
CD80 Expression on Tumor Cells Alters Tumor Microenvironment and Efficacy of Cancer Immunotherapy by CTLA-4 Blockade


Abstract
Simple Summary
Abstract
1. Introduction
2. Results
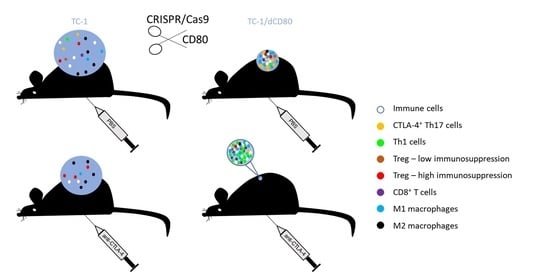
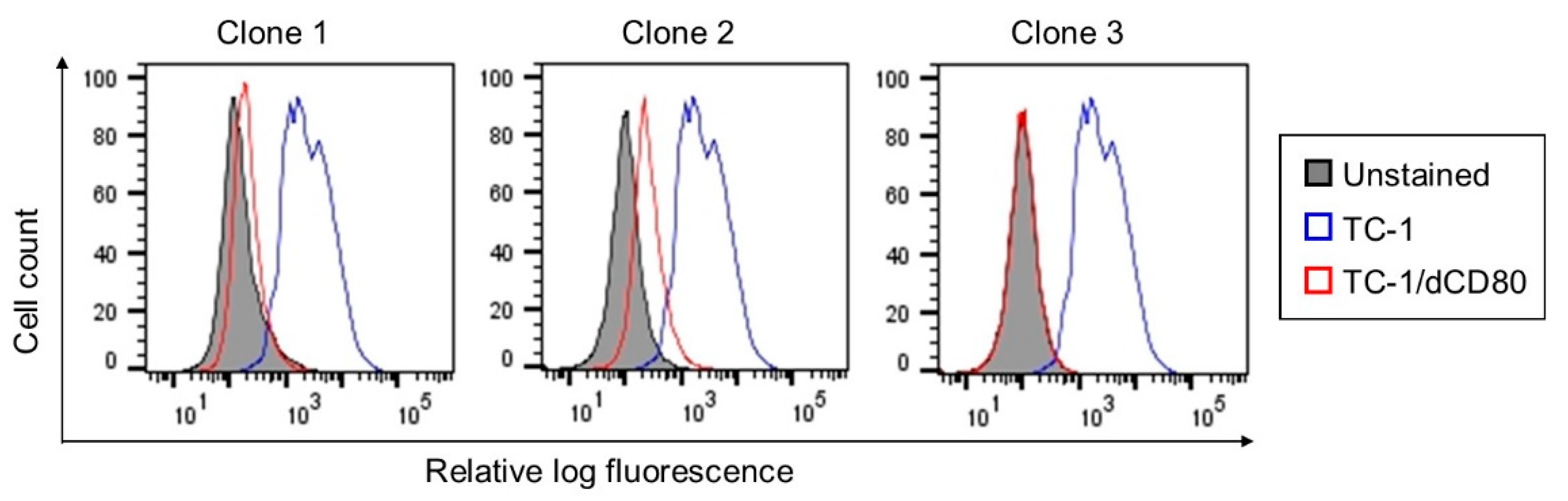
2.1. CD80 Deactivation Reduced Tumor Growth
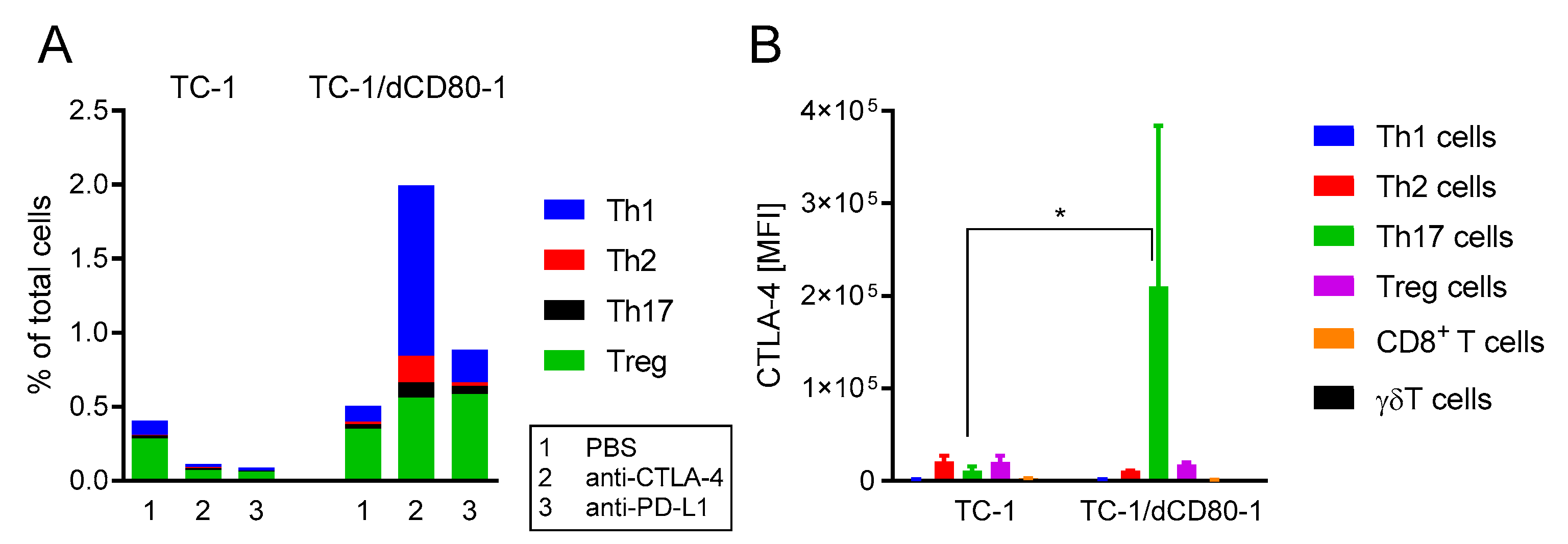
2.2. CD80 Deactivation Altered Immune Reactions and Sensitivity to CTLA-4 Blockade
2.3. CD80 Deactivation and CTLA-4 Blockade Altered Tumor Microenvironment
2.4. CD80 Deactivation Resulted in Increased CTL4-4 Expression on Th17 T Cells and Enhanced Frequency of Th1 Cells in Tumors after CTLA-4 Blockade
2.5. CD80 Deactivation and CTLA-4 Blockade Affected Heterogeneity of Tumor-Infiltrating Treg Cells
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Cells and CD80 Deactivation
4.3. Animal Experiments
4.4. Stimulation of Cells Isolated from Tumors
4.5. Flow Cytometry
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lenschow, D.J.; Su, G.H.; Zuckerman, L.A.; Nabavi, N.; Jellis, C.L.; Gray, G.S.; Miller, J.; Bluestone, J.A. Expression and Functional Significance of an Additional Ligand for CTLA-4. Proc. Natl. Acad. Sci. USA 1993, 90, 11054–11058. [Google Scholar] [CrossRef] [PubMed]
- Sansom, D.M. CD28, CTLA-4 and their ligands: Who does what and to whom? Immunology 2000, 101, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Lindsten, T.; Lee, K.P.; Harris, E.S.; Petryniak, B.; Craighead, N.; Reynolds, P.J.; Lombard, D.B.; Freeman, G.J.; Nadler, L.M.; Gray, G.S. Characterization of CTLA-4 structure and expression on human T cells. J. Immunol. 1993, 151, 3489–3499. [Google Scholar]
- Feng, X.-Y.; Lu, L.; Wang, K.-F.; Zhu, B.-Y.; Wen, X.-Z.; Peng, R.-Q.; Ding, Y.; Li, D.-D.; Li, J.-J.; Li, Y.; et al. Low expression of CD80 predicts for poor prognosis in patients with gastric adenocarcinoma. Future Oncol. Lond. Engl. 2019, 15, 473–483. [Google Scholar] [CrossRef]
- Ganesan, P.L.; Alexander, S.I.; Watson, D.; Logan, G.J.; Zhang, G.Y.; Alexander, I.E. Robust anti-tumor immunity and memory in rag-1-deficient mice following adoptive transfer of cytokine-primed splenocytes and tumor CD80 expression. Cancer Immunol. Immunother. 2007, 56, 1955–1965. [Google Scholar] [CrossRef]
- Tirapu, I.; Huarte, E.; Guiducci, C.; Arina, A.; Zaratiegui, M.; Murillo, O.; Gonzalez, A.; Berasain, C.; Berraondo, P.; Fortes, P.; et al. Low surface expression of B7-1 (CD80) is an immunoescape mechanism of colon carcinoma. Cancer Res. 2006, 66, 2442–2450. [Google Scholar] [CrossRef]
- Ostrand-Rosenberg, S.; Horn, L.A.; Alvarez, J.A. Novel strategies for inhibiting PD-1 pathway-mediated immune suppression while simultaneously delivering activating signals to tumor-reactive T cells. Cancer Immunol. Immunother. 2015, 64, 1287–1293. [Google Scholar] [CrossRef]
- Chaudhri, A.; Xiao, Y.; Klee, A.N.; Wang, X.; Zhu, B.; Freeman, G.J. PD-L1 binds to B7-1 only in cis on the same cell surface. Cancer Immunol. Res. 2018, 6, 921–929. [Google Scholar] [CrossRef]
- Zhao, Y.; Lee, C.K.; Lin, C.-H.; Gassen, R.B.; Xu, X.; Huang, Z.; Xiao, C.; Bonorino, C.; Lu, L.-F.; Bui, J.D.; et al. PD-L1:CD80 cis-heterodimer triggers the co-stimulatory receptor CD28 while repressing the inhibitory PD-1 and CTLA-4 pathways. Immunity 2019, 51, 1059–1073.e9. [Google Scholar] [CrossRef]
- Mayoux, M.; Roller, A.; Pulko, V.; Sammicheli, S.; Chen, S.; Sum, E.; Jost, C.; Fransen, M.F.; Buser, R.B.; Kowanetz, M.; et al. Dendritic cells dictate responses to PD-L1 blockade cancer immunotherapy. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- Liu, A.; Hu, P.; Khawli, L.A.; Epstein, A.L. B7.1/NHS76: A new costimulator fusion protein for the immunotherapy of solid tumors. J. Immunother. 2006, 29, 425–435. [Google Scholar] [CrossRef]
- Mikami, N.; Kawakami, R.; Chen, K.Y.; Sugimoto, A.; Ohkura, N.; Sakaguchi, S. Epigenetic conversion of conventional T Cells into regulatory T cells by CD28 signal deprivation. Proc. Natl. Acad. Sci. USA 2020, 117, 12258–12268. [Google Scholar] [CrossRef] [PubMed]
- Kalekar, L.A.; Schmiel, S.E.; Nandiwada, S.L.; Lam, W.Y.; Barsness, L.O.; Zhang, N.; Stritesky, G.L.; Malhotra, D.; Pauken, K.E.; Linehan, J.L.; et al. CD4+ T cell anergy prevents autoimmunity and generates regulatory T cell precursors. Nat. Immunol. 2016, 17, 304–314. [Google Scholar] [CrossRef] [PubMed]
- Pletinckx, K.; Vaeth, M.; Schneider, T.; Beyersdorf, N.; Hünig, T.; Berberich-Siebelt, F.; Lutz, M.B. Immature dendritic cells convert anergic nonregulatory T Cells into Foxp3−IL-10+ regulatory T cells by engaging CD28 and CTLA-4. Eur. J. Immunol. 2015, 45, 480–491. [Google Scholar] [CrossRef]
- Ronchetti, S.; Ricci, E.; Petrillo, M.G.; Cari, L.; Migliorati, G.; Nocentini, G.; Riccardi, C. Glucocorticoid-induced tumour necrosis factor receptor-related protein: A key marker of functional regulatory T cells. J. Immunol. Res. 2015, 2015, 171520. [Google Scholar] [CrossRef]
- Sun, B.; Liu, M.; Cui, M.; Li, T. Granzyme b-expressing treg cells are enriched in colorectal cancer and present the potential to eliminate autologous t conventional cells. Immunol. Lett. 2020, 217, 7–14. [Google Scholar] [CrossRef]
- Di Virgilio, F.; Sarti, A.C.; Falzoni, S.; De Marchi, E.; Adinolfi, E. Extracellular ATP and P2 purinergic signalling in the tumour microenvironment. Nat. Rev. Cancer 2018, 18, 601–618. [Google Scholar] [CrossRef] [PubMed]
- de Leve, S.; Wirsdörfer, F.; Jendrossek, V. Targeting the immunomodulatory CD73/adenosine system to improve the therapeutic gain of radiotherapy. Front. Immunol. 2019, 10, 698. [Google Scholar] [CrossRef]
- Wing, K.; Onishi, Y.; Prieto-Martin, P.; Yamaguchi, T.; Miyara, M.; Fehervari, Z.; Nomura, T.; Sakaguchi, S. CTLA-4 control over Foxp3+ regulatory T cell function. Science 2008, 322, 271–275. [Google Scholar] [CrossRef]
- Hou, T.Z.; Qureshi, O.S.; Wang, C.J.; Baker, J.; Young, S.P.; Walker, L.S.K.; Sansom, D.M. A transendocytosis model of CTLA-4 function predicts its suppressive behavior on regulatory T Cells. J. Immunol. Baltim. Md 1950 2015, 194, 2148–2159. [Google Scholar] [CrossRef]
- Sugár, I.P.; Das, J.; Jayaprakash, C.; Sealfon, S.C. Multiscale modeling of complex formation and CD80 depletion during immune synapse development. Biophys. J. 2017, 112, 997–1009. [Google Scholar] [CrossRef] [PubMed]
- Rudd, C.E. CTLA-4 co-receptor impacts on the function of treg and CD8+ T-cell subsets. Eur. J. Immunol. 2009, 39, 687–690. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-T.; Workman, C.J.; Flies, D.; Pan, X.; Marson, A.L.; Zhou, G.; Hipkiss, E.L.; Ravi, S.; Kowalski, J.; Levitsky, H.I.; et al. Role of LAG-3 in regulatory T cells. Immunity 2004, 21, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Maruhashi, T.; Sugiura, D.; Okazaki, I.; Okazaki, T. LAG-3: From molecular functions to clinical applications. J. Immunother. Cancer 2020, 8, e001014. [Google Scholar] [CrossRef]
- Hansen, W.; Hutzler, M.; Abel, S.; Alter, C.; Stockmann, C.; Kliche, S.; Albert, J.; Sparwasser, T.; Sakaguchi, S.; Westendorf, A.M.; et al. Neuropilin 1 deficiency on CD4+Foxp3+ regulatory t cells impairs mouse melanoma growth. J. Exp. Med. 2012, 209, 2001–2016. [Google Scholar] [CrossRef] [PubMed]
- Arce Vargas, F.; Furness, A.J.S.; Litchfield, K.; Joshi, K.; Rosenthal, R.; Ghorani, E.; Solomon, I.; Lesko, M.H.; Ruef, N.; Roddie, C.; et al. Fc effector function contributes to the activity of human anti-CTLA-4 antibodies. Cancer Cell 2018, 33, 649–663.e4. [Google Scholar] [CrossRef] [PubMed]
- Kamada, T.; Togashi, Y.; Tay, C.; Ha, D.; Sasaki, A.; Nakamura, Y.; Sato, E.; Fukuoka, S.; Tada, Y.; Tanaka, A.; et al. PD-1+ regulatory t cells amplified by PD-1 blockade promote hyperprogression of cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 9999–10008. [Google Scholar] [CrossRef]
- Tan, C.L.; Kuchroo, J.R.; Sage, P.T.; Liang, D.; Francisco, L.M.; Buck, J.; Thaker, Y.R.; Zhang, Q.; McArdel, S.L.; Juneja, V.R.; et al. PD-1 restraint of regulatory T cell suppressive activity is critical for immune tolerance. J. Exp. Med. 2021, 218, e20182232. [Google Scholar] [CrossRef]
- Cameron, F.; Whiteside, G.; Perry, C. Ipilimumab. Drugs 2011, 71, 1093–1104. [Google Scholar] [CrossRef]
- Kwok, G.; Yau, T.C.C.; Chiu, J.W.; Tse, E.; Kwong, Y.-L. Pembrolizumab (Keytruda). Hum. Vaccines Immunother. 2016, 12, 2777–2789. [Google Scholar] [CrossRef]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef]
- Sul, J.; Blumenthal, G.M.; Jiang, X.; He, K.; Keegan, P.; Pazdur, R. FDA approval summary: Pembrolizumab for the treatment of patients with metastatic non-small cell lung cancer whose tumors express programmed death-ligand 1. Oncologist 2016, 21, 643–650. [Google Scholar] [CrossRef]
- Barrueto, L.; Caminero, F.; Cash, L.; Makris, C.; Lamichhane, P.; Deshmukh, R.R. Resistance to checkpoint inhibition in cancer immunotherapy. Transl. Oncol. 2020, 13, 100738. [Google Scholar] [CrossRef]
- Janousková, O.; Síma, P.; Kunke, D. Combined suicide gene and immunostimulatory gene therapy using AAV-mediated gene transfer to HPV-16 transformed mouse cell: Decrease of oncogenicity and induction of protection. Int. J. Oncol. 2003, 22, 569–577. [Google Scholar] [CrossRef]
- Bodor, J.N.; Boumber, Y.; Borghaei, H. Biomarkers for immune checkpoint inhibition in non-small cell lung cancer (NSCLC). Cancer 2020, 126, 260–270. [Google Scholar] [CrossRef]
- Bernsen, M.R.; Håkansson, L.; Gustafsson, B.; Krysander, L.; Rettrup, B.; Ruiter, D.; Håkansson, A. On the biological relevance of MHC Class II and B7 expression by tumour cells in melanoma metastases. Br. J. Cancer 2003, 88, 424–431. [Google Scholar] [CrossRef]
- Marchiori, C.; Scarpa, M.; Kotsafti, A.; Morgan, S.; Fassan, M.; Guzzardo, V.; Porzionato, A.; Angriman, I.; Ruffolo, C.; Sut, S.; et al. Epithelial CD80 promotes immune surveillance of colonic preneoplastic lesions and its expression is increased by oxidative stress through STAT3 in colon cancer cells. J. Exp. Clin. Cancer Res. 2019, 38, 190. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yang, Y.; Inoue, H.; Mori, M.; Akiyoshi, T. The expression of costimulatory molecules CD80 and CD86 in human carcinoma cell lines: Its regulation by interferon γ and interleukin-10. Cancer Immunol. Immunother. 1996, 43, 213–219. [Google Scholar] [CrossRef]
- Madonna, G.; Ballesteros-Merino, C.; Feng, Z.; Bifulco, C.; Capone, M.; Giannarelli, D.; Mallardo, D.; Simeone, E.; Grimaldi, A.M.; Caracò, C.; et al. PD-L1 Expression with immune-infiltrate evaluation and outcome prediction in melanoma patients treated with ipilimumab. OncoImmunology 2018, 7, e1405206. [Google Scholar] [CrossRef]
- Bogen, B.; Fauskanger, M.; Haabeth, O.A.; Tveita, A. CD4+ T Cells indirectly kill tumor cells via induction of cytotoxic macrophages in mouse models. Cancer Immunol. Immunother. 2019, 68, 1865–1873. [Google Scholar] [CrossRef]
- Eisel, D.; Das, K.; Dickes, E.; König, R.; Osen, W.; Eichmüller, S.B. Cognate interaction with CD4+ T cells instructs tumor-associated macrophages to acquire m1-like phenotype. Front. Immunol. 2019, 10, 219. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Chhatar, S.; Mishra, A.; Lal, G. Natural killer T cell activation increases INOS+CD206- M1 macrophage and controls the growth of solid tumor. J. Immunother. Cancer 2019, 7, 208. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.V.; Gibson, H.M.; Aufiero, B.M.; Wilson, A.J.; Hafner, M.S.; Mi, Q.-S.; Wong, H.K. Differential CTLA-4 expression in human CD4+ versus CD8+ T cells is associated with increased NFAT1 and inhibition of CD4+ proliferation. Genes Immun. 2014, 15, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Krummey, S.M.; Hartigan, C.R.; Liu, D.; Ford, M.L. CD28-dependent CTLA-4 expression fine-tunes the activation of human Th17 cells. iScience 2020, 23, 100912. [Google Scholar] [CrossRef]
- Beyranvand Nejad, E.; van der Sluis, T.C.; van Duikeren, S.; Yagita, H.; Janssen, G.M.; van Veelen, P.A.; Melief, C.J.M.; van der Burg, S.H.; Arens, R. Tumor eradication by cisplatin is sustained by CD80/86-mediated costimulation of CD8+ T cells. Cancer Res. 2016, 76, 6017–6029. [Google Scholar] [CrossRef]
- Liu, Z.; Zhou, H.; Wang, W.; Fu, Y.-X.; Zhu, M. A Novel dendritic cell targeting HPV16 E7 synthetic vaccine in combination with PD-L1 blockade elicits therapeutic antitumor immunity in mice. Oncoimmunology 2016, 5, e1147641. [Google Scholar] [CrossRef]
- van Hooren, L.; Sandin, L.C.; Moskalev, I.; Ellmark, P.; Dimberg, A.; Black, P.; Tötterman, T.H.; Mangsbo, S.M. Local checkpoint inhibition of CTLA-4 as a monotherapy or in combination with anti-PD1 prevents the growth of murine bladder cancer. Eur. J. Immunol. 2017, 47, 385–393. [Google Scholar] [CrossRef]
- Fiegle, E.; Doleschel, D.; Koletnik, S.; Rix, A.; Weiskirchen, R.; Borkham-Kamphorst, E.; Kiessling, F.; Lederle, W. Dual CTLA-4 and PD-L1 blockade inhibits tumor growth and liver metastasis in a highly aggressive orthotopic mouse model of colon cancer. Neoplasia 2019, 21, 932–944. [Google Scholar] [CrossRef]
- Baweja, A.; Mar, N. Metastatic penile squamous cell carcinoma with dramatic response to combined checkpoint blockade with ipilimumab and nivolumab. J. Oncol. Pharm. Pract. 2021, 27, 212–215. [Google Scholar] [CrossRef]
- Parikh, N.D.; Marshall, A.; Betts, K.A.; Song, J.; Zhao, J.; Yuan, M.; Wu, A.; Huff, K.D.; Kim, R. Network meta-analysis of nivolumab plus ipilimumab in the second-line setting for advanced hepatocellular carcinoma. J. Comp. Eff. Res. 2021. [Google Scholar] [CrossRef]
- Garrett-Thomson, S.C.; Massimi, A.; Fedorov, E.V.; Bonanno, J.B.; Scandiuzzi, L.; Hillerich, B.; Iii, R.D.S.; Love, J.D.; Garforth, S.J.; Guha, C.; et al. Mechanistic dissection of the PD-L1:B7-1 co-inhibitory immune complex. PLoS ONE 2020, 15, e0233578. [Google Scholar] [CrossRef]
- Vackova, J.; Piatakova, A.; Polakova, I.; Smahel, M. Abrogation of IFN-γ signaling may not worsen sensitivity to PD-1/PD-L1 blockade. Int. J. Mol. Sci. 2020, 21, 1806. [Google Scholar] [CrossRef]
- Spranger, S.; Koblish, H.K.; Horton, B.; Scherle, P.A.; Newton, R.; Gajewski, T.F. Mechanism of tumor rejection with doublets of CTLA-4, PD-1/PD-L1, or IDO blockade involves restored IL-2 production and proliferation of CD8+ T cells directly within the tumor microenvironment. J. Immunother. Cancer 2014, 2, 3. [Google Scholar] [CrossRef]
- Ahn, E.; Araki, K.; Hashimoto, M.; Li, W.; Riley, J.L.; Cheung, J.; Sharpe, A.H.; Freeman, G.J.; Irving, B.A.; Ahmed, R. Role of PD-1 during effector CD8 T cell differentiation. Proc. Natl. Acad. Sci. USA 2018, 115, 4749–4754. [Google Scholar] [CrossRef]
- Siddiqui, I.; Schaeuble, K.; Chennupati, V.; Fuertes Marraco, S.A.; Calderon-Copete, S.; Pais Ferreira, D.; Carmona, S.J.; Scarpellino, L.; Gfeller, D.; Pradervand, S.; et al. Intratumoral Tcf1+PD-1+CD8+ T cells with stem-like properties promote tumor control in response to vaccination and checkpoint blockade immunotherapy. Immunity 2019, 50, 195–211.e10. [Google Scholar] [CrossRef]
- Sanseviero, E.; O’Brien, E.M.; Karras, J.R.; Shabaneh, T.B.; Aksoy, B.A.; Xu, W.; Zheng, C.; Yin, X.; Xu, X.; Karakousis, G.C.; et al. Anti-CTLA-4 activates intratumoral NK cells and combined with IL15/IL15Rα complexes enhances tumor control. Cancer Immunol. Res. 2019, 7, 1371–1380. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Hogg, G.D.; DeNardo, D.G. Rethinking Immune checkpoint blockade: Beyond the T cell. J. Immunother. Cancer 2021, 9, e001460. [Google Scholar] [CrossRef]
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef]
- Tosolini, M.; Kirilovsky, A.; Mlecnik, B.; Fredriksen, T.; Mauger, S.; Bindea, G.; Berger, A.; Bruneval, P.; Fridman, W.-H.; Pagès, F.; et al. Clinical impact of different classes of infiltrating t cytotoxic and helper cells (Th1, Th2, Treg, Th17) in patients with colorectal cancer. Cancer Res. 2011, 71, 1263–1271. [Google Scholar] [CrossRef]
- De Simone, V.; Franzè, E.; Ronchetti, G.; Colantoni, A.; Fantini, M.C.; Di Fusco, D.; Sica, G.S.; Sileri, P.; MacDonald, T.T.; Pallone, F.; et al. Th17-type cytokines, IL-6 and TNF-α synergistically activate STAT3 and NF-KB to promote colorectal cancer cell growth. Oncogene 2015, 34, 3493–3503. [Google Scholar] [CrossRef]
- Kaewkangsadan, V.; Verma, C.; Eremin, J.M.; Cowley, G.; Ilyas, M.; Eremin, O. Crucial contributions by t lymphocytes (effector, regulatory, and checkpoint inhibitor) and cytokines (TH1, TH2, and TH17) to a pathological complete response induced by neoadjuvant chemotherapy in women with breast cancer. J. Immunol. Res. 2016, 2016, 4757405. [Google Scholar] [CrossRef]
- Ohue, Y.; Nishikawa, H. Regulatory T (Treg) cells in cancer: Can treg cells be a new therapeutic target? Cancer Sci. 2019, 110, 2080–2089. [Google Scholar] [CrossRef]
- Liu, D.; Xing, S.; Wang, W.; Huang, X.; Lin, H.; Chen, Y.; Lan, K.; Chen, L.; Luo, F.; Qin, S.; et al. Prognostic value of serum soluble interleukin-23 receptor and related T-helper 17 cell cytokines in non-small cell lung carcinoma. Cancer Sci. 2020, 111, 1093–1102. [Google Scholar] [CrossRef]
- Walunas, T.L.; Bluestone, J.A. CTLA-4 Regulates tolerance induction and t cell differentiation in vivo. J. Immunol. 1998, 160, 3855–3860. [Google Scholar] [PubMed]
- Gao, J.; Shi, L.Z.; Zhao, H.; Chen, J.; Xiong, L.; He, Q.; Chen, T.; Roszik, J.; Bernatchez, C.; Woodman, S.E.; et al. Loss of IFN-γ pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell 2016, 167, 397–404.e9. [Google Scholar] [CrossRef]
- Wei, S.C.; Levine, J.H.; Cogdill, A.P.; Zhao, Y.; Anang, N.-A.A.S.; Andrews, M.C.; Sharma, P.; Wang, J.; Wargo, J.A.; Pe’er, D.; et al. Distinct cellular mechanisms underlie anti-CTLA-4 and anti-PD-1 checkpoint blockade. Cell 2017, 170, 1120–1133.e17. [Google Scholar] [CrossRef]
- Zhang, M.; Wu, Y.; Bastian, D.; Iamsawat, S.; Chang, J.; Daenthanasanmak, A.; Nguyen, H.D.; Schutt, S.; Dai, M.; Chen, F.; et al. Inducible T-cell co-stimulator impacts chronic graft-versus-host disease by regulating both pathogenic and regulatory t cells. Front. Immunol. 2018, 9, 1461. [Google Scholar] [CrossRef]
- Li, D.-Y.; Xiong, X.-Z. ICOS+ tregs: A functional subset of tregs in immune diseases. Front. Immunol. 2020, 11, 2104. [Google Scholar] [CrossRef] [PubMed]
- Ricco, M.L.; Ronin, E.; Collares, D.; Divoux, J.; Grégoire, S.; Wajant, H.; Gomes, T.; Grinberg-Bleyer, Y.; Baud, V.; Marodon, G.; et al. Tumor necrosis factor receptor family costimulation increases regulatory T-cell activation and function via NF-ΚB. Eur. J. Immunol. 2020, 50, 972–985. [Google Scholar] [CrossRef]
- Ikebuchi, R.; Fujimoto, M.; Nakanishi, Y.; Okuyama, H.; Moriya, T.; Kusumoto, Y.; Tomura, M. Functional phenotypic diversity of regulatory T cells remaining in inflamed skin. Front. Immunol. 2019, 10, 1098. [Google Scholar] [CrossRef]
- Predina, J.; Eruslanov, E.; Judy, B.; Kapoor, V.; Cheng, G.; Wang, L.-C.; Sun, J.; Moon, E.K.; Fridlender, Z.G.; Albelda, S.; et al. Changes in the local tumor microenvironment in recurrent cancers may explain the failure of vaccines after surgery. Proc. Natl. Acad. Sci. USA 2013, 110, E415–E424. [Google Scholar] [CrossRef]
- Lin, K.-Y.; Guarnieri, F.G.; Staveley-O’Carroll, K.F.; Levitsky, H.I.; August, J.T.; Pardoll, D.M.; Wu, T.-C. Treatment of established tumors with a novel vaccine that enhances major histocompatibility class II presentation of tumor antigen. Cancer Res. 1996, 56, 21–26. [Google Scholar] [PubMed]
- Van Gassen, S.; Callebaut, B.; Van Helden, M.J.; Lambrecht, B.N.; Demeester, P.; Dhaene, T.; Saeys, Y. FlowSOM: Using self-organizing maps for visualization and interpretation of cytometry data. Cytol. J. Int. Soc. Anal. Cytol. 2015, 87, 636–645. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antigen | Conjugate | Clone | Source | Staining | Panels | |||
|---|---|---|---|---|---|---|---|---|
| CD11b | BV421 | M1/70 | BioLegend | Surface | 1 | |||
| CD11c | APC-Cy7 | N418 | BioLegend | Surface | 1 | |||
| CD25 | APC | PC61.5 | eBiosciences | Surface | 1 | 1 | 1 | |
| CD3 | APC-Cy7 | 145-2C11 | BioLegend | Surface | 1 | 1 | 1 | |
| CD317 | APC | 927 | BioLegend | Surface | 1 | |||
| CD4 | BV5101/PerCP-Cy5.52 | RM4-5 | BioLegend | Surface | 1 | 2 | 2 | |
| CD45 | Alexa Fluor 700 | 30-F11 | BioLegend | Surface | 1 | 1 | 1 | 1 |
| CD73 | BV605 | TY/11.8 | BioLegend | Surface | 1 | |||
| CD8 | FITC | 53-6.7 | BD Biosciences | Surface | 1 | 1 | ||
| CTLA-4 | PE/Dazzle594 | UC10-4B9 | BioLegend | Surface | 1 | 1 | ||
| F4/80 | BV510 | BM8 | BioLegend | Surface | 1 | |||
| Foxp3 | PE | FJK-16s | eBiosciences | Nuclear | 1 | 1 | 1 | |
| GITR | FITC | DTA-1 | BioLegend | Surface | 1 | |||
| Granzyme B | PE/Cy7 | NGZB | BioLegend | Intracellular | 1 | 1 | ||
| ICOS | BV650 | 7E.17G9 | BioLegend | Surface | 1 | |||
| IFN-γ | BV421 | XMG1.2 | BioLegend | Intracellular | 1 | |||
| IL-17A | BV650 | TC11-18H10.1 | BioLegend | Intracellular | 1 | |||
| IL-4 | BV786 | 11B11 | BD Biosciences | Intracellular | 1 | |||
| Lag3 | BV785 | C9B7W | BioLegend | Surface | 1 | |||
| Ly6C | BV786 | HK1.4 | BioLegend | Surface | 1 | |||
| Ly6G | FITC | 1A8 | BioLegend | Surface | 1 | |||
| MHC-II | PE-Cy7 | 114.15.2 | BioLegend | Surface | 1 | |||
| NK1.1 | BV650 | PK136 | BioLegend | Surface | 1 | |||
| Nrp-1 | BV 421 | 3E12 | BioLegend | Surface | 1 | |||
| PD-1 | PE-Cy71/PE2 | 29F.1A12 | BioLegend | Surface | 1 | 2 | ||
| PD-L1 | PE-DAZZLE 594 1/BV650 2 | 10F.9G2 | BioLegend | Surface | 1 | 2 | ||
| TCR γ/δ | BV605 | GL3 | BioLegend | Surface | 1 | 1 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vackova, J.; Polakova, I.; Johari, S.D.; Smahel, M. CD80 Expression on Tumor Cells Alters Tumor Microenvironment and Efficacy of Cancer Immunotherapy by CTLA-4 Blockade. Cancers 2021, 13, 1935. https://doi.org/10.3390/cancers13081935
Vackova J, Polakova I, Johari SD, Smahel M. CD80 Expression on Tumor Cells Alters Tumor Microenvironment and Efficacy of Cancer Immunotherapy by CTLA-4 Blockade. Cancers. 2021; 13(8):1935. https://doi.org/10.3390/cancers13081935
Chicago/Turabian StyleVackova, Julie, Ingrid Polakova, Shweta Dilip Johari, and Michal Smahel. 2021. "CD80 Expression on Tumor Cells Alters Tumor Microenvironment and Efficacy of Cancer Immunotherapy by CTLA-4 Blockade" Cancers 13, no. 8: 1935. https://doi.org/10.3390/cancers13081935
APA StyleVackova, J., Polakova, I., Johari, S. D., & Smahel, M. (2021). CD80 Expression on Tumor Cells Alters Tumor Microenvironment and Efficacy of Cancer Immunotherapy by CTLA-4 Blockade. Cancers, 13(8), 1935. https://doi.org/10.3390/cancers13081935