey, A.; Jilani, I.; Giles, F.; Estey, E.; Freireich, E.; Keating, M.; Kantarjian, H.; et al. Relative increase in leukemia-specific DNA in peripheral blood plasma from patients with acute myeloid leukemia and myelodysplasia. Blood 2004, 103, 2799–2801. [Google Scholar] [CrossRef]
- Gao, Y.J.; He, Y.J.; Yang, Z.L.; Shao, H.Y.; Zuo, Y.; Bai, Y.; Chen, H.; Chen, X.C.; Qin, F.X.; Tan, S.; et al. Increased integrity of circulating cell-free DNA in plasma of patients with acute leukemia. Clin. Chem. Lab. Med. 2010, 48, 1651–1656. [Google Scholar] [CrossRef]
- Iriyama, C.; Tomita, A.; Hoshino, H.; Adachi-Shirahata, M.; Furukawa-Hibi, Y.; Yamada, K.; Kiyoi, H.; Naoe, T. Using peripheral blood circulating DNAs to detect CpG global methylation status and genetic mutations in patients with myelodysplastic syndrome. Biochem. Biophys. Res. Commun. 2012, 419, 662–669. [Google Scholar] [CrossRef] [PubMed]
- Quan, J.; Gao, Y.; Yang, Z.; Chen, H.; Xian, J.; Zhang, S.; Zou, Q.; Zhang, L. Quantitative detection of circulating nucleophosmin mutations DNA in the plasma of patients with acute myeloid leukemia. Int. J. Med. Sci. 2015, 12, 17–22. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Albitar, F.; Ma, W.; Diep, K.; De Dios, I.; Agersborg, S.; Thangavelu, M.; Brodie, S.; Albitar, M. Deep Sequencing of Cell-Free Peripheral Blood DNA as a Reliable Method for Confirming the Diagnosis of Myelodysplastic Syndrome. Genet. Test. Mol. Biomark. 2016, 20, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Tomita, A.; Nakamura, F.; Iriyama, C.; Shirahata-Adachi, M.; Shimada, K.; Akashi, A.; Ishikawa, Y.; Kaneda, N.; Kiyoi, H. Peripheral blood cell-free DNA is an alternative tumor DNA source reflecting disease status in myelodysplastic syndromes. Cancer Sci. 2016, 107, 1329–1337. [Google Scholar] [CrossRef]
- Zhao, P.; Qin, J.; Liu, W.; Zhu, Q.; Fan, T.; Xiao, H.; Wang, J.; Huang, G.; Xiaomei, H. Using circulating tumor DNA to monitor myelodysplastic syndromes status. Hematol. Oncol. 2019, 37, 531–533. [Google Scholar] [CrossRef]
- Yeh, P.; Hunter, T.; Sinha, D.; Ftouni, S.; Wallach, E.; Jiang, D.; Chan, Y.C.; Wong, S.Q.; Silva, M.J.; Vedururu, R.; et al. Circulating tumour DNA reflects treatment response and clonal evolution in chronic lymphocytic leukaemia. Nat. Commun. 2017, 8, 1–7. [Google Scholar] [CrossRef]
- Klco, J.M.; Miller, C.A.; Griffith, M.; Petti, A.; Spencer, D.H.; Ketkar-Kulkarni, S.; Wartman, L.D.; Christopher, M.; Lamprecht, T.L.; Helton, N.M.; et al. Association between mutation clearance after induction therapy and outcomes in acute myeloid leukemia. JAMA J. Am. Med. Assoc. 2015, 314, 811–822. [Google Scholar] [CrossRef]
- Morita, K.; Kantarjian, H.M.; Wang, F.; Yan, Y.; Bueso-Ramos, C.; Sasaki, K.; Issa, G.C.; Wang, S.; Jorgensen, J.; Song, X.; et al. Clearance of somatic mutations at remission and the risk of relapse in acute myeloid leukemia. J. Clin. Oncol. 2018, 36, 1788–1797. [Google Scholar] [CrossRef]
- Jongen-Lavrencic, M.; Grob, T.; Hanekamp, D.; Kavelaars, F.G.; al Hinai, A.; Zeilemaker, A.; Erpelinck-Verschueren, C.A.J.; Gradowska, P.L.; Meijer, R.; Cloos, J.; et al. Molecular Minimal Residual Disease in Acute Myeloid Leukemia. N. Engl. J. Med. 2018, 378, 1189–1199. [Google Scholar] [CrossRef]
- Thomas, E.; Storb, R.; Clift, R.A.; Fefer, A.; Johnson, F.L.; Neiman, P.E.; Lerner, K.G.; Glucksberg, H.; Buckner, C.D. Bone-marrow transplantation (first of two parts). N. Engl. J. Med. 1975, 292, 832–843. [Google Scholar] [CrossRef]
- Araki, D.; Wood, B.L.; Othus, M.; Radich, J.P.; Halpern, A.B.; Zhou, Y.; Mielcarek, M.; Estey, E.H.; Appelbaum, F.R.; Walter, R.B. Allogeneic Hematopoietic Cell Transplantation for Acute Myeloid Leukemia: Time to Move Toward a Minimal Residual Disease–Based Definition of Complete Remission? J. Clin. Oncol. 2021, 34. [Google Scholar] [CrossRef]
- Schmid, C.; Labopin, M.; Nagler, A.; Niederwieser, D.; Castagna, L.; Tabrizi, R.; Stadler, M.; Cornelissen, J.; Vorlicek, J.; Socie, G.; et al. Treatment, risk factors, and outcome of adults with relapsed AML after reduced intensity conditioning for allogeneic stem cell transplantation. Blood J. Am. Soc. Hematol. 2012, 119, 1599–1606. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Othus, M.; Araki, D.; Wood, B.L.; Radich, J.P.; Halpern, A.B.; Mielcarek, M.; Estey, E.H.; Appelbaum, F.R.; Walter, R.B. Pre- and post-transplant quanti fi cation of measurable (‘minimal’) residual disease via multiparameter fl ow cytometry in adult acute myeloid leukemia. Leukemia 2016, 1456–1464. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Yokoyama, K.; Yusa, N.; Ogawa, M.; Takei, T.; Kobayashi, A.; Ito, M.; Shimizu, E.; Kasajima, R.; Wada, Y.; et al. Circulating tumor DNA dynamically predicts response and/or relapse in patients with hematological malignancies. Int. J. Hematol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Yokoyama, K.; Shimizu, E.; Yusa, N.; Kondoh, K.; Ogawa, M.; Takei, T.; Kobayashi, A.; Ito, M.; Isobe, M.; et al. Prognostic impact of circulating tumor DNA status post–allogeneic hematopoietic stem cell transplantation in AML and MDS. Blood 2019, 133, 2682–2695. [Google Scholar] [CrossRef] [PubMed]
- Short, N.J.; Patel, K.P.; Albitar, M.; Franquiz, M.; Luthra, R.; Kanagal-Shamanna, R.; Wang, F.; Assi, R.; Montalban-Bravo, G.; Matthews, J.; et al. Targeted next-generation sequencing of circulating cell-free DNA vs bone marrow in patients with acute myeloid leukemia. Blood Adv. 2020, 4, 1670–1677. [Google Scholar] [CrossRef]
- Zhong, L.; Chen, J.; Huang, X.; Li, Y.; Jiang, T. Monitoring immunoglobulin heavy chain and t-cell receptor gene rearrangement in cfDNA as minimal residual disease detection for patients with acute myeloid leukemia. Oncol. Lett. 2018, 16, 2279–2288. [Google Scholar] [CrossRef]
- Christenson, E.S.; Dalton, W.B.; Chu, D.; Waters, I.; Cravero, K.; Zabransky, D.J.; DeZern, A.E.; Park, B.H. Single-nucleotide polymorphism leading to false allelic fraction by droplet digital PCR. Clin. Chem. 2017, 63, 1370–1376. [Google Scholar] [CrossRef]
- Buscarlet, M.; Provost, S.; Zada, Y.F.; Barhdadi, A.; Bourgoin, V.; Lépine, G.; Mollica, L.; Szuber, N.; Dubé, M.-P.; Busque, L. DNMT3A and TET2 dominate clonal hematopoiesis and demonstrate benign phenotypes and different genetic predispositions. Blood 2017, 130, 753–762. [Google Scholar] [CrossRef]
- Nagase, R.; Inoue, D.; Pastore, A.; Fujino, T.; Hou, H.A.; Yamasaki, N.; Goyama, S.; Saika, M.; Kanai, A.; Sera, Y.; et al. Expression of mutant Asxl1 perturbs hematopoiesis and promotes susceptibility to leukemic transformation. J. Exp. Med. 2018, 215, 1729–1747. [Google Scholar] [CrossRef]
- Chan, H.T.; Nagayama, S.; Chin, Y.M.; Otaki, M.; Hayashi, R.; Kiyotani, K.; Fukunaga, Y.; Ueno, M.; Nakamura, Y.; Low, S.K. Clinical significance of clonal hematopoiesis in the interpretation of blood liquid biopsy. Mol. Oncol. 2020, 14, 1719–1730. [Google Scholar] [CrossRef]
- Chan, H.T.; Chin, Y.M.; Nakamura, Y.; Low, S.K. Clonal hematopoiesis in liquid biopsy: From biological noise to valuable clinical implications. Cancers 2020, 12, 2277. [Google Scholar] [CrossRef]
- Zeidan, A.M.; Ridinger, M.; Lin, T.L.; Becker, P.S.; Schiller, G.J.; Patel, P.A.; Spira, A.I.; Tsai, M.L.; Samuëlsz, E.; Silberman, S.L.; et al. A Phase Ib Study of Onvansertib, a Novel Oral PLK1 Inhibitor, in Combination Therapy for Patients with Relapsed or Refractory Acute Myeloid Leukemia. Clin. Cancer Res. 2020, 26, 6132–6140. [Google Scholar] [CrossRef] [PubMed]
- Yeh, P.; Dickinson, M.; Ftouni, S.; Hunter, T.; Sinha, D.; Wong, S.Q.; Agarwal, R.; Vedururu, R.; Doig, K.; Fong, C.Y.; et al. Molecular disease monitoring using circulating tumor DNA in myelodysplastic syndromes. Blood 2017, 129, 1685–1690. [Google Scholar] [CrossRef]
- Frickhofen, N.; Müller, E.; Sandherr, M.; Binder, T.; Bangerter, M.; Wiest, C.; Enz, M.; Heimpel, H. Rearranged Ig heavy chain DNA is detectable in cell-free blood samples of patients with B-cell neoplasia. Blood 1997, 90, 4953–4960. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Huang, W.F. Better detection of Ig heavy chain and TCRγ gene rearrangement in plasma cell-free DNA from patients with non-Hodgkin Lymphoma. Neoplasma 2010, 57, 507–511. [Google Scholar] [CrossRef] [PubMed][Green Version]
- He, J.; Wu, J.; Jiao, Y.; Wagner-Johnston, N.; Ambinder, R.F.; Diaz, L.A.; Kinzler, K.W.; Vogelstein, B.; Papadopoulos, N. IgH gene rearrangements as plasma biomarkers in non-Hodgkin’s Lymphoma patients. Oncotarget 2011, 2, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Armand, P.; Oki, Y.; Neuberg, D.S.; Faham, M.; Cummings, C.; Klinger, M.; Weng, L.; Bhattar, S.; Lacasce, A.S.; Jacobsen, E.D.; et al. Detection of circulating tumour DNA in patients with aggressive B-cell non-Hodgkin lymphoma. Br. J. Haematol. 2013, 163, 123–126. [Google Scholar] [CrossRef]
- Kurtz, D.M.; Green, M.R.; Bratman, S.V.; Scherer, F.; Liu, C.L.; Kunder, C.A.; Takahashi, K.; Glover, C.; Keane, C.; Kihira, S.; et al. Noninvasive monitoring of diffuse large B-cell lymphoma by immunoglobulin high-throughput sequencing. Blood 2015, 125, 3679–3687. [Google Scholar] [CrossRef]
- Roschewski, M.; Dunleavy, K.; Pittaluga, S.; Moorhead, M.; Pepin, F.; Kong, K.; Shovlin, M.; Jaffe, E.S.; Staudt, L.M.; Lai, C.; et al. Circulating tumour DNA and CT monitoring in patients with untreated diffuse large B-cell lymphoma: A correlative biomarker study. Lancet Oncol. 2015, 16, 541–549. [Google Scholar] [CrossRef]
- Sarkozy, C.; Huet, S.; Carlton, V.E.H.; Fabiani, B.; Delmer, A.; Jardin, F.; Delfau-Larue, M.-H.; Hacini, M.; Ribrag, V.; Guidez, S.; et al. The prognostic value of clonal heterogeneity and quantitative assessment of plasma circulating clonal IG-VDJ sequences at diagnosis in patients with follicular lymphoma. Oncotarget 2017, 8, 8765–8774. [Google Scholar] [CrossRef] [PubMed]
- Wagner-Johnston, N.D.; Lensing, S.; Noy, A.; Ratner, L.; Henry, D.; Lee, J.Y.; Silver, S.; Faham, M.; Ambinder, R.F. High frequency of identical clonal immunoglobulin DNA in pre-treatment tumor and plasma from untreated patients with HIV-associated lymphoma: Prospective multicenter trial of the AIDS malignancies consortium (AMC 064). Leuk. Lymphoma 2017, 58, 2939–2942. [Google Scholar] [CrossRef]
- Kumar, A.; Bantilan, K.S.; Jacob, A.P.; Park, A.; Schoninger, S.F.; Sauter, C.; Ulaner, G.A.; Casulo, C.; Faham, M.; Kong, K.A.; et al. Noninvasive Monitoring of Mantle Cell Lymphoma by Immunoglobulin Gene Next-Generation Sequencing in a Phase 2 Study of Sequential Chemoradioimmunotherapy Followed by Autologous Stem-Cell Rescue. Clin. Lymphoma Myeloma Leuk. 2021. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, W.; Han, X.; Gan, Y.; Qian, L.; Zhang, Y.; Zhang, C.; Wang, Y.; Guan, Y.; Yang, L.; et al. Circulating tumor DNA by high-throughput sequencing of T cell receptor monitored treatment response and predicted treatment failure in T cell lymphomas. Int. J. Lab. Hematol. 2021, 1–9. [Google Scholar] [CrossRef]
- Hosny, G.; Farahat, N.; Hainaut, P. TP53 mutations in circulating free DNA from Egyptian patients with non-Hodgkin’s lymphoma. Cancer Lett. 2009, 275, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Camus, V.; Sarafan-Vasseur, N.; Bohers, E.; Dubois, S.; Mareschal, S.; Bertrand, P.; Viailly, P.-J.; Ruminy, P.; Maingonnat, C.; Lemasle, E.; et al. Digital PCR for quantification of recurrent and potentially actionable somatic mutations in circulating free DNA from patients with diffuse large B-cell lymphoma. Leuk. Lymphoma 2016, 57, 2171–2179. [Google Scholar] [CrossRef] [PubMed]
- Camus, V.; Stamatoullas, A.; Mareschal, S.; Viailly, P.-J.; Sarafan-Vasseur, N.; Bohers, E.; Dubois, S.; Picquenot, J.M.; Ruminy, P.; Maingonnat, C.; et al. Detection and prognostic value of recurrent exportin 1 mutations in tumor and cell-free circulating DNA of patients with classical Hodgkin lymphoma. Haematologica 2016, 101, 1094–1101. [Google Scholar] [CrossRef]
- Watanabe, J.; Natsumeda, M.; Kanemaru, Y.; Okada, M.; Oishi, M.; Kakita, A.; Fujii, Y. Comparison of circulating tumor DNA between body fluids in patients with primary central nervous system lymphoma. Leuk. Lymphoma 2019, 60, 3587–3589. [Google Scholar] [CrossRef]
- Hickmann, A.K.; Frick, M.; Hadaschik, D.; Battke, F.; Bittl, M.; Ganslandt, O.; Biskup, S.; Döcker, D. Molecular tumor analysis and liquid biopsy: A feasibility investigation analyzing circulating tumor DNA in patients with central nervous system lymphomas. BMC Cancer 2019, 19, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Nagy, Á.; Bátai, B.; Balogh, A.; Illés, S.; Mikala, G.; Nagy, N.; Kiss, L.; Kotmayer, L.; Matolcsy, A.; Alpár, D.; et al. Quantitative analysis and monitoring of ezh2 mutations using liquid biopsy in follicular lymphoma. Genes 2020, 11, 785. [Google Scholar] [CrossRef] [PubMed]
- Scherer, F.; Kurtz, D.M.; Newman, A.M.; Stehr, H.; Craig, A.F.M.; Esfahani, M.S.; Lovejoy, A.F.; Chabon, J.J.; Klass, D.M.; Liu, C.L.; et al. Distinct biological subtypes and patterns of genome evolution in lymphoma revealed by circulating tumor DNA. Sci. Transl. Med. 2016, 8. [Google Scholar] [CrossRef]
- Kurtz, D.M.; Scherer, F.; Jin, M.C.; Soo, J.; Craig, A.F.M.; Esfahani, M.S.; Chabon, J.J.; Stehr, H.; Liu, C.L.; Tibshirani, R.; et al. Circulating tumor DNA measurements as early outcome predictors in diffuse large B-cell lymphoma. J. Clin. Oncol. 2018, 36, 2845–2853. [Google Scholar] [CrossRef]
- Rossi, D.; Diop, F.; Spaccarotella, E.; Monti, S.; Zanni, M.; Rasi, S.; Deambrogi, C.; Spina, V.; Bruscaggin, A.; Favini, C.; et al. Diffuse large B-cell lymphoma genotyping on the liquid biopsy. Blood 2017, 129, 1947–1957. [Google Scholar] [CrossRef] [PubMed]
- Spina, V.; Bruscaggin, A.; Cuccaro, A.; Martini, M.; Di Trani, M.; Forestieri, G.; Manzoni, M.; Condoluci, A.; Arribas, A.; Terzi-Di-Bergamo, L.; et al. Circulating tumor DNA reveals genetics, clonal evolution and residual disease in classical Hodgkin lymphoma. Blood 2018. [Google Scholar] [CrossRef]
- Bohers, E.; Viailly, P.J.; Dubois, S.; Bertrand, P.; Maingonnat, C.; Mareschal, S.; Ruminy, P.; Picquenot, J.M.; Bastard, C.; Desmots, F.; et al. Somatic mutations of cell-free circulating DNA detected by next-generation sequencing reflect the genetic changes in both germinal center B-cell-like and activated B-cell-like diffuse large B-cell lymphomas at the time of diagnosis. Haematologica 2015, 100, e280–e284. [Google Scholar] [CrossRef]
- Rivas-Delgado, A.; Nadeu, F.; Enjuanes, A.; Casanueva-Eliceiry, S.; Mozas, P.; Magnano, L.; Castrejón de Anta, N.; Rovira, J.; Dlouhy, I.; Martín, S.; et al. Mutational Landscape and Tumor Burden Assessed by Cell-free DNA in Diffuse Large B-Cell Lymphoma in a Population-Based Study. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2021, 27, 513–521. [Google Scholar] [CrossRef]
- Sun, P.; Chen, C.; Xia, Y.; Wang, Y.; Liu, P.P.; Bi, X.W.; Shao, Y.W.; Ou, Q.X.; Wu, X.; Yang, H.; et al. Mutation profiling of malignant lymphoma by next-generation sequencing of circulating cell-free DNA. J. Cancer 2019, 10, 323–331. [Google Scholar] [CrossRef]
- Desch, A.K.; Hartung, K.; Botzen, A.; Brobeil, A.; Rummel, M.; Kurch, L.; Georgi, T.; Jox, T.; Bielack, S.; Burdach, S.; et al. Genotyping circulating tumor DNA of pediatric Hodgkin lymphoma. Leukemia 2020, 34, 151–166. [Google Scholar] [CrossRef]
- Suehara, Y.; Sakata-Yanagimoto, M.; Hattori, K.; Kusakabe, M.; Nanmoku, T.; Sato, T.; Noguchi, M.; Chiba, S. Mutations found in cell-free DNAs of patients with malignant lymphoma at remission can derive from clonal hematopoiesis. Cancer Sci. 2019, 110, 3375–3381. [Google Scholar] [CrossRef]
- Blombery, P.A.; Ryland, G.L.; Markham, J.; Guinto, J.; Wall, M.; McBean, M.; Jones, K.; Thompson, E.R.; Cameron, D.L.; Papenfuss, A.T.; et al. Detection of clinically relevant early genomic lesions in B-cell malignancies from circulating tumour DNA using a single hybridisation-based next generation sequencing assay. Br. J. Haematol. 2018, 183, 146–149. [Google Scholar] [CrossRef]
- Bessi, L.; Viailly, P.J.; Bohers, E.; Ruminy, P.; Maingonnat, C.; Bertrand, P.; Vasseur, N.S.; Beaussire, L.; Cornic, M.; Etancelin, P.; et al. Somatic mutations of cell-free circulating DNA detected by targeted next-generation sequencing and digital droplet PCR in classical Hodgkin lymphoma. Leuk. Lymphoma 2019, 60, 498–502. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Su, H.; Song, Y.; Jiang, W.; Sun, X.; Qian, W.; Zhang, W.; Gao, Y.; Jin, Z.; Zhou, J.; et al. Circulating tumor DNA predicts response in Chinese patients with relapsed or refractory classical hodgkin lymphoma treated with sintilimab. EBioMedicine 2020, 54. [Google Scholar] [CrossRef] [PubMed]
- Rushton, C.K.; Arthur, S.E.; Alcaide, M.; Cheung, M.; Jiang, A.; Coyle, K.M.; Cleary, K.L.S.; Thomas, N.; Hilton, L.K.; Michaud, N.; et al. Genetic and evolutionary patterns of treatment resistance in relapsed B-cell lymphoma. Blood Adv. 2020, 4, 2886–2898. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Yang, C.; Zhao, X.; Le, J.; Wu, G.; Wei, J.; Liang, Y.; Qian, W. Genotyping on ctdna identifies shifts in mutation spectrum between newly diagnosed and relapse/refractory dlbcl. Onco. Targets. Ther. 2020, 13, 10797–10806. [Google Scholar] [CrossRef]
- Li, Q.; Zhang, W.; Li, J.; Xiong, J.; Liu, J.; Chen, T.; Wen, Q.; Zeng, Y.; Gao, L.; Gao, L.; et al. Plasma circulating tumor DNA assessment reveals KMT2D as a potential poor prognostic factor in extranodal NK/T-cell lymphoma. Biomark. Res. 2020, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.-H.; Kim, Y.J.; Lee, D.; Cho, D.; Ko, Y.H.; Cho, J.; Park, W.-Y.; Park, D.; Kim, S.J.; Kim, W.S. Analysis of circulating tumor DNA by targeted ultra-deep sequencing across various non-Hodgkin lymphoma subtypes. Leuk. Lymphoma 2019, 60, 2237–2246. [Google Scholar] [CrossRef]
- Camus, V.; Viennot, M.; Lequesne, J.; Viailly, P.J.; Bohers, E.; Bessi, L.; Marcq, B.; Etancelin, P.; Dubois, S.; Picquenot, J.M.; et al. Targeted genotyping of circulating tumor DNA for classical Hodgkin lymphoma monitoring: A prospective study. Haematologica 2021, 106, 154–162. [Google Scholar] [CrossRef]
- Chen, F.; Pang, D.; Guo, H.; Jiang, X.; Liu, S.; Huang, L.; Wei, X.; Liang, Z.; Wang, X.; Li, W. Clinicopathological characteristics and mutational profiling of adult t-cell lymphoblastic lymphoma in a chinese population. Cancer Manag. Res. 2020, 12, 3003–3012. [Google Scholar] [CrossRef]
- Grommes, C.; Tang, S.S.; Wolfe, J.; Kaley, T.J.; Daras, M.; Pentsova, E.I.; Piotrowski, A.F.; Stone, J.; Lin, A.; Nolan, C.P.; et al. Phase 1b trial of an ibrutinib-based combination therapy in recurrent/refractory CNS lymphoma. Blood 2019, 133, 436–445. [Google Scholar] [CrossRef]
- Bobillo, S.; Crespo, M.; Escudero, L.; Mayor, R.; Raheja, P.; Carpio, C.; Rubio-perez, C.; Tazón-vega, B.; Palacio, C.; Carabia, J.; et al. System involvement of B-cell lymphomas. Haematologica 2021, 106, 513–521. [Google Scholar]
- Chen, F.; Pang, D.; Guo, H.; Ou, Q.; Wu, X.; Jiang, X.; Wei, X.; Liu, S.; Huang, L.; Liang, Z.; et al. Clinical outcomes of newly diagnosed primary CNS lymphoma treated with ibrutinib-based combination therapy: A real-world experience of off-label ibrutinib use. Cancer Med. 2020, 9, 8676–8684. [Google Scholar] [CrossRef]
- Wang, X.; Gao, Y.; Shan, C.; Lai, M.; He, H.; Bai, B.; Ping, L.; Rong, Q.; Ai, R.; Wen, L.; et al. Association of circulating tumor DNA from the cerebrospinal fluid with high-risk CNS involvement in patients with diffuse large B-cell lymphoma. Clin. Transl. Med. 2021, 11, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Fontanilles, M.; Marguet, F.; Bohers, É.; Viailly, P.J.; Dubois, S.; Bertrand, P.; Camus, V.; Mareschal, S.; Ruminy, P.; Maingonnat, C.; et al. Non-invasive detection of somatic mutations using nextgeneration sequencing in primary central nervous system lymphoma. Oncotarget 2017, 8, 48157–48168. [Google Scholar] [CrossRef] [PubMed]
- Hattori, K.; Sakata-Yanagimoto, M.; Suehara, Y.; Yokoyama, Y.; Kato, T.; Kurita, N.; Nishikii, H.; Obara, N.; Takano, S.; Ishikawa, E.; et al. Clinical significance of disease-specific MYD88 mutations in circulating DNA in primary central nervous system lymphoma. Cancer Sci. 2018, 109, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, R.; Chan, Y.C.; Tam, C.S.; Hunter, T.; Vassiliadis, D.; Teh, C.E.; Thijssen, R.; Yeh, P.; Wong, S.Q.; Ftouni, S.; et al. Dynamic molecular monitoring reveals that SWI–SNF mutations mediate resistance to ibrutinib plus venetoclax in mantle cell lymphoma. Nat. Med. 2019, 25, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Laird, P.W. Cancer epigenetics comes of age. Nat. Genet. 1999, 21, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M.; Corn, P.G.; Baylin, S.B.; Herman, J.G. A gene hypermethylation profile of human cancer. Cancer Res. 2001, 61, 3225–3229. [Google Scholar]
- Kawano, S.; Miller, C.W.; Gombart, A.F.; Bartram, C.R.; Matsuo, Y.; Asou, H.; Sakashita, A.; Said, J.; Tatsumi, E.; Koeffler, H.P. Loss of p73 gene expression in leukemias/lymphomas due to hypermethylation. Blood 1999, 94, 1113–1120. [Google Scholar] [CrossRef]
- Deligezer, U.; Yaman, F.; Erten, N.; Dalay, N. Frequent copresence of methylated DNA and fragmented nucleosomal DNA in plasma of lymphoma patients. Clin. Chim. Acta 2003, 335, 89–94. [Google Scholar] [CrossRef]
- Shi, H.; Guo, J.; Duff, D.J.; Rahmatpanah, F.; Chitima-Matsiga, R.; Al-Kuhlani, M.; Taylor, K.H.; Sjahputera, O.; Andreski, M.; Wooldridge, J.E.; et al. Discovery of novel epigenetic markers in non-Hodgkin’s lymphoma. Carcinogenesis 2007, 28, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Hohaus, S.; Giachelia, M.; Massini, G.; Mansueto, G.; Vannata, B.; Bozzoli, V.; Criscuolo, M.; D’Alò, F.; Martini, M.; Larocca, L.M.; et al. Cell-free circulating DNA in Hodgkin’s and non-Hodgkin’s lymphomas. Ann. Oncol. 2009, 20, 1408–1413. [Google Scholar] [CrossRef] [PubMed]
- Mussolin, L.; Burnelli, R.; Pillon, M.; Carraro, E.; Farruggia, P.; Todesco, A.; Mascarin, M.; Rosolen, A. Plasma cell-free DNA in paediatric lymphomas. J. Cancer 2013, 4, 323–329. [Google Scholar] [CrossRef]
- Li, M.; Jia, Y.; Xu, J.; Cheng, X.; Xu, C. Assessment of the circulating cell-free DNA marker association with diagnosis and prognostic prediction in patients with lymphoma: A single-center experience. Ann. Hematol. 2017, 96, 1343–1351. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Tang, W.; Huang, L.; Hou, N.; Wu, J.; Cheng, X.; Ma, D.; Qian, P.; Shen, Q.; Guo, W.; et al. The analysis of cell-free DNA concentrations and integrity in serum of initial and treated of lymphoma patients. Clin. Biochem. 2019, 63, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Delfau-Larue, M.H.; Van Der Gucht, A.; Dupuis, J.; Jais, J.P.; Nel, I.; Beldi-Ferchiou, A.; Hamdane, S.; Benmaad, I.; Laboure, G.; Verret, B.; et al. Total metabolic tumor volume, circulating tumor cells, cell-free DNA: Distinct prognostic value in follicular lymphoma. Blood Adv. 2018, 2, 807–816. [Google Scholar] [CrossRef]
- Kurtz, D.M.; Esfahani, M.S.; Scherer, F.; Soo, J.; Jin, M.C.; Liu, C.L.; Newman, A.M.; Dührsen, U.; Hüttmann, A.; Casasnovas, O.; et al. Dynamic Risk Profiling Using Serial Tumor Biomarkers for Personalized Outcome Prediction. Cell 2019, 178, 699–713.e19. [Google Scholar] [CrossRef]
- Rajkumar, S.V.; Dimopoulos, M.A.; Palumbo, A.; Blade, J.; Merlini, G.; Mateos, M.V.; Kumar, S.; Hillengass, J.; Kastritis, E.; Richardson, P.; et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014, 15, e538–e548. [Google Scholar] [CrossRef]
- Bahlis, N.J. Darwinian evolution and tiding clones in multiple myeloma. Blood 2012, 120, 927–928. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Lopez, J.; Lahuerta, J.J.; Pepin, F.; González, M.; Barrio, S.; Ayala, R.; Puig, N.; Montalban, M.A.; Paiva, B.; Weng, L.; et al. Prognostic value of deep sequencing method for minimal residual disease detection in multiple myeloma. Blood 2014, 123, 3073–3079. [Google Scholar] [CrossRef]
- Sata, H.; Shibayama, H.; Maeda, I.; Habuchi, Y.; Nakatani, E.; Fukushima, K.; Fujita, J.; Ezoe, S.; Tadokoro, S.; Maeda, T.; et al. Quantitative polymerase chain reaction analysis with allele-specific oligonucleotide primers for individual IgH VDJ regions to evaluate tumor burden in myeloma patients. Exp. Hematol. 2015, 43, 374–381.e2. [Google Scholar] [CrossRef]
- Oberle, A.; Brandt, A.; Voigtlaender, M.; Thiele, B.; Radloff, J.; Schulenkorf, A.; Alawi, M.; Akyüz, N.; März, M.; Ford, C.T.; et al. Monitoring multiple myeloma by next-generation sequencing of V(D)J rearrangements from circulating myeloma cells and cell-free myeloma DNA. Haematologica 2017, 102, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Mazzotti, Ć.; Buisson, L.; Maheo, S.; Perrot, A.; Chretien, M.L.; Leleu, X.; Hulin, C.; Manier, S.; Hebraud, B.; Roussel, M.; et al. Myeloma MRD by deep sequencing from circulating tumor DNA does not correlate with results obtained in the bone marrow. Blood Adv. 2018, 2, 2811–2813. [Google Scholar] [CrossRef] [PubMed]
- Biancon, G.; Gimondi, S.; Vendramin, A.; Carniti, C.; Corradini, P. Noninvasive Molecular Monitoring in Multiple Myeloma Patients Using Cell-Free Tumor DNA: A Pilot Study. J. Mol. Diagn. 2018, 20, 859–870. [Google Scholar] [CrossRef]
- Kis, O.; Kaedbey, R.; Chow, S.; Danesh, A.; Dowar, M.; Li, T.; Li, Z.; Liu, J.; Mansour, M.; Masih-Khan, E.; et al. Circulating tumour DNA sequence analysis as an alternative to multiple myeloma bone marrow aspirates. Nat. Commun. 2017, 8, 1–11. [Google Scholar] [CrossRef]
- Rustad, E.H.; Coward, E.; Skytøen, E.R.; Misund, K.; Holien, T.; Standal, T.; Børset, M.; Beisvag, V.; Myklebost, O.; Meza-Zepeda, L.A.; et al. Monitoring multiple myeloma by quantification of recurrent mutations in serum. Haematologica 2017, 102, 1266–1272. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Huang, H.J.; Ma, J.; Wang, Y.; Cao, Z.; Karlin-Neumann, G.; Janku, F.; Liu, Z. RAS/RAF mutations in tumor samples and cell-free DNA from plasma and bone marrow aspirates in multiple myeloma patients. J. Cancer 2020, 11, 3543–3550. [Google Scholar] [CrossRef]
- Mithraprabhu, S.; Khong, T.; Ramachandran, M.; Chow, A.W.S.; Klarica, D.; Mai, L.; Walsh, S.; Broemeling, D.; Marziali, A.; Wiggin, M.; et al. Mutational Characterisation and Tracking Disease Progression Using Circulating Cell-Free Tumor DNA in Multiple Myeloma Patients. Blood 2016, 128, 3280. [Google Scholar] [CrossRef]
- Mithraprabhu, S.; Khong, T.; Ramachandran, M.; Chow, A.; Klarica, D.; Mai, L.; Walsh, S.; Broemeling, D.; Marziali, A.; Wiggin, M.; et al. Circulating tumour DNA analysis demonstrates spatial mutational heterogeneity that coincides with disease relapse in myeloma. Leukemia 2017, 31, 1695–1705. [Google Scholar] [CrossRef]
- Mithraprabhu, S.; Morley, R.; Khong, T.; Kalff, A.; Bergin, K.; Hocking, J.; Savvidou, I.; Bowen, K.M.; Ramachandran, M.; Choi, K.; et al. Monitoring tumour burden and therapeutic response through analysis of circulating tumour DNA and extracellular RNA in multiple myeloma patients. Leukemia 2019, 33, 2022–2033. [Google Scholar] [CrossRef]
- Manzoni, M.; Pompa, A.; Fabris, S.; Pelizzoni, F.; Ciceri, G.; Seia, M.; Ziccheddu, B.; Bolli, N.; Corradini, P.; Baldini, L.; et al. Limits and Applications of Genomic Analysis of Circulating Tumor DNA as a Liquid Biopsy in Asymptomatic Forms of Multiple Myeloma. HemaSphere 2020, 4, e402. [Google Scholar] [CrossRef] [PubMed]
- Long, X.; Xu, Q.; Lou, Y.; Li, C.; Gu, J.; Cai, H.; Wang, D.; Xu, J.; Li, T.; Zhou, X.; et al. The utility of non-invasive liquid biopsy for mutational analysis and minimal residual disease assessment in extramedullary multiple myeloma. Br. J. Haematol. 2020, 189, e45–e48. [Google Scholar] [CrossRef]
- Guo, G.; Raje, N.S.; Seifer, C.; Kloeber, J.; Isenhart, R.; Ha, G.; Yee, A.J.; O’Donnell, E.K.; Tai, Y.T.; Richardson, P.G.; et al. Genomic discovery and clonal tracking in multiple myeloma by cell-free DNA sequencing. Leukemia 2018, 32, 1838–1841. [Google Scholar] [CrossRef] [PubMed]
- Manier, S.; Park, J.; Capelletti, M.; Bustoros, M.; Freeman, S.S.; Ha, G.; Rhoades, J.; Liu, C.J.; Huynh, D.; Reed, S.C.; et al. Whole-exome sequencing of cell-free DNA and circulating tumor cells in multiple myeloma. Nat. Commun. 2018, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Lenaerts, L.; Vandenberghe, P.; Brison, N.; Che, H.; Neofytou, M.; Verheecke, M.; Leemans, L.; Maggen, C.; Dewaele, B.; Dehaspe, L.; et al. Genomewide copy number alteration screening of circulating plasma DNA: Potential for the detection of incipient tumors. Ann. Oncol. 2019, 30, 85–95. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).