
Identification and Validation of an Immune-Associated RNA-Binding Proteins Signature to Predict Clinical Outcomes and Therapeutic Responses in Glioma Patients


Abstract
Simple Summary
Abstract
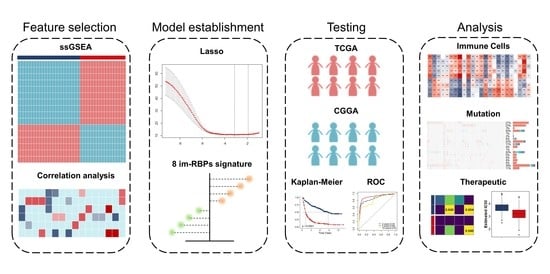
1. Introduction
2. Materials and Methods
2.1. Data Acquisition
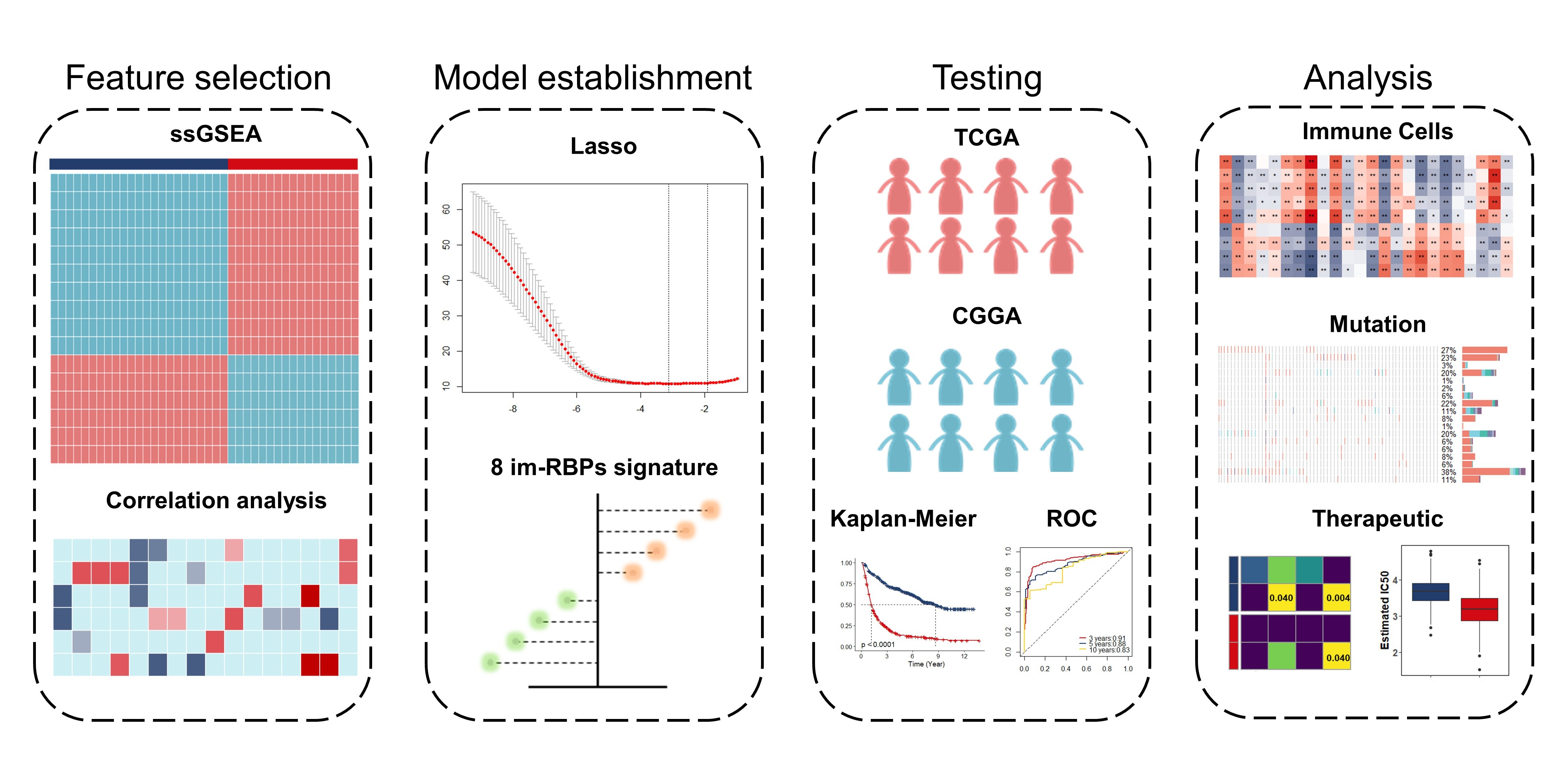
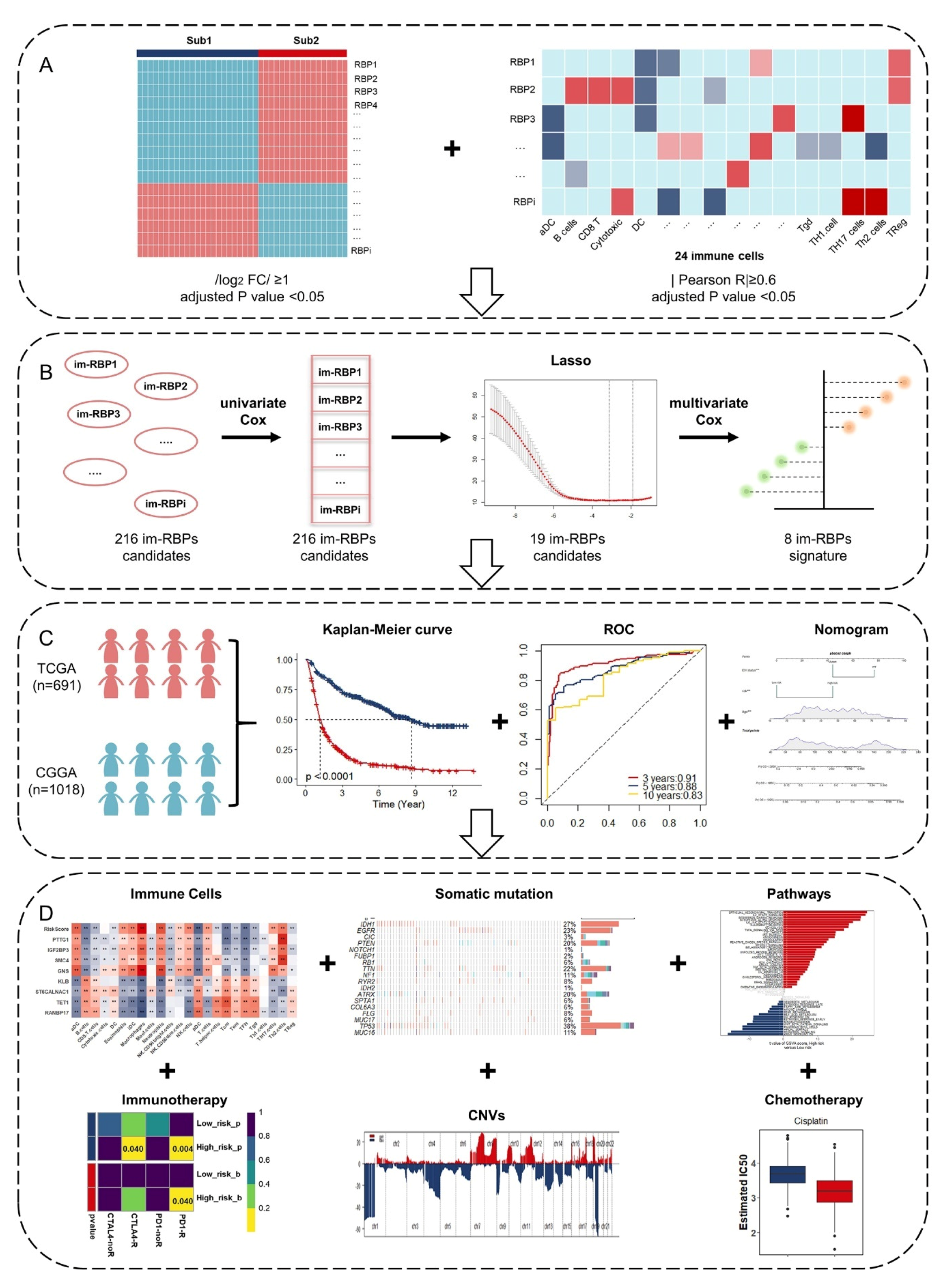
2.2. Identification of Glioma Immune Subtypes Based on ssGSEA Score
2.3. Identification of Immune-Associated RBPs in Glioma
2.4. Risk-Based Modeling
2.5. Pathway Enrichment Analysis
2.6. Immuno-/Chemotherapeutic Response Prediction
2.7. Statistical Analysis
3. Results
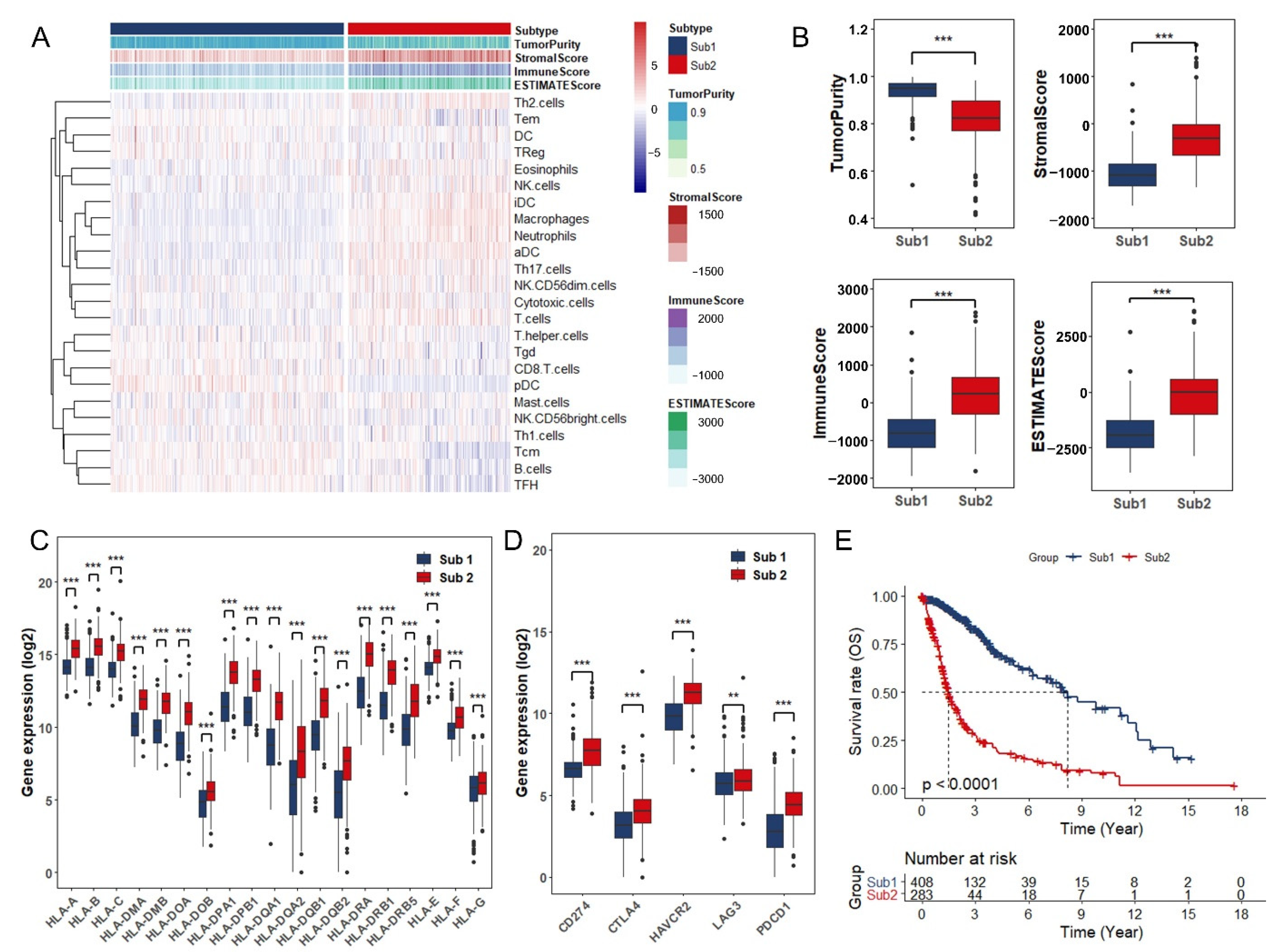
3.1. Identification of Glioma Immune Subtypes Based on Infiltration of Immune Cells
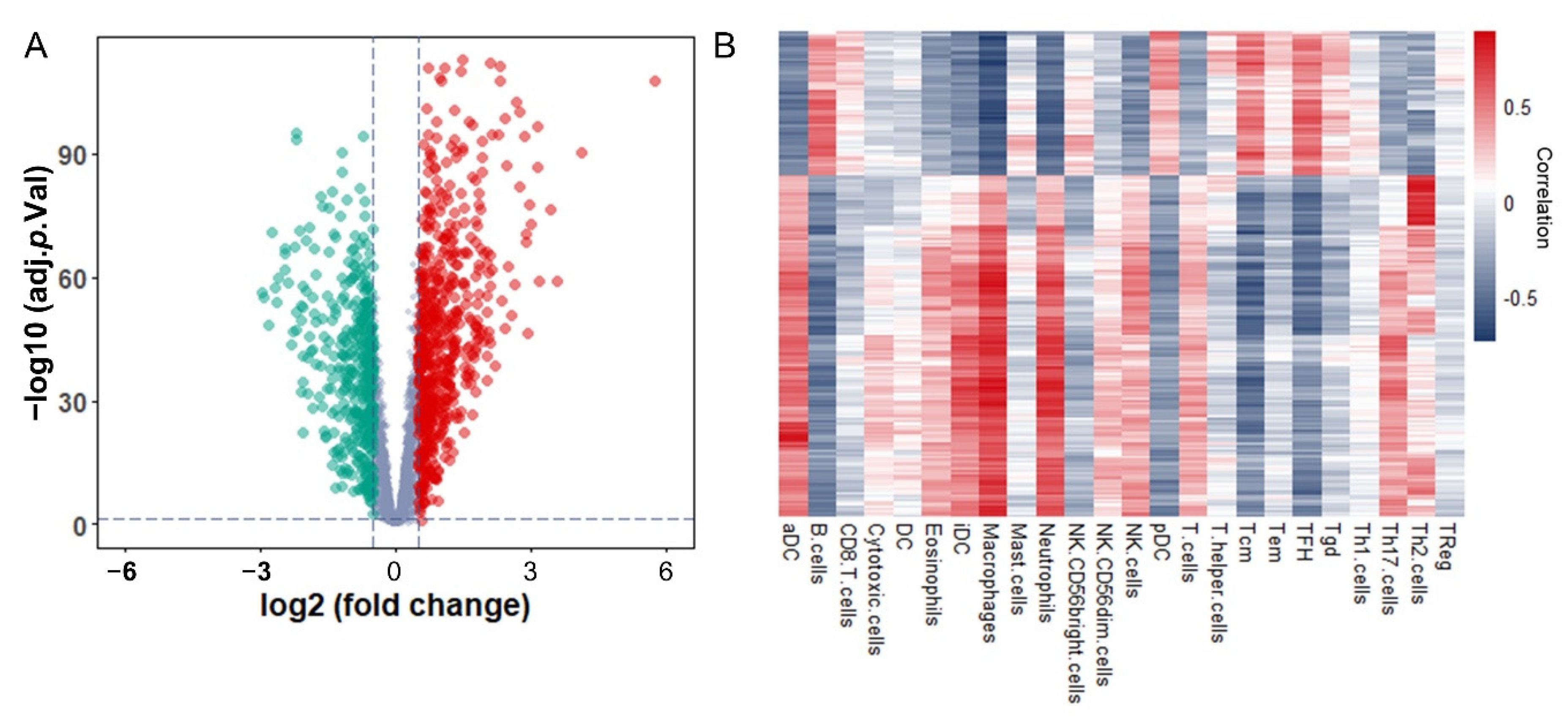
3.2. Identification and Functional Enrichment Analysis of Immune-Associated RBPs in Glioma Patients
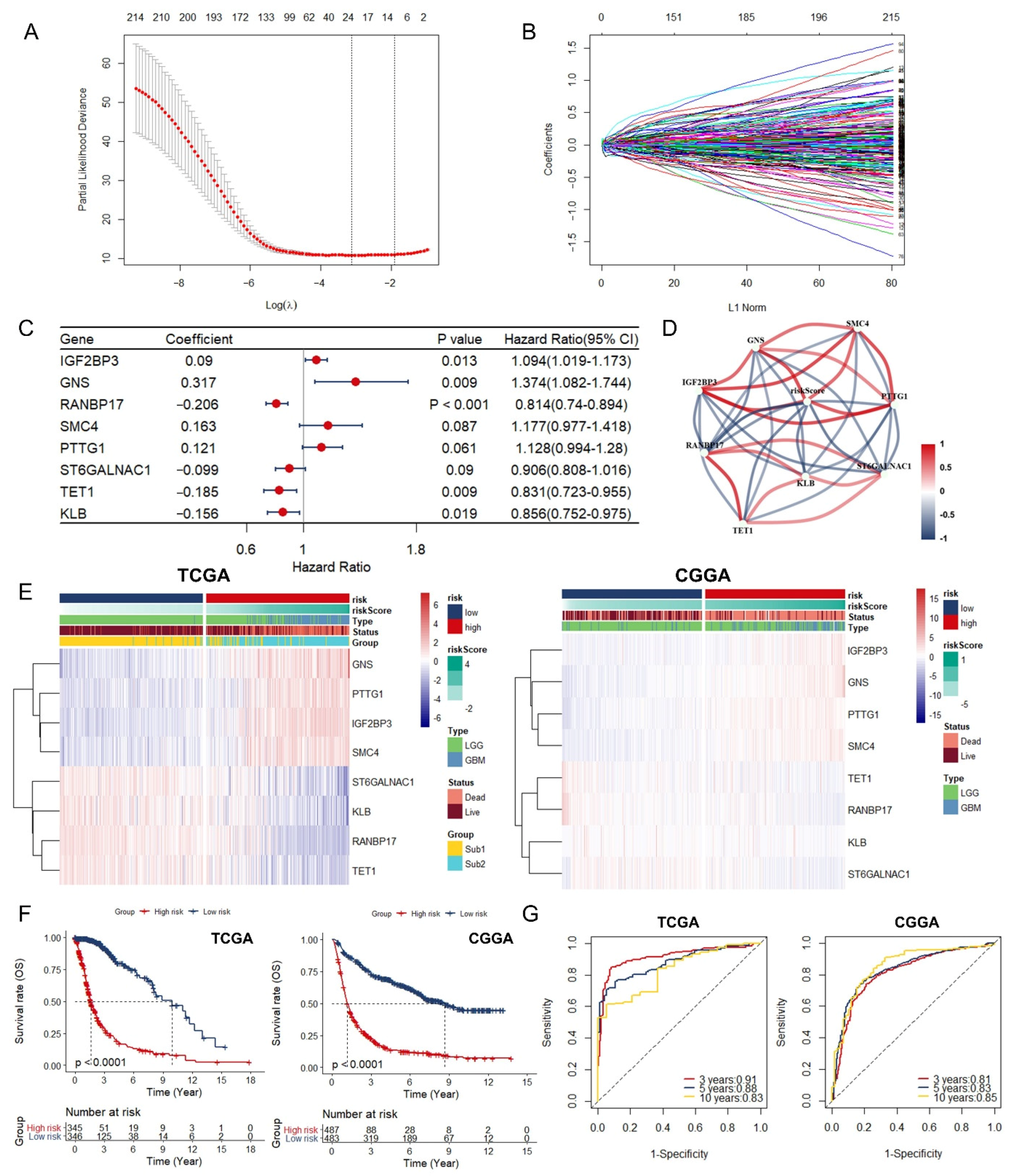
3.3. Identification and Assessment of an Immune-Associated RBPs Prognostic Signature for Overall Survival in Glioma.
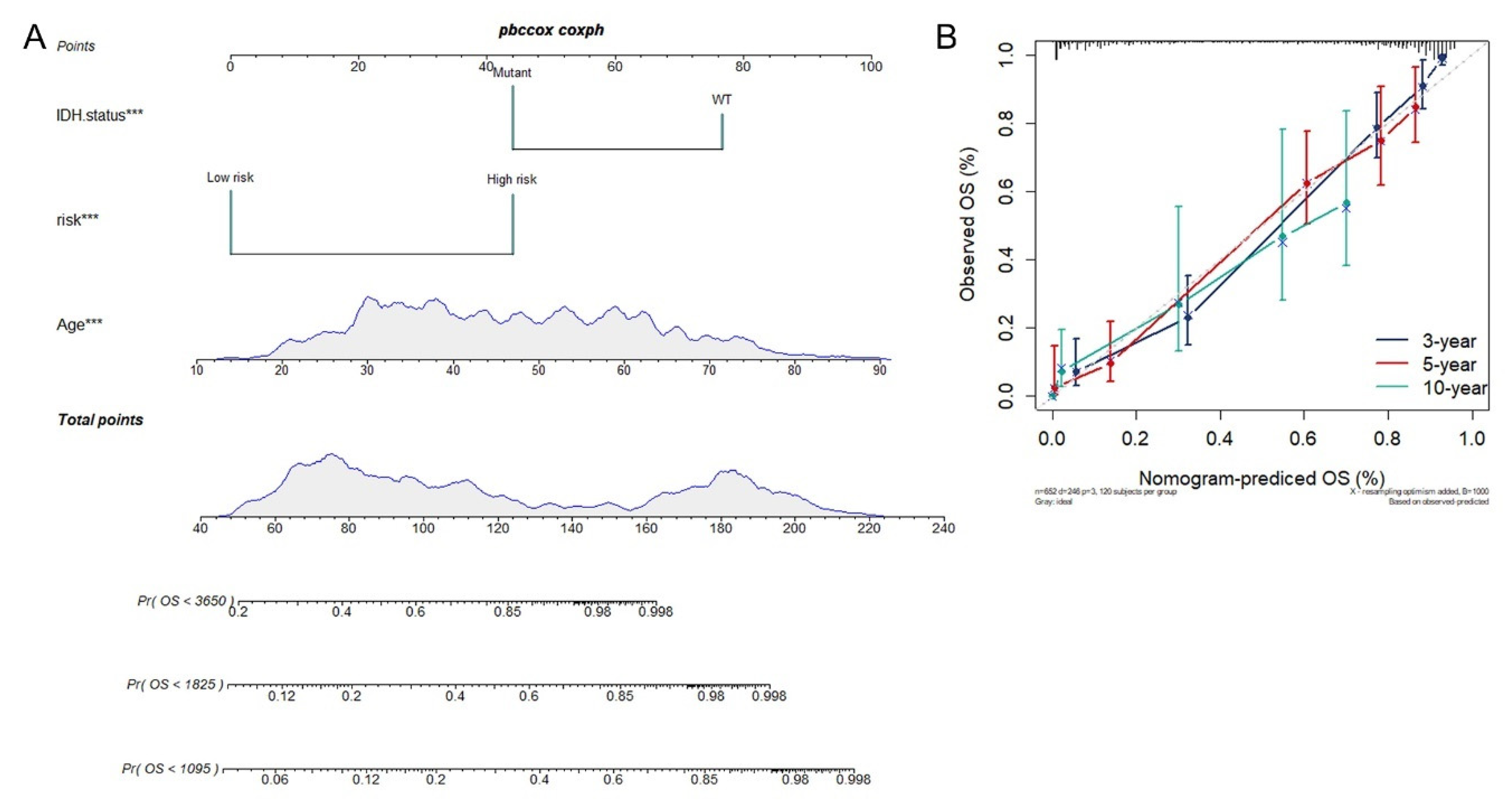
3.4. Construction of Integrated Model to Predict Survival of Glioma Patients
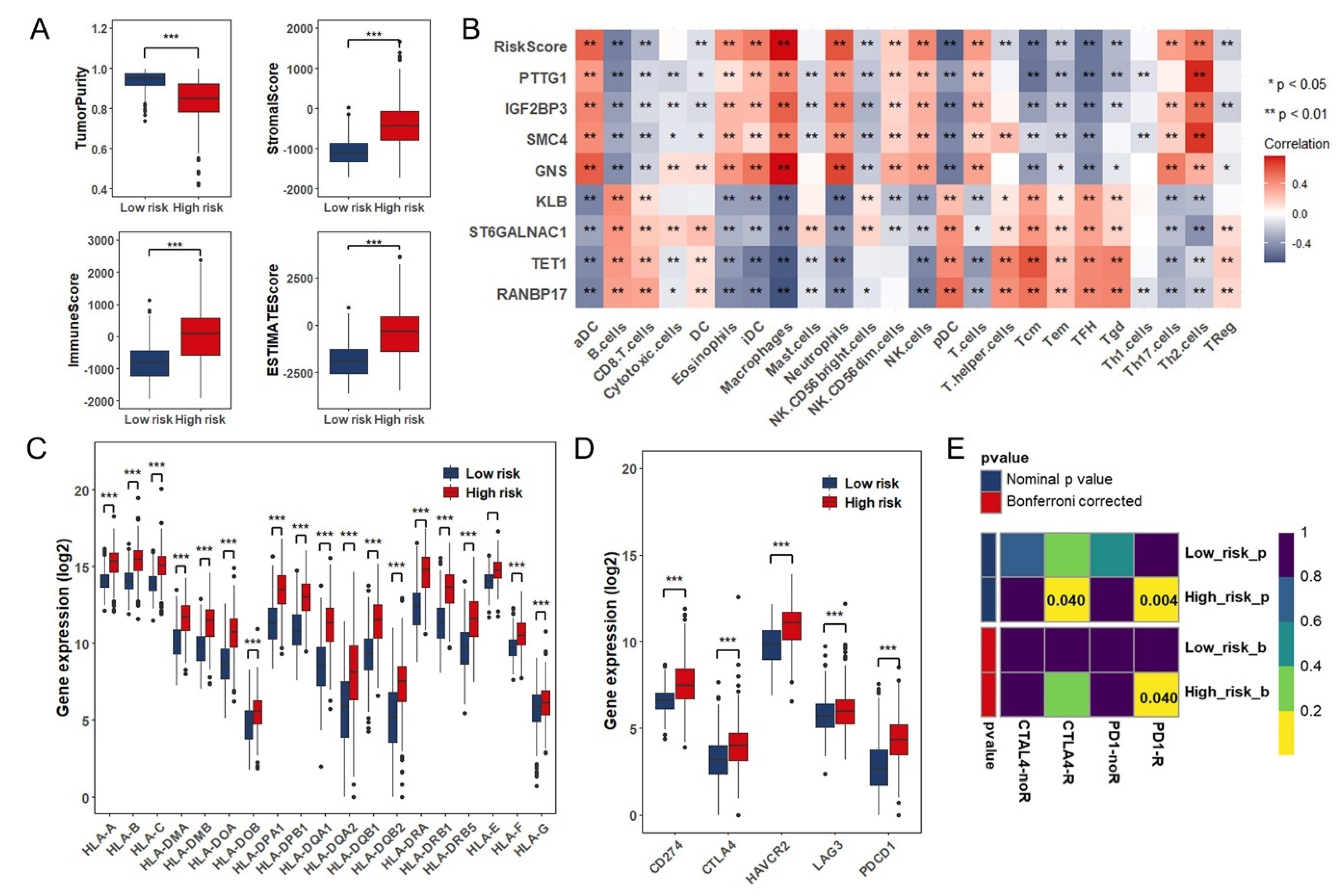
3.5. Estimation of Tumor-Infiltrating Immune Cells and Prediction of Therapeutic Response to Immune Checkpoint Inhibitors
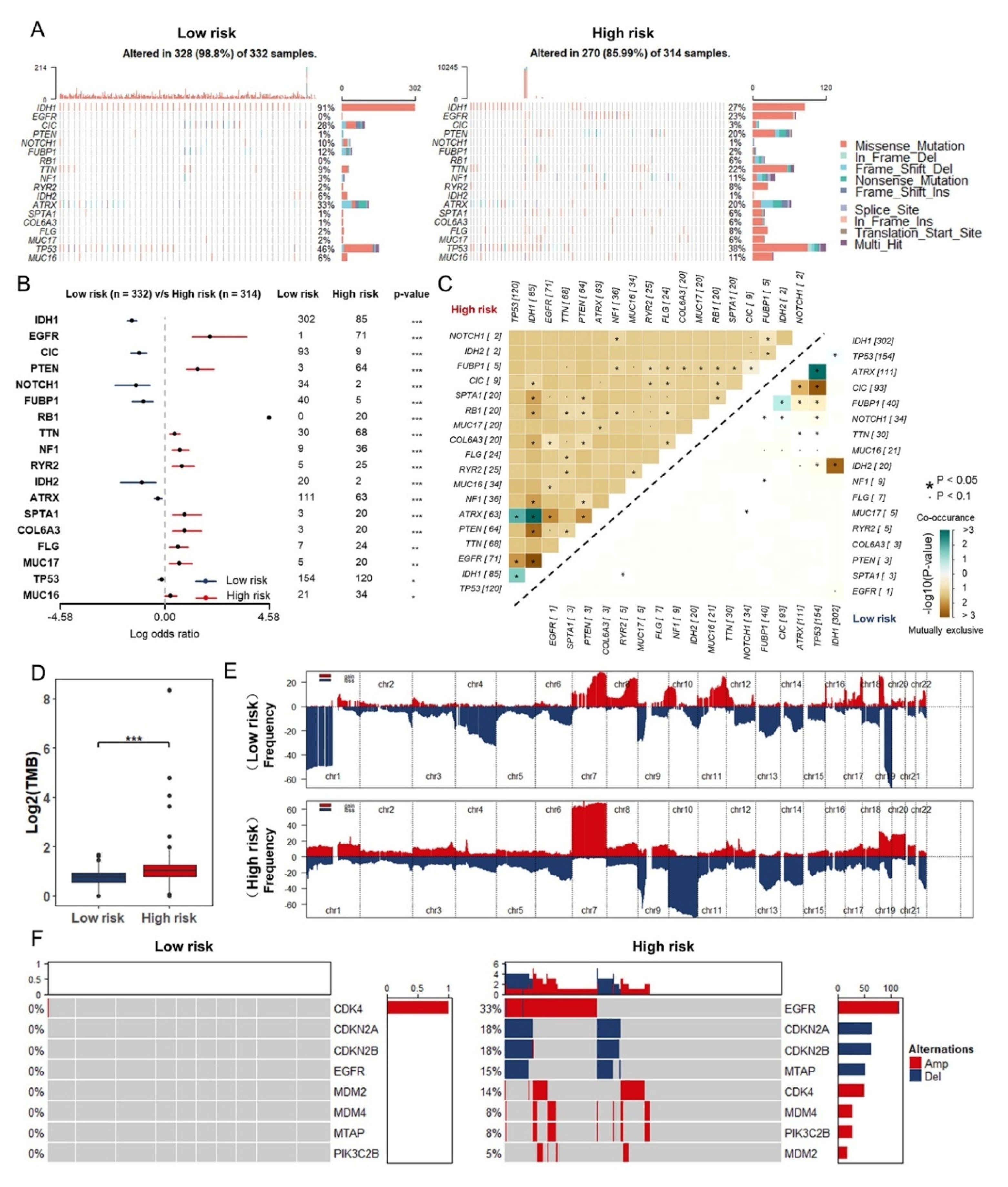
3.6. Somatic Mutation and Copy Number Variations in Different Risk Groups
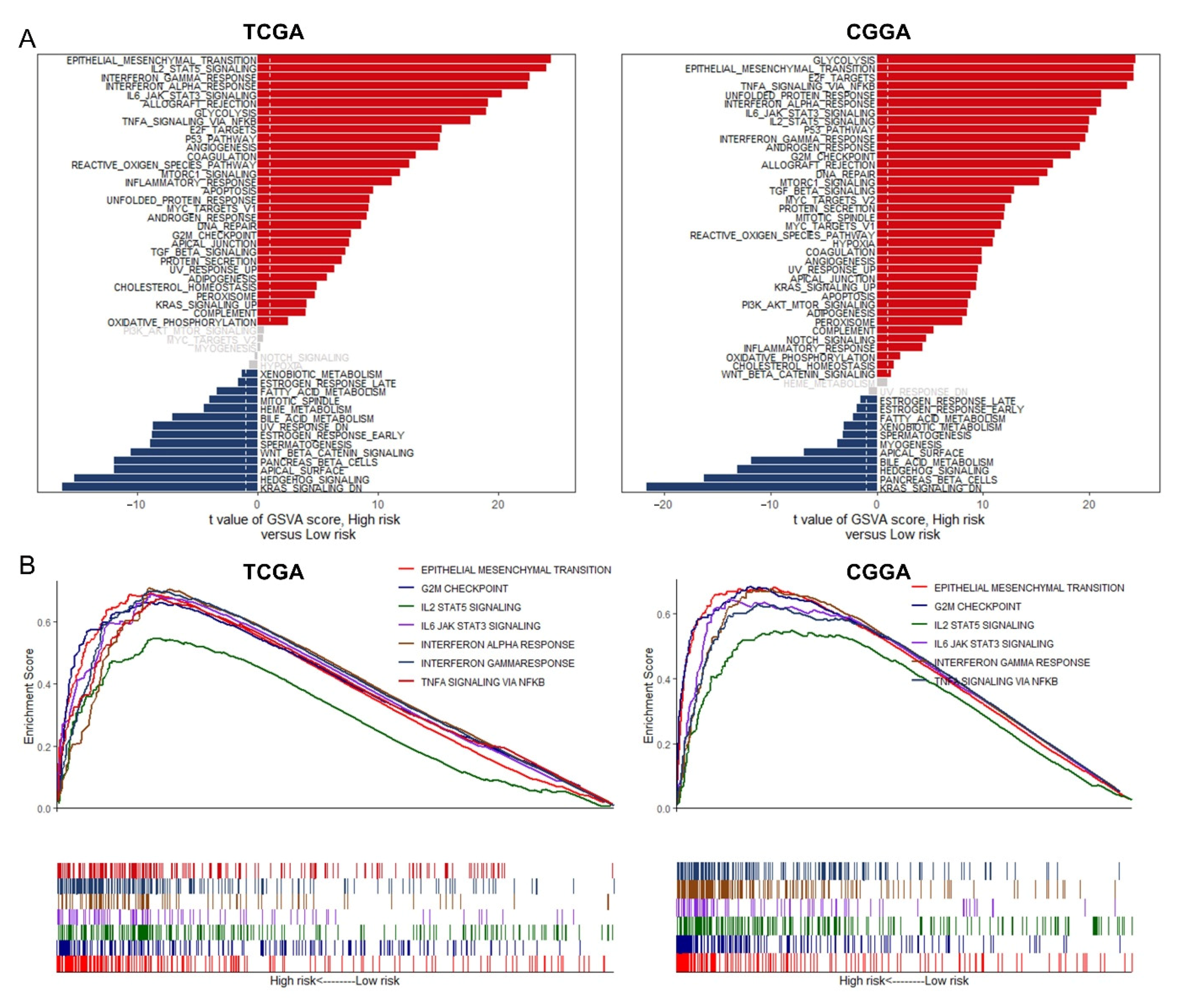
3.7. GSVA and GSEA for Different Risk Groups
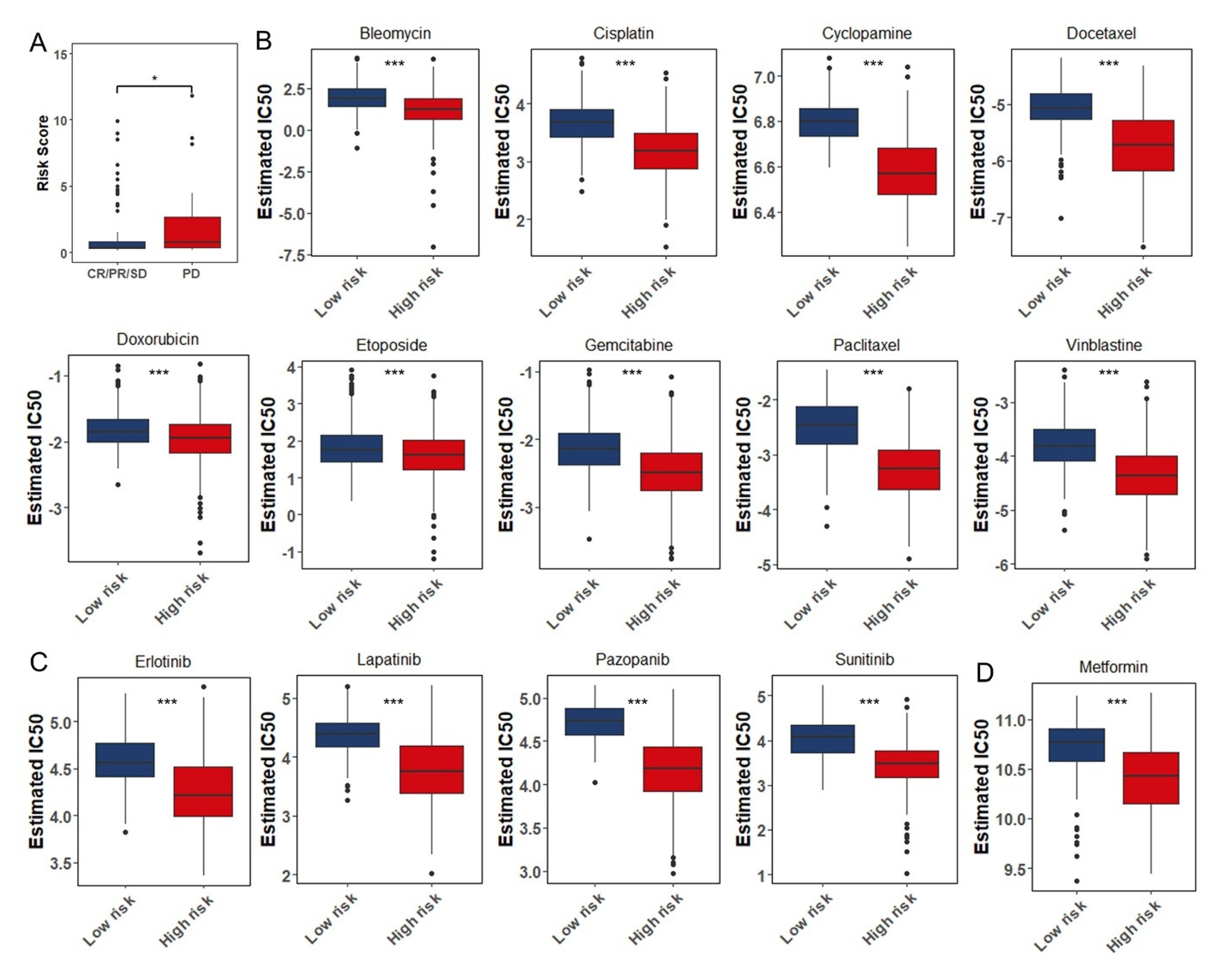
3.8. The Role of the Eight Immune-Associated RBPs Signature in Predicting the Sensitivity to Chemotherapeutic Agents
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ostrom, Q.T.; Patil, N.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. Corrigendum to: CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2013–2017. Neuro-Oncology 2020, 22, iv1–iv96. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; Von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef]
- Heimberger, A.B.; Abou-Ghazal, M.; Reina-Ortiz, C.; Yang, D.S.; Sun, W.; Qiao, W.; Hiraoka, N.; Fuller, G.N. Incidence and Prognostic Impact of FoxP3+ Regulatory T Cells in Human Gliomas. Clin. Cancer Res. 2008, 14, 5166–5172. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Liu, B.; Sun, Q.; Xiong, X.; Chen, Q. Immune Checkpoint Targeted Therapy in Glioma: Status and Hopes. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Jiang, X.; Tan, J.; Xin, Z.; Zhou, Q.; Zhan, C.; Fu, X.; Wu, Z.; Guo, Y.; Jiang, Z.; et al. Development and Validation of a Tumor Mutation Burden-Related Immune Prognostic Model for Lower-Grade Glioma. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Marisetty, A.; Schrand, B.; Gabrusiewicz, K.; Hashimoto, Y.; Ott, M.; Grami, Z.; Kong, L.Y.; Ling, X.; Caruso, H.G.; et al. Osteopontin mediates glioblastoma-associated macrophage infiltration and is a potential therapeutic target. J. Clin. Investig. 2018, 129, 137–149. [Google Scholar] [CrossRef]
- Bloch, O.; Crane, C.A.; Kaur, R.; Safaee, M.; Rutkowski, M.J.; Parsa, A.T. Gliomas Promote Immunosuppression through Induction of B7-H1 Expression in Tumor-Associated Macrophages. Clin. Cancer Res. 2013, 19, 3165–3175. [Google Scholar] [CrossRef]
- Gerstberger, S.; Hafner, M.; Tuschl, T. A census of human RNA-binding proteins. Nat. Rev. Genet. 2014, 15, 829–845. [Google Scholar] [CrossRef]
- Cheung, H.C.; Hai, T.; Zhu, W.; Baggerly, K.A.; Tsavachidis, S.; Krahe, R.; Cote, G.J. Splicing factors PTBP1 and PTBP2 promote proliferation and migration of glioma cell lines. Brain 2009, 132, 2277–2288. [Google Scholar] [CrossRef]
- Kosti, A.; De Araujo, P.R.; Li, W.Q.; Guardia, G.D.A.; Chiou, J.; Yi, C.; Ray, D.; Meliso, F.; Li, Y.M.; Delambre, T.; et al. The RNA-binding protein SERBP1 functions as a novel oncogenic factor in glioblastoma by bridging cancer metabolism and epigenetic regulation. Genome Biol. 2020, 21, 1–32. [Google Scholar] [CrossRef]
- Lan, Y.; Lou, J.; Hu, J.; Yu, Z.; Lyu, W.; Zhang, B. Downregulation of SNRPG induces cell cycle arrest and sensitizes human glioblastoma cells to temozolomide by targeting Myc through a p53-dependent signaling pathway. Cancer Biol. Med. 2020, 17, 112–131. [Google Scholar] [PubMed]
- Koso, H.; Yi, H.; Sheridan, P.; Miyano, S.; Ino, Y.; Todo, T.; Watanabe, S. Identification of RNA-Binding Protein LARP4B as a Tumor Suppressor in Glioma. Cancer Res. 2016, 76, 2254–2264. [Google Scholar] [CrossRef] [PubMed]
- Dreyfuss, G.; Kim, V.N.; Kataoka, N. Messenger-RNA-binding proteins and the messages they carry. Nat. Rev. Mol. Cell Biol. 2002, 3, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Kafasla, P.; Skliris, A.; Kontoyiannis, D.L. Post-transcriptional coordination of immunological responses by RNA-binding proteins. Nat. Immunol. 2014, 15, 492–502. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.; Díaz-Muñoz, M.D. RNA-binding proteins control gene expression and cell fate in the immune system. Nat. Immunol. 2018, 19, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Tang, W.; Yuan, J.; Qiang, B.; Han, W.; Peng, X. Integrated Analysis of RNA-Binding Proteins in Glioma. Cancers 2020, 12, 892. [Google Scholar] [CrossRef]
- Bindea, G.; Mlecnik, B.; Tosolini, M.; Kirilovsky, A.; Waldner, M.; Obenauf, A.C.; Angell, H.; Fredriksen, T.; Lafontaine, L.; Berger, A.; et al. Spatiotemporal Dynamics of Intratumoral Immune Cells Reveal the Immune Landscape in Human Cancer. Immunity 2013, 39, 782–795. [Google Scholar] [CrossRef]
- Zuo, S.; Wei, M.; Wang, S.; Dong, J.; Wei, J. Pan-Cancer Analysis of Immune Cell Infiltration Identifies a Prognostic Immune-Cell Characteristic Score (ICCS) in Lung Adenocarcinoma. Front. Immunol. 2020, 11, 1218. [Google Scholar] [CrossRef]
- Zhang, L.; Zhao, Y.; Dai, Y.; Cheng, J.N.; Gong, Z.; Feng, Y.; Sun, C.; Jia, Q.; Zhu, B. Immune Landscape of Colorectal Cancer Tumor Microenvironment from Different Primary Tumor Location. Front. Immunol. 2018, 9, 1578. [Google Scholar] [CrossRef]
- Finotello, F.; Trajanoski, Z. Quantifying tumor-infiltrating immune cells from transcriptomics data. Cancer Immunol. Immunother. 2018, 67, 1031–1040. [Google Scholar] [CrossRef]
- Aran, D.; Hu, Z.; Butte, A.J. xCell: Digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017, 18, 220. [Google Scholar] [CrossRef] [PubMed]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-Seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef]
- Wilkerson, M.D.; Hayes, D.N. ConsensusClusterPlus: A class discovery tool with confidence assessments and item tracking. Bioinformarics 2010, 26, 1572–1573. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Zhang, T.; Guo, W.; Zhou, C.; Cui, X.; Gao, L.; Cai, C.; Xu, Y. Integrative Analysis of Multi-Omics Identified the Prognostic Biomarkers in Acute Myelogenous Leukemia. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef]
- Yoshihara, K.; Shahmoradgoli, M.; Martínez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Treviño, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Lu, X.; Jiang, L.; Zhang, L.; Zhu, Y.; Hu, W.; Wang, J.; Ruan, X.; Xu, Z.; Meng, X.; Gao, J.; et al. Immune Signature-Based Subtypes of Cervical Squamous Cell Carcinoma Tightly Associated with Human Papillomavirus Type 16 Expression, Molecular Features, and Clinical Outcome. Neoplasia 2019, 21, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Ma, J.; Liu, X.; Liu, Z. Development and validation of a novel stem cell subtype for bladder cancer based on stem genomic profiling. Stem Cell Res. Ther. 2020, 11, 1–15. [Google Scholar] [CrossRef]
- Hoshida, Y.; Brunet, J.P.; Tamayo, P.; Golub, T.R.; Mesirov, J.P. Subclass Mapping: Identifying Common Subtypes in Independent Disease Data Sets. PLoS ONE 2007, 2, e1195. [Google Scholar] [CrossRef]
- Reardon, D.A.; Gokhale, P.C.; Klein, S.R.; Ligon, K.L.; Rodig, S.J.; Ramkissoon, S.H.; Jones, K.L.; Conway, A.S.; Liao, X.; Zhou, J.; et al. Glioblastoma Eradication Following Immune Checkpoint Blockade in an Orthotopic, Immunocompetent Model. Cancer Immunol. Res. 2016, 4, 124–135. [Google Scholar] [CrossRef]
- Roh, W.; Chen, P.L.; Reuben, A.; Spencer, C.N.; Prieto, P.A.; Miller, J.P.; Gopalakrishnan, V.; Wang, F.; Cooper, Z.A.; Reddy, S.M.; et al. Integrated molecular analysis of tumor biopsies on sequential CTLA-4 and PD-1 blockade reveals markers of response and resistance. Sci. Transl. Med. 2017, 9, eaah3560. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The Somatic Genomic Landscape of Glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Frattini, V.; Trifonov, V.; Chan, J.M.; Castano, A.; Lia, M.; Abate, F.; Keir, S.T.; Ji, A.X.; Zoppoli, P.; Niola, F.; et al. The integrated landscape of driver genomic alterations in glioblastoma. Nat. Genet. 2013, 45, 1141–1149. [Google Scholar] [CrossRef]
- Cimino, P.J.; McFerrin, L.; Wirsching, H.G.; Arora, S.; Bolouri, H.; Rabadan, R.; Weller, M.; Holland, E.C. Copy number profiling across glioblastoma populations has implications for clinical trial design. Neuro Oncol. 2018, 20, 1368–1373. [Google Scholar] [CrossRef] [PubMed]
- Brat, D.J.; Verhaak, R.G.W.; Aldape, K.D.; Yung, W.K.A.; Salama, S.R.; Cooper, L.A.D.; Rheinbay, E.; Miller, C.R.; Vitucci, M.; Morozova, O.; et al. Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N. Engl. J. Med. 2017, 372, 2481–2498. [Google Scholar] [CrossRef]
- Van Meir, E.G.; Hadjipanayis, C.G.; Norden, A.D.; Shu, H.K.; Wen, P.Y.; Olson, J.J. Exciting new advances in neuro-oncology: The avenue to a cure for malignant glioma. CA Cancer J. Clin. 2010, 60, 166–193. [Google Scholar] [CrossRef] [PubMed]
- Borisov, N.; Sorokin, M.; Tkachev, V.; Garazha, A.; Buzdin, A. Cancer gene expression profiles associated with clinical outcomes to chemotherapy treatments. BMC Med Genom. 2020, 13, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Dolecek, T.A.; Propp, J.M.; Stroup, N.E.; Kruchko, C. CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2005-2009. Neuro-Oncology 2012, 14, v1–v49. [Google Scholar] [CrossRef]
- Young, R.M.; Jamshidi, A.; Davis, G.; Sherman, J.H. Current trends in the surgical management and treatment of adult glioblastoma. Ann. Transl. Med. 2015, 3, 121. [Google Scholar]
- Emens, L.A.; Ascierto, P.A.; Darcy, P.K.; Demaria, S.; Eggermont, A.M.; Redmond, W.L.; Seliger, B.; Marincola, F.M. Cancer immunotherapy: Opportunities and challenges in the rapidly evolving clinical landscape. Eur. J. Cancer 2017, 81, 116–129. [Google Scholar] [CrossRef] [PubMed]
- Lunde, B.M.; Moore, C.; Varani, G. RNA-binding proteins: Modular design for efficient function. Nat. Rev. Mol. Cell Biol. 2007, 8, 479–490. [Google Scholar] [CrossRef]
- Mancarella, C.; Scotlandi, K. IGF2BP3 From Physiology to Cancer: Novel Discoveries, Unsolved Issues, and Future Perspectives. Front. Cell Dev. Biol. 2020, 7, 363. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, X.; Guan, G.; Zhao, W.; Zhuang, M. A Risk Classification System with Five-Gene for Survival Prediction of Glioblastoma Patients. Front. Neurol. 2019, 10, 745. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cao, X. The origin and function of tumor-associated macrophages. Cell. Mol. Immunol. 2015, 12, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Becht, E.; Giraldo, N.A.; Germain, C.; De Reyniès, A.; Laurent-Puig, P.; Zucman-Rossi, J.; Dieu-Nosjean, M.C.; Sautès-Fridman, C.; Fridman, W.H. Immune Contexture, Immunoscore, and Malignant Cell Molecular Subgroups for Prognostic and Theranostic Classifications of Cancers. Adv. Immunol. 2016, 130, 95–190. [Google Scholar] [CrossRef]
- Zhou, W.; Ke, S.Q.; Huang, Z.; Flavahan, W.A.; Fang, X.; Paul, J.; Wu, L.; Sloan, A.E.; McLendon, R.E.; Li, X.; et al. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat. Cell Biol. 2015, 17, 170–182. [Google Scholar] [CrossRef]
- Fujita, M.; Scheurer, M.E.; Decker, S.A.; McDonald, H.A.; Kohanbash, G.; Kastenhuber, E.R.; Kato, H.; Bondy, M.L.; Ohlfest, J.R.; Okada, H. Role of Type 1 IFNs in Antiglioma Immunosurveillance: Using Mouse Studies to Guide Examination of Novel Prognostic Markers in Humans. Clin. Cancer Res. 2010, 16, 3409–3419. [Google Scholar] [CrossRef]
- Colombo, M.P.; Piconese, S. Regulatory T-cell inhibition versus depletion: The right choice in cancer immunotherapy. Nat. Rev. Cancer 2007, 7, 880–887. [Google Scholar] [CrossRef]
- Klebanoff, C.A.; Gattinoni, L.; Torabi-Parizi, P.; Kerstann, K.; Cardones, A.R.; Finkelstein, S.E.; Palmer, D.C.; Antony, P.A.; Hwang, S.T.; Rosenberg, S.A.; et al. Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proc. Natl. Acad. Sci. USA 2005, 102, 9571–9576. [Google Scholar] [CrossRef]
- Maire, C.L.; Ligon, K.L. Molecular pathologic diagnosis of epidermal growth factor receptor. Neuro-Oncology 2014, 16, viii1–viii6. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Bonde, A.K.; Tischler, V.; Kumar, S.; Soltermann, A.; Schwendener, R.A. Intratumoral macrophages contribute to epithelial-mesenchymal transition in solid tumors. BMC Cancer 2012, 12, 35. [Google Scholar] [CrossRef]
- Liu, C.Y.; Xu, J.Y.; Shi, X.Y.; Huang, W.; Ruan, T.Y.; Xie, P.; Ding, J.L. M2-polarized tumor-associated macrophages promoted epithelial–mesenchymal transition in pancreatic cancer cells, partially through TLR4/IL-10 signaling pathway. Lab. Investig. 2013, 93, 844–854. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, S.; Sho, M.; Yamato, I.; Yoshiji, H.; Wakatsuki, K.; Nishiwada, S.; Yagita, H.; Nakajima, Y. Simultaneous blockade of programmed death 1 and vascular endothelial growth factor receptor 2 (VEGFR2) induces synergistic anti-tumour effect in vivo. Clin Exp Immunol. 2013, 172, 500–506. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Univariate Analysis | Multivariate Analysis | |||
|---|---|---|---|---|
| HR (95% CI) | p Value | HR (95% CI) | p Value | |
| Age ≥ Median vs. < Median) | 2.891 (2.38–3.511) | <0.001 | 1.818 (1.186–2.788) | 0.006 |
| Sex (Male vs. Female) | 1.189 (1.001–1.412) | 0.049 | 1.262 (0.917–1.737) | 0.154 |
| Grade (WHO IV vs. WHO II~III) | 4.729 (3.913–5.716) | <0.001 | 1.402 (0.904–2.173) | 0.131 |
| ATRX.status (WT vs. Mutant) | 1.911 (1.528–2.389) | <0.001 | 1.095 (0.623–1.924) | 0.754 |
| IDH.status (WT vs. Mutant) | 5.032 (4.12–6.146) | <0.001 | 2.28 (1.256–4.139) | 0.007 |
| MGMT.promoter (Methylated vs. Unmethylated) | 2.333 (1.924–2.829) | <0.001 | 0.854 (0.573–1.272) | 0.436 |
| TERT.promoter (WT vs. Mutant) | 0.588 (0.44–0.785) | <0.001 | 0.948 (0.558–1.612) | 0.845 |
| Immune subtype (Sub2 vs. Sub1) | 3.262 (2.708–3.929) | <0.001 | 1.414 (0.912–2.195) | 0.122 |
| Risk (High vs. Low) | 4.335 (3.468–5.421) | <0.001 | 1.897 (1.147–3.138) | 0.013 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, R.; Li, Y.; Liu, Q.; Shu, M. Identification and Validation of an Immune-Associated RNA-Binding Proteins Signature to Predict Clinical Outcomes and Therapeutic Responses in Glioma Patients. Cancers 2021, 13, 1730. https://doi.org/10.3390/cancers13071730
Tian R, Li Y, Liu Q, Shu M. Identification and Validation of an Immune-Associated RNA-Binding Proteins Signature to Predict Clinical Outcomes and Therapeutic Responses in Glioma Patients. Cancers. 2021; 13(7):1730. https://doi.org/10.3390/cancers13071730
Chicago/Turabian StyleTian, Ruotong, Yimin Li, Qian Liu, and Minfeng Shu. 2021. "Identification and Validation of an Immune-Associated RNA-Binding Proteins Signature to Predict Clinical Outcomes and Therapeutic Responses in Glioma Patients" Cancers 13, no. 7: 1730. https://doi.org/10.3390/cancers13071730
APA StyleTian, R., Li, Y., Liu, Q., & Shu, M. (2021). Identification and Validation of an Immune-Associated RNA-Binding Proteins Signature to Predict Clinical Outcomes and Therapeutic Responses in Glioma Patients. Cancers, 13(7), 1730. https://doi.org/10.3390/cancers13071730