
Benign Tumors in Long-Term Survivors of Retinoblastoma

 , , , ,
, , , ,
Abstract
Simple Summary
Abstract
1. Introduction
2. Methods
2.1. Study Population and Data Collection
2.2. Subsequent Neoplasm Ascertainment
2.3. Statistical Analysis
3. Results
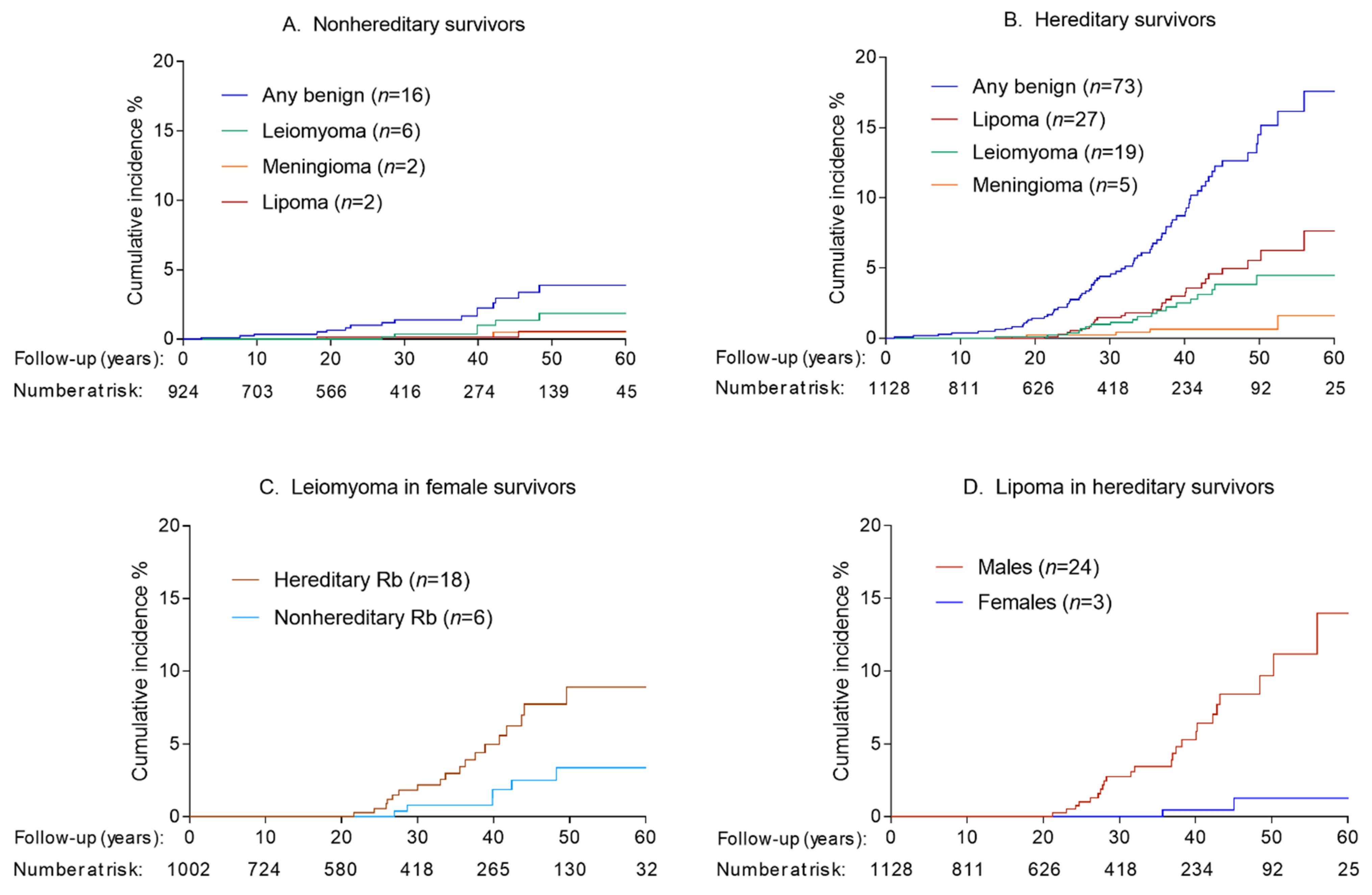
3.1. Incidence of Benign Tumors
3.2. Risk Factors for Benign Tumors
3.3. SMNs and Subsequent Risk for Benign Tumors
3.4. Benign Tumors and Subsequent Risk for SMNs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wong, J.R.; Morton, L.M.; Tucker, M.A.; Abramson, D.H.; Seddon, J.M.; Sampson, J.N.; Kleinerman, R.A. Risk of subsequent malignant neoplasms in long-term hereditary retinoblastoma survivors after chemotherapy and radiotherapy. J. Clin. Oncol. 2014, 32, 3284–3290. [Google Scholar] [CrossRef]
- Marees, T.; Moll, A.C.; Imhof, S.M.; de Boer, M.R.; Ringens, P.J.; van Leeuwen, F.E. Risk of second malignancies in survivors of retinoblastoma: More than 40 years of follow-up. J. Natl. Cancer Inst. 2008, 100, 1771–1779. [Google Scholar] [CrossRef] [PubMed]
- MacCarthy, A.; Bayne, A.M.; Brownbill, P.A.; Bunch, K.J.; Diggens, N.L.; Draper, G.J.; Hawkins, M.M.; Jenkinson, H.C.; Kingston, J.E.; Stiller, C.A.; et al. Second and subsequent tumours among 1927 retinoblastoma patients diagnosed in Britain 1951–2004. Br. J. Cancer 2013, 108, 2455–2463. [Google Scholar] [CrossRef]
- Temming, P.; Arendt, M.; Viehmann, A.; Eisele, L.; Le Guin, C.H.; Schundeln, M.M.; Biewald, E.; Astrahantseff, K.; Wieland, R.; Bornfeld, N.; et al. Incidence of second cancers after radiotherapy and systemic chemotherapy in heritable retinoblastoma survivors: A report from the German reference center. Pediatr. Blood Cancer 2017, 64, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Kleinerman, R.A.; Tucker, M.A.; Sigel, B.S.; Abramson, D.H.; Seddon, J.M.; Morton, L.M. Patterns of Cause-Specific Mortality among 2053 Survivors of Retinoblastoma, 1914–2016. J. Natl. Cancer Inst. 2019, 111, 961–969. [Google Scholar] [CrossRef]
- Marees, T.; van Leeuwen, F.E.; de Boer, M.R.; Imhof, S.M.; Ringens, P.J.; Moll, A.C. Cancer mortality in long-term survivors of retinoblastoma. Eur. J. Cancer 2009, 45, 3245–3253. [Google Scholar] [CrossRef]
- Li, F.P.; Abramson, D.H.; Tarone, R.E.; Kleinerman, R.A.; Fraumeni, J.F., Jr.; Boice, J.D., Jr. Hereditary retinoblastoma, lipoma, and second primary cancers. J. Natl. Cancer Inst. 1997, 89, 83–84. [Google Scholar] [CrossRef]
- Genuardi, M.; Klutz, M.; Devriendt, K.; Caruso, D.; Stirpe, M.; Lohmann, D.R. Multiple lipomas linked to an RB1 gene mutation in a large pedigree with low penetrance retinoblastoma. Eur. J. Hum. Genet. 2001, 9, 690–694. [Google Scholar] [CrossRef]
- Tenna, S.; Barone, M.; Gigliofiorito, P.; Persichetti, P. Retinoblastoma plus lipomatosis: An autosomal dominant syndrome. Arch. Plast. Surg. 2014, 41, 785–787. [Google Scholar] [CrossRef][Green Version]
- Rada, R. Sleep and Quality of Life in Head and Neck Neoplasm. In Sleep and Quality of Life in Clinical Medicine; Verster, J.C., Pandi-Perumal, S.R., Streiner, D.L., Eds.; Humana Press: Totowa, NJ, USA, 2008; pp. 483–488. [Google Scholar]
- Guler, O.; Mutlu, S.; Mahirogullari, M. Giant lipoma of the back affecting quality of life. Ann. Med. Surg. 2015, 4, 279–282. [Google Scholar] [CrossRef] [PubMed]
- Stewart, E.A. Clinical practice. Uterine fibroids. N. Engl. J. Med. 2015, 372, 1646–1655. [Google Scholar] [CrossRef]
- Bowers, D.C.; Moskowitz, C.S.; Chou, J.F.; Mazewski, C.M.; Neglia, J.P.; Armstrong, G.T.; Leisenring, W.M.; Robison, L.L.; Oeffinger, K.C. Morbidity and Mortality Associated With Meningioma After Cranial Radiotherapy: A Report From the Childhood Cancer Survivor Study. J. Clin. Oncol. 2017, 35, 1570–1576. [Google Scholar] [CrossRef]
- World Health Organization. International Classification of Diseases for Oncology, 3rd ed.; World Health Organization: Geneva, Switzerland, 2013. [Google Scholar]
- Kleinerman, R.A.; Schonfeld, S.J.; Sigel, B.S.; Wong-Siegel, J.R.; Gilbert, E.S.; Abramson, D.H.; Seddon, J.M.; Tucker, M.A.; Morton, L.M. Bone and Soft-Tissue Sarcoma Risk in Long-Term Survivors of Hereditary Retinoblastoma Treated With Radiation. J. Clin. Oncol. 2019, 37, 3436–3445. [Google Scholar] [CrossRef]
- Coviello, V.; Boggess, M. Cumulative incidence estimation in the presence of competing risks. Stata J. 2004, 4, 103–112. [Google Scholar] [CrossRef]
- Kok Judith, L.J. Incidence of and Risk Factors for Histologically Confirmed Solid Benign Tumors among Long-term Survivors of Childhood Cancer. JAMA Oncol. 2019, 5, 671–680. [Google Scholar]
- Kok, J.L.; Teepen, J.C.; van Leeuwen, F.E.; Tissing, W.J.E.; Neggers, S.; van der Pal, H.J.; Loonen, J.J.; Bresters, D.; Versluys, B.; van den Heuvel-Eibrink, M.M.; et al. Risk of benign meningioma after childhood cancer in the DCOG-LATER cohort: Contributions of radiation dose, exposed cranial volume, and age. Neuro Oncol. 2019, 21, 392–403. [Google Scholar] [CrossRef]
- Kleinerman, R.A.; Schonfeld, S.J.; Tucker, M.A. Sarcomas in hereditary retinoblastoma. Clin. Sarcoma Res. 2012, 2, 15. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.J.; Marino-Enriquez, A.; Fletcher, C.D.; Hornick, J.L. Loss of retinoblastoma protein expression in spindle cell/pleomorphic lipomas and cytogenetically related tumors: An immunohistochemical study with diagnostic implications. Am. J. Surg. Pathol. 2012, 36, 1119–1128. [Google Scholar] [CrossRef]
- Agaimy, A. Anisometric cell lipoma: Insight from a case series and review of the literature on adipocytic neoplasms in survivors of retinoblastoma suggest a role for RB1 loss and possible relationship to fat-predominant (“fat-only”) spindle cell lipoma. Ann. Diagn. Pathol. 2017, 29, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Dahlen, A.; Debiec-Rychter, M.; Pedeutour, F.; Domanski, H.A.; Hoglund, M.; Bauer, H.C.; Rydholm, A.; Sciot, R.; Mandahl, N.; Mertens, F. Clustering of deletions on chromosome 13 in benign and low-malignant lipomatous tumors. Int. J. Cancer 2003, 103, 616–623. [Google Scholar] [CrossRef]
- Johnson, C.N.; Ha, A.S.; Chen, E.; Davidson, D. Lipomatous Soft-tissue Tumors. J. Am. Acad. Orthop. Surg. 2018, 26, 779–788. [Google Scholar] [CrossRef]
- Michal, M.; Agaimy, A.; Contreras, A.L.; Svajdler, M.; Kazakov, D.V.; Steiner, P.; Grossmann, P.; Martinek, P.; Hadravsky, L.; Michalova, K.; et al. Dysplastic Lipoma: A Distinctive Atypical Lipomatous Neoplasm With Anisocytosis, Focal Nuclear Atypia, p53 Overexpression, and a Lack of MDM2 Gene Amplification by FISH.; A Report of 66 Cases Demonstrating Occasional Multifocality and a Rare Association With Retinoblastoma. Am. J. Surg. Pathol. 2018, 42, 1530–1540. [Google Scholar] [CrossRef]
- Downes, E.; Sikirica, V.; Gilabert-Estelles, J.; Bolge, S.C.; Dodd, S.L.; Maroulis, C.; Subramanian, D. The burden of uterine fibroids in five European countries. Eur. J. Obstet. Gynecol. Reprod. Biol. 2010, 152, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Francis, J.H.; Kleinerman, R.A.; Seddon, J.M.; Abramson, D.H. Increased risk of secondary uterine leiomyosarcoma in hereditary retinoblastoma. Gynecol. Oncol. 2012, 124, 254–259. [Google Scholar] [CrossRef] [PubMed]
- Hodge, J.C.; Morton, C.C. Genetic heterogeneity among uterine leiomyomata: Insights into malignant progression. Hum. Mol. Genet. 2007, 16, R7–R13. [Google Scholar] [CrossRef] [PubMed]
- Dolecek, T.A.; Dressler, E.V.; Thakkar, J.P.; Liu, M.; Al-Qaisi, A.; Villano, J.L. Epidemiology of meningiomas post-Public Law 107–206: The Benign Brain Tumor Cancer Registries Amendment Act. Cancer 2015, 121, 2400–2410. [Google Scholar] [CrossRef]
- Neglia, J.P.; Robison, L.L.; Stovall, M.; Liu, Y.; Packer, R.J.; Hammond, S.; Yasui, Y.; Kasper, C.E.; Mertens, A.C.; Donaldson, S.S.; et al. New primary neoplasms of the central nervous system in survivors of childhood cancer: A report from the Childhood Cancer Survivor Study. J. Natl. Cancer Inst. 2006, 98, 1528–1537. [Google Scholar] [CrossRef]
- Taylor, A.J.; Little, M.P.; Winter, D.L.; Sugden, E.; Ellison, D.W.; Stiller, C.A.; Stovall, M.; Frobisher, C.; Lancashire, E.R.; Reulen, R.C.; et al. Population-based risks of CNS tumors in survivors of childhood cancer: The British Childhood Cancer Survivor Study. J. Clin. Oncol. 2010, 28, 5287–5293. [Google Scholar] [CrossRef] [PubMed]
- Munier, F.L.; Beck-Popovic, M.; Chantada, G.L.; Cobrinik, D.; Kivela, T.T.; Lohmann, D.; Maeder, P.; Moll, A.C.; Carcaboso, A.M.; Moulin, A.; et al. Conservative management of retinoblastoma: Challenging orthodoxy without compromising the state of metastatic grace. “Alive, with good vision and no comorbidity”. Prog. Retin. Eye Res. 2019, 73, 100764. [Google Scholar] [CrossRef]


{kind=link}
| Hereditary * Retinoblastoma | Nonhereditary Retinoblastoma | |||
|---|---|---|---|---|
| N | % | N | % | |
| Total | 1128 | 100% | 924 | 100% |
| Laterality | ||||
| Unilateral | 29 | 2.6% | 924 | 100% |
| Bilateral | 1099 | 97.4% | 0 | 0% |
| Sex | ||||
| Male | 578 | 51.2% | 472 | 51.1% |
| Female | 550 | 48.8% | 452 | 48.9% |
| Age at retinoblastoma diagnosis | ||||
| 0–11 months | 661 | 58.6% | 200 | 21.6% |
| 12–23 months | 303 | 26.9% | 268 | 29.0% |
| ≥24 months | 164 | 14.5% | 456 | 49.4% |
| Calendar year of retinoblastoma diagnosis | ||||
| 1914–1959 | 304 | 27.0% | 212 | 22.9% |
| 1960–1969 | 297 | 26.3% | 224 | 24.2% |
| 1970–1979 | 248 | 22.0% | 204 | 22.1% |
| 1980–2006 | 279 | 24.7% | 284 | 30.7% |
| Family history of retinoblastoma | ||||
| Yes | 237 | 21.0% | 0 | 0.0% |
| No/unknown | 891 | 79.0% | 924 | 100.0% |
| Treatment for retinoblastoma | ||||
| Surgery only | 90 | 8.0% | 636 | 68.8% |
| Radiotherapy, no chemotherapy | 534 | 47.3% | 97 | 10.5% |
| Radiotherapy and chemotherapy | 435 | 38.6% | 86 | 9.3% |
| Chemotherapy, no radiotherapy | 38 | 3.4% | 58 | 6.3% |
| Other/unknown | 31 | 2.7% | 47 | 5.1% |
| First Benign Tumor | Second Benign Tumor | Subsequent Benign Tumors | N People |
|---|---|---|---|
| Lipoma | 16 | ||
| Lipoma | Lipoma | 6 | |
| Lipoma | Lipoma | 3rd Lipoma | 2 |
| Lipoma | Lipoma | 3rd & 4th Lipoma | 1 |
| Lipoma | Lipoma | 3rd, 4th & 5th Lipoma | 1 |
| Lipoma | Neurilemoma | 1 | |
| Leiomyoma | 17 | ||
| Leiomyoma | Leiomyoma | 2 | |
| Meningioma | 4 | ||
| Meningioma | Meningioma | 1 | |
| Fibroma | 2 | ||
| Fibroma | Fibroma | 1 | |
| Fibroma | Breast (noninvasive) | 1 | |
| Breast (noninvasive) | 4 | ||
| Thyroid adenoma | Leiomyoma | 1 | |
| Thyroid adenoma | Lipoma | 1 | |
| Sebaceous adenoma | 1 | ||
| Cutaneous adnexal tumor | Lipoma | 1 | |
| Hemangioma | Hemangioma | 1 | |
| Astrocytoma | 1 | ||
| Sinus myxoma | 1 | ||
| Lymphangioma | 1 | ||
| Hemangioma | 1 | ||
| Fibrous histiocytoma | 1 | ||
| Ovarian adenoma | 1 | ||
| Tongue papilloma | 1 | ||
| Colorectal, unspecified | 1 | ||
| Head and neck, unspecified | 1 | ||
| Any Benign | Lipoma | Leiomyoma | |||||
|---|---|---|---|---|---|---|---|
| N Total | N Cases | HR (95% CI) ‡ | N Cases | HR (95% CI) ‡ | N Cases | HR (95% CI) ‡ | |
| Total | 1128 | 73 | 27 | 19 | |||
| Sex | |||||||
| Male | 578 | 35 | 1.0 (Ref) | 24 | 1.0 (Ref) | 1 | 1.0 (Ref) |
| Female | 550 | 38 | 1.1 (0.7–1.8) | 3 | 0.1 (0.0–0.4) * | 18 | 21.1 (2.8–161.0) * |
| Family history of retinoblastoma | |||||||
| No/Unknown | 891 | 60 | 1.0 (Ref) | 21 | 1.0 (Ref) | 18 | 1.0 (Ref) |
| Yes | 237 | 13 | 1.1 (0.6–2.1) | 6 | 1.3 (0.5–3.4) | 1 | 0.4 (0.1–3.1) |
| Age at RB diagnosis | |||||||
| 0–11 months | 661 | 39 | 1.0 (Ref) | 12 | 1.0 (Ref) | 9 | 1.0 (Ref) |
| 12–23 months | 303 | 18 | 0.7 (0.4–1.3) | 9 | 1.2 (0.5–2.9) | 5 | 0.6 (0.2–1.9) |
| ≥24 months | 164 | 16 | 1.4 (0.8–2.6) | 6 | 1.9 (0.7–5.2) | 5 | 1.1 (0.3–3.5) |
| Year of RB diagnosis | |||||||
| 1914–1959 | 304 | 34 | 1.0 (Ref) | 15 | 1.0 (Ref) | 7 | 1.0 (Ref) |
| 1960–1969 | 297 | 26 | 0.7 (0.4–1.2) | 9 | 0.5 (0.2–1.2) | 7 | 1.1 (0.4–3.2) |
| 1970–1979 | 248 | 10 | 0.6 (0.3–1.3) | 3 | 0.4 (0.1–1.4) | 5 | 3.0 (0.9–10.0) |
| 1980–2006 | 279 | 3 | 0.5 (0.1–1.6) | 0 | – | 0 | − |
| Treatment for retinoblastoma | |||||||
| Radiation, no chemotherapy | 534 | 33 | 1.0 (Ref) | 12 | 1.0 (Ref) | 5 | 1.0 (Ref) |
| Radiation and chemotherapy | 435 | 35 | 1.4 (0.9–2.3) | 13 | 1.5 (0.7–3.4) | 12 | 2.7 (0.9–8.0) |
| Other † | 159 | 5 | 0.6 (0.2–1.5) | 2 | 0.6 (0.1–2.6) | 2 | 1.5 (0.3–8.0) |
| Prior malignancy | |||||||
| No | 932 | 54 | 1.0 (Ref) | 18 | 1.0 (Ref) | 13 | 1.0 (Ref) |
| Yes | 196 | 19 | 3.5 (2.0–6.1) * | 9 | 3.7 (1.6–8.7) * | 6 | 6.4 (2.2–18.7) * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
van Hoefen Wijsard, M.; Schonfeld, S.J.; van Leeuwen, F.E.; Moll, A.C.; Fabius, A.W.; Abramson, D.H.; Seddon, J.M.; Francis, J.H.; Tucker, M.A.; Kleinerman, R.A.; et al. Benign Tumors in Long-Term Survivors of Retinoblastoma. Cancers 2021, 13, 1773. https://doi.org/10.3390/cancers13081773
van Hoefen Wijsard M, Schonfeld SJ, van Leeuwen FE, Moll AC, Fabius AW, Abramson DH, Seddon JM, Francis JH, Tucker MA, Kleinerman RA, et al. Benign Tumors in Long-Term Survivors of Retinoblastoma. Cancers. 2021; 13(8):1773. https://doi.org/10.3390/cancers13081773
Chicago/Turabian Stylevan Hoefen Wijsard, Milo, Sara J. Schonfeld, Flora E. van Leeuwen, Annette C. Moll, Armida W. Fabius, David H. Abramson, Johanna M. Seddon, Jasmine H. Francis, Margaret A. Tucker, Ruth A. Kleinerman, and et al. 2021. "Benign Tumors in Long-Term Survivors of Retinoblastoma" Cancers 13, no. 8: 1773. https://doi.org/10.3390/cancers13081773
APA Stylevan Hoefen Wijsard, M., Schonfeld, S. J., van Leeuwen, F. E., Moll, A. C., Fabius, A. W., Abramson, D. H., Seddon, J. M., Francis, J. H., Tucker, M. A., Kleinerman, R. A., & Morton, L. M. (2021). Benign Tumors in Long-Term Survivors of Retinoblastoma. Cancers, 13(8), 1773. https://doi.org/10.3390/cancers13081773