
From Proteomic Mapping to Invasion-Metastasis-Cascade Systemic Biomarkering and Targeted Drugging of Mutant BRAF-Dependent Human Cutaneous Melanomagenesis


 ,
,  , , , ,
, , , ,  , , ,
, , ,  ,
,  and
and  add
Show full author list
add
Show full author list
Abstract
Simple Summary
Abstract
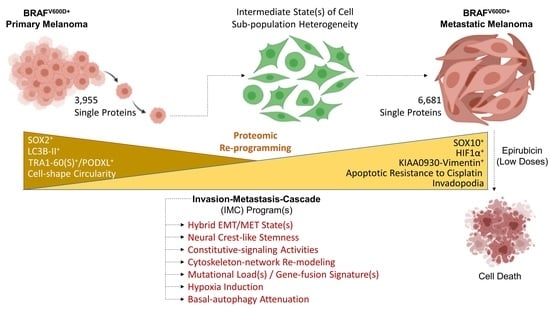
1. Introduction
2. Materials and Methods
2.1. Antibodies, Drugs, and Chemicals
2.2. Cell Lines—Culture Conditions
2.3. Protein Extraction—Tryptic Peptide Generation
2.4. LC-MS/MS—Data Analysis
2.5. Bioinformatics Platforms
2.6. Immunofluorescence
2.7. F-Actin Staining
2.8. Lysosomal Staining
2.9. Scratch Wound—Wound Healing Assay
2.10. Cell Viability—MTT Assay
2.11. Western Blotting
2.12. Molecular Modeling
3. Results and Discussion
3.1. Functional Mapping of WM115 Proteomic Landscape Unveils Diverse Pathways Being Engaged in Signal Transduction, Drug Metabolism/Resistance, Cell Death, and Cytoskeleton Re-modeling
3.2. WM115 and WM266-4 Human Melanoma Cells Are Subjected to Hybrid Epithelial-to-Mesenchymal Transition (EMT)/Mesenchymal-to-Epithelial Transition (MET) Programs
3.3. TGF-β Signaling Controls WM115 and WM266-4 Cell Motility In Vitro
3.4. WM115 and WM266-4 Melanoma Cells Feature Different Neural Crest-Like Stemness Signatures
3.5. Cytoskeleton Architecture Re-Modeling of Melanoma Cells during the IMC Process
3.6. Molecular Modeling of a KIAA0930-VIM Gene-Fusion Product Exclusively Identified in WM266-4 Cells
3.7. Signal Transduction SignatuRes. in BRAFV600D-Dependent Melanoma Undergoing Metastasis
3.8. Apoptotic and Autophagic Sub-Routines in BRAFV600D Primary and Metastatic Melanoma Cells
3.9. Targeted Drugging Efficacy against WM115 and WM266-4 Melanoma Cells Depends on Their IMC States
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lo, J.A.; Fisher, D.E. The melanoma revolution: From UV carcinogenesis to a new era in therapeutics. Science 2014, 346, 945–949. [Google Scholar] [CrossRef]
- Yang, K.; Oak, A.S.W.; Slominski, R.M.; Brozyna, A.A.; Slominski, A.T. Current Molecular Markers of Melanoma and Treatment Targets. Int. J. Mol. Sci. 2020, 21, 3535. [Google Scholar] [CrossRef]
- Surveillance, Epidemiology, and End Results (SEER) Program (National Cancer Institute). Cancer Stat Facts: Melanoma of the Skin 2020, Reports on Cancer. Available online: https://seer.cancer.gov/statfacts/html/melan.html (accessed on 15 November 2020).
- Chin, L. The genetics of malignant melanoma: Lessons from mouse and man. Nat. Rev. Cancer 2003, 3, 559–570. [Google Scholar] [CrossRef]
- Chin, L.; Garraway, L.A.; Fisher, D.E. Malignant melanoma: Genetics and therapeutics in the genomic era. Genes Dev. 2006, 20, 2149–2182. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, P.; Chin, L. Genetics and genomics of melanoma. Expert Rev. Dermatol. 2009, 4, 131. [Google Scholar] [CrossRef]
- Shain, A.H.; Bastian, B.C. From melanocytes to melanomas. Nat. Rev. Cancer 2016, 16, 345–358. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Hodi, F.S.; Fisher, D.E. From genes to drugs: Targeted strategies for melanoma. Nat. Rev. Cancer 2012, 12, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Shtivelman, E.; Davies, M.Q.; Hwu, P.; Yang, J.; Lotem, M.; Oren, M.; Flaherty, K.T.; Fisher, D.E. Pathways and therapeutic targets in melanoma. Oncotarget 2014, 5, 1701–1752. [Google Scholar] [CrossRef] [PubMed]
- Dhomen, N.; Marais, R. New insight into BRAF mutations in cancer. Curr. Opin. Genet. Dev. 2007, 17, 31–39. [Google Scholar] [CrossRef]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Ackermann, J.; Frutschi, M.; Kaloulis, K.; McKee, T.; Trumpp, A.; Beermann, F. Metastasizing melanoma formation caused by expression of activated N-RasQ61K on an INK4a-deficient background. Cancer Res. 2005, 65, 4005–4011. [Google Scholar] [CrossRef]
- Shain, A.H.; Yeh, I.; Kovalyshyn, I.; Sriharan, A.; Talevich, E.; Gagnon, A.; Dummer, R.; North, J.; Pincus, L.; Ruben, B.; et al. The Genetic Evolution of Melanoma from Precursor Lesions. N. Engl. J. Med. 2015, 373, 1926–1936. [Google Scholar] [CrossRef] [PubMed]
- Werner-Klein, M.; Scheitler, S.; Hoffmann, M.; Hodak, I.; Dietz, K.; Lehnert, P.; Naimer, V.; Polzer, B.; Treitschke, S.; Werno, C.; et al. Genetic alterations driving metastatic colony formation are acquired outside of the primary tumour in melanoma. Nat. Commun. 2018, 9, 595. [Google Scholar] [CrossRef] [PubMed]
- Dankort, D.; Curley, D.P.; Cartlidge, R.A.; Nelson, B.; Karnezis, A.N.; Damsky, W.E., Jr.; You, M.J.; DePinho, R.A.; McMahon, M.; Bosenberg, M. Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nat. Genet. 2009, 41, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Viros, A.; Sanchez-Laorden, B.; Pedersen, M.; Furney, S.J.; Rae, J.; Hogan, K.; Ejiama, S.; Girotti, M.R.; Cook, M.; Dhomen, N.; et al. Ultraviolet radiation accelerates BRAF-driven melanomagenesis by targeting TP53. Nature 2014, 511, 478–482. [Google Scholar] [CrossRef]
- Bollag, G.; Tsai, J.; Zhang, J.; Zhang, C.; Ibrahim, P.; Nolop, K.; Hirth, P. Vemurafenib: The first drug approved for BRAF-mutant cancer. Nat. Rev. Drug Discov. 2012, 11, 873–886. [Google Scholar] [CrossRef]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef]
- Hauschild, A.; Grob, J.J.; Demidov, L.V.; Jouary, T.; Gutzmer, R.; Millward, M.; Rutkowski, P.; Blank, C.U.; Miller, W.H., Jr.; Kaempgen, E.; et al. Dabrafenib in BRAF-mutated metastatic melanoma: A multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012, 380, 358–365. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Infante, J.R.; Daud, A.; Gonzalez, R.; Kefford, R.F.; Sosman, J.; Hamid, O.; Schuchter, L.; Cebon, J.; Ibrahim, N.; et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N. Engl. J. Med. 2012, 367, 1694–1703. [Google Scholar] [CrossRef]
- Luke, J.J.; Flaherty, K.T.; Ribas, A.; Long, G.V. Targeted agents and immunotherapies: Optimizing outcomes in melanoma. Nat. Rev. Clin. Oncol. 2017, 14, 463–482. [Google Scholar] [CrossRef]
- Trunzer, K.; Pavlick, A.C.; Schuchter, L.; Gonzalez, R.; McArthur, G.A.; Hutson, T.E.; Moschos, S.J.; Flaherty, K.T.; Kim, K.B.; Weber, J.S.; et al. Pharmacodynamic effects and mechanisms of resistance to vemurafenib in patients with metastatic melanoma. J. Clin. Oncol. 2013, 31, 1767–1774. [Google Scholar] [CrossRef] [PubMed]
- Holderfield, M.; Deuker, M.M.; McCormick, F.; McMahon, M. Targeting RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nat. Rev. Cancer 2014, 14, 455–467. [Google Scholar] [CrossRef] [PubMed]
- Nazarian, R.; Shi, H.; Wang, Q.; Kong, X.; Koya, R.C.; Lee, H.; Chen, Z.; Lee, M.K.; Attar, N.; Sazegar, H.; et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010, 468, 973–977. [Google Scholar] [CrossRef]
- Sun, C.; Wang, L.; Huang, S.; Heynen, G.J.; Prahallad, A.; Robert, C.; Haanen, J.; Blank, C.; Wesseling, J.; Willems, S.M.; et al. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature 2014, 508, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Poulikakos, P.I.; Persaud, Y.; Janakiraman, M.; Kong, X.; Ng, C.; Moriceau, G.; Shi, H.; Atefi, M.; Titz, B.; Gabay, M.T.; et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature 2011, 480, 387–390. [Google Scholar] [CrossRef]
- Straussman, R.; Morikawa, T.; Shee, K.; Barzily-Rokni, M.; Qian, Z.R.; Du, J.; Davis, A.; Mongare, M.M.; Gould, J.; Frederick, D.T.; et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 2012, 487, 500–504. [Google Scholar] [CrossRef] [PubMed]
- Johannessen, C.M.; Boehm, J.S.; Kim, S.Y.; Thomas, S.R.; Wardwell, L.; Johnson, L.A.; Emery, C.M.; Stransky, N.; Cogdill, A.P.; Barretina, J.; et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature 2010, 468, 968–972. [Google Scholar] [CrossRef]
- Van Allen, E.M.; Wagle, N.; Sucker, A.; Treacy, D.J.; Johannessen, C.M.; Goetz, E.M.; Place, C.S.; Taylor-Weiner, A.; Whittaker, S.; Kryukov, G.V.; et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov. 2014, 4, 94–109. [Google Scholar] [CrossRef]
- Paraiso, K.H.; Xiang, Y.; Rebecca, V.W.; Abel, E.V.; Chen, Y.A.; Munko, A.C.; Wood, E.; Fedorenko, I.V.; Sondak, V.K.; Anderson, A.R.; et al. PTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expression. Cancer Res. 2011, 71, 2750–2760. [Google Scholar] [CrossRef]
- Valastyan, S.; Weinberg, R.A. Tumor metastasis: Molecular insights and evolving paradigms. Cell 2011, 147, 275–292. [Google Scholar] [CrossRef]
- Talmadge, J.E.; Fidler, I.J. AACR centennial series: The biology of cancer metastasis: Historical perspective. Cancer Res. 2010, 70, 5649–5669. [Google Scholar] [CrossRef] [PubMed]
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging Biological Principles of Metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef] [PubMed]
- Braeuer, R.R.; Watson, I.R.; Wu, C.J.; Mobley, A.K.; Kamiya, T.; Shoshan, E.; Bar-Eli, M. Why is melanoma so metastatic? Pigment. Cell Melanoma Res. 2014, 27, 19–36. [Google Scholar] [CrossRef]
- Konstantakou, E.G.; Velentzas, A.D.; Anagnostopoulos, A.K.; Litou, Z.I.; Konstandi, O.A.; Giannopoulou, A.F.; Anastasiadou, E.; Voutsinas, G.E.; Tsangaris, G.T.; Stravopodis, D.J. Deep-proteome mapping of WM-266-4 human metastatic melanoma cells: From oncogenic addiction to druggable targets. PLoS ONE 2017, 12, e0171512. [Google Scholar] [CrossRef]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Pastushenko, I.; Blanpain, C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 2019, 29, 212–226. [Google Scholar] [CrossRef]
- Gupta, P.B.; Pastushenko, I.; Skibinski, A.; Blanpain, C.; Kuperwasser, C. Phenotypic Plasticity: Driver of Cancer Initiation, Progression, and Therapy Resistance. Cell Stem Cell 2019, 24, 65–78. [Google Scholar] [CrossRef]
- Derynck, R.; Weinberg, R.A. EMT and Cancer: More Than Meets the Eye. Dev. Cell 2019, 49, 313–316. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Wellner, U.; Schubert, J.; Burk, U.C.; Schmalhofer, O.; Zhu, F.; Sonntag, A.; Waldvogel, B.; Vannier, C.; Darling, D.; zur Hausen, A.; et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat. Cell Biol. 2009, 11, 1487–1495. [Google Scholar] [CrossRef]
- Krebs, A.M.; Mitschke, J.; Lasierra Losada, M.; Schmalhofer, O.; Boerries, M.; Busch, H.; Boettcher, M.; Mougiakakos, D.; Reichardt, W.; Bronsert, P.; et al. The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat. Cell Biol. 2017, 19, 518–529. [Google Scholar] [CrossRef]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Herlyn, M.; Balaban, G.; Bennicelli, J.; Guerry, D.t.; Halaban, R.; Herlyn, D.; Elder, D.E.; Maul, G.G.; Steplewski, Z.; Nowell, P.C.; et al. Primary melanoma cells of the vertical growth phase: Similarities to metastatic cells. J. Natl. Cancer Inst. 1985, 74, 283–289. [Google Scholar]
- Herlyn, M.; Thurin, J.; Balaban, G.; Bennicelli, J.L.; Herlyn, D.; Elder, D.E.; Bondi, E.; Guerry, D.; Nowell, P.; Clark, W.H.; et al. Characteristics of cultured human melanocytes isolated from different stages of tumor progression. Cancer Res. 1985, 45, 5670–5676. [Google Scholar] [PubMed]
- Konstantakou, E.G.; Velentzas, A.D.; Anagnostopoulos, A.K.; Giannopoulou, A.F.; Anastasiadou, E.; Papassideri, I.S.; Voutsinas, G.E.; Tsangaris, G.T.; Stravopodis, D.J. Unraveling the human protein atlas of metastatic melanoma in the course of ultraviolet radiation-derived photo-therapy. J. Proteom. 2018, 188, 119–138. [Google Scholar] [CrossRef] [PubMed]
- Anagnostopoulos, A.K.; Stravopodis, D.J.; Tsangaris, G.T. Yield of 6000 proteins by 1D nLC-MS/MS without pre-fractionation. J. Chromatogr. B Analyt. Technol. BioMed. Life Sci. 2017, 1047, 92–96. [Google Scholar] [CrossRef] [PubMed]
- UniProt, C. UniProt: A hub for protein information. Nucleic Acids Res. 2015, 43, D204–D212. [Google Scholar] [CrossRef]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Huang da, W.; Sherman, B.T.; Zheng, X.; Yang, J.; Imamichi, T.; Stephens, R.; Lempicki, R.A. Extracting biological meaning from large gene lists with DAVID. Curr. Protoc. Bioinform. 2009, 13. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. Data, information, knowledge and principle: Back to metabolism in KEGG. Nucleic Acids Res. 2014, 42, D199–D205. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Muruganujan, A.; Casagrande, J.T.; Thomas, P.D. Large-scale gene function analysis with the PANTHER classification system. Nat. Protoc. 2013, 8, 1551–1566. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Muruganujan, A.; Thomas, P.D. PANTHER in 2013: Modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. 2013, 41, D377–D386. [Google Scholar] [CrossRef]
- Zhao, M.; Kong, L.; Liu, Y.; Qu, H. dbEMT: An epithelial-mesenchymal transition associated gene resource. Sci. Rep. 2015, 5, 11459. [Google Scholar] [CrossRef]
- Zhao, M.; Liu, Y.; Zheng, C.; Qu, H. dbEMT 2.0: An updated database for epithelial-mesenchymal transition genes with experimentally verified information and precalculated regulation information for cancer metastasis. J. Genet. Genomics 2019, 46, 595–597. [Google Scholar] [CrossRef]
- Liu, Y.; Li, Z.; Lu, J.; Zhao, M.; Qu, H. CMGene: A literature-based database and knowledge resource for cancer metastasis genes. J. Genet. Genomics 2017, 44, 277–279. [Google Scholar] [CrossRef]
- Edgar, R.; Mazor, Y.; Rinon, A.; Blumenthal, J.; Golan, Y.; Buzhor, E.; Livnat, I.; Ben-Ari, S.; Lieder, I.; Shitrit, A.; et al. LifeMap Discovery: The embryonic development, stem cells, and regenerative medicine research portal. PLoS ONE 2013, 8, e66629. [Google Scholar] [CrossRef]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehar, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Tsherniak, A.; Vazquez, F.; Montgomery, P.G.; Weir, B.A.; Kryukov, G.; Cowley, G.S.; Gill, S.; Harrington, W.F.; Pantel, S.; Krill-Burger, J.M.; et al. Defining a Cancer Dependency Map. Cell 2017, 170, 564–576.e516. [Google Scholar] [CrossRef] [PubMed]
- McFarland, J.M.; Ho, Z.V.; Kugener, G.; Dempster, J.M.; Montgomery, P.G.; Bryan, J.G.; Krill-Burger, J.M.; Green, T.M.; Vazquez, F.; Boehm, J.S.; et al. Improved estimation of cancer dependencies from large-scale RNAi screens using model-based normalization and data integration. Nat. Commun. 2018, 9, 4610. [Google Scholar] [CrossRef] [PubMed]
- Ghandi, M.; Huang, F.W.; Jane-Valbuena, J.; Kryukov, G.V.; Lo, C.C.; McDonald, E.R., 3rd; Barretina, J.; Gelfand, E.T.; Bielski, C.M.; Li, H.; et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 2019, 569, 503–508. [Google Scholar] [CrossRef]
- Konstantakou, E.G.; Voutsinas, G.E.; Velentzas, A.D.; Basogianni, A.S.; Paronis, E.; Balafas, E.; Kostomitsopoulos, N.; Syrigos, K.N.; Anastasiadou, E.; Stravopodis, D.J. 3-BrPA eliminates human bladder cancer cells with highly oncogenic signatures via engagement of specific death programs and perturbation of multiple signaling and metabolic determinants. Mol. Cancer 2015, 14, 135. [Google Scholar] [CrossRef] [PubMed]
- Karkoulis, P.K.; Stravopodis, D.J.; Margaritis, L.H.; Voutsinas, G.E. 17-Allylamino-17-demethoxygeldanamycin induces downregulation of critical Hsp90 protein clients and results in cell cycle arrest and apoptosis of human urinary bladder cancer cells. BMC Cancer 2010, 10, 481. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef]
- Yang, J.; Roy, A.; Zhang, Y. Protein-ligand binding site recognition using complementary binding-specific substructure comparison and sequence profile alignment. Bioinformatics 2013, 29, 2588–2595. [Google Scholar] [CrossRef]
- Vajda, S.; Yueh, C.; Beglov, D.; Bohnuud, T.; Mottarella, S.E.; Xia, B.; Hall, D.R.; Kozakov, D. New additions to the ClusPro server motivated by CAPRI. Proteins 2017, 85, 435–444. [Google Scholar] [CrossRef]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro web server for protein-protein docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef]
- Kozakov, D.; Beglov, D.; Bohnuud, T.; Mottarella, S.E.; Xia, B.; Hall, D.R.; Vajda, S. How good is automated protein docking? Proteins 2013, 81, 2159–2166. [Google Scholar] [CrossRef]
- Porter, K.A.; Xia, B.; Beglov, D.; Bohnuud, T.; Alam, N.; Schueler-Furman, O.; Kozakov, D. ClusPro PeptiDock: Efficient global docking of peptide recognition motifs using FFT. Bioinformatics 2017, 33, 3299–3301. [Google Scholar] [CrossRef]
- Lensink, M.F.; Velankar, S.; Wodak, S.J. Modeling protein-protein and protein-peptide complexes: CAPRI 6th edition. Proteins 2017, 85, 359–377. [Google Scholar] [CrossRef]
- Vila, I.K.; Yao, Y.; Kim, G.; Xia, W.; Kim, H.; Kim, S.J.; Park, M.K.; Hwang, J.P.; Gonzalez-Billalabeitia, E.; Hung, M.C.; et al. A UBE2O-AMPKalpha2 Axis that Promotes Tumor Initiation and Progression Offers Opportunities for Therapy. Cancer Cell 2017, 31, 208–224. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.; Chen, D.; Ao, J.; Zhang, W.; Yi, J.; Ren, X.; Fei, J.; Li, F.; Niu, M.; Chen, H.; et al. Transcriptional suppression of AMPKalpha1 promotes breast cancer metastasis upon oncogene activation. Proc. Natl. Acad. Sci. USA 2020, 117, 8013–8021. [Google Scholar] [CrossRef]
- Dong, L.; You, S.; Zhang, Q.; Osuka, S.; Devi, N.S.; Kaluz, S.; Ferguson, J.H.; Yang, H.; Chen, G.; Wang, B.; et al. Arylsulfonamide 64B Inhibits Hypoxia/HIF-Induced Expression of c-Met and CXCR4 and Reduces Primary Tumor Growth and Metastasis of Uveal Melanoma. Clin. Cancer Res. 2019, 25, 2206–2218. [Google Scholar] [CrossRef]
- Zhao, X.; Gao, S.; Ren, H.; Sun, W.; Zhang, H.; Sun, J.; Yang, S.; Hao, J. Hypoxia-inducible factor-1 promotes pancreatic ductal adenocarcinoma invasion and metastasis by activating transcription of the actin-bundling protein fascin. Cancer Res. 2014, 74, 2455–2464. [Google Scholar] [CrossRef] [PubMed]
- Bhoumik, A.; Gangi, L.; Ronai, Z. Inhibition of melanoma growth and metastasis by ATF2-derived peptides. Cancer Res. 2004, 64, 8222–8230. [Google Scholar] [CrossRef]
- Wencong, M.; Jinghan, W.; Yong, Y.; Jianyang, A.; Bin, L.; Qingbao, C.; Chen, L.; Xiaoqing, J. FOXK1 Promotes Proliferation and Metastasis of Gallbladder Cancer by Activating AKT/mTOR Signaling Pathway. Front. Oncol. 2020, 10, 545. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, G.; Liu, M.; Xiang, L.; Xiao, Y.; Zhu, H.; Wu, X.; Peng, Y.; Zhang, W.; Jiang, P.; et al. The FOXK1-CCDC43 Axis Promotes the Invasion and Metastasis of Colorectal Cancer Cells. Cell. Physiol. Biochem. 2018, 51, 2547–2563. [Google Scholar] [CrossRef]
- Huang, X.; Xiang, L.; Li, Y.; Zhao, Y.; Zhu, H.; Xiao, Y.; Liu, M.; Wu, X.; Wang, Z.; Jiang, P.; et al. Snail/FOXK1/Cyr61 Signaling Axis Regulates the Epithelial-Mesenchymal Transition and Metastasis in Colorectal Cancer. Cell. Physiol. Biochem. 2018, 47, 590–603. [Google Scholar] [CrossRef]
- Manolopoulou, M.; Guo, Q.; Malito, E.; Schilling, A.B.; Tang, W.J. Molecular basis of catalytic chamber-assisted unfolding and cleavage of human insulin by human insulin-degrading enzyme. J. Biol. Chem. 2009, 284, 14177–14188. [Google Scholar] [CrossRef]
- Meerson, A.; Yehuda, H. Leptin and insulin up-regulate miR-4443 to suppress NCOA1 and TRAF4, and decrease the invasiveness of human colon cancer cells. BMC Cancer 2016, 16, 882. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Li, J.; Fu, S.; Ren, P.; Tang, J.; Wang, N.; Shi, X.; Wu, J.; Lin, S. Up-regulation of Insulin-like Growth Factor Binding Protein-3 Is Associated with Brain Metastasis in Lung Adenocarcinoma. Mol. Cells 2019, 42, 321–332. [Google Scholar] [CrossRef]
- Bao, L.; Liu, H.; You, B.; Gu, M.; Shi, S.; Shan, Y.; Li, L.; Chen, J.; You, Y. Overexpression of IGFBP3 is associated with poor prognosis and tumor metastasis in nasopharyngeal carcinoma. Tumour Biol. 2016, 37, 15043–15052. [Google Scholar] [CrossRef] [PubMed]
- Navarro, R.; Tapia-Galisteo, A.; Martin-Garcia, L.; Tarin, C.; Corbacho, C.; Gomez-Lopez, G.; Sanchez-Tirado, E.; Campuzano, S.; Gonzalez-Cortes, A.; Yanez-Sedeno, P.; et al. TGF-beta-induced IGFBP-3 is a key paracrine factor from activated pericytes that promotes colorectal cancer cell migration and invasion. Mol. Oncol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Levy-Strumpf, N.; Kimchi, A. Death associated proteins (DAPs): From gene identification to the analysis of their apoptotic and tumor suppressive functions. Oncogene 1998, 17, 3331–3340. [Google Scholar] [CrossRef] [PubMed]
- Deiss, L.P.; Feinstein, E.; Berissi, H.; Cohen, O.; Kimchi, A. Identification of a novel serine/threonine kinase and a novel 15-kD protein as potential mediators of the gamma interferon-induced cell death. Genes Dev. 1995, 9, 15–30. [Google Scholar] [CrossRef]
- Santos, M.; Maia, L.L.; Silva, C.V.; Peterle, G.T.; Mercante, A.M.; Nunes, F.D.; Carvalho, M.B.; Tajara, E.H.; Louro, I.D.; Silva-Conforti, A.M. DAP1 high expression increases risk of lymph node metastases in squamous cell carcinoma of the oral cavity. Genet. Mol. Res. 2015, 14, 10515–10523. [Google Scholar] [CrossRef]
- Koren, I.; Reem, E.; Kimchi, A. Autophagy gets a brake: DAP1, a novel mTOR substrate, is activated to suppress the autophagic process. Autophagy 2010, 6, 1179–1180. [Google Scholar] [CrossRef] [PubMed]
- Koren, I.; Reem, E.; Kimchi, A. DAP1, a novel substrate of mTOR, negatively regulates autophagy. Curr. Biol. 2010, 20, 1093–1098. [Google Scholar] [CrossRef]
- Wang, Z.; Jiang, H.; Chen, S.; Du, F.; Wang, X. The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell 2012, 148, 228–243. [Google Scholar] [CrossRef] [PubMed]
- Jiao, D.; Cai, Z.; Choksi, S.; Ma, D.; Choe, M.; Kwon, H.J.; Baik, J.Y.; Rowan, B.G.; Liu, C.; Liu, Z.G. Necroptosis of tumor cells leads to tumor necrosis and promotes tumor metastasis. Cell Res. 2018, 28, 868–870. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, K.; Basnet, H.; Kaygusuz, Y.; Laughney, A.M.; He, L.; Sharma, R.; O’Rourke, K.P.; Reuter, V.P.; Huang, Y.H.; Turkekul, M.; et al. L1CAM defines the regenerative origin of metastasis-initiating cells in colorectal cancer. Nat. Cancer 2020, 1, 28–45. [Google Scholar] [CrossRef]
- Altevogt, P.; Ben-Ze’ev, A.; Gavert, N.; Schumacher, U.; Schafer, H.; Sebens, S. Recent insights into the role of L1CAM in cancer initiation and progression. Int. J. Cancer 2020, 147. [Google Scholar] [CrossRef]
- Thies, A.; Schachner, M.; Moll, I.; Berger, J.; Schulze, H.J.; Brunner, G.; Schumacher, U. Overexpression of the cell adhesion molecule L1 is associated with metastasis in cutaneous malignant melanoma. Eur. J. Cancer 2002, 38, 1708–1716. [Google Scholar] [CrossRef]
- Wolterink, S.; Moldenhauer, G.; Fogel, M.; Kiefel, H.; Pfeifer, M.; Luttgau, S.; Gouveia, R.; Costa, J.; Endell, J.; Moebius, U.; et al. Therapeutic antibodies to human L1CAM: Functional characterization and application in a mouse model for ovarian carcinoma. Cancer Res. 2010, 70, 2504–2515. [Google Scholar] [CrossRef]
- Cho, S.; Park, I.; Kim, H.; Jeong, M.S.; Lim, M.; Lee, E.S.; Kim, J.H.; Kim, S.; Hong, H.J. Generation, characterization and preclinical studies of a human anti-L1CAM monoclonal antibody that cross-reacts with rodent L1CAM. MAbs 2016, 8, 414–425. [Google Scholar] [CrossRef]
- Brozyna, A.A.; Jozwicki, W.; Carlson, J.A.; Slominski, A.T. Melanogenesis affects overall and disease-free survival in patients with stage III and IV melanoma. Hum. Pathol. 2013, 44, 2071–2074. [Google Scholar] [CrossRef]
- Brozyna, A.A.; Jozwicki, W.; Roszkowski, K.; Filipiak, J.; Slominski, A.T. Melanin content in melanoma metastases affects the outcome of radiotherapy. Oncotarget 2016, 7, 17844–17853. [Google Scholar] [CrossRef] [PubMed]
- Brozyna, A.A.; VanMiddlesworth, L.; Slominski, A.T. Inhibition of melanogenesis as a radiation sensitizer for melanoma therapy. Int. J. Cancer 2008, 123, 1448–1456. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.; Zbytek, B.; Slominski, R. Inhibitors of melanogenesis increase toxicity of cyclophosphamide and lymphocytes against melanoma cells. Int. J. Cancer 2009, 124, 1470–1477. [Google Scholar] [CrossRef] [PubMed]
- Mort, R.L.; Jackson, I.J.; Patton, E.E. The melanocyte lineage in development and disease. Development 2015, 142, 620–632. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.Y.; Fisher, D.E. Melanocyte biology and skin pigmentation. Nature 2007, 445, 843–850. [Google Scholar] [CrossRef]
- Park, H.Y.; Kosmadaki, M.; Yaar, M.; Gilchrest, B.A. Cellular mechanisms regulating human melanogenesis. Cell. Mol. Life Sci. 2009, 66, 1493–1506. [Google Scholar] [CrossRef]
- Pillaiyar, T.; Manickam, M.; Jung, S.H. Downregulation of melanogenesis: Drug discovery and therapeutic options. Drug Discov. Today 2017, 22, 282–298. [Google Scholar] [CrossRef]
- Argast, G.M.; Croy, C.H.; Couts, K.L.; Zhang, Z.; Litman, E.; Chan, D.C.; Ahn, N.G. Plexin B1 is repressed by oncogenic B-Raf signaling and functions as a tumor suppressor in melanoma cells. Oncogene 2009, 28, 2697–2709. [Google Scholar] [CrossRef]
- Zimmerer, R.M.; Korn, P.; Demougin, P.; Kampmann, A.; Kokemuller, H.; Eckardt, A.M.; Gellrich, N.C.; Tavassol, F. Functional features of cancer stem cells in melanoma cell lines. Cancer Cell Int. 2013, 13, 78. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. Emt: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed]
- De Craene, B.; Berx, G. Regulatory networks defining EMT during cancer initiation and progression. Nat. Rev. Cancer 2013, 13, 97–110. [Google Scholar] [CrossRef]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; Van Keymeulen, A.; Brown, D.; Moers, V.; Lemaire, S.; et al. Identification of the tumour transition states occurring during EMT. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Kroger, C.; Afeyan, A.; Mraz, J.; Eaton, E.N.; Reinhardt, F.; Khodor, Y.L.; Thiru, P.; Bierie, B.; Ye, X.; Burge, C.B.; et al. Acquisition of a hybrid E/M state is essential for tumorigenicity of basal breast cancer cells. Proc. Natl. Acad. Sci. USA 2019, 116, 7353–7362. [Google Scholar] [CrossRef] [PubMed]
- Williams, E.D.; Gao, D.; Redfern, A.; Thompson, E.W. Controversies around epithelial-mesenchymal plasticity in cancer metastasis. Nat. Rev. Cancer 2019, 19, 716–732. [Google Scholar] [CrossRef] [PubMed]
- Ocana, O.H.; Corcoles, R.; Fabra, A.; Moreno-Bueno, G.; Acloque, H.; Vega, S.; Barrallo-Gimeno, A.; Cano, A.; Nieto, M.A. Metastatic colonization requires the repression of the epithelial-mesenchymal transition inducer Prrx1. Cancer Cell 2012, 22, 709–724. [Google Scholar] [CrossRef]
- Takano, S.; Reichert, M.; Bakir, B.; Das, K.K.; Nishida, T.; Miyazaki, M.; Heeg, S.; Collins, M.A.; Marchand, B.; Hicks, P.D.; et al. Prrx1 isoform switching regulates pancreatic cancer invasion and metastatic colonization. Genes Dev. 2016, 30, 233–247. [Google Scholar] [CrossRef]
- Lv, Z.D.; Yang, Z.C.; Liu, X.P.; Jin, L.Y.; Dong, Q.; Qu, H.L.; Li, F.N.; Kong, B.; Sun, J.; Zhao, J.J.; et al. Silencing of Prrx1b suppresses cellular proliferation, migration, invasion and epithelial-mesenchymal transition in triple-negative breast cancer. J. Cell Mol. Med. 2016, 20, 1640–1650. [Google Scholar] [CrossRef]
- Revenco, T.; Nicodeme, A.; Pastushenko, I.; Sznurkowska, M.K.; Latil, M.; Sotiropoulou, P.A.; Dubois, C.; Moers, V.; Lemaire, S.; de Maertelaer, V.; et al. Context Dependency of Epithelial-to-Mesenchymal Transition for Metastasis. Cell Rep. 2019, 29, 1458–1468.e1453. [Google Scholar] [CrossRef]
- Barker, H.E.; Cox, T.R.; Erler, J.T. The rationale for targeting the LOX family in cancer. Nat. Rev. Cancer 2012, 12, 540–552. [Google Scholar] [CrossRef]
- Erler, J.T.; Bennewith, K.L.; Nicolau, M.; Dornhofer, N.; Kong, C.; Le, Q.T.; Chi, J.T.; Jeffrey, S.S.; Giaccia, A.J. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature 2006, 440, 1222–1226. [Google Scholar] [CrossRef]
- Erler, J.T.; Giaccia, A.J. Lysyl oxidase mediates hypoxic control of metastasis. Cancer Res. 2006, 66, 10238–10241. [Google Scholar] [CrossRef]
- Barker, H.E.; Chang, J.; Cox, T.R.; Lang, G.; Bird, D.; Nicolau, M.; Evans, H.R.; Gartland, A.; Erler, J.T. LOXL2-mediated matrix remodeling in metastasis and mammary gland involution. Cancer Res. 2011, 71, 1561–1572. [Google Scholar] [CrossRef] [PubMed]
- Schietke, R.; Warnecke, C.; Wacker, I.; Schodel, J.; Mole, D.R.; Campean, V.; Amann, K.; Goppelt-Struebe, M.; Behrens, J.; Eckardt, K.U.; et al. The lysyl oxidases LOX and LOXL2 are necessary and sufficient to repress E-cadherin in hypoxia: Insights into cellular transformation processes mediated by HIF-1. J. Biol. Chem. 2010, 285, 6658–6669. [Google Scholar] [CrossRef] [PubMed]
- Moustakas, A.; Heldin, C.H. Mechanisms of TGFbeta-Induced Epithelial-Mesenchymal Transition. J. Clin. Med. 2016, 5, 63. [Google Scholar] [CrossRef]
- Dhasarathy, A.; Phadke, D.; Mav, D.; Shah, R.R.; Wade, P.A. The transcription factors Snail and Slug activate the transforming growth factor-beta signaling pathway in breast cancer. PLoS ONE 2011, 6, e26514. [Google Scholar] [CrossRef]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef] [PubMed]
- Colak, S.; Ten Dijke, P. Targeting TGF-beta Signaling in Cancer. Trends Cancer 2017, 3, 56–71. [Google Scholar] [CrossRef]
- Pickup, M.; Novitskiy, S.; Moses, H.L. The roles of TGFbeta in the tumour microenvironment. Nat. Rev. Cancer 2013, 13, 788–799. [Google Scholar] [CrossRef]
- Shiou, S.R.; Datta, P.K.; Dhawan, P.; Law, B.K.; Yingling, J.M.; Dixon, D.A.; Beauchamp, R.D. Smad4-dependent regulation of urokinase plasminogen activator secretion and RNA stability associated with invasiveness by autocrine and paracrine transforming growth factor-beta. J. Biol. Chem. 2006, 281, 33971–33981. [Google Scholar] [CrossRef]
- Sarkar, A.; Hochedlinger, K. The sox family of transcription factors: Versatile regulators of stem and progenitor cell fate. Cell Stem Cell 2013, 12, 15–30. [Google Scholar] [CrossRef]
- Arnold, K.; Sarkar, A.; Yram, M.A.; Polo, J.M.; Bronson, R.; Sengupta, S.; Seandel, M.; Geijsen, N.; Hochedlinger, K. Sox2(+) adult stem and progenitor cells are important for tissue regeneration and survival of mice. Cell Stem Cell 2011, 9, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Keckesova, Z.; Donaher, J.L.; Shibue, T.; Tischler, V.; Reinhardt, F.; Itzkovitz, S.; Noske, A.; Zurrer-Hardi, U.; Bell, G.; et al. Slug and Sox9 cooperatively determine the mammary stem cell state. Cell 2012, 148, 1015–1028. [Google Scholar] [CrossRef] [PubMed]
- Mertelmeyer, S.; Weider, M.; Baroti, T.; Reiprich, S.; Frob, F.; Stolt, C.C.; Wagner, K.U.; Wegner, M. The transcription factor Sox10 is an essential determinant of branching morphogenesis and involution in the mouse mammary gland. Sci. Rep. 2020, 10, 17807. [Google Scholar] [CrossRef]
- Motohashi, T.; Kawamura, N.; Watanabe, N.; Kitagawa, D.; Goshima, N.; Kunisada, T. Sox10 functions as an inducer of the direct conversion of keratinocytes into neural crest cells. Stem Cells Dev. 2020, 29, 1510–1519. [Google Scholar] [CrossRef]
- Yu, H.; Lee, H.; Herrmann, A.; Buettner, R.; Jove, R. Revisiting STAT3 signalling in cancer: New and unexpected biological functions. Nat. Rev. Cancer 2014, 14, 736–746. [Google Scholar] [CrossRef] [PubMed]
- Marotta, L.L.; Almendro, V.; Marusyk, A.; Shipitsin, M.; Schemme, J.; Walker, S.R.; Bloushtain-Qimron, N.; Kim, J.J.; Choudhury, S.A.; Maruyama, R.; et al. The JAK2/STAT3 signaling pathway is required for growth of CD44(+)CD24(-) stem cell-like breast cancer cells in human tumors. J. Clin. Investig. 2011, 121, 2723–2735. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, A.; Herrmann, A.; Cherryholmes, G.; Kowolik, C.; Buettner, R.; Pal, S.; Yu, H.; Muller-Newen, G.; Jove, R. Loss of androgen receptor expression promotes a stem-like cell phenotype in prostate cancer through STAT3 signaling. Cancer Res. 2014, 74, 1227–1237. [Google Scholar] [CrossRef] [PubMed]
- Diener, J.; Sommer, L. Reemergence of neural crest stem cell-like states in melanoma during disease progression and treatment. Stem Cells Transl. Med. 2021, 10, 522–533. [Google Scholar] [CrossRef]
- Simoes-Costa, M.; Bronner, M.E. Establishing neural crest identity: A gene regulatory recipe. Development 2015, 142, 242–257. [Google Scholar] [CrossRef]
- Leucci, E.; Vendramin, R.; Spinazzi, M.; Laurette, P.; Fiers, M.; Wouters, J.; Radaelli, E.; Eyckerman, S.; Leonelli, C.; Vanderheyden, K.; et al. Melanoma addiction to the long non-coding RNA SAMMSON. Nature 2016, 531, 518–522. [Google Scholar] [CrossRef]
- Li, X.; Li, M.; Chen, J.; Dai, H.; Wang, L.; Xiong, Y.; Zhong, Y.; Zhang, L. SAMMSON drives the self-renewal of liver tumor initiating cells through EZH2-dependent Wnt/beta-catenin activation. Oncotarget 2017, 8, 103785–103796. [Google Scholar] [CrossRef]
- Sens, P.; Plastino, J. Membrane tension and cytoskeleton organization in cell motility. J. Phys. Condens. Matter 2015, 27, 273103. [Google Scholar] [CrossRef]
- Wu, P.H.; Gilkes, D.M.; Phillip, J.M.; Narkar, A.; Cheng, T.W.; Marchand, J.; Lee, M.H.; Li, R.; Wirtz, D. Single-cell morphology encodes metastatic potential. Sci. Adv. 2020, 6, eaaw6938. [Google Scholar] [CrossRef]
- Gupta, G.P.; Massague, J. Cancer metastasis: Building a framework. Cell 2006, 127, 679–695. [Google Scholar] [CrossRef]
- Yin, Z.; Sadok, A.; Sailem, H.; McCarthy, A.; Xia, X.; Li, F.; Garcia, M.A.; Evans, L.; Barr, A.R.; Perrimon, N.; et al. A screen for morphological complexity identifies regulators of switch-like transitions between discrete cell shapes. Nat. Cell Biol. 2013, 15, 860–871. [Google Scholar] [CrossRef] [PubMed]
- Wirtz, D.; Konstantopoulos, K.; Searson, P.C. The physics of cancer: The role of physical interactions and mechanical forces in metastasis. Nat. Rev. Cancer 2011, 11, 512–522. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.H.; Phillip, J.M.; Khatau, S.B.; Chen, W.C.; Stirman, J.; Rosseel, S.; Tschudi, K.; Van Patten, J.; Wong, M.; Gupta, S.; et al. Evolution of cellular morpho-phenotypes in cancer metastasis. Sci. Rep. 2015, 5, 18437. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, R.A.; Delic, S.; Herrmann, H.; Snider, N.T. Vimentin on the move: New developments in cell migration. F1000Research 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; He, Y.; Zhang, P.; Wang, J.; Fan, C.; Yang, L.; Xiong, F.; Zhang, S.; Gong, Z.; Nie, S.; et al. LncRNAs regulate the cytoskeleton and related Rho/ROCK signaling in cancer metastasis. Mol. Cancer 2018, 17, 77. [Google Scholar] [CrossRef] [PubMed]
- Nurnberg, A.; Kitzing, T.; Grosse, R. Nucleating actin for invasion. Nat. Rev. Cancer 2011, 11, 177–187. [Google Scholar] [CrossRef]
- Fife, C.M.; McCarroll, J.A.; Kavallaris, M. Movers and shakers: Cell cytoskeleton in cancer metastasis. Br. J. Pharmacol. 2014, 171, 5507–5523. [Google Scholar] [CrossRef]
- Akhshi, T.K.; Wernike, D.; Piekny, A. Microtubules and actin crosstalk in cell migration and division. Cytoskeleton 2014, 71, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Krause, M.; Gautreau, A. Steering cell migration: Lamellipodium dynamics and the regulation of directional persistence. Nat. Rev. Mol. Cell Biol. 2014, 15, 577–590. [Google Scholar] [CrossRef]
- Campellone, K.G.; Welch, M.D. A nucleator arms race: Cellular control of actin assembly. Nat. Rev. Mol. Cell Biol. 2010, 11, 237–251. [Google Scholar] [CrossRef] [PubMed]
- Rotty, J.D.; Wu, C.; Bear, J.E. New insights into the regulation and cellular functions of the ARP2/3 complex. Nat. Rev. Mol. Cell Biol. 2013, 14, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Chanez-Paredes, S.; Montoya-Garcia, A.; Schnoor, M. Cellular and pathophysiological consequences of Arp2/3 complex inhibition: Role of inhibitory proteins and pharmacological compounds. Cell. Mol. Life Sci. 2019, 76, 3349–3361. [Google Scholar] [CrossRef]
- Hirao, N.; Sato, S.; Gotoh, T.; Maruoka, M.; Suzuki, J.; Matsuda, S.; Shishido, T.; Tani, K. NESH (Abi-3) is present in the Abi/WAVE complex but does not promote c-Abl-mediated phosphorylation. FEBS Lett. 2006, 580, 6464–6470. [Google Scholar] [CrossRef]
- Frose, J.; Chen, M.B.; Hebron, K.E.; Reinhardt, F.; Hajal, C.; Zijlstra, A.; Kamm, R.D.; Weinberg, R.A. Epithelial-Mesenchymal Transition Induces Podocalyxin to Promote Extravasation via Ezrin Signaling. Cell Rep. 2018, 24, 962–972. [Google Scholar] [CrossRef]
- Fehon, R.G.; McClatchey, A.I.; Bretscher, A. Organizing the cell cortex: The role of ERM proteins. Nat. Rev. Mol. Cell Biol. 2010, 11, 276–287. [Google Scholar] [CrossRef]
- Schopperle, W.M.; DeWolf, W.C. The TRA-1-60 and TRA-1-81 human pluripotent stem cell markers are expressed on podocalyxin in embryonal carcinoma. Stem Cells 2007, 25, 723–730. [Google Scholar] [CrossRef]
- Toyoda, H.; Nagai, Y.; Kojima, A.; Kinoshita-Toyoda, A. Podocalyxin as a major pluripotent marker and novel keratan sulfate proteoglycan in human embryonic and induced pluripotent stem cells. Glycoconj. J. 2017, 34, 817–823. [Google Scholar] [CrossRef]
- Rambow, F.; Rogiers, A.; Marin-Bejar, O.; Aibar, S.; Femel, J.; Dewaele, M.; Karras, P.; Brown, D.; Chang, Y.H.; Debiec-Rychter, M.; et al. Toward Minimal Residual Disease-Directed Therapy in Melanoma. Cell 2018, 174, 843–855.e819. [Google Scholar] [CrossRef]
- Ebisawa, M.; Umemiya, H.; Ohta, K.; Fukasawa, H.; Kawachi, E.; Christoffel, G.; Gronemeyer, H.; Tsuji, M.; Hashimoto, Y.; Shudo, K.; et al. Retinoid X receptor-antagonistic diazepinylbenzoic acids. Chem. Pharm. Bull. 1999, 47, 1778–1786. [Google Scholar] [CrossRef] [PubMed]
- Gan, P.P.; Kavallaris, M. Tubulin-targeted drug action: Functional significance of class ii and class IVb beta-tubulin in vinca alkaloid sensitivity. Cancer Res. 2008, 68, 9817–9824. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, S.H.; Guo, X.L. New insights into Vinca alkaloids resistance mechanism and circumvention in lung cancer. BioMed. Pharmacother. 2017, 96, 659–666. [Google Scholar] [CrossRef] [PubMed]
- Badano, J.L.; Teslovich, T.M.; Katsanis, N. The centrosome in human genetic disease. Nat. Rev. Genet. 2005, 6, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Gonczy, P. Towards a molecular architecture of centriole assembly. Nat. Rev. Mol. Cell Biol. 2012, 13, 425–435. [Google Scholar] [CrossRef]
- Conduit, P.T.; Wainman, A.; Raff, J.W. Centrosome function and assembly in animal cells. Nat. Rev. Mol. Cell Biol. 2015, 16, 611–624. [Google Scholar] [CrossRef] [PubMed]
- Dammermann, A.; Merdes, A. Assembly of centrosomal proteins and microtubule organization depends on PCM-1. J. Cell Biol. 2002, 159, 255–266. [Google Scholar] [CrossRef]
- Kubo, A.; Tsukita, S. Non-membranous granular organelle consisting of PCM-1: Subcellular distribution and cell-cycle-dependent assembly/disassembly. J. Cell Sci. 2003, 116, 919–928. [Google Scholar] [CrossRef] [PubMed]
- Ballabio, A.; Bonifacino, J.S. Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat. Rev. Mol. Cell Biol. 2020, 21, 101–118. [Google Scholar] [CrossRef] [PubMed]
- Eisner, V.; Picard, M.; Hajnoczky, G. Mitochondrial dynamics in adaptive and maladaptive cellular stress responses. Nat. Cell Biol. 2018, 20, 755–765. [Google Scholar] [CrossRef]
- Cabukusta, B.; Neefjes, J. Mechanisms of lysosomal positioning and movement. Traffic 2018, 19, 761–769. [Google Scholar] [CrossRef]
- Pu, J.; Guardia, C.M.; Keren-Kaplan, T.; Bonifacino, J.S. Mechanisms and functions of lysosome positioning. J. Cell Sci. 2016, 129, 4329–4339. [Google Scholar] [CrossRef]
- Ji, X.; Mukherjee, S.; Landi, M.T.; Bosse, Y.; Joubert, P.; Zhu, D.; Gorlov, I.; Xiao, X.; Han, Y.; Gorlova, O.; et al. Protein-altering germline mutations implicate novel genes related to lung cancer development. Nat. Commun. 2020, 11, 2220. [Google Scholar] [CrossRef] [PubMed]
- Hossain, D.; Shih, S.Y.; Xiao, X.; White, J.; Tsang, W.Y. Cep44 functions in centrosome cohesion by stabilizing rootletin. J. Cell Sci. 2020, 133. [Google Scholar] [CrossRef]
- Atorino, E.S.; Hata, S.; Funaya, C.; Neuner, A.; Schiebel, E. CEP44 ensures the formation of bona fide centriole wall, a requirement for the centriole-to-centrosome conversion. Nat. Commun. 2020, 11, 903. [Google Scholar] [CrossRef]
- Murphy, T.L.; Tussiwand, R.; Murphy, K.M. Specificity through cooperation: BATF-IRF interactions control immune-regulatory networks. Nat. Rev. Immunol. 2013, 13, 499–509. [Google Scholar] [CrossRef]
- Vinson, C.; Myakishev, M.; Acharya, A.; Mir, A.A.; Moll, J.R.; Bonovich, M. Classification of human B-ZIP proteins based on dimerization properties. Mol. Cell Biol. 2002, 22, 6321–6335. [Google Scholar] [CrossRef]
- Landschulz, W.H.; Johnson, P.F.; McKnight, S.L. The leucine zipper: A hypothetical structure common to a new class of DNA binding proteins. Science 1988, 240, 1759–1764. [Google Scholar] [CrossRef]
- Turner, R.; Tjian, R. Leucine repeats and an adjacent DNA binding domain mediate the formation of functional cFos-cJun heterodimers. Science 1989, 243, 1689–1694. [Google Scholar] [CrossRef] [PubMed]
- Remo, A.; Li, X.; Schiebel, E.; Pancione, M. The Centrosome Linker and Its Role in Cancer and Genetic Disorders. Trends Mol. Med. 2020, 26, 380–393. [Google Scholar] [CrossRef] [PubMed]
- Trevor, K.T.; McGuire, J.G.; Leonova, E.V. Association of vimentin intermediate filaments with the centrosome. J. Cell Sci. 1995, 108 (Pt 1), 343–356. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Ferguson, F.M.; Gray, N.S. Kinase inhibitors: The road ahead. Nat. Rev. Drug Discov. 2018, 17, 353–377. [Google Scholar] [CrossRef] [PubMed]
- Hoxhaj, G.; Manning, B.D. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef]
- Janku, F.; Yap, T.A.; Meric-Bernstam, F. Targeting the PI3K pathway in cancer: Are we making headway? Nat. Rev. Clin. Oncol. 2018, 15, 273–291. [Google Scholar] [CrossRef]
- Liu, R.; Chen, Y.; Liu, G.; Li, C.; Song, Y.; Cao, Z.; Li, W.; Hu, J.; Lu, C.; Liu, Y. PI3K/AKT pathway as a key link modulates the multidrug resistance of cancers. Cell Death Dis. 2020, 11, 797. [Google Scholar] [CrossRef]
- Hur, E.M.; Zhou, F.Q. GSK3 signalling in neural development. Nat. Rev. Neurosci. 2010, 11, 539–551. [Google Scholar] [CrossRef]
- Augello, G.; Emma, M.R.; Cusimano, A.; Azzolina, A.; Montalto, G.; McCubrey, J.A.; Cervello, M. The Role of GSK-3 in Cancer Immunotherapy: GSK-3 Inhibitors as a New Frontier in Cancer Treatment. Cells 2020, 9, 1427. [Google Scholar] [CrossRef] [PubMed]
- Lavoie, H.; Gagnon, J.; Therrien, M. ERK signalling: A master regulator of cell behaviour, life and fate. Nat. Rev. Mol. Cell Biol. 2020, 21, 607–632. [Google Scholar] [CrossRef]
- Samatar, A.A.; Poulikakos, P.I. Targeting RAS-ERK signalling in cancer: Promises and challenges. Nat. Rev. Drug Discov. 2014, 13, 928–942. [Google Scholar] [CrossRef]
- Kolch, W. Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nat. Rev. Mol. Cell Biol. 2005, 6, 827–837. [Google Scholar] [CrossRef]
- Caunt, C.J.; Sale, M.J.; Smith, P.D.; Cook, S.J. MEK1 and MEK2 inhibitors and cancer therapy: The long and winding road. Nat. Rev. Cancer 2015, 15, 577–592. [Google Scholar] [CrossRef] [PubMed]
- Nikolic, I.; Leiva, M.; Sabio, G. The role of stress kinases in metabolic disease. Nat. Rev. Endocrinol. 2020, 16, 697–716. [Google Scholar] [CrossRef]
- Wagner, E.F.; Nebreda, A.R. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat. Rev. Cancer 2009, 9, 537–549. [Google Scholar] [CrossRef] [PubMed]
- Mossmann, D.; Park, S.; Hall, M.N. mTOR signalling and cellular metabolism are mutual determinants in cancer. Nat. Rev. Cancer 2018, 18, 744–757. [Google Scholar] [CrossRef]
- Kim, J.; Guan, K.L. mTOR as a central hub of nutrient signalling and cell growth. Nat. Cell Biol. 2019, 21, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef] [PubMed]
- Bugter, J.M.; Fenderico, N.; Maurice, M.M. Mutations and mechanisms of WNT pathway tumour suppressors in cancer. Nat. Rev. Cancer 2020. [Google Scholar] [CrossRef] [PubMed]
- McCrea, P.D.; Gottardi, C.J. Beyond beta-catenin: Prospects for a larger catenin network in the nucleus. Nat. Rev. Mol. Cell Biol. 2016, 17, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Niehrs, C. The complex world of WNT receptor signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 767–779. [Google Scholar] [CrossRef] [PubMed]
- Moroishi, T.; Hansen, C.G.; Guan, K.L. The emerging roles of YAP and TAZ in cancer. Nat. Rev. Cancer 2015, 15, 73–79. [Google Scholar] [CrossRef]
- Moya, I.M.; Halder, G. Hippo-YAP/TAZ signalling in organ regeneration and regenerative medicine. Nat. Rev. Mol. Cell Biol. 2019, 20, 211–226. [Google Scholar] [CrossRef]
- Johnson, R.; Halder, G. The two faces of Hippo: Targeting the Hippo pathway for regenerative medicine and cancer treatment. Nat. Rev. Drug Discov. 2014, 13, 63–79. [Google Scholar] [CrossRef]
- Cho, J.H.; Robinson, J.P.; Arave, R.A.; Burnett, W.J.; Kircher, D.A.; Chen, G.; Davies, M.A.; Grossmann, A.H.; VanBrocklin, M.W.; McMahon, M.; et al. AKT1 Activation Promotes Development of Melanoma Metastases. Cell Rep. 2015, 13, 898–905. [Google Scholar] [CrossRef]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP/TAZ at the Roots of Cancer. Cancer Cell 2016, 29, 783–803. [Google Scholar] [CrossRef]
- Yan, F.; Qian, M.; He, Q.; Zhu, H.; Yang, B. The posttranslational modifications of Hippo-YAP pathway in cancer. Biochim Biophys. Acta Gen. Subj. 2020, 1864, 129397. [Google Scholar] [CrossRef]
- Shao, D.D.; Xue, W.; Krall, E.B.; Bhutkar, A.; Piccioni, F.; Wang, X.; Schinzel, A.C.; Sood, S.; Rosenbluh, J.; Kim, J.W.; et al. KRAS and YAP1 converge to regulate EMT and tumor survival. Cell 2014, 158, 171–184. [Google Scholar] [CrossRef]
- Cubillos-Ruiz, J.R.; Bettigole, S.E.; Glimcher, L.H. Tumorigenic and Immunosuppressive Effects of Endoplasmic Reticulum Stress in Cancer. Cell 2017, 168, 692–706. [Google Scholar] [CrossRef] [PubMed]
- Urra, H.; Dufey, E.; Avril, T.; Chevet, E.; Hetz, C. Endoplasmic Reticulum Stress and the Hallmarks of Cancer. Trends Cancer 2016, 2, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.K.; Chiu, C.C.; Dahms, H.U.; Chou, C.K.; Cheng, C.M.; Chang, W.T.; Cheng, K.C.; Wang, H.D.; Lin, I.L. Unfolded Protein Response (UPR) in Survival, Dormancy, Immunosuppression, Metastasis, and Treatments of Cancer Cells. Int. J. Mol. Sci. 2019, 20, 2518. [Google Scholar] [CrossRef] [PubMed]
- Pommier, A.; Anaparthy, N.; Memos, N.; Kelley, Z.L.; Gouronnec, A.; Yan, R.; Auffray, C.; Albrengues, J.; Egeblad, M.; Iacobuzio-Donahue, C.A.; et al. Unresolved endoplasmic reticulum stress engenders immune-resistant, latent pancreatic cancer metastases. Science 2018, 360. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.X.; Sokol, E.S.; Del Vecchio, C.A.; Sanduja, S.; Claessen, J.H.; Proia, T.A.; Jin, D.X.; Reinhardt, F.; Ploegh, H.L.; Wang, Q.; et al. Epithelial-to-mesenchymal transition activates PERK-eIF2alpha and sensitizes cells to endoplasmic reticulum stress. Cancer Discov. 2014, 4, 702–715. [Google Scholar] [CrossRef] [PubMed]
- Dey, S.; Sayers, C.M.; Verginadis, I.I.; Lehman, S.L.; Cheng, Y.; Cerniglia, G.J.; Tuttle, S.W.; Feldman, M.D.; Zhang, P.J.; Fuchs, S.Y.; et al. ATF4-dependent induction of heme oxygenase 1 prevents anoikis and promotes metastasis. J. Clin. Investig. 2015, 125, 2592–2608. [Google Scholar] [CrossRef]
- Kosmopoulou, M.; Giannopoulou, A.F.; Iliou, A.; Benaki, D.; Panagiotakis, A.; Velentzas, A.D.; Konstantakou, E.G.; Papassideri, I.S.; Mikros, E.; Stravopodis, D.J.; et al. Human Melanoma-Cell Metabolic Profiling: Identification of Novel Biomarkers Indicating Metastasis. Int. J. Mol. Sci. 2020, 21, 2436. [Google Scholar] [CrossRef]
- Tasdogan, A.; Faubert, B.; Ramesh, V.; Ubellacker, J.M.; Shen, B.; Solmonson, A.; Murphy, M.M.; Gu, Z.; Gu, W.; Martin, M.; et al. Metabolic heterogeneity confers differences in melanoma metastatic potential. Nature 2020, 577, 115–120. [Google Scholar] [CrossRef]
- Payen, V.L.; Mina, E.; Van Hee, V.F.; Porporato, P.E.; Sonveaux, P. Monocarboxylate transporters in cancer. Mol. Metab. 2020, 33, 48–66. [Google Scholar] [CrossRef]
- Payen, V.L.; Hsu, M.Y.; Radecke, K.S.; Wyart, E.; Vazeille, T.; Bouzin, C.; Porporato, P.E.; Sonveaux, P. Monocarboxylate Transporter MCT1 Promotes Tumor Metastasis Independently of Its Activity as a Lactate Transporter. Cancer Res. 2017, 77, 5591–5601. [Google Scholar] [CrossRef]
- Zhao, Z.; Wu, M.S.; Zou, C.; Tang, Q.; Lu, J.; Liu, D.; Wu, Y.; Yin, J.; Xie, X.; Shen, J.; et al. Downregulation of MCT1 inhibits tumor growth, metastasis and enhances chemotherapeutic efficacy in osteosarcoma through regulation of the NF-kappaB pathway. Cancer Lett. 2014, 342, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Gray, A.L.; Coleman, D.T.; Shi, R.; Cardelli, J.A. Monocarboxylate transporter 1 contributes to growth factor-induced tumor cell migration independent of transporter activity. Oncotarget 2016, 7, 32695–32706. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bosshart, P.D.; Charles, R.P.; Garibsingh, R.A.; Schlessinger, A.; Fotiadis, D. SLC16 Family: From Atomic Structure to Human Disease. Trends Biochem. Sci. 2020, 46, 28–40. [Google Scholar] [CrossRef]
- Valenca, I.; Pertega-Gomes, N.; Vizcaino, J.R.; Henrique, R.M.; Lopes, C.; Baltazar, F.; Ribeiro, D. Localization of MCT2 at peroxisomes is associated with malignant transformation in prostate cancer. J. Cell Mol. Med. 2015, 19, 723–733. [Google Scholar] [CrossRef]
- Pinheiro, C.; Penna, V.; Morais-Santos, F.; Abrahao-Machado, L.F.; Ribeiro, G.; Curcelli, E.C.; Olivieri, M.V.; Morini, S.; Valenca, I.; Ribeiro, D.; et al. Characterization of monocarboxylate transporters (MCTs) expression in soft tissue sarcomas: Distinct prognostic impact of MCT1 sub-cellular localization. J. Transl. Med. 2014, 12, 118. [Google Scholar] [CrossRef]
- Latif, A.; Chadwick, A.L.; Kitson, S.J.; Gregson, H.J.; Sivalingam, V.N.; Bolton, J.; McVey, R.J.; Roberts, S.A.; Marshall, K.M.; Williams, K.J.; et al. Monocarboxylate Transporter 1 (MCT1) is an independent prognostic biomarker in endometrial cancer. BMC Clin. Pathol. 2017, 17, 27. [Google Scholar] [CrossRef]
- Jansen, S.; Pantaleon, M.; Kaye, P.L. Characterization and regulation of monocarboxylate cotransporters Slc16a7 and Slc16a3 in preimplantation mouse embryos. Biol. Reprod. 2008, 79, 84–92. [Google Scholar] [CrossRef]
- Yu, S.; Wu, Y.; Li, C.; Qu, Z.; Lou, G.; Guo, X.; Ji, J.; Li, N.; Guo, M.; Zhang, M.; et al. Comprehensive analysis of the SLC16A gene family in pancreatic cancer via integrated bioinformatics. Sci. Rep. 2020, 10, 7315. [Google Scholar] [CrossRef]
- Marchiq, I.; Pouyssegur, J. Hypoxia, cancer metabolism and the therapeutic benefit of targeting lactate/H(+) symporters. J. Mol. Med. 2016, 94, 155–171. [Google Scholar] [CrossRef]
- Lee, P.; Chandel, N.S.; Simon, M.C. Cellular adaptation to hypoxia through hypoxia inducible factors and beyond. Nat. Rev. Mol. Cell Biol. 2020, 21, 268–283. [Google Scholar] [CrossRef] [PubMed]
- Ullah, M.S.; Davies, A.J.; Halestrap, A.P. The plasma membrane lactate transporter MCT4, but not MCT1, is up-regulated by hypoxia through a HIF-1alpha-dependent mechanism. J. Biol. Chem. 2006, 281, 9030–9037. [Google Scholar] [CrossRef] [PubMed]
- Chipurupalli, S.; Kannan, E.; Tergaonkar, V.; D’Andrea, R.; Robinson, N. Hypoxia Induced ER Stress Response as an Adaptive Mechanism in Cancer. Int. J. Mol. Sci. 2019, 20, 749. [Google Scholar] [CrossRef] [PubMed]
- Percio, S.; Coltella, N.; Grisanti, S.; Bernardi, R.; Pattini, L. A HIF-1 network reveals characteristics of epithelial-mesenchymal transition in acute promyelocytic leukemia. Genome Med. 2014, 6, 84. [Google Scholar] [CrossRef]
- Higgins, D.F.; Kimura, K.; Bernhardt, W.M.; Shrimanker, N.; Akai, Y.; Hohenstein, B.; Saito, Y.; Johnson, R.S.; Kretzler, M.; Cohen, C.D.; et al. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J. Clin. Investig. 2007, 117, 3810–3820. [Google Scholar] [CrossRef] [PubMed]
- Barriga, E.H.; Maxwell, P.H.; Reyes, A.E.; Mayor, R. The hypoxia factor Hif-1alpha controls neural crest chemotaxis and epithelial to mesenchymal transition. J. Cell Biol. 2013, 201, 759–776. [Google Scholar] [CrossRef]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Schodel, J.; Ratcliffe, P.J. Mechanisms of hypoxia signalling: New implications for nephrology. Nat. Rev. Nephrol. 2019, 15, 641–659. [Google Scholar] [CrossRef]
- Gonzalez, F.J.; Xie, C.; Jiang, C. The role of hypoxia-inducible factors in metabolic diseases. Nat. Rev. Endocrinol. 2018, 15, 21–32. [Google Scholar] [CrossRef]
- Pezzuto, A.; Carico, E. Role of HIF-1 in Cancer Progression: Novel Insights. A Review. Curr. Mol. Med. 2018, 18, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Denko, N.C. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat. Rev. Cancer 2008, 8, 705–713. [Google Scholar] [CrossRef]
- Norman, J.T.; Clark, I.M.; Garcia, P.L. Hypoxia promotes fibrogenesis in human renal fibroblasts. Kidney Int. 2000, 58, 2351–2366. [Google Scholar] [CrossRef]
- Levine, A.J. p53: 800 million years of evolution and 40 years of discovery. Nat. Rev. Cancer 2020, 20, 471–480. [Google Scholar] [CrossRef]
- Bykov, V.J.N.; Eriksson, S.E.; Bianchi, J.; Wiman, K.G. Targeting mutant p53 for efficient cancer therapy. Nat. Rev. Cancer 2018, 18, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Long, J.S.; Crighton, D.; O’Prey, J.; Mackay, G.; Zheng, L.; Palmer, T.M.; Gottlieb, E.; Ryan, K.M. Extracellular adenosine sensing-a metabolic cell death priming mechanism downstream of p53. Mol. Cell 2013, 50, 394–406. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.M.; Attardi, L.D. The role of apoptosis in cancer development and treatment response. Nat. Rev. Cancer 2005, 5, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Roos, W.P.; Thomas, A.D.; Kaina, B. DNA damage and the balance between survival and death in cancer biology. Nat. Rev. Cancer 2016, 16, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, B.A.; El-Deiry, W.S. Targeting apoptosis in cancer therapy. Nat. Rev. Clin. Oncol. 2020, 17, 395–417. [Google Scholar] [CrossRef] [PubMed]
- Bonner, W.M.; Redon, C.E.; Dickey, J.S.; Nakamura, A.J.; Sedelnikova, O.A.; Solier, S.; Pommier, Y. GammaH2AX and cancer. Nat. Rev. Cancer 2008, 8, 957–967. [Google Scholar] [CrossRef]
- Puisieux, A.; Brabletz, T.; Caramel, J. Oncogenic roles of EMT-inducing transcription factors. Nat. Cell Biol. 2014, 16, 488–494. [Google Scholar] [CrossRef] [PubMed]
- Okada, H.; Mak, T.W. Pathways of apoptotic and non-apoptotic death in tumour cells. Nat. Rev. Cancer 2004, 4, 592–603. [Google Scholar] [CrossRef]
- Nakayama, M.; Hong, C.P.; Oshima, H.; Sakai, E.; Kim, S.J.; Oshima, M. Loss of wild-type p53 promotes mutant p53-driven metastasis through acquisition of survival and tumor-initiating properties. Nat. Commun. 2020, 11, 2333. [Google Scholar] [CrossRef]
- Zhang, Y.; Xiong, S.; Liu, B.; Pant, V.; Celii, F.; Chau, G.; Elizondo-Fraire, A.C.; Yang, P.; You, M.J.; El-Naggar, A.K.; et al. Somatic Trp53 mutations differentially drive breast cancer and evolution of metastases. Nat. Commun. 2018, 9, 3953. [Google Scholar] [CrossRef]
- Campbell, H.; Fleming, N.; Roth, I.; Mehta, S.; Wiles, A.; Williams, G.; Vennin, C.; Arsic, N.; Parkin, A.; Pajic, M.; et al. 133p53 isoform promotes tumour invasion and metastasis via interleukin-6 activation of JAK-STAT and RhoA-ROCK signalling. Nat. Commun. 2018, 9, 254. [Google Scholar] [CrossRef]
- McCarthy, N. Metastasis: Understanding the prowess of mutant p53. Nat. Rev. Cancer 2014, 14, 385. [Google Scholar] [CrossRef] [PubMed]
- Weissmueller, S.; Manchado, E.; Saborowski, M.; Morris, J.P.t.; Wagenblast, E.; Davis, C.A.; Moon, S.H.; Pfister, N.T.; Tschaharganeh, D.F.; Kitzing, T.; et al. Mutant p53 drives pancreatic cancer metastasis through cell-autonomous PDGF receptor beta signaling. Cell 2014, 157, 382–394. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.; Vousden, K.H. p53 mutations in cancer. Nat. Cell Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Stracquadanio, G.; Wang, X.; Wallace, M.D.; Grawenda, A.M.; Zhang, P.; Hewitt, J.; Zeron-Medina, J.; Castro-Giner, F.; Tomlinson, I.P.; Goding, C.R.; et al. The importance of p53 pathway genetics in inherited and somatic cancer genomes. Nat. Rev. Cancer 2016, 16, 251–265. [Google Scholar] [CrossRef] [PubMed]
- Hafner, A.; Bulyk, M.L.; Jambhekar, A.; Lahav, G. The multiple mechanisms that regulate p53 activity and cell fate. Nat. Rev. Mol. Cell Biol. 2019, 20, 199–210. [Google Scholar] [CrossRef]
- Kelland, L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584. [Google Scholar] [CrossRef]
- Rottenberg, S.; Disler, C.; Perego, P. The rediscovery of platinum-based cancer therapy. Nat. Rev. Cancer 2020, 21, 37–50. [Google Scholar] [CrossRef]
- Bode, A.M.; Dong, Z. Post-translational modification of p53 in tumorigenesis. Nat. Rev. Cancer 2004, 4, 793–805. [Google Scholar] [CrossRef] [PubMed]
- Shieh, S.Y.; Ikeda, M.; Taya, Y.; Prives, C. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 1997, 91, 325–334. [Google Scholar] [CrossRef]
- Rogakou, E.P.; Boon, C.; Redon, C.; Bonner, W.M. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J. Cell Biol. 1999, 146, 905–916. [Google Scholar] [CrossRef]
- Rogakou, E.P.; Nieves-Neira, W.; Boon, C.; Pommier, Y.; Bonner, W.M. Initiation of DNA fragmentation during apoptosis induces phosphorylation of H2AX histone at serine 139. J. Biol. Chem. 2000, 275, 9390–9395. [Google Scholar] [CrossRef]
- Liu, Y.; Tseng, M.; Perdreau, S.A.; Rossi, F.; Antonescu, C.; Besmer, P.; Fletcher, J.A.; Duensing, S.; Duensing, A. Histone H2AX is a mediator of gastrointestinal stromal tumor cell apoptosis following treatment with imatinib mesylate. Cancer Res. 2007, 67, 2685–2692. [Google Scholar] [CrossRef]
- Lu, C.; Zhu, F.; Cho, Y.Y.; Tang, F.; Zykova, T.; Ma, W.Y.; Bode, A.M.; Dong, Z. Cell apoptosis: Requirement of H2AX in DNA ladder formation, but not for the activation of caspase-3. Mol. Cell 2006, 23, 121–132. [Google Scholar] [CrossRef]
- Lu, C.; Shi, Y.; Wang, Z.; Song, Z.; Zhu, M.; Cai, Q.; Chen, T. Serum starvation induces H2AX phosphorylation to regulate apoptosis via p38 MAPK pathway. FEBS Lett. 2008, 582, 2703–2708. [Google Scholar] [CrossRef]
- Pabla, N.; Huang, S.; Mi, Q.S.; Daniel, R.; Dong, Z. ATR-Chk2 signaling in p53 activation and DNA damage response during cisplatin-induced apoptosis. J. Biol. Chem. 2008, 283, 6572–6583. [Google Scholar] [CrossRef] [PubMed]
- Bedoui, S.; Herold, M.J.; Strasser, A. Emerging connectivity of programmed cell death pathways and its physiological implications. Nat. Rev. Mol. Cell Biol. 2020, 21, 678–695. [Google Scholar] [CrossRef]
- Igney, F.H.; Krammer, P.H. Death and anti-death: Tumour resistance to apoptosis. Nat. Rev. Cancer 2002, 2, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Marino, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-consumption: The interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2014, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.C.; Cullen, S.P.; Martin, S.J. Apoptosis: Controlled demolition at the cellular level. Nat. Rev. Mol. Cell Biol. 2008, 9, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Riedl, S.J.; Salvesen, G.S. The apoptosome: Signalling platform of cell death. Nat. Rev. Mol. Cell Biol. 2007, 8, 405–413. [Google Scholar] [CrossRef]
- Riedl, S.J.; Shi, Y. Molecular mechanisms of caspase regulation during apoptosis. Nat. Rev. Mol. Cell Biol. 2004, 5, 897–907. [Google Scholar] [CrossRef]
- Hersey, P.; Zhang, X.D. How melanoma cells evade trail-induced apoptosis. Nat. Rev. Cancer 2001, 1, 142–150. [Google Scholar] [CrossRef]
- Nguyen, T.; Zhang, X.D.; Hersey, P. Relative resistance of fresh isolates of melanoma to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis. Clin. Cancer Res. 2001, 7, 966s–973s. [Google Scholar]
- Zhang, X.D.; Franco, A.; Myers, K.; Gray, C.; Nguyen, T.; Hersey, P. Relation of TNF-related apoptosis-inducing ligand (TRAIL) receptor and FLICE-inhibitory protein expression to TRAIL-induced apoptosis of melanoma. Cancer Res. 1999, 59, 2747–2753. [Google Scholar]
- Twomey, J.D.; Zhang, B. Circulating Tumor Cells Develop Resistance to TRAIL-Induced Apoptosis Through Autophagic Removal of Death Receptor 5: Evidence from an In Vitro Model. Cancers 2019, 11, 94. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.D.; Franco, A.V.; Nguyen, T.; Gray, C.P.; Hersey, P. Differential localization and regulation of death and decoy receptors for TNF-related apoptosis-inducing ligand (TRAIL) in human melanoma cells. J. Immunol. 2000, 164, 3961–3970. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Marsters, S.; Ye, X.; Luis, E.; Gonzalez, L.; Ashkenazi, A. E-cadherin couples death receptors to the cytoskeleton to regulate apoptosis. Mol. Cell 2014, 54, 987–998. [Google Scholar] [CrossRef]
- Takahashi, K.; Takeda, K.; Saiki, I.; Irimura, T.; Hayakawa, Y. Functional roles of tumor necrosis factor-related apoptosis-inducing ligand-DR5 interaction in B16F10 cells by activating the nuclear factor-kappaB pathway to induce metastatic potential. Cancer Sci. 2013, 104, 558–562. [Google Scholar] [CrossRef]
- Hoogwater, F.J.; Nijkamp, M.W.; Smakman, N.; Steller, E.J.; Emmink, B.L.; Westendorp, B.F.; Raats, D.A.; Sprick, M.R.; Schaefer, U.; Van Houdt, W.J.; et al. Oncogenic K-Ras turns death receptors into metastasis-promoting receptors in human and mouse colorectal cancer cells. Gastroenterology 2010, 138, 2357–2367. [Google Scholar] [CrossRef]
- Sun, S.Y. Understanding the Role of the Death Receptor 5/FADD/caspase-8 Death Signaling in Cancer Metastasis. Mol. Cell Pharmacol. 2011, 3, 31–34. [Google Scholar]
- von Karstedt, S.; Walczak, H. An unexpected turn of fortune: Targeting TRAIL-Rs in KRAS-driven cancer. Cell Death Discov. 2020, 6, 14. [Google Scholar] [CrossRef] [PubMed]
- von Karstedt, S.; Conti, A.; Nobis, M.; Montinaro, A.; Hartwig, T.; Lemke, J.; Legler, K.; Annewanter, F.; Campbell, A.D.; Taraborrelli, L.; et al. Cancer cell-autonomous TRAIL-R signaling promotes KRAS-driven cancer progression, invasion, and metastasis. Cancer Cell 2015, 27, 561–573. [Google Scholar] [CrossRef]
- Kroemer, G.; Levine, B. Autophagic cell death: The story of a misnomer. Nat. Rev. Mol. Cell Biol. 2008, 9, 1004–1010. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef]
- White, E. Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer 2012, 12, 401–410. [Google Scholar] [CrossRef]
- Janku, F.; McConkey, D.J.; Hong, D.S.; Kurzrock, R. Autophagy as a target for anticancer therapy. Nat. Rev. Clin. Oncol. 2011, 8, 528–539. [Google Scholar] [CrossRef] [PubMed]
- Rybstein, M.D.; Bravo-San Pedro, J.M.; Kroemer, G.; Galluzzi, L. The autophagic network and cancer. Nat. Cell Biol. 2018, 20, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef] [PubMed]
- Boya, P.; Reggiori, F.; Codogno, P. Emerging regulation and functions of autophagy. Nat. Cell Biol. 2013, 15, 713–720. [Google Scholar] [CrossRef]
- Mathew, R.; Karantza-Wadsworth, V.; White, E. Role of autophagy in cancer. Nat. Rev. Cancer 2007, 7, 961–967. [Google Scholar] [CrossRef]
- Kondo, Y.; Kanzawa, T.; Sawaya, R.; Kondo, S. The role of autophagy in cancer development and response to therapy. Nat. Rev. Cancer 2005, 5, 726–734. [Google Scholar] [CrossRef]
- Chavez-Dominguez, R.; Perez-Medina, M.; Lopez-Gonzalez, J.S.; Galicia-Velasco, M.; Aguilar-Cazares, D. The Double-Edge Sword of Autophagy in Cancer: From Tumor Suppression to Pro-tumor Activity. Front. Oncol. 2020, 10, 578418. [Google Scholar] [CrossRef]
- Guo, J.Y.; Chen, H.Y.; Mathew, R.; Fan, J.; Strohecker, A.M.; Karsli-Uzunbas, G.; Kamphorst, J.J.; Chen, G.; Lemons, J.M.; Karantza, V.; et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011, 25, 460–470. [Google Scholar] [CrossRef]
- Karsli-Uzunbas, G.; Guo, J.Y.; Price, S.; Teng, X.; Laddha, S.V.; Khor, S.; Kalaany, N.Y.; Jacks, T.; Chan, C.S.; Rabinowitz, J.D.; et al. Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov. 2014, 4, 914–927. [Google Scholar] [CrossRef]
- Lee, C.S.; Lee, L.C.; Yuan, T.L.; Chakka, S.; Fellmann, C.; Lowe, S.W.; Caplen, N.J.; McCormick, F.; Luo, J. MAP kinase and autophagy pathways cooperate to maintain RAS mutant cancer cell survival. Proc. Natl. Acad. Sci. USA 2019, 116, 4508–4517. [Google Scholar] [CrossRef]
- Wen, X.; Klionsky, D.J. At a glance: A history of autophagy and cancer. Semin. Cancer Biol. 2020, 66, 3–11. [Google Scholar] [CrossRef]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef]
- Ichimura, Y.; Kirisako, T.; Takao, T.; Satomi, Y.; Shimonishi, Y.; Ishihara, N.; Mizushima, N.; Tanida, I.; Kominami, E.; Ohsumi, M.; et al. A ubiquitin-like system mediates protein lipidation. Nature 2000, 408, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Kabeya, Y.; Mizushima, N.; Yamamoto, A.; Oshitani-Okamoto, S.; Ohsumi, Y.; Yoshimori, T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J. Cell Sci. 2004, 117, 2805–2812. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I.; Ueno, T.; Kominami, E. Human light chain 3/MAP1LC3B is cleaved at its carboxyl-terminal Met121 to expose Gly120 for lipidation and targeting to autophagosomal membranes. J. Biol. Chem. 2004, 279, 47704–47710. [Google Scholar] [CrossRef] [PubMed]
- La Belle Flynn, A.; Calhoun, B.C.; Sharma, A.; Chang, J.C.; Almasan, A.; Schiemann, W.P. Autophagy inhibition elicits emergence from metastatic dormancy by inducing and stabilizing Pfkfb3 expression. Nat. Commun. 2019, 10, 3668. [Google Scholar] [CrossRef]
- Clem, B.F.; O’Neal, J.; Tapolsky, G.; Clem, A.L.; Imbert-Fernandez, Y.; Kerr, D.A., 2nd; Klarer, A.C.; Redman, R.; Miller, D.M.; Trent, J.O.; et al. Targeting 6-phosphofructo-2-kinase (PFKFB3) as a therapeutic strategy against cancer. Mol. Cancer Ther. 2013, 12, 1461–1470. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Chen, Y.; Zhu, Y. The molecular basis of targeting PFKFB3 as a therapeutic strategy against cancer. Oncotarget 2017, 8, 62793–62802. [Google Scholar] [CrossRef]
- Zhu, W.; Ye, L.; Zhang, J.; Yu, P.; Wang, H.; Ye, Z.; Tian, J. PFK15, a Small Molecule Inhibitor of PFKFB3, Induces Cell Cycle Arrest, Apoptosis and Inhibits Invasion in Gastric Cancer. PLoS ONE 2016, 11, e0163768. [Google Scholar] [CrossRef]
- Giaccia, A.; Siim, B.G.; Johnson, R.S. HIF-1 as a target for drug development. Nat. Rev. Drug Discov. 2003, 2, 803–811. [Google Scholar] [CrossRef]
- Karoulia, Z.; Gavathiotis, E.; Poulikakos, P.I. New perspectives for targeting RAF kinase in human cancer. Nat. Rev. Cancer 2017, 17, 676–691. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, J.S.; Anderson, B.D.; Beight, D.W.; Campbell, R.M.; Jones, M.L.; Herron, D.K.; Lampe, J.W.; McCowan, J.R.; McMillen, W.T.; Mort, N.; et al. Synthesis and activity of new aryl- and heteroaryl-substituted pyrazole inhibitors of the transforming growth factor-beta type I receptor kinase domain. J. Med. Chem. 2003, 46, 3953–3956. [Google Scholar] [CrossRef] [PubMed]
- Willems, E.; Cabral-Teixeira, J.; Schade, D.; Cai, W.; Reeves, P.; Bushway, P.J.; Lanier, M.; Walsh, C.; Kirchhausen, T.; Izpisua Belmonte, J.C.; et al. Small molecule-mediated TGF-beta type II receptor degradation promotes cardiomyogenesis in embryonic stem cells. Cell Stem Cell 2012, 11, 242–252. [Google Scholar] [CrossRef] [PubMed]
- Rosado-Olivieri, E.A.; Aigha, I.I.; Kenty, J.H.; Melton, D.A. Identification of a LIF-Responsive, Replication-Competent Subpopulation of Human beta Cells. Cell Metab. 2020, 31, 327–338.e326. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Zhan, M.; Guo, M.; Yuan, H.; Wang, Y.; Zhang, Y.; Zhang, W.; Chen, S.; de The, H.; Chen, Z.; et al. Yolk sac-derived Pdcd11-positive cells modulate zebrafish microglia differentiation through the NF-kappaB-Tgfbeta1 pathway. Cell Death Differ. 2020. [Google Scholar] [CrossRef]
- Kavallaris, M. Microtubules and resistance to tubulin-binding agents. Nat. Rev. Cancer 2010, 10, 194–204. [Google Scholar] [CrossRef]
- Jordan, M.A.; Wilson, L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 2004, 4, 253–265. [Google Scholar] [CrossRef]
- Gigant, B.; Wang, C.; Ravelli, R.B.; Roussi, F.; Steinmetz, M.O.; Curmi, P.A.; Sobel, A.; Knossow, M. Structural basis for the regulation of tubulin by vinblastine. Nature 2005, 435, 519–522. [Google Scholar] [CrossRef]
- Moudi, M.; Go, R.; Yien, C.Y.; Nazre, M. Vinca alkaloids. Int. J. Prev. Med. 2013, 4, 1231–1235. [Google Scholar]
- Chagas, C.M.; Alisaraie, L. Metabolites of Vinca Alkaloid Vinblastine: Tubulin Binding and Activation of Nausea-Associated Receptors. ACS Omega 2019, 4, 9784–9799. [Google Scholar] [CrossRef] [PubMed]
- Tamura, D.; Arao, T.; Nagai, T.; Kaneda, H.; Aomatsu, K.; Fujita, Y.; Matsumoto, K.; De Velasco, M.A.; Kato, H.; Hayashi, H.; et al. Slug increases sensitivity to tubulin-binding agents via the downregulation of betaIII and betaIVa-tubulin in lung cancer cells. Cancer Med. 2013, 2, 144–154. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Lippard, S.J. Cellular processing of platinum anticancer drugs. Nat. Rev. Drug Discov. 2005, 4, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Gentilin, E.; Simoni, E.; Candito, M.; Cazzador, D.; Astolfi, L. Cisplatin-Induced Ototoxicity: Updates on Molecular Targets. Trends Mol. Med. 2019, 25, 1123–1132. [Google Scholar] [CrossRef]
- Yu, D.; Gu, J.; Chen, Y.; Kang, W.; Wang, X.; Wu, H. Current Strategies to Combat Cisplatin-Induced Ototoxicity. Front. Pharmacol. 2020, 11, 999. [Google Scholar] [CrossRef]
- Nogales, E.; Wolf, S.G.; Khan, I.A.; Luduena, R.F.; Downing, K.H. Structure of tubulin at 6.5 A and location of the taxol-binding site. Nature 1995, 375, 424–427. [Google Scholar] [CrossRef]
- Schiff, P.B.; Horwitz, S.B. Taxol stabilizes microtubules in mouse fibroblast cells. Proc. Natl. Acad. Sci. USA 1980, 77, 1561–1565. [Google Scholar] [CrossRef]
- Freedman, H.; Huzil, J.T.; Luchko, T.; Luduena, R.F.; Tuszynski, J.A. Identification and characterization of an intermediate taxol binding site within microtubule nanopores and a mechanism for tubulin isotype binding selectivity. J. Chem. Inf. Model. 2009, 49, 424–436. [Google Scholar] [CrossRef]
- Kavallaris, M.; Kuo, D.Y.; Burkhart, C.A.; Regl, D.L.; Norris, M.D.; Haber, M.; Horwitz, S.B. Taxol-resistant epithelial ovarian tumors are associated with altered expression of specific beta-tubulin isotypes. J. Clin. Investig. 1997, 100, 1282–1293. [Google Scholar] [CrossRef]
- Kavallaris, M.; Burkhart, C.A.; Horwitz, S.B. Antisense oligonucleotides to class III beta-tubulin sensitize drug-resistant cells to Taxol. Br. J. Cancer 1999, 80, 1020–1025. [Google Scholar] [CrossRef]
- Hari, M.; Yang, H.; Zeng, C.; Canizales, M.; Cabral, F. Expression of class III beta-tubulin reduces microtubule assembly and confers resistance to paclitaxel. Cell Motil. Cytoskelet. 2003, 56, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Kamath, K.; Wilson, L.; Cabral, F.; Jordan, M.A. BetaIII-tubulin induces paclitaxel resistance in association with reduced effects on microtubule dynamic instability. J. Biol. Chem. 2005, 280, 12902–12907. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.M.; Cao, D.; Itami, A.; Pour, P.M.; Hruban, R.H.; Maitra, A.; Ouellette, M.M. Class III beta-tubulin, a marker of resistance to paclitaxel, is overexpressed in pancreatic ductal adenocarcinoma and intraepithelial neoplasia. Histopathology 2007, 51, 539–546. [Google Scholar] [CrossRef]
- Wenger, R.H.; Stiehl, D.P.; Camenisch, G. Integration of oxygen signaling at the consensus HRE. Sci. STKE 2005, 2005, re12. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, S.; Carter, C.; Lynch, M.; Lowinger, T.; Dumas, J.; Smith, R.A.; Schwartz, B.; Simantov, R.; Kelley, S. Discovery and development of sorafenib: A multikinase inhibitor for treating cancer. Nat. Rev. Drug Discov. 2006, 5, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Gild, M.L.; Bullock, M.; Robinson, B.G.; Clifton-Bligh, R. Multikinase inhibitors: A new option for the treatment of thyroid cancer. Nat. Rev. Endocrinol. 2011, 7, 617–624. [Google Scholar] [CrossRef]
- Rudalska, R.; Dauch, D.; Longerich, T.; McJunkin, K.; Wuestefeld, T.; Kang, T.W.; Hohmeyer, A.; Pesic, M.; Leibold, J.; von Thun, A.; et al. In vivo RNAi screening identifies a mechanism of sorafenib resistance in liver cancer. Nat. Med. 2014, 20, 1138–1146. [Google Scholar] [CrossRef]
- Wei, L.; Lee, D.; Law, C.T.; Zhang, M.S.; Shen, J.; Chin, D.W.; Zhang, A.; Tsang, F.H.; Wong, C.L.; Ng, I.O.; et al. Genome-wide CRISPR/Cas9 library screening identified PHGDH as a critical driver for Sorafenib resistance in HCC. Nat. Commun. 2019, 10, 4681. [Google Scholar] [CrossRef]
- Llovet, J.M.; Montal, R.; Sia, D.; Finn, R.S. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 2018, 15, 599–616. [Google Scholar] [CrossRef]
- Capeloa, T.; Benyahia, Z.; Zampieri, L.X.; Blackman, M.; Sonveaux, P. Metabolic and non-metabolic pathways that control cancer resistance to anthracyclines. Semin. Cell Dev. Biol. 2020, 98, 181–191. [Google Scholar] [CrossRef]
- Marinello, J.; Delcuratolo, M.; Capranico, G. Anthracyclines as Topoisomerase II Poisons: From Early Studies to New Perspectives. Int. J. Mol. Sci. 2018, 19, 3480. [Google Scholar] [CrossRef]
- Meredith, A.M.; Dass, C.R. Increasing role of the cancer chemotherapeutic doxorubicin in cellular metabolism. J. Pharm. Pharmacol. 2016, 68, 729–741. [Google Scholar] [CrossRef]
- Stravopodis, D.J.; Karkoulis, P.K.; Konstantakou, E.G.; Melachroinou, S.; Lampidonis, A.D.; Anastasiou, D.; Kachrilas, S.; Messini-Nikolaki, N.; Papassideri, I.S.; Aravantinos, G.; et al. Grade-dependent effects on cell cycle progression and apoptosis in response to doxorubicin in human bladder cancer cell lines. Int. J. Oncol. 2009, 34, 137–160. [Google Scholar]
- Herrmann, J. Adverse cardiac effects of cancer therapies: Cardiotoxicity and arrhythmia. Nat. Rev. Cardiol. 2020, 17, 474–502. [Google Scholar] [CrossRef]
- Palmieri, C.; Krell, J.; James, C.R.; Harper-Wynne, C.; Misra, V.; Cleator, S.; Miles, D. Rechallenging with anthracyclines and taxanes in metastatic breast cancer. Nat. Rev. Clin. Oncol. 2010, 7, 561–574. [Google Scholar] [CrossRef]
- Minotti, G.; Menna, P.; Salvatorelli, E.; Cairo, G.; Gianni, L. Anthracyclines: Molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol. Rev. 2004, 56, 185–229. [Google Scholar] [CrossRef]
- Chittaranjan, S.; Bortnik, S.; Dragowska, W.H.; Xu, J.; Abeysundara, N.; Leung, A.; Go, N.E.; DeVorkin, L.; Weppler, S.A.; Gelmon, K.; et al. Autophagy inhibition augments the anticancer effects of epirubicin treatment in anthracycline-sensitive and -resistant triple-negative breast cancer. Clin. Cancer Res. 2014, 20, 3159–3173. [Google Scholar] [CrossRef]
- Sun, W.L.; Chen, J.; Wang, Y.P.; Zheng, H. Autophagy protects breast cancer cells from epirubicin-induced apoptosis and facilitates epirubicin-resistance development. Autophagy 2011, 7, 1035–1044. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Naiditch, J.; Czurylo, M.; Jie, C.; Lautz, T.; Clark, S.; Jafari, N.; Qiu, Y.; Chu, F.; Madonna, M.B. Differential effect of long-term drug selection with doxorubicin and vorinostat on neuroblastoma cells with cancer stem cell characteristics. Cell Death Dis. 2013, 4, e740. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.C.; Lima, N.D.S.; Sarian, L.O.; Matheu, A.; Ribeiro, M.L.; Derchain, S.F.M. Exosome-mediated breast cancer chemoresistance via miR-155 transfer. Sci. Rep. 2018, 8, 829. [Google Scholar] [CrossRef] [PubMed]
- Balahmar, R.M.; Boocock, D.J.; Coveney, C.; Ray, S.; Vadakekolathu, J.; Regad, T.; Ali, S.; Sivasubramaniam, S. Identification and characterisation of NANOG+/ OCT-4(high)/SOX2+ doxorubicin-resistant stem-like cells from transformed trophoblastic cell lines. Oncotarget 2018, 9, 7054–7065. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Wang, L.; Wang, J.; Chen, T.; Li, H.; Zhang, K.; Chen, J.; Zhen, S.; Tuluhong, D.; Li, J.; et al. microRNA-129-5p suppresses Adriamycin resistance in breast cancer by targeting SOX2. Arch Biochem. Biophys. 2018, 651, 52–60. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WM115 | WM266-4 | ||||||
|---|---|---|---|---|---|---|---|
| IF (phenotype) | Global Expression (100%) | Absence of Expression | Expression in Sub-Populations (%) | Global Expression (100%) | Absence of Expression | Expression in Sub-Populations (%) | |
| Proteins | |||||||
| Vimentin | + | + | |||||
| N-Cadherin | 66 | + | |||||
| E-Cadherin | + | + | |||||
| ZEB1 | 77 | 85 | |||||
| ZEB2 | 14 | 80 | |||||
| SLUG | 63 (54: nuclear strong signal) | 69 (3: nuclear strong signal) | |||||
| p63α | + | + | |||||
| PRRX1 | 77 | 70 | |||||
| Keratin-5 | + | + | |||||
| Keratin-8/18 | + | + | |||||
| PDGFRβ | 74 | 76 | |||||
| LOXL2 | 40 | 46 | |||||
| p-SMAD2 | + | + | |||||
| SOX2 | + | 10 | |||||
| SOX9 | + | + | |||||
| SOX10 | 2 | 11 | |||||
| p-STAT3 | 87 | 87 | |||||
| PCM1 | 83 | + | |||||
| TRA1-60(S) | + | 1.7 (strong signal) | + | ||||
| p-AKT | + | + | |||||
| p-GSK3β | + | + | |||||
| p-ERK1/2 | + | + | |||||
| p-p38 | 90 | + | |||||
| p-S6 | + | + | |||||
| β-Catenin | + | + | |||||
| YAP | 98 | 68 | |||||
| ATF4 | + | + | |||||
| CHOP | + | + | |||||
| MCT1 | 97 | + | |||||
| MCT4 | 95 | + | |||||
| HIF1α | + | 66 | |||||
| p-p53Ser15 | 7 | 7 | |||||
| p-p53Ser37 | 31 | 42 | |||||
| p-H2AX | 48 | 44 | |||||
| a-Caspase-3 | + | + | |||||
| DR5 | + | + | |||||
| ATG12 | + | + | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giannopoulou, A.F.; Velentzas, A.D.; Anagnostopoulos, A.K.; Agalou, A.; Papandreou, N.C.; Katarachia, S.A.; Koumoundourou, D.G.; Konstantakou, E.G.; Pantazopoulou, V.I.; Delis, A.; et al. From Proteomic Mapping to Invasion-Metastasis-Cascade Systemic Biomarkering and Targeted Drugging of Mutant BRAF-Dependent Human Cutaneous Melanomagenesis. Cancers 2021, 13, 2024. https://doi.org/10.3390/cancers13092024
Giannopoulou AF, Velentzas AD, Anagnostopoulos AK, Agalou A, Papandreou NC, Katarachia SA, Koumoundourou DG, Konstantakou EG, Pantazopoulou VI, Delis A, et al. From Proteomic Mapping to Invasion-Metastasis-Cascade Systemic Biomarkering and Targeted Drugging of Mutant BRAF-Dependent Human Cutaneous Melanomagenesis. Cancers. 2021; 13(9):2024. https://doi.org/10.3390/cancers13092024
Chicago/Turabian StyleGiannopoulou, Aikaterini F., Athanassios D. Velentzas, Athanasios K. Anagnostopoulos, Adamantia Agalou, Nikos C. Papandreou, Stamatia A. Katarachia, Dimitra G. Koumoundourou, Eumorphia G. Konstantakou, Vasiliki I. Pantazopoulou, Anastasios Delis, and et al. 2021. "From Proteomic Mapping to Invasion-Metastasis-Cascade Systemic Biomarkering and Targeted Drugging of Mutant BRAF-Dependent Human Cutaneous Melanomagenesis" Cancers 13, no. 9: 2024. https://doi.org/10.3390/cancers13092024
APA StyleGiannopoulou, A. F., Velentzas, A. D., Anagnostopoulos, A. K., Agalou, A., Papandreou, N. C., Katarachia, S. A., Koumoundourou, D. G., Konstantakou, E. G., Pantazopoulou, V. I., Delis, A., Michailidi, M. T., Valakos, D., Chatzopoulos, D., Syntichaki, P., Iconomidou, V. A., Tsitsilonis, O. E., Papassideri, I. S., Voutsinas, G. E., Hatzopoulos, P., ... Stravopodis, D. J. (2021). From Proteomic Mapping to Invasion-Metastasis-Cascade Systemic Biomarkering and Targeted Drugging of Mutant BRAF-Dependent Human Cutaneous Melanomagenesis. Cancers, 13(9), 2024. https://doi.org/10.3390/cancers13092024