
Analysis of Intra-Tumoral Macrophages and T Cells in Non-Small Cell Lung Cancer (NSCLC) Indicates a Role for Immune Checkpoint and CD200-CD200R Interactions

 ,
,  ,
,  and
and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
3. Results
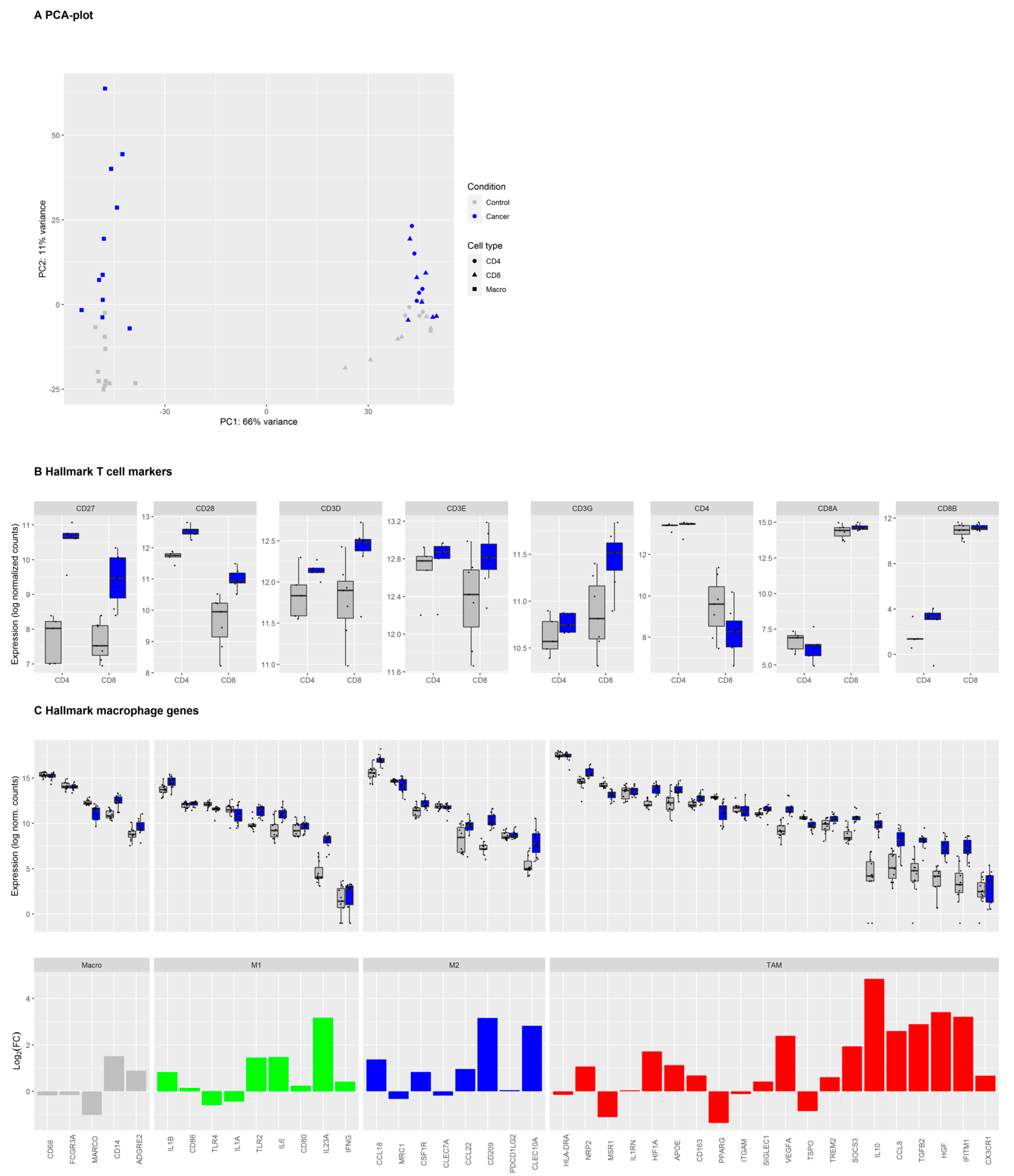
3.1. Immune Cells in NSCLC and Healthy Tissue Express Lineage-Specific Genes and Increased Expression of TAM-Associated Genes in Macrophages from the Tumor
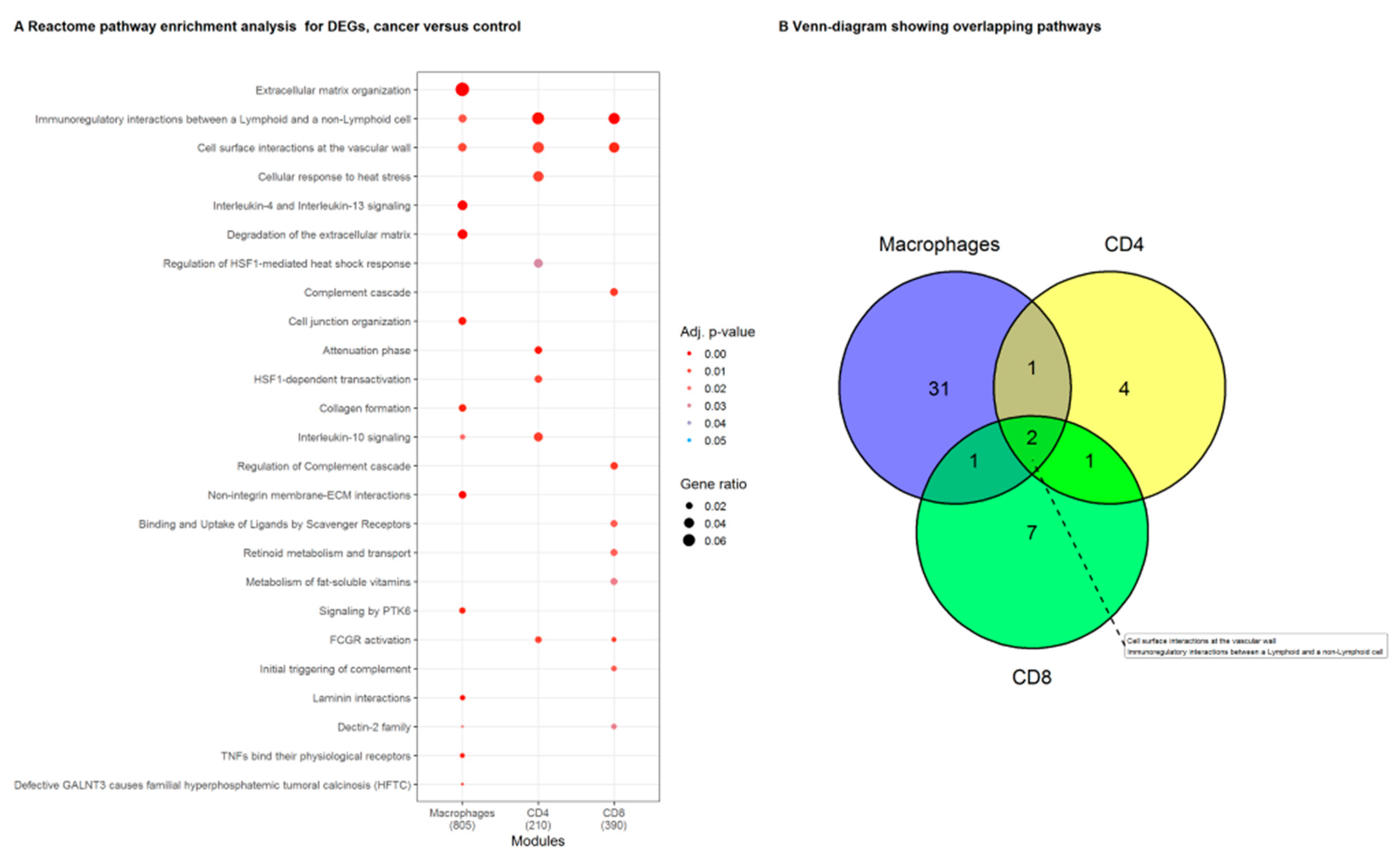
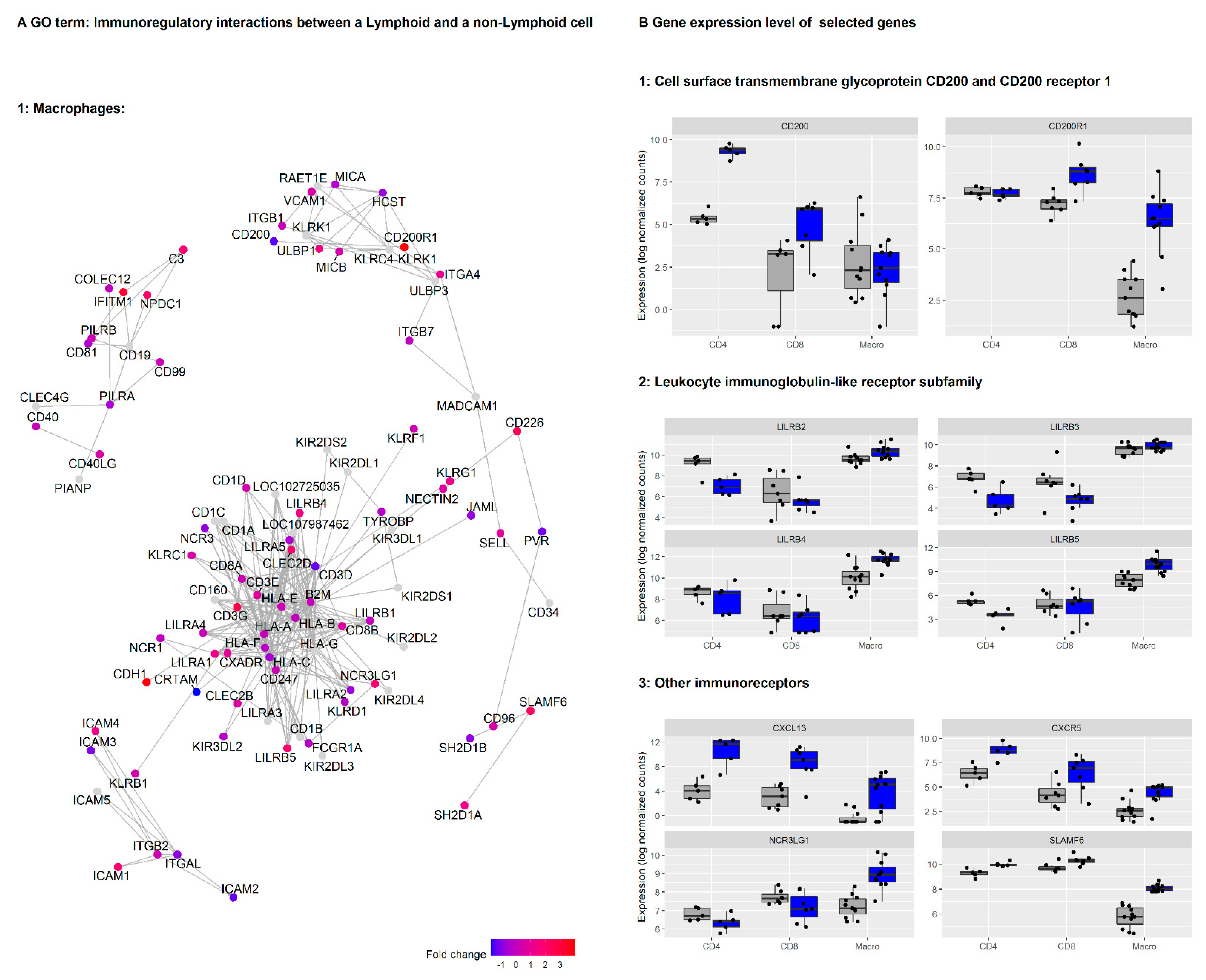
3.2. Genes in the Reactome Pathway «Immunoregulatory Interactions between Lymphoid and Non-Lymphoid Cells» Are Highly Regulated in NSCLC Macrophages, CD4+ and CD8+ T Cells
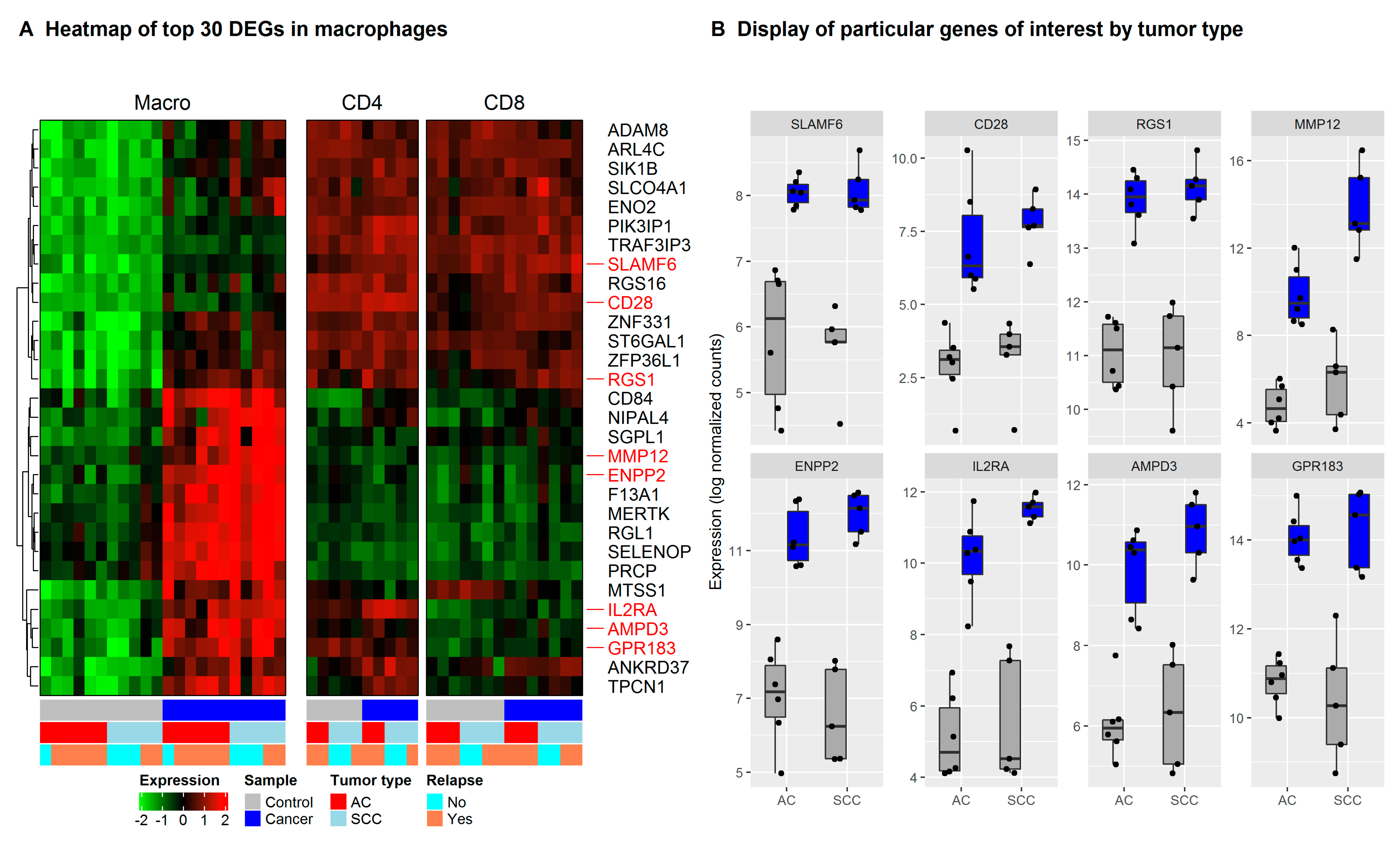
3.3. Network Clustering Analysis Identifies Networks of Genes with Highly Correlated Expression Profiles
3.4. CD200/CD200R Pathway and Inhibitory Leukocyte Immunoglobulin-Like Receptors (LILRs) Represent Possible Mechanisms for Macrophage-Induced Immunosuppression/Innate Checkpoints
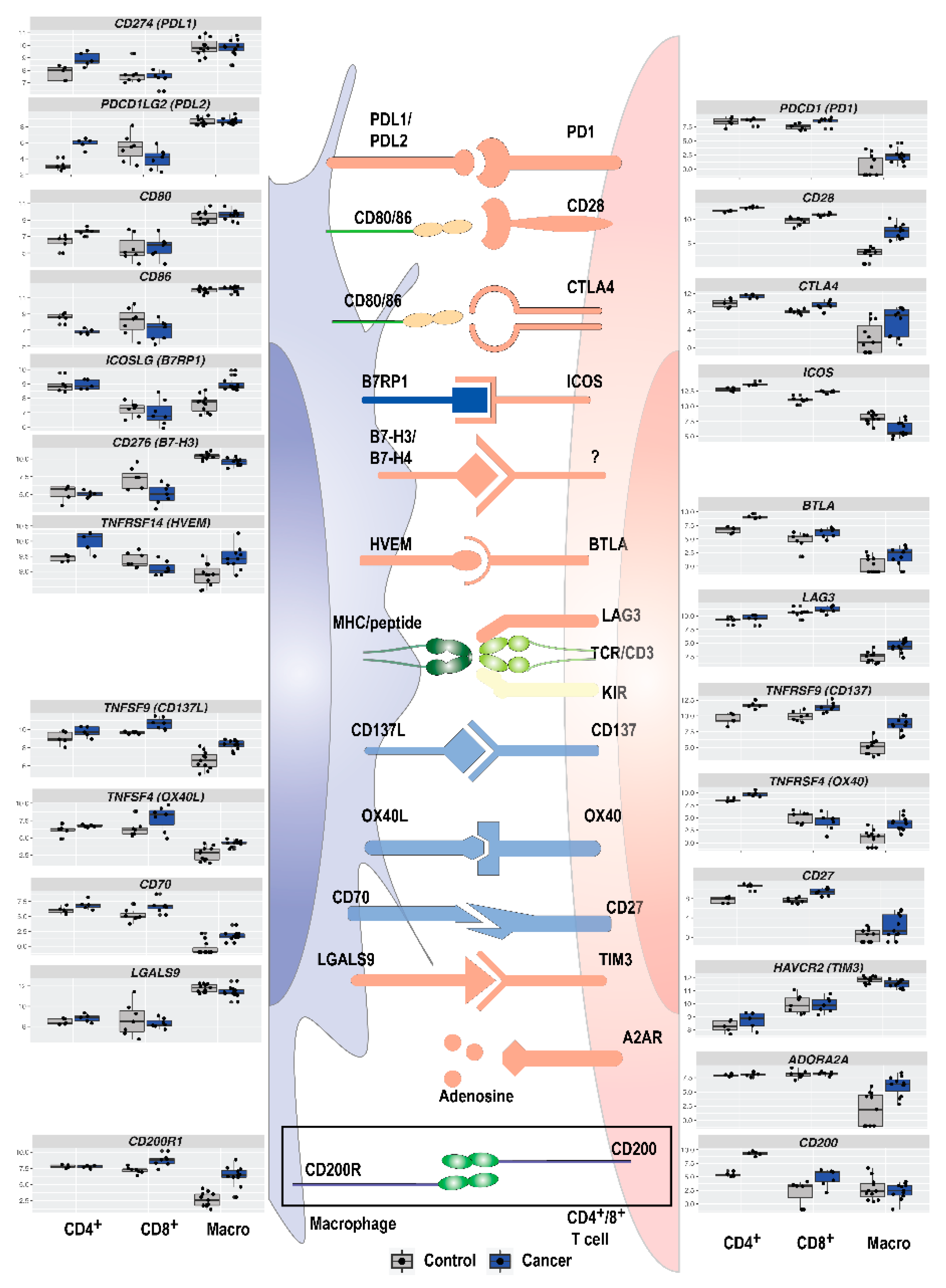
3.5. Immune Checkpoint Expression in TAMs and T Cells in NSCLC
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Schabath, M.B.; Cote, M.L. Cancer Progress and Priorities: Lung Cancer. Cancer Epidemiol. Biomark. Prev. 2019, 28, 1563–1579. [Google Scholar] [CrossRef]
- Xia, L.; Liu, Y.; Wang, Y. PD-1/PD-L1 Blockade Therapy in Advanced Non-Small-Cell Lung Cancer: Current Status and Future Directions. Oncologist 2019, 24, S31–S41. [Google Scholar] [CrossRef]
- Berghmans, T.; Dingemans, A.-M.; Hendriks, L.E.L.; Cadranel, J. Immunotherapy for nonsmall cell lung cancer: A new therapeutic algorithm. Eur. Respir. J. 2020, 55, 1901907. [Google Scholar] [CrossRef] [PubMed]
- Gettinger, S.; Horn, L.; Jackman, D.; Spigel, D.; Antonia, S.; Hellmann, M.; Powderly, J.; Heist, R.; Sequist, L.V.; Smith, D.C.; et al. Five-Year Follow-Up of Nivolumab in Previously Treated Advanced Non-Small-Cell Lung Cancer: Results from the CA209-003 Study. J. Clin. Oncol. 2018, 36, 1675–1684. [Google Scholar] [CrossRef] [PubMed]
- Nadal, E.; Massuti, B.; Dómine, M.; García-Campelo, R.; Cobo, M.; Felip, E. Immunotherapy with checkpoint inhibitors in non-small cell lung cancer: Insights from long-term survivors. Cancer Immunol. Immunother. 2019, 68, 341–352. [Google Scholar] [CrossRef]
- Grigg, C.; Rizvi, N.A. PD-L1 biomarker testing for non-small cell lung cancer: Truth or fiction? J. Immunother. Cancer 2016, 4, 48. [Google Scholar] [CrossRef]
- Qin, S.; Xu, L.; Yi, M.; Yu, S.; Wu, K.; Luo, S. Novel immune checkpoint targets: Moving beyond PD-1 and CTLA-4. Mol. Cancer 2019, 18, 155. [Google Scholar] [CrossRef]
- Di Virgilio, F.; Sarti, A.C.; Falzoni, S.; De Marchi, E.; Adinolfi, E. Extracellular ATP and P2 purinergic signalling in the tumour microenvironment. Nat. Rev. Cancer 2018, 18, 601–618. [Google Scholar] [CrossRef] [PubMed]
- Chiang, A.C.; Herbst, R.S. Frontline immunotherapy for NSCLC—The tale of the tail. Nat. Rev. Clin. Oncol. 2020, 17, 73–74. [Google Scholar] [CrossRef]
- Doroshow, D.B.; Sanmamed, M.F.; Hastings, K.; Politi, K.; Rimm, D.L.; Chen, L.; Melero, I.; Schalper, K.A.; Herbst, R.S. Immunotherapy in Non–Small Cell Lung Cancer: Facts and Hopes. Clin. Cancer Res. 2019, 25, 4592. [Google Scholar] [CrossRef] [PubMed]
- Munn, D.H.; Bronte, V. Immune suppressive mechanisms in the tumor microenvironment. Curr. Opin. Immunol. 2016, 39, 1–6. [Google Scholar] [CrossRef]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896. [Google Scholar] [CrossRef]
- Feng, M.; Jiang, W.; Kim, B.Y.S.; Zhang, C.C.; Fu, Y.-X.; Weissman, I.L. Phagocytosis checkpoints as new targets for cancer immunotherapy. Nat. Rev. Cancer 2019, 19, 568–586. [Google Scholar] [CrossRef] [PubMed]
- Tøndell, A.; Wahl, S.G.F.; Sponaas, A.M.; Sørhaug, S.; Børset, M.; Haug, M. Ectonucleotidase CD39 and Checkpoint Signalling Receptor Programmed Death 1 are Highly Elevated in Intratumoral Immune Cells in Non-small-cell Lung Cancer. Transl. Oncol. 2020, 13, 17–24. [Google Scholar] [CrossRef]
- Cekic, C.; Day, Y.-J.; Sag, D.; Linden, J. Myeloid expression of Adenosine A(2A) receptor suppresses T and NK cell responses in the solid tumor microenvironment. Cancer Res. 2014, 74, 7250–7259. [Google Scholar] [CrossRef] [PubMed]
- Leone, R.D.; Emens, L.A. Targeting adenosine for cancer immunotherapy. J. Immunother. Cancer 2018, 6, 57. [Google Scholar] [CrossRef]
- Kang, X.; Kim, J.; Deng, M.; John, S.; Chen, H.; Wu, G.; Phan, H.; Zhang, C.C. Inhibitory leukocyte immunoglobulin-like receptors: Immune checkpoint proteins and tumor sustaining factors. Cell Cycle 2016, 15, 25–40. [Google Scholar] [CrossRef]
- Ries, C.H.; Cannarile, M.A.; Hoves, S.; Benz, J.; Wartha, K.; Runza, V.; Rey-Giraud, F.; Pradel, L.P.; Feuerhake, F.; Klaman, I.; et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell 2014, 25, 846–859. [Google Scholar] [CrossRef] [PubMed]
- Travis, W.D.; Brambilla, E.; Burke, A.; Marx, A.; Nicholson, A.G.; International Agency for Research on Cancer (Eds.) WHO Classification of Tumours of the Lung, Pleura, Thymus and Heart; International Agency for Research on Cancer: Lyon, France, 2015. [Google Scholar]
- Quatromoni, J.G.; Singhal, S.; Bhojnagarwala, P.; Hancock, W.W.; Albelda, S.M.; Eruslanov, E. An optimized disaggregation method for human lung tumors that preserves the phenotype and function of the immune cells. J. Leukoc. Biol. 2015, 97, 201–209. [Google Scholar] [CrossRef]
- Bharat, A.; Bhorade, S.M.; Morales-Nebreda, L.; McQuattie-Pimentel, A.C.; Soberanes, S.; Ridge, K.; DeCamp, M.M.; Mestan, K.K.; Perlman, H.; Budinger, G.R.S.; et al. Flow Cytometry Reveals Similarities Between Lung Macrophages in Humans and Mice. Am. J. Respir. Cell Mol. Biol. 2016, 54, 147–149. [Google Scholar] [CrossRef] [PubMed]
- Mitsi, E.; Kamng’ona, R.; Rylance, J.; Solórzano, C.; Jesus Reiné, J.; Mwandumba, H.C.; Ferreira, D.M.; Jambo, K.C. Human alveolar macrophages predominately express combined classical M1 and M2 surface markers in steady state. Respir. Res. 2018, 19, 66. [Google Scholar] [CrossRef] [PubMed]
- Haque, A.S.M.R.; Moriyama, M.; Kubota, K.; Ishiguro, N.; Sakamoto, M.; Chinju, A.; Mochizuki, K.; Sakamoto, T.; Kaneko, N.; Munemura, R.; et al. CD206+ tumor-associated macrophages promote proliferation and invasion in oral squamous cell carcinoma via EGF production. Sci. Rep. 2019, 9, 14611. [Google Scholar] [CrossRef] [PubMed]
- Davidov, V.; Jensen, G.; Mai, S.; Chen, S.-H.; Pan, P.-Y. Analyzing One Cell at a TIME: Analysis of Myeloid Cell Contributions in the Tumor Immune Microenvironment. Front. Immunol. 2020, 11, 1842. [Google Scholar] [CrossRef]
- Dutertre, C.-A.; Becht, E.; Irac, S.E.; Khalilnezhad, A.; Narang, V.; Khalilnezhad, S.; Ng, P.Y.; van den Hoogen, L.L.; Leong, J.Y.; Lee, B.; et al. Single-Cell Analysis of Human Mononuclear Phagocytes Reveals Subset-Defining Markers and Identifies Circulating Inflammatory Dendritic Cells. Immunity 2019, 51, 573–589.e578. [Google Scholar] [CrossRef]
- clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Győrffy, B.; Surowiak, P.; Budczies, J.; Lánczky, A. Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non-small-cell lung cancer. PLoS ONE 2013, 8, e82241. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Maeda, D.; Yoshida, M.; Umakoshi, M.; Nanjo, H.; Shiraishi, K.; Saito, M.; Kohno, T.; Konno, H.; Saito, H.; et al. The intratumoral distribution influences the prognostic impact of CD68- and CD204-positive macrophages in non-small cell lung cancer. Lung Cancer 2018, 123, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Shipley, J.M.; Wesselschmidt, R.L.; Kobayashi, D.K.; Ley, T.J.; Shapiro, S.D. Metalloelastase is required for macrophage-mediated proteolysis and matrix invasion in mice. Proc. Natl. Acad. Sci. USA 1996, 93, 3942. [Google Scholar] [CrossRef]
- Knowlden, S.; Georas, S.N. The autotaxin-LPA axis emerges as a novel regulator of lymphocyte homing and inflammation. J. Immunol. 2014, 192, 851–857. [Google Scholar] [CrossRef] [PubMed]
- Myers, K.V.; Amend, S.R.; Pienta, K.J. Targeting Tyro3, Axl and MerTK (TAM receptors): Implications for macrophages in the tumor microenvironment. Mol. Cancer 2019, 18, 94. [Google Scholar] [CrossRef] [PubMed]
- Porrello, A.; Leslie, P.L.; Harrison, E.B.; Gorentla, B.K.; Kattula, S.; Ghosh, S.K.; Azam, S.H.; Holtzhausen, A.; Chao, Y.L.; Hayward, M.C.; et al. Factor XIIIA—Expressing inflammatory monocytes promote lung squamous cancer through fibrin cross-linking. Nat. Commun. 2018, 9, 1988. [Google Scholar] [CrossRef]
- Alvisi, G.; Brummelman, J.; Puccio, S.; Mazza, E.M.; Tomada, E.P.; Losurdo, A.; Zanon, V.; Peano, C.; Colombo, F.S.; Scarpa, A.; et al. IRF4 instructs effector Treg differentiation and immune suppression in human cancer. J. Clin. Investig. 2020, 130, 3137–3150. [Google Scholar] [CrossRef]
- Bercovici, N.; Guérin, M.V.; Trautmann, A.; Donnadieu, E. The Remarkable Plasticity of Macrophages: A Chance to Fight Cancer. Front. Immunol. 2019, 10, 1563. [Google Scholar] [CrossRef]
- Modjeski, K.L.; Levy, S.C.; Ture, S.K.; Field, D.J.; Shi, G.; Ko, K.; Zhu, Q.; Morrell, C.N. Glutamate Receptor Interacting Protein 1 Regulates CD4(+) CTLA-4 Expression and Transplant Rejection. Am. J. Transplant. 2016, 16, 1383–1393. [Google Scholar] [CrossRef]
- Improgo, M.R.D.; Scofield, M.D.; Tapper, A.R.; Gardner, P.D. From smoking to lung cancer: The CHRNA5/A3/B4 connection. Oncogene 2010, 29, 4874–4884. [Google Scholar] [CrossRef] [PubMed]
- Janghorban, M.; Xin, L.; Rosen, J.M.; Zhang, X.H.F. Notch Signaling as a Regulator of the Tumor Immune Response: To Target or Not To Target? Front. Immunol. 2018, 9, 1649. [Google Scholar] [CrossRef]
- Vahl, J.M.; Friedrich, J.; Mittler, S.; Trump, S.; Heim, L.; Kachler, K.; Balabko, L.; Fuhrich, N.; Geppert, C.-I.; Trufa, D.I.; et al. Interleukin-10-regulated tumour tolerance in non-small cell lung cancer. Br. J. Cancer 2017, 117, 1644–1655. [Google Scholar] [CrossRef]
- Wang, X.; Iyer, A.; Lyons, A.B.; Körner, H.; Wei, W. Emerging Roles for G-protein Coupled Receptors in Development and Activation of Macrophages. Front. Immunol. 2019, 10, 2031. [Google Scholar] [CrossRef]
- Liu, J.Q.; Hu, A.; Zhu, J.; Yu, J.; Talebian, F.; Bai, X.F. CD200-CD200R Pathway in the Regulation of Tumor Immune Microenvironment and Immunotherapy. Adv. Exp. Med. Biol. 2020, 1223, 155–165. [Google Scholar] [CrossRef] [PubMed]
- van der Touw, W.; Chen, H.M.; Pan, P.Y.; Chen, S.H. LILRB receptor-mediated regulation of myeloid cell maturation and function. Cancer Immunol. Immunother. CII 2017, 66, 1079–1087. [Google Scholar] [CrossRef] [PubMed]
- Izraely, S.; Ben-Menachem, S.; Sagi-Assif, O.; Meshel, T.; Marzese, D.M.; Ohe, S.; Zubrilov, I.; Pasmanik-Chor, M.; Hoon, D.S.B.; Witz, I.P. ANGPTL4 promotes the progression of cutaneous melanoma to brain metastasis. Oncotarget 2017, 8, 75778–75796. [Google Scholar] [CrossRef]
- Vijayan, D.; Young, A.; Teng, M.W.L.; Smyth, M.J. Targeting immunosuppressive adenosine in cancer. Nat. Rev. Cancer 2017, 17, 709–724. [Google Scholar] [CrossRef]
- Birbrair, A. Tumor Microenvironment: Signaling Pathways—Part A.; Springer: Berlin/Heidelberg, Germany, 2020. [Google Scholar]
- Khan, M.; Arooj, S.; Wang, H. NK Cell-Based Immune Checkpoint Inhibition. Front. Immunol. 2020, 11, 167. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, K.; Suzuki, Y.; Inoue, Y.; Tsuchiya, K.; Karayama, M.; Iwashita, Y.; Kahyo, T.; Kawase, A.; Tanahashi, M.; Ogawa, H.; et al. CD200 and CD200R1 are differentially expressed and have differential prognostic roles in non-small cell lung cancer. Oncoimmunology 2020, 9, 1746554. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Kim, H.K.; Lee, K.; Hong, Y.; Cho, J.H.; Choi, J.W.; Lee, J.-I.; Suh, Y.-L.; Ku, B.M.; Eum, H.H.; et al. Single-cell RNA sequencing demonstrates the molecular and cellular reprogramming of metastatic lung adenocarcinoma. Nat. Commun. 2020, 11, 2285. [Google Scholar] [CrossRef]
- Deng, M.; Gui, X.; Kim, J.; Xie, L.; Chen, W.; Li, Z.; He, L.; Chen, Y.; Chen, H.; Luo, W.; et al. LILRB4 signalling in leukaemia cells mediates T cell suppression and tumour infiltration. Nature 2018, 562, 605–609. [Google Scholar] [CrossRef]
- de Goeje, P.L.; Bezemer, K.; Heuvers, M.E.; Dingemans, A.C.; Groen, H.J.; Smit, E.F.; Hoogsteden, H.C.; Hendriks, R.W.; Aerts, J.G.; Hegmans, J.P. Immunoglobulin-like transcript 3 is expressed by myeloid-derived suppressor cells and correlates with survival in patients with non-small cell lung cancer. Oncoimmunology 2015, 4, e1014242. [Google Scholar] [CrossRef]
- Zhang, P.; Yu, S.; Li, H.; Liu, C.; Li, J.; Lin, W.; Gao, A.; Wang, L.; Gao, W.; Sun, Y. ILT4 drives B7-H3 expression via PI3K/AKT/mTOR signalling and ILT4/B7-H3 co-expression correlates with poor prognosis in non-small cell lung cancer. FEBS Lett. 2015, 589, 2248–2256. [Google Scholar] [CrossRef]
- Sun, Y.; Liu, J.; Gao, P.; Wang, Y.; Liu, C. Expression of Ig-like transcript 4 inhibitory receptor in human non-small cell lung cancer. Chest 2008, 134, 783–788. [Google Scholar] [CrossRef]
- Fouquet, G.; Marcq, I.; Debuysscher, V.; Bayry, J.; Rabbind Singh, A.; Bengrine, A.; Nguyen-Khac, E.; Naassila, M.; Bouhlal, H. Signaling lymphocytic activation molecules Slam and cancers: Friends or foes? Oncotarget 2018, 9, 16248–16262. [Google Scholar] [CrossRef]
- Kazanietz, M.G.; Durando, M.; Cooke, M. CXCL13 and Its Receptor CXCR5 in Cancer: Inflammation, Immune Response, and Beyond. Front. Endocrinol. 2019, 10, 471. [Google Scholar] [CrossRef] [PubMed]
- Tan, P.; Shi, M.; Lai, L.; Tang, Z.; Xie, N.; Xu, H.; Wei, Q.; Zhang, X.; Yang, L.; Wu, L. Regulative role of the CXCL13-CXCR5 axis in the tumor microenvironment. Precis. Clin. Med. 2018, 1, 49–56. [Google Scholar] [CrossRef]
- Di Virgilio, F.; Adinolfi, E. Extracellular purines, purinergic receptors and tumor growth. Oncogene 2017, 36, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Cassetta, L.; Fragkogianni, S.; Sims, A.H.; Swierczak, A.; Forrester, L.M.; Zhang, H.; Soong, D.Y.H.; Cotechini, T.; Anur, P.; Lin, E.Y.; et al. Human Tumor-Associated Macrophage and Monocyte Transcriptional Landscapes Reveal Cancer-Specific Reprogramming, Biomarkers, and Therapeutic Targets. Cancer Cell 2019, 35, 588–602.e510. [Google Scholar] [CrossRef]
- Anichini, A.; Perotti, V.E.; Sgambelluri, F.; Mortarini, R. Immune Escape Mechanisms in Non Small Cell Lung Cancer. Cancers 2020, 12, 3605. [Google Scholar] [CrossRef]
- McGranahan, N.; Swanton, C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell 2017, 168, 613–628. [Google Scholar] [CrossRef]
- McGranahan, N.; Rosenthal, R.; Hiley, C.T.; Rowan, A.J.; Watkins, T.B.K.; Wilson, G.A.; Birkbak, N.J.; Veeriah, S.; Van Loo, P.; Herrero, J.; et al. Allele-Specific HLA Loss and Immune Escape in Lung Cancer Evolution. Cell 2017, 171, 1259–1271.e1211. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Age | Smoker | Tumor | Subtype | PDL1 | pTNM | Stage | Relapse | DFS | Immunotherapy | PFS | Samples Included (Macro/CD4/CD8) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 70–74 | former | AC | solid | 1 | pT2bN2-3M0 | IIIA | Yes | 273 | pembrolizumab | 14 | Yes/No/No |
| 2 | 75–79 | former | AC | acinary | 0 | pT2bN1M0 | IIB | Yes | 891 | Yes/Yes/Yes | ||
| 3 | 70–74 | former | AC | micropapillary | 1 | pT1cN0M0 | IA3 | No | Yes/No/No | |||
| 4 | 75–79 | current | SCC | keratinizing SCC | 15 | pT3pN0M0 | IIB | No | Yes/Yes/Yes | |||
| 5 | 70–74 | current | AC | acinary | 30 | pT1cN0M1a | IVA | Yes | 127 | atezolizumab | >570 * | Yes/No/Yes |
| 6 | 80–84 | former | SCC | non-keratinizing SCC | 0 | pT2aN1M0 | IIB | Yes | 388 | Yes/No/Yes | ||
| 7 | 70–74 | former | SCC | non-keratinizing SCC | 0 | pT3N0M0 | IIB | No | Yes/Yes/No | |||
| 8 | 80–84 | former | SCC | keratinizing SCC | 0 | pT2aN0M0 | IB | No | Yes/No/Yes | |||
| 9 | 70–74 | former | AC | micropapillary | 0 | pT3N2M0 | IIIB | Yes | 655 | Yes/Yes/Yes | ||
| 10 | 75–79 | former | SCC | keratinizing SCC | 0 | pT2bN0M0 | IIA | Yes | 176 | Yes/Yes/Yes | ||
| 11 | 55–59 | current | AC | cribriform | 0 | pT2aN2M0 | IIIA | Yes | 942 | Yes/No/No |
| Gene Symbol | Gene Name | Fold-Change * (Macrophages) | Fold-Change * (CD4+ T Cells) | Fold-Change * (CD8+ T Cells) |
|---|---|---|---|---|
| ADORA | Adenosine receptors | Gene family | ||
| AGPAT4 | Phospholipid acyltransferase | 0.58 | −1.47 | −1.81 |
| ANGPTL4 | Angiopoietin-like protein family 4 | 2.41 | # | # |
| APOE | Apolipoprotein E | 1.13 | # | # |
| B7RP1 (CD275, ICOSLG) | ICOS ligand | 1.33 | # | # |
| BTLA (CD272) | B-lymphocyte and T-lymphocyte attenuator | 2.17 | 2.38 | # |
| CCL18 | Chemokine (C-C motif) ligand 18 | 1.38 | # | # |
| CCL8 | Chemokine (C-C motif) ligand 8 | 2.59 | −2.27 | # |
| CHRNA5 | Neuronal acetylcholine receptor subunit alpha-5 acetylcholine receptor subunit | # | # | # |
| CSF1R | Colony stimulating factor 1 receptor | # | −1.76 | # |
| CTLA-4 | cytotoxic T lymphocyte protein 4 | 2.51 | # | # |
| CXCL13 | Chemokine (C-X-C motif) ligand 13 | 6.41 | 6.66 | 5.94 |
| CXCR5 | C-X-C chemokine receptor type 5 | 1.77 | 2.31 | 2.26 |
| DC-SIGN (CD209) | Dendritic cell-specific intercellular adhesion molecule-3-Grabbing non-integrin | 3.15 | # | # |
| ENPP2 | Ectonucleotide pyrophosphatase/phosphodiesterase family member 2 | 4.56 | # | # |
| ENTPD1 (CD39) | Ectonucleoside triphosphate diphosphohydrolase-1 | 1.37 | 2.73 | 2 |
| F13A1 | Coagulation factor XIII A chain | 5.07 | # | # |
| FCGR3A (CD16a) | Low affinity immunoglobulin gamma Fc region receptor III-A | # | −1.77 | −1.9 |
| FOXP3 | Forkhead box P3 | 3.23 | 2.79 | 1.92 |
| GRIP1 | Glutamate receptor-interacting protein 1 | 7.98 | # | # |
| HGF | Hepatocyte growth factor | 3.41 | # | # |
| HIF1A | Hypoxia-inducible factor 1-alpha | 1.72 | # | # |
| HLA-DR | HLA class II histocompatibility antigen, DR alpha chain | # | # | −1.21 |
| HVEM (TNFRSF14) | Tumor necrosis factor receptor superfamily member 14 | 0.66 | # | # |
| ICOS (CD278) | Inducible T-cell co-stimulator | −1.54 | # | 1.29 |
| IFITM1 | Interferon-induced transmembrane protein 1 | 3.21 | # | # |
| IFNG | Interferon gamma | # | # | 1.03 |
| IL10 | Interleukin 10 | 4.83 | # | 1.99 |
| IL1A | Interleukin 1 alpha | # | # | # |
| IL1B | Interleukin 1 beta | # | −1.95 | # |
| IL1RN | Interleukin-1 receptor antagonist | # | # | −1.38 |
| IL1RN | Interleukin 1 Receptor Antagonist | # | # | −1.38 |
| IL23A | Interleukin-23 subunit alpha | 3.16 | # | # |
| IL2RA (CD25) | Interleukin-2 receptor alpha chain | 5.1 | 1.99 | # |
| IL6 | Interleukin 6 | 1.47 | # | # |
| IRF4 (MUM1) | Interferon regulatory factor 4 | 2.48 | # | # |
| ITGAM (CD11B, Mac-1) | Integrin alpha M | # | −2.19 | −2.08 |
| LILRA | Leukocyte immunoglobulin-like receptor subfamily A | Gene family | ||
| LILRB | Leukocyte immunoglobulin-like receptor subfamily B | Gene family | ||
| MAGEH1 | Melanoma-associated antigen H1 | 0.96 | 3.42 | # |
| MARCO | Macrophage receptor with collagenous structure | −1.01 | −2.44 | −2.85 |
| MERTK | Proto-oncogene tyrosine-protein kinase MER | - | # | # |
| MMP12 | Matrix metalloproteinase-12 | 7.79 | # | # |
| MRC1 (CD206) | Mannose receptor C-type 1 | # | # | −2.28 |
| MSR1 (CD204) | Macrophage scavenger receptor 1 | −1.10 | # | −2.29 |
| NOTCH3 | Neurogenic locus notch homolog protein 3 | 4.21 | # | 4.3 |
| NRP2 | Neuropilin 2 | 1.07 | # | −1.47 |
| NT5E (CD73) | 5′-nucleotidase | 3.26 | # | # |
| OX40 (CD134, TNFRSF4) | Tumor necrosis factor receptor superfamily, member 4 | 2.98 | # | # |
| OX40L (CD252, TNFSF4) | Tumor necrosis factor (ligand) superfamily, member 4 | 1.37 | # | 1.62 |
| PDCD1 (PD-1) | programmed cell death protein 1 | 1.4 | # | # |
| PDCD1LG2 | Programmed cell death 1 ligand 2 | # | 2.85 | −1.81 |
| PD-L1 (CD274) | programmed cell death protein ligand 1 | # | # | # |
| PPARG | Peroxisome proliferator-activated receptor gamma | −1.36 | # | −2.3 |
| SIGLEC1 (CD169) | Sialo-adhesin | # | # | −2.01 |
| SLAMF | signaling lymphocytic activation molecule family | Gene family | ||
| SOCS3 | Suppressor of cytokine signaling 3 | 1.94 | # | # |
| TGFB2 | Transforming growth factor-beta 2 | 2.89 | # | # |
| TLR2 | Toll-like receptor 2 | 1.45 | # | # |
| VEGFA | Vascular endothelial growth factor A | 2.39 | # | # |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tøndell, A.; Subbannayya, Y.; Wahl, S.G.F.; Flatberg, A.; Sørhaug, S.; Børset, M.; Haug, M. Analysis of Intra-Tumoral Macrophages and T Cells in Non-Small Cell Lung Cancer (NSCLC) Indicates a Role for Immune Checkpoint and CD200-CD200R Interactions. Cancers 2021, 13, 1788. https://doi.org/10.3390/cancers13081788
Tøndell A, Subbannayya Y, Wahl SGF, Flatberg A, Sørhaug S, Børset M, Haug M. Analysis of Intra-Tumoral Macrophages and T Cells in Non-Small Cell Lung Cancer (NSCLC) Indicates a Role for Immune Checkpoint and CD200-CD200R Interactions. Cancers. 2021; 13(8):1788. https://doi.org/10.3390/cancers13081788
Chicago/Turabian StyleTøndell, Anders, Yashwanth Subbannayya, Sissel Gyrid Freim Wahl, Arnar Flatberg, Sveinung Sørhaug, Magne Børset, and Markus Haug. 2021. "Analysis of Intra-Tumoral Macrophages and T Cells in Non-Small Cell Lung Cancer (NSCLC) Indicates a Role for Immune Checkpoint and CD200-CD200R Interactions" Cancers 13, no. 8: 1788. https://doi.org/10.3390/cancers13081788
APA StyleTøndell, A., Subbannayya, Y., Wahl, S. G. F., Flatberg, A., Sørhaug, S., Børset, M., & Haug, M. (2021). Analysis of Intra-Tumoral Macrophages and T Cells in Non-Small Cell Lung Cancer (NSCLC) Indicates a Role for Immune Checkpoint and CD200-CD200R Interactions. Cancers, 13(8), 1788. https://doi.org/10.3390/cancers13081788