
Functional Characterization of Circulating Tumor Cells (CTCs) from Metastatic ER+/HER2− Breast Cancer Reveals Dependence on HER2 and FOXM1 for Endocrine Therapy Resistance and Tumor Cell Survival: Implications for Treatment of ER+/HER2− Breast Cancer

 ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Results
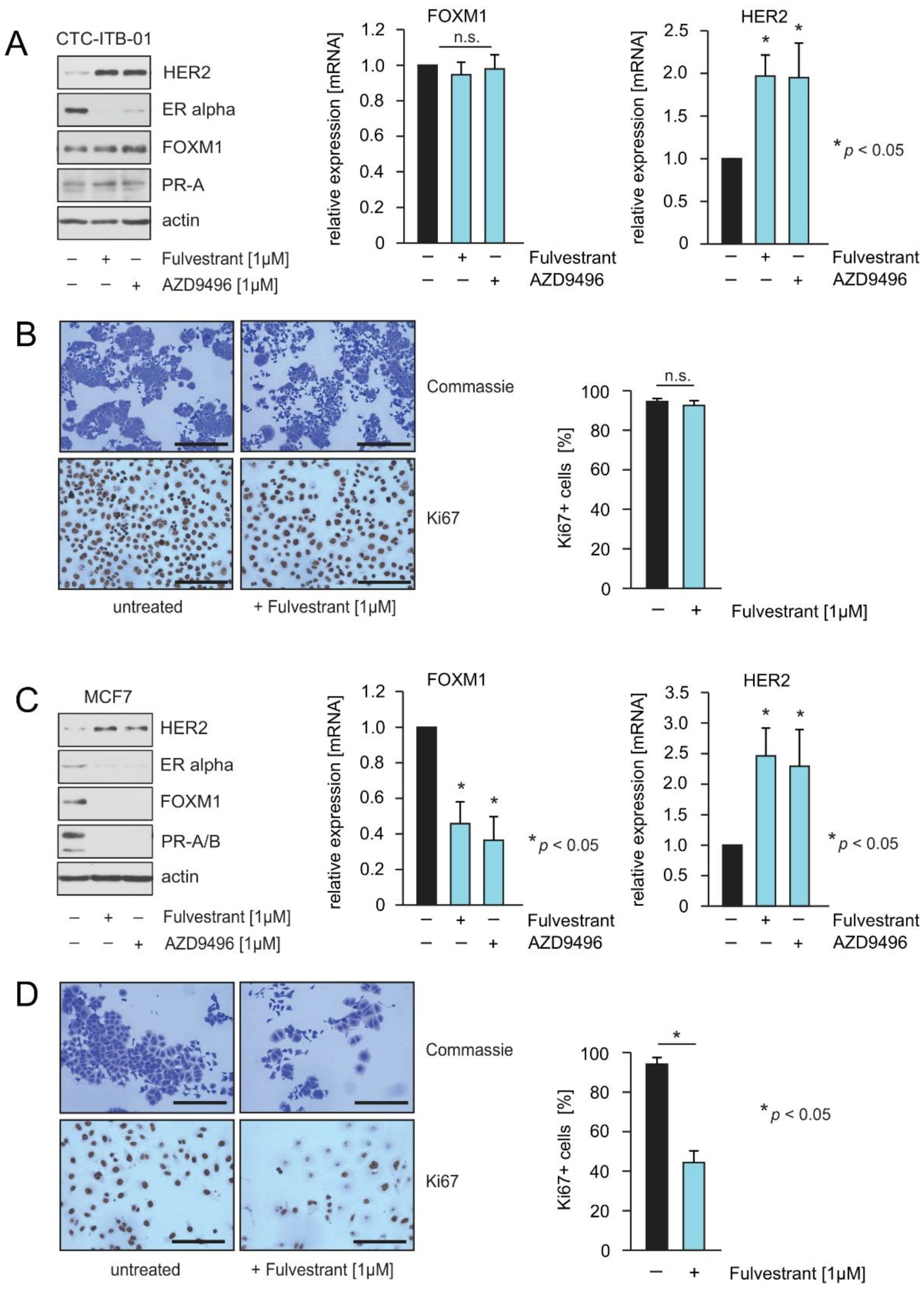
2.1. CTC-ITB-01 Cells Established from a Patient with Metastatic ER+/HER2− Breast Cancer Show No Response to Pharmacological Inhibition of ER alpha
2.2. The ERα-Degrader Fulvestrant and AZD9496 Inhibit Cell Cycle Progression and FOXM1 Expression in MCF7 but Not in CTC-ITB-01 Cells
2.3. Knockdown Experiments Reveal a Role for HER2 in FOXM1 Expression, Growth and Survival in CTC-ITB-01 Cells
2.4. Pharmacological HER2 Inhibition Has Different Effects in CTC-ITB-01 and MCF7 Cells
2.5. Treatment of CTC-ITB-01 Cells with the Proteasome Inhibitor Carfilzomib Causes Inhibition of ER alpha, FOXM1 and HER2 Expression and Induction of Cell Death
2.6. HER2 and FOXM1 Expression within CTC-ITB-01 Cells Depends on NFkB-Signaling
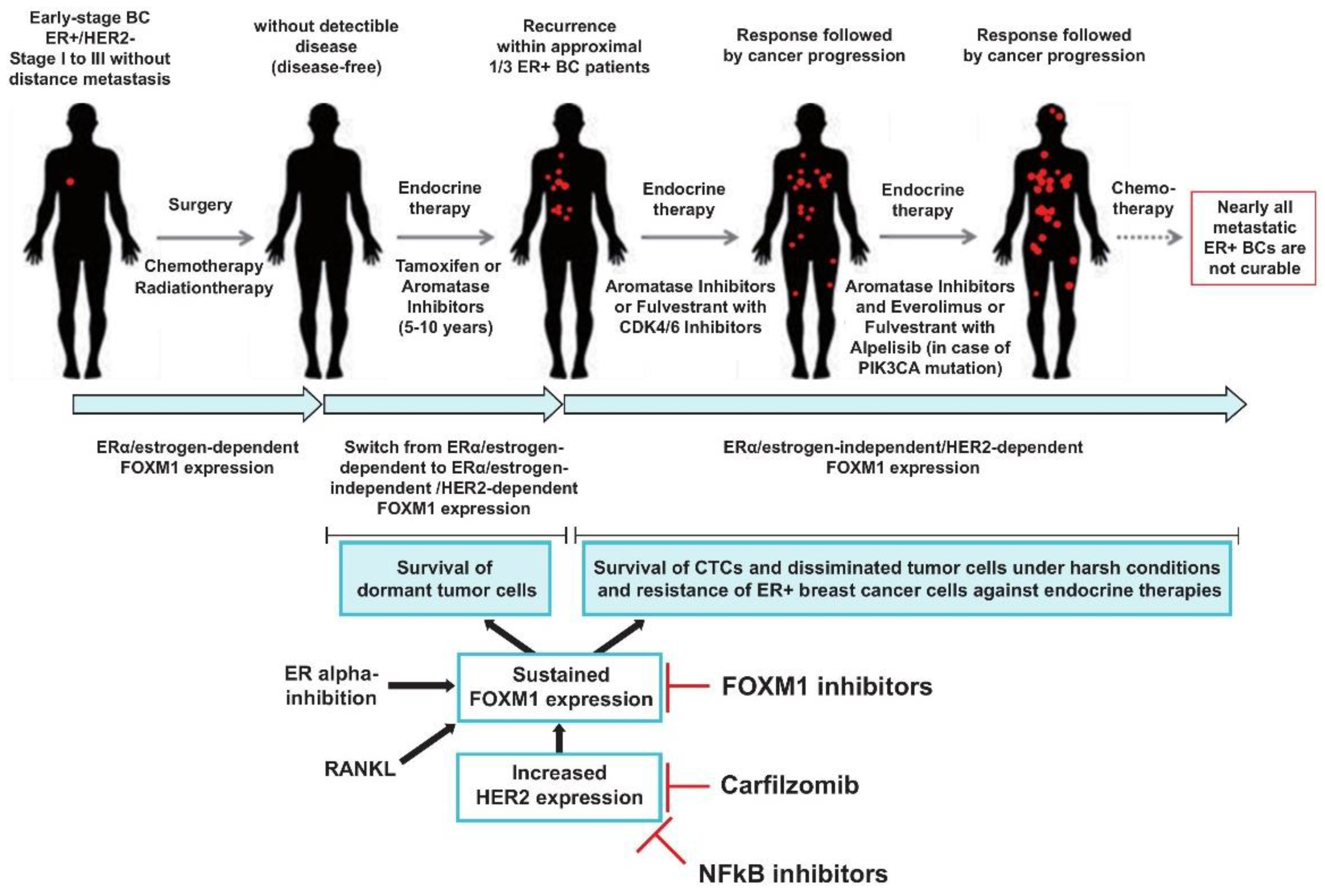
3. Discussion
4. Materials and Methods
4.1. Cultivation of CTC-ITB-01 and Established MCF7 Cells
4.2. Viral Transduction
4.3. Other Methods
4.4. HER2 CISH and Immunohistochemical Staining and Immunofluorescence
4.5. Cell Cycle Analysis
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jordan:, V.C.; Brodie, A.M. Development and evolution of therapies targeted to the estrogen receptor for the treatment and prevention of breast cancer. Steroids 2007, 72, 7–25. [Google Scholar] [CrossRef] [PubMed]
- Jordan, V.C.; O’Malley, B.W. Selective estrogen-receptor modulators and antihormonal resistance in breast cancer. J. Clin. Oncol. 2007, 25, 5815–5824. [Google Scholar] [CrossRef]
- Bense, R.D.; Qiu, S.Q.; de Vries, E.G.E.; Schröder, C.P.; Fehrmann, R.S.N. Considering the biology of late recurrences in selecting patients for extended enocrine therapy in breast cancer. Cancer Treat. Rev. 2018, 70, 118–126. [Google Scholar] [CrossRef]
- Jordan, V.C. Selective estrogen receptor modulation: Concept and consequences in cancer. Cancer Cell 2004, 5, 207–213. [Google Scholar] [CrossRef]
- Bergamaschi, A.; Madak-Erdogan, Z.; Kim, Y.J.; Choi, Y.L.; Lu, H.; Katzenellenbogen, B.S. The forkhead transcription factor FOXM1 promotes endocrine resistance and invasiveness in estrogen receptor-positive breast cancer by expansion of stem-like cancer cells. Breast Cancer Res. 2014, 16, 436. [Google Scholar] [CrossRef]
- Raychaudhuri, P.; Park, H.J. FoxM1: A master regulator of tumor metastasis. Cancer Res. 2011, 71, 4329–4333. [Google Scholar] [CrossRef]
- Rani, A.; Stebbing, J.; Giamas, G.; Murphy, J. Endocrine resistance in hormone receptor positive breast cancer-from mechanism to therapy. Front. Endocrinol. 2019, 10, 245. [Google Scholar] [CrossRef] [PubMed]
- Fan, P.; Jordan, V.C. New insights into acquired endocrine resistance of breast cancer. Cancer Drug Res. 2019, 2, 198–209. [Google Scholar] [CrossRef] [PubMed]
- Giltnane, J.M.; Hutchinson, K.E.; Stricker, T.P.; Formisano, L.; Young, C.D.; Estrada, M.V.; Nixon, M.J.; Du, L.; Sanchez, V.; Gonzalez Ericsson, P.; et al. Genomic profiling of ER. Sci. Transl. Med. 2017, 9, 7993. [Google Scholar] [CrossRef] [PubMed]
- Banerji, S.; Cibulskis, K.; Rangel-Escareno, C.; Brown, K.K.; Carter, S.L.; Frederick, A.M.; Lawrence, M.S.; Sivachenko, A.Y.; Sougnez, C.; Zou, L.; et al. Sequence analysis of mutations and translocations across breast cancer subtypes. Nature 2012, 486, 405–409. [Google Scholar] [CrossRef]
- Griffith, O.L.; Spies, N.C.; Anurag, M.; Griffith, M.; Luo, J.; Tu, D.; Yeo, B.; Kunisaki, J.; Miller, C.A.; Krysiak, K.; et al. The prognostic effects of somatic mutations in ER-positive breast cancer. Nat. Commun. 2018, 9, 3476. [Google Scholar] [CrossRef]
- Curtis, C.; Shah, S.P.; Chin, S.F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef]
- Yates, L.R.; Knappskog, S.; Wedge, D.; Farmery, J.H.R.; Gonzalez, S.; Martincorena, I.; Alexandrov, L.B.; Van Loo, P.; Haugland, H.K.; Lilleng, P.K.; et al. Genomic evolution of breast cancer metastasis and relapse. Cancer Cell 2017, 32, 169–184. [Google Scholar] [CrossRef]
- Whittle, J.R.; Lewis, M.T.; Lindeman, G.J.; Visvader, J.E. Patient-derived xenograft models of breast cancer and their predictive power. Breast Cancer Res. 2015, 17, 17. [Google Scholar] [CrossRef]
- Li, S.; Shen, D.; Shao, J.; Crowder, R.; Liu, W.; Prat, A.; He, X.; Liu, S.; Hoog, J.; Lu, C.; et al. Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer derived xenografts. Cell Rep. 2013, 4, 1116–1130. [Google Scholar] [CrossRef] [PubMed]
- Jeselsohn, R.; Yelensky, R.; Buchwalter, G.; Frampton, G.; Meric-Bernstam, F.; Gonzalez-Angulo, A.M.; Ferrer-Lozano, J.; Perez-Fidalgo, J.A.; Cristofanilli, M.; Gomez, H.; et al. Emergence of constitutively active estrogen receptor-α mutations in pretreated advanced estrogen receptor-positive breast cancer. Clin. Cancer Res. 2014, 20, 1757–1767. [Google Scholar] [CrossRef]
- Toy, W.; Shen, Y.; Won, H.; Green, B.; Sakr, R.A.; Will, M.; Li, Z.; Gala, K.; Fanning, S.; King, T.A.; et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat. Genet. 2013, 45, 1439–1445. [Google Scholar] [CrossRef]
- Keller, L.; Pantel, K. Unravelling tumour heterogeneity by single-cell profiling of circulating tumour cells. Nat. Rev. Cancer 2019, 19, 553–567. [Google Scholar] [CrossRef]
- Pantel, K.; Alix-Panabières, C. Liquid biopsy and minimal residual disease—Latest advances and implications for cure. Nat. Rev. Clin. Oncol. 2019, 16, 409–424. [Google Scholar] [CrossRef] [PubMed]
- Alix-Panabières, C.; Pantel, K. Challenges in circulating tumour cell research. Nat. Rev. Cancer. 2014, 14, 623–631. [Google Scholar] [CrossRef]
- Kang, Y.; Pantel, K. Tumor cell dissemination: Emerging biological insights from animal models and cancer patients. Cancer Cell 2013, 23, 573–581. [Google Scholar] [CrossRef]
- Koch, C.; Kuske, A.; Joosse, S.A.; Yigit, G.; Sflomos, G.; Thaler, S.; Smit, D.J.; Borgman, K.; Gärtner, S.; Mohammadi, P.M.; et al. Characterization of circulating breast cancer cells with tumorigenic and metastatic capacity. EMBO Mol. Med. 2020, 12, e11908. [Google Scholar] [CrossRef] [PubMed]
- Toy, W.; Weir, H.; Razavi, P.; Lawson, P.; Goeppert, A.U.; Mazzola, A.M.; Smith, A.; Wilson, J.; Morrow, C.; Wong, W.L.; et al. Activating ESR1 mutations differentially affect the efficacy of ER antagonists. Cancer Discov. 2017, 7, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Millour, J.; Constantinidou, D.; Stavropoulou, A.V.; Wilson, M.S.; Myatt, S.S.; Kwok, J.M.; Sivanandan, K.; Coombes, R.C.; Medema, R.H.; Hartman, J.; et al. FOXM1 is a transcriptional target of ERalpha and has a critical role in breast cancer endocrine sensitivity and resistance. Oncogene 2010, 29, 2983–2995. [Google Scholar] [CrossRef]
- Petz, L.N.; Ziegler, Y.S.; Loven, M.A.; Nardulli, A.M. Estrogen receptor alpha and activating protein-1 mediate estrogen responsiveness of the progesterone receptor gene in MCF-7 breast cancer cells. Endocrinology 2002, 143, 4583–4591. [Google Scholar] [CrossRef]
- Kastner, P.; Krust, A.; Turcotte, B.; Stropp, U.; Tora, L.; Gronemeyer, H.; Chambon, P. Two distinct estrogen-regulated promoters generate transcripts encoding the two functionally different human progesterone receptor forms A and B. EMBO J. 1990, 9, 1603–1614. [Google Scholar] [CrossRef]
- Osborne, C.K.; Schiff, R. Mechanisms of endocrine resistance in breast cancer. Annu. Rev. Med. 2011, 62, 233–247. [Google Scholar] [CrossRef]
- Nicholson, R.I.; Hutcheson, I.R.; Britton, D.; Knowlden, J.M.; Jones, H.E.; Harper, M.E.; Hiscox, S.E.; Barrow, D.; Gee, J.M.W. Growth factor signalling networks in breast cancer and resistance to endocrine agents: New therapeutic strategies. J. Steroid. Biochem. Mol. Biol. 2005, 93, 257–262. [Google Scholar] [CrossRef]
- Madureira, P.A.; Varshochi, R.; Constantinidou, D.; Francis, R.E.; Coombes, R.C.; Yao, K.M.; Lam, E.W. The Forkhead box M1 protein regulates the transcription of the estrogen receptor alpha in breast cancer cells. J. Biol. Chem. 2006, 281, 25167–25176. [Google Scholar] [CrossRef]
- Shou, J.; Massarweh, S.; Osborne, C.K.; Wakeling, A.E.; Ali, S.; Weiss, H.; Schiff, R. Mechanisms of tamoxifen resistance: Increased estrogen receptor-HER2/neu cross-talk in ER/HER2-positive breast cancer. J. Natl. Cancer Inst. 2004, 96, 926–935. [Google Scholar] [CrossRef]
- Osborne, C.K.; Schiff, R. Growth factor receptor cross-talk with estrogen receptor as a mechanism for tamoxifen resistance in breast cancer. Breast 2003, 12, 362–367. [Google Scholar] [CrossRef]
- Wang, Y.C.; Morrison, G.; Gillihan, R.; Guo, J.; Ward, R.M.; Fu, X.; Botero, M.F.; Healy, N.A.; Hilsenbeck, S.G.; Phillips, G.L.; et al. Different mechanisms for resistance to trastuzumab versus lapatinib in HER2-positive breast cancers--role of estrogen receptor and HER2 reactivation. Breast Cancer Res. 2011, 13, R121. [Google Scholar] [CrossRef]
- Burstein, H.J.; Cirrincione, C.T.; Barry, W.T.; Chew, H.K.; Tolaney, S.M.; Lake, D.E.; Ma, C.; Blackwell, K.L.; Winer, E.P.; Hudis, C.A. Endocrine therapy with or without inhibition of epidermal growth factor receptor and human epidermal growth factor receptor 2: A randomized, double-blind, placebo-controlled phase III trial of fulvestrant with or without lapatinib for postmenopausal women with hormone receptor-positive advanced breast cancer-CALGB 40302 (Alliance). J. Clin. Oncol. 2014, 32, 3959–3966. [Google Scholar]
- Thaler, S.; Thiede, G.; Hengstler, J.G.; Schad, A.; Schmidt, M.; Sleeman, J.P. The proteasome inhibitor Bortezomib (Velcade) as potential inhibitor of estrogen receptor-positive breast cancer. Int. J. Cancer 2015, 137, 686–697. [Google Scholar] [CrossRef]
- Thaler, S.; Schmidt, M.; Roßwag, S.; Thiede, G.; Schad, A.; Sleeman, J.P. Proteasome inhibitors prevent bi-directional HER2/estrogen-receptor cross-talk leading to cell death in endocrine and lapatinib-resistant HER2+/ER+ breast cancer cells. Oncotarget 2017, 8, 72281–72301. [Google Scholar] [CrossRef]
- Halasi, M.; Gartel, A.L. Targeting FOXM1 in cancer. Biochem. Pharmacol. 2013, 85, 644–652. [Google Scholar] [CrossRef]
- Gartel, A.L. A new target for proteasome inhibitors: FOXM1. Expert Opin. Investig. Drugs 2010, 19, 235–242. [Google Scholar] [CrossRef]
- Giuliano, M.; Shaikh, A.; Lo, H.C.; Arpino, G.; De Placido, S.; Zhang, X.H.; Cristofanilli, M.; Schiff, R.; Trivedi, M.V. Perspective on circulating tumor cell clusters: Why it takes a village to metastasize. Cancer Res. 2018, 78, 845–852. [Google Scholar] [CrossRef]
- Baldwin, A.S. Control of oncogenesis and cancer therapy resistance by the transcription factor NF-kappaB. J. Clin. Investig. 2001, 107, 241–246. [Google Scholar] [CrossRef]
- Riggins, R.B.; Zwart, A.; Nehra, R.; Clark, R. The nuclear factor kappa B inhibitor parthenolide restoresICI 182,780 (Faslodex; Fulvestrant)-induced apoptosis in antiestrogen-resistant breast cancer cells. Mol. Cancer Ther. 2005, 4, 33–41. [Google Scholar]
- Gasparian, A.V.; Guryanova, O.A.; Chebotaev, D.V.; Shishkin, A.A.; Yemelyanov, A.Y.; Budunova, I.V. Targeting transcription factor NFkappaB: Comparative analysis of proteasome and IKK inhibitors. Cell Cycle 2009, 8, 1559–1566. [Google Scholar] [CrossRef]
- Kandel, E.S. NFkappaB inhibition and more: A side-by-side comparison of the inhibitors of IKK and proteasome. Cell Cycle. 2009, 8, 1819–1820. [Google Scholar] [CrossRef] [PubMed]
- Halasi, M.; Gartel, A.L. A novel mode of FoxM1 regulation: Positive auto-regulatory loop. Cell Cycle. 2009, 8, 1966–1967. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.M.; Kim, M.H.; Kim, B.H.; Jung, B.H.; Kim, Y.S.; Park, H.J.; Hong, J.T.; Min, K.R.; Kim, Y. Inhibitory action of novel aromatic diamine compound on lipopolysaccharide-induced nuclear translocation of NF-kappaB without affecting IkappaB degradation. FEBS Lett. 2004, 571, 50–54. [Google Scholar] [CrossRef]
- Guan, J.; Zhou, W.; Hafner, M.; Blake, R.A.; Chalouni, C.; Chen, I.P.; De Bruyn, T.; Giltnane, J.M.; Hartman, S.J.; Heidersbach, A.; et al. Therapeutic ligands antagonize estrogen receptor function by impairing its mobility. Cell 2019, 178, 949–963. [Google Scholar] [CrossRef]
- Francis, R.E.; Myatt, S.S.; Krol, J.; Hartman, J.; Peck, B.; McGovern, U.B.; Wang, J.; Guest, S.K.; Filipovic, A.; Gojis, O.; et al. FoxM1 is a downstream target and marker of HER2 overexpression in breast cancer. Int. J. Oncol. 2009, 35, 57–68. [Google Scholar]
- Leary, A.F.; Drury, S.; Detre, S.; Pancholi, S.; Lykkesfeldt, A.E.; Martin, L.A.; Dowsett, M.; Johnston, S.R. Lapatinib restores hormone sensitivity with differential effects on estrogen receptor signaling in cell models of human epidermal growth factor receptor 2-negative breast cancer with acquired endocrine resistance. Clin. Cancer Res. 2010, 16, 1486–1497. [Google Scholar] [CrossRef] [PubMed]
- Frasor, J.; El-Shennawy, L.; Stender, J.D.; Kastrati, I. NFkB affects estrogen receptor expression and activity in breast cancer through multiple mechanisms. Mol. Cell Endocrinol. 2015, 418, 235–239. [Google Scholar] [CrossRef]
- Fan, P.; Abderrahman, B.; Chai, T.S.; Yerrum, S.; Jordan, V.C. Targeting Peroxisome Proliferator-Activated Receptor γ to Increase Estrogen-Induced Apoptosis in Estrogen-Deprived Breast Cancer Cells. Mol. Cancer Ther. 2018, 17, 2732–2745. [Google Scholar] [CrossRef]
- Sas, L.; Lardon, F.; Vermeulen, P.B.; Hauspy, J.; Van Dam, P.; Pauwels, P.; Dirix, L.Y.; Van Laere, S.J. The interaction between ER and NFkB in resistance to endocrine therapy. Breast Cancer Res. 2012, 14, 212. [Google Scholar] [CrossRef]
- Jin, B.; Wang, C.; Li, J.; Du, X.; Ding, K.; Pan, J. Anthalmintic Niclosamide Disrupts the interplay of p65 and FOXM1/beta-catenin and eradicates leukemia stem cells in chronic myelogenous leukemia. Clin. Cancer Res. 2017, 23, 789–803. [Google Scholar] [CrossRef] [PubMed]
- Arora, R.; Yates, C.; Gary, B.D.; McClellan, S.; Tan, M.; Xi, Y.; Reed, E.; Piazza, G.A.; Owen, L.B.; Dean-Colomb, W. Panepoxydone targets NF-kB and FOXM1 to inhibit proliferation, induce apoptosis and reverse epithelial to mesenchymal transition in breast cancer. PLoS ONE 2014, 9, e98370. [Google Scholar] [CrossRef]
- Yuan, Q.; Wen, M.; Xu, C.; Chen, A.; Qiu, Y.B.; Cao, J.G.; Zhang, J.S.; Song, Z.W. 8-bromo-7-methoxychrysin targets NF-kB and FoxM1 to inhibit lung cancer stem cells induced by pro-inflammatory factors. J. Cancer. 2019, 10, 5244–5255. [Google Scholar] [CrossRef]
- Ithimakin, S.; Day, K.C.; Malik, F.; Zen, Q.; Dawsey, S.J.; Bersano-Begey, T.F.; Quraishi, A.A.; Ignatoski, K.W.; Daignault, S.; Davis, A.; et al. HER2 drives luminal breast cancer stem cells in the absence of HER2 amplification: Implications for efficacy of adjuvant trastuzumab. Cancer Res. 2013, 73, 1635–1646. [Google Scholar] [CrossRef] [PubMed]
- Bates, N.P.; Hurst, H.C. An intron 1 enhancer element mediates oestrogen-induced suppression of ERBB2 expression. Oncogene 1997, 15, 473–481. [Google Scholar] [CrossRef]
- Newman, S.P.; Bates, N.P.; Vernimmen, D.; Parker, M.G.; Hurst, H.C. Cofactor competition between the ligand-bound oestrogen receptor and an intron 1 enhancer leads to oestrogen repression of ERBB2 expression in breast cancer. Oncogene 2000, 19, 490–497. [Google Scholar] [CrossRef]
- Garee, J.P.; Chien, C.D.; Li, J.V.; Wellstein, A.; Riegel, A.T. Regulation of HER2 oncogene transcription by a multifunctional coactivator/corepressor complex. Mol. Endocrinol. 2014, 28, 846–859. [Google Scholar] [CrossRef]
- Ziegler, Y.; Laws, M.J.; Sanabria Guillen, V.; Kim, S.H.; Dey, P.; Smith, B.P.; Gong, P.; Bindman, N.; Zhao, Y.; Carlson, K.; et al. Suppression of FOXM1 activities and breast cancer growth in vitro and in vivo by a new class of compounds. NPJ Breast Cancer 2019, 5, 45. [Google Scholar] [CrossRef]
- Jordan, N.V.; Bardia, A.; Wittner, B.S.; Benes, C.; Ligorio, M.; Zheng, Y.; Yu, M.; Sundaresan, T.K.; Licausi, J.A.; Desai, R.; et al. HER2 expression identifies dynamic functional states within circulating breast cancer cells. Nature 2016, 537, 102–106. [Google Scholar] [CrossRef]
- Schochter, F.; Friedl, T.W.P.; deGregorio, A.; Krause, S.; Huober, J.; Rack, B.; Janni, W. Are Circulating Tumor Cells (CTCs) Ready for Clinical Use in Breast Cancer? An Overview of Completed and Ongoing Trials Using CTCs for Clinical Treatment Decisions. Cells 2019, 8, 1412. [Google Scholar] [CrossRef] [PubMed]
- Thaler, S.; Schmidt, M.; Schad, A.; Sleeman, J.P. RASSF1A inhibits estrogen receptor alpha expression and estrogen-independent signalling: Implications for breast cancer development. Oncogene 2012, 31, 4912–4922. [Google Scholar] [CrossRef] [PubMed][Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roßwag, S.; Cotarelo, C.L.; Pantel, K.; Riethdorf, S.; Sleeman, J.P.; Schmidt, M.; Thaler, S. Functional Characterization of Circulating Tumor Cells (CTCs) from Metastatic ER+/HER2− Breast Cancer Reveals Dependence on HER2 and FOXM1 for Endocrine Therapy Resistance and Tumor Cell Survival: Implications for Treatment of ER+/HER2− Breast Cancer. Cancers 2021, 13, 1810. https://doi.org/10.3390/cancers13081810
Roßwag S, Cotarelo CL, Pantel K, Riethdorf S, Sleeman JP, Schmidt M, Thaler S. Functional Characterization of Circulating Tumor Cells (CTCs) from Metastatic ER+/HER2− Breast Cancer Reveals Dependence on HER2 and FOXM1 for Endocrine Therapy Resistance and Tumor Cell Survival: Implications for Treatment of ER+/HER2− Breast Cancer. Cancers. 2021; 13(8):1810. https://doi.org/10.3390/cancers13081810
Chicago/Turabian StyleRoßwag, Sven, Cristina L. Cotarelo, Klaus Pantel, Sabine Riethdorf, Jonathan P. Sleeman, Marcus Schmidt, and Sonja Thaler. 2021. "Functional Characterization of Circulating Tumor Cells (CTCs) from Metastatic ER+/HER2− Breast Cancer Reveals Dependence on HER2 and FOXM1 for Endocrine Therapy Resistance and Tumor Cell Survival: Implications for Treatment of ER+/HER2− Breast Cancer" Cancers 13, no. 8: 1810. https://doi.org/10.3390/cancers13081810
APA StyleRoßwag, S., Cotarelo, C. L., Pantel, K., Riethdorf, S., Sleeman, J. P., Schmidt, M., & Thaler, S. (2021). Functional Characterization of Circulating Tumor Cells (CTCs) from Metastatic ER+/HER2− Breast Cancer Reveals Dependence on HER2 and FOXM1 for Endocrine Therapy Resistance and Tumor Cell Survival: Implications for Treatment of ER+/HER2− Breast Cancer. Cancers, 13(8), 1810. https://doi.org/10.3390/cancers13081810