
Colorectal Cancer and Immunity: From the Wet Lab to Individuals



Abstract
:Simple Summary
Abstract
1. Introduction
2. In Vitro Models for Immunotherapy Studies
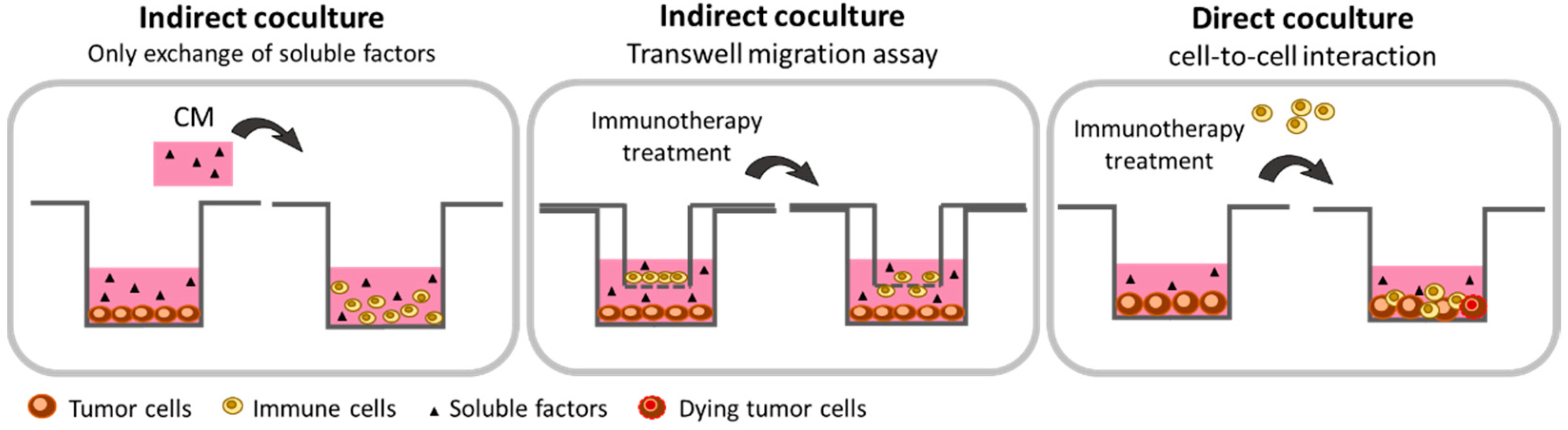
2.1. 2-Dimensional Methods
2.1.1. Cancer Cells: Secretome Assessment
2.1.2. Co-Culture with Paracrine Interaction: The Transwell Technology
2.1.3. Co-Culture with Direct Cell-to-Cell Interaction
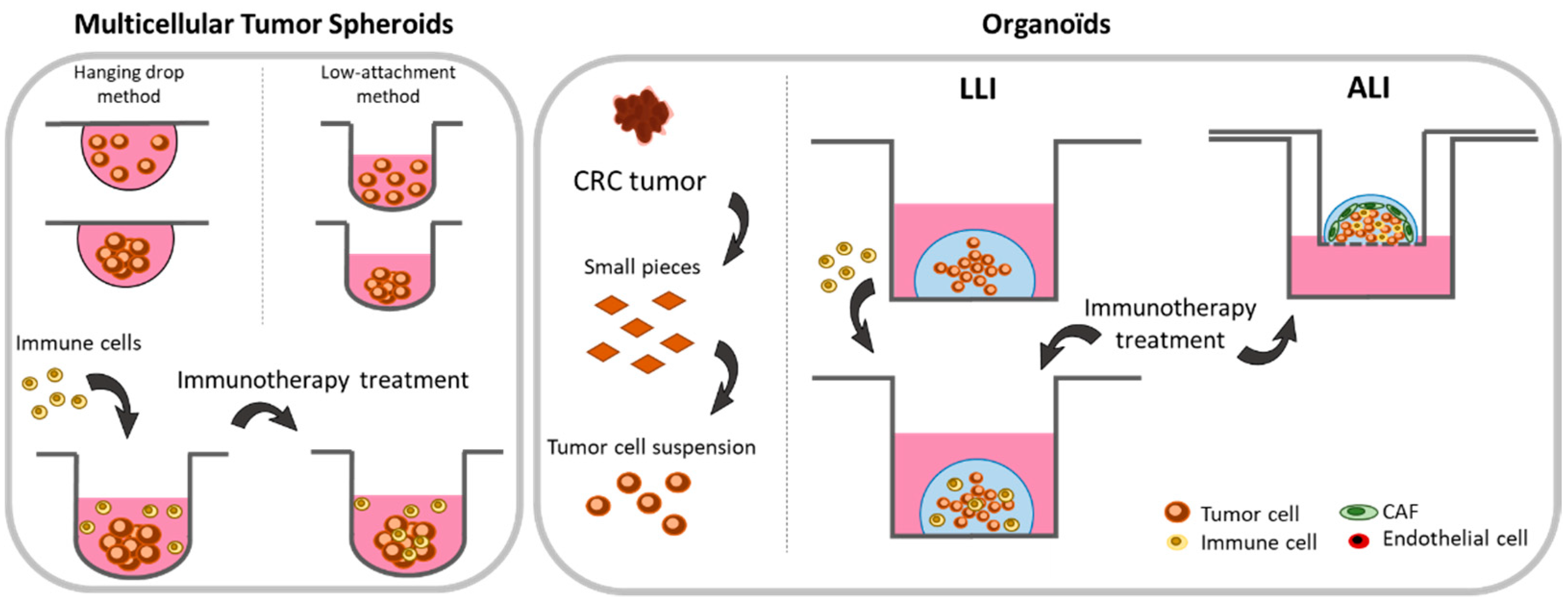
2.2. 3-Dimensional Methods
2.2.1. Spheroids
2.2.2. Organoids
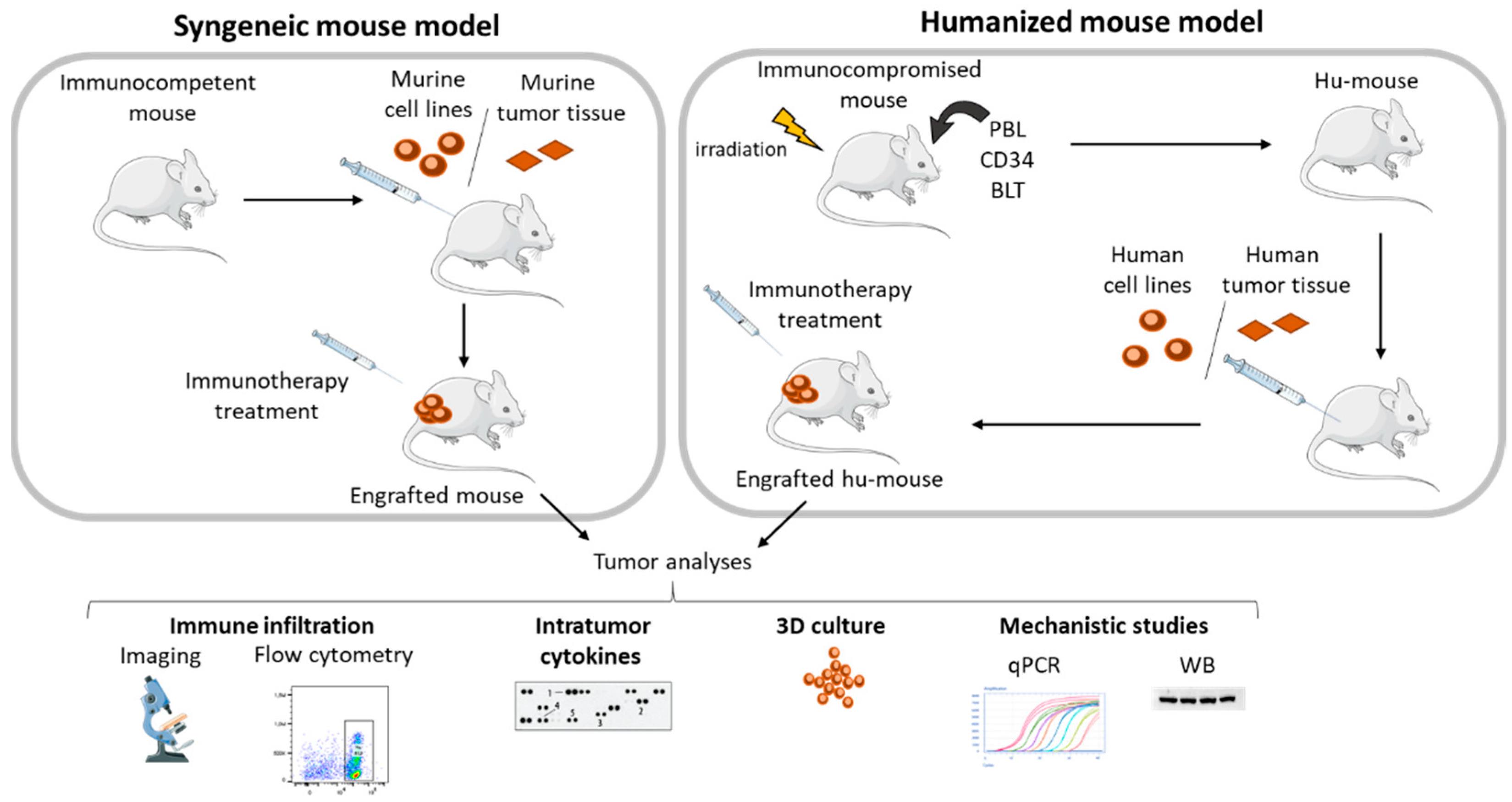
3. In Vivo CRC Models for Immunotherapy Studies
3.1. Syngeneic Models
3.2. Humanized Mouse Model
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koi, M.; Carethers, J.M. The Colorectal Cancer Immune Microenvironment and Approach to Immunotherapies. Future Oncol. 2017, 13, 1633–1647. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Sobrero, A.; Lonardi, S.; Rosati, G.; Di Bartolomeo, M.; Ronzoni, M.; Pella, N.; Scartozzi, M.; Banzi, M.; Zampino, M.G.; Pasini, F.; et al. FOLFOX or CAPOX in Stage II to III Colon Cancer: Efficacy Results of the Italian Three or Six Colon Adjuvant Trial. J. Clin. Oncol. 2018, 36, 1478–1485. [Google Scholar] [CrossRef] [PubMed]
- André, T.; Meyerhardt, J.; Iveson, T.; Sobrero, A.; Yoshino, T.; Souglakos, I.; Grothey, A.; Niedzwiecki, D.; Saunders, M.; Labianca, R.; et al. Effect of Duration of Adjuvant Chemotherapy for Patients with Stage III Colon Cancer (IDEA Collaboration): Final Results from a Prospective, Pooled Analysis of Six Randomised, Phase 3 Trials. Lancet Oncol. 2020, 21, 1620–1629. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Cervantes, A.; Adam, R.; Sobrero, A.; Van Krieken, J.H.; Aderka, D.; Aranda Aguilar, E.; Bardelli, A.; Benson, A.; Bodoky, G.; et al. ESMO Consensus Guidelines for the Management of Patients with Metastatic Colorectal Cancer. Ann. Oncol. 2016, 27, 1386–1422. [Google Scholar] [CrossRef]
- Pagès, F.; Berger, A.; Camus, M.; Sanchez-Cabo, F.; Costes, A.; Molidor, R.; Mlecnik, B.; Kirilovsky, A.; Nilsson, M.; Damotte, D.; et al. Effector Memory T Cells, Early Metastasis, and Survival in Colorectal Cancer. N. Engl. J. Med. 2005, 353, 2654–2666. [Google Scholar] [CrossRef]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pagès, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, Density, and Location of Immune Cells within Human Colorectal Tumors Predict Clinical Outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [Green Version]
- Roelands, J.; Kuppen, P.J.K.; Vermeulen, L.; Maccalli, C.; Decock, J.; Wang, E.; Marincola, F.M.; Bedognetti, D.; Hendrickx, W. Immunogenomic Classification of Colorectal Cancer and Therapeutic Implications. Int. J. Mol. Sci. 2017, 18, 2229. [Google Scholar] [CrossRef] [Green Version]
- Galon, J.; Pagès, F.; Marincola, F.M.; Angell, H.K.; Thurin, M.; Lugli, A.; Zlobec, I.; Berger, A.; Bifulco, C.; Botti, G.; et al. Cancer Classification Using the Immunoscore: A Worldwide Task Force. J. Transl. Med. 2012, 10, 205. [Google Scholar] [CrossRef]
- Pagès, F.; Mlecnik, B.; Marliot, F.; Bindea, G.; Ou, F.-S.; Bifulco, C.; Lugli, A.; Zlobec, I.; Rau, T.T.; Berger, M.D.; et al. International Validation of the Consensus Immunoscore for the Classification of Colon Cancer: A Prognostic and Accuracy Study. Lancet 2018, 391, 2128–2139. [Google Scholar] [CrossRef]
- Ciardiello, D.; Vitiello, P.P.; Cardone, C.; Martini, G.; Troiani, T.; Martinelli, E.; Ciardiello, F. Immunotherapy of Colorectal Cancer: Challenges for Therapeutic Efficacy. Cancer Treat. Rev. 2019, 76, 22–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, R.; Rousseau, B.; Vidal, J.; Colle, R.; Diaz, L.A.; André, T. Immune Checkpoint Inhibition in Colorectal Cancer: Microsatellite Instability and Beyond. Target. Oncol. 2020, 15, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Marmorino, F.; Boccaccino, A.; Germani, M.M.; Falcone, A.; Cremolini, C. Immune Checkpoint Inhibitors in PMMR Metastatic Colorectal Cancer: A Tough Challenge. Cancers 2020, 12, 2317. [Google Scholar] [CrossRef] [PubMed]
- Jass, J.R. Classification of Colorectal Cancer Based on Correlation of Clinical, Morphological and Molecular Features. Histopathology 2007, 50, 113–130. [Google Scholar] [CrossRef]
- Bindea, G.; Mlecnik, B.; Tosolini, M.; Kirilovsky, A.; Waldner, M.; Obenauf, A.C.; Angell, H.; Fredriksen, T.; Lafontaine, L.; Berger, A.; et al. Spatiotemporal Dynamics of Intratumoral Immune Cells Reveal the Immune Landscape in Human Cancer. Immunity 2013, 39, 782–795. [Google Scholar] [CrossRef] [Green Version]
- McGranahan, N.; Furness, A.J.S.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal Neoantigens Elicit T Cell Immunoreactivity and Sensitivity to Immune Checkpoint Blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef] [Green Version]
- Mouw, K.W.; Goldberg, M.S.; Konstantinopoulos, P.A.; D’Andrea, A.D. DNA Damage and Repair Biomarkers of Immunotherapy Response. Cancer Discov. 2017, 7, 675–693. [Google Scholar] [CrossRef] [Green Version]
- Germano, G.; Lamba, S.; Rospo, G.; Barault, L.; Magrì, A.; Maione, F.; Russo, M.; Crisafulli, G.; Bartolini, A.; Lerda, G.; et al. Inactivation of DNA Repair Triggers Neoantigen Generation and Impairs Tumour Growth. Nature 2017, 552, 116–120. [Google Scholar] [CrossRef]
- Llosa, N.J.; Cruise, M.; Tam, A.; Wicks, E.C.; Hechenbleikner, E.M.; Taube, J.M.; Blosser, R.L.; Fan, H.; Wang, H.; Luber, B.S.; et al. The Vigorous Immune Microenvironment of Microsatellite Instable Colon Cancer Is Balanced by Multiple Counter-Inhibitory Checkpoints. Cancer Discov. 2015, 5, 43–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becht, E.; de Reyniès, A.; Giraldo, N.A.; Pilati, C.; Buttard, B.; Lacroix, L.; Selves, J.; Sautès-Fridman, C.; Laurent-Puig, P.; Fridman, W.H. Immune and Stromal Classification of Colorectal Cancer Is Associated with Molecular Subtypes and Relevant for Precision Immunotherapy. Clin. Cancer Res. 2016, 22, 4057–4066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malesci, A.; Laghi, L.; Bianchi, P.; Delconte, G.; Randolph, A.; Torri, V.; Carnaghi, C.; Doci, R.; Rosati, R.; Montorsi, M.; et al. Reduced Likelihood of Metastases in Patients with Microsatellite-Unstable Colorectal Cancer. Clin. Cancer Res. 2007, 13, 3831–3839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boland, C.R.; Goel, A. Microsatellite Instability in Colorectal Cancer. Gastroenterology 2010, 138, 2073–2087.e3. [Google Scholar] [CrossRef]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The Consensus Molecular Subtypes of Colorectal Cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- Angelova, M.; Charoentong, P.; Hackl, H.; Fischer, M.L.; Snajder, R.; Krogsdam, A.M.; Waldner, M.J.; Bindea, G.; Mlecnik, B.; Galon, J.; et al. Characterization of the Immunophenotypes and Antigenomes of Colorectal Cancers Reveals Distinct Tumor Escape Mechanisms and Novel Targets for Immunotherapy. Genome Biol. 2015, 16, 64. [Google Scholar] [CrossRef]
- Courau, T.; Nehar-Belaid, D.; Florez, L.; Levacher, B.; Vazquez, T.; Brimaud, F.; Bellier, B.; Klatzmann, D. TGF-β and VEGF Cooperatively Control the Immunotolerant Tumor Environment and the Efficacy of Cancer Immunotherapies. JCI Insight 2016, 1, e85974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Cañellas, A.; Hernando-Momblona, X.; et al. TGFβ Drives Immune Evasion in Genetically Reconstituted Colon Cancer Metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef] [Green Version]
- Ebert, P.J.R.; Cheung, J.; Yang, Y.; McNamara, E.; Hong, R.; Moskalenko, M.; Gould, S.E.; Maecker, H.; Irving, B.A.; Kim, J.M.; et al. MAP Kinase Inhibition Promotes T Cell and Anti-Tumor Activity in Combination with PD-L1 Checkpoint Blockade. Immunity 2016, 44, 609–621. [Google Scholar] [CrossRef] [Green Version]
- Kanterman, J.; Sade-Feldman, M.; Biton, M.; Ish-Shalom, E.; Lasry, A.; Goldshtein, A.; Hubert, A.; Baniyash, M. Adverse Immunoregulatory Effects of 5FU and CPT11 Chemotherapy on Myeloid-Derived Suppressor Cells and Colorectal Cancer Outcomes. Cancer Res. 2014, 74, 6022–6035. [Google Scholar] [CrossRef] [Green Version]
- Emens, L.A.; Middleton, G. The Interplay of Immunotherapy and Chemotherapy: Harnessing Potential Synergies. Cancer Immunol. Res. 2015, 3, 436–443. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunological Effects of Conventional Chemotherapy and Targeted Anticancer Agents. Cancer Cell 2015, 28, 690–714. [Google Scholar] [CrossRef] [Green Version]
- Terme, M.; Pernot, S.; Marcheteau, E.; Sandoval, F.; Benhamouda, N.; Colussi, O.; Dubreuil, O.; Carpentier, A.F.; Tartour, E.; Taieb, J. VEGFA-VEGFR Pathway Blockade Inhibits Tumor-Induced Regulatory T-Cell Proliferation in Colorectal Cancer. Cancer Res. 2013, 73, 539–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Wei, Y.; Fang, W.; Lu, C.; Chen, J.; Cui, G.; Diao, H. Cetuximab Enhanced the Cytotoxic Activity of Immune Cells during Treatment of Colorectal Cancer. Cell. Physiol. Biochem. 2017, 44, 1038–1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raimondi, C.; Nicolazzo, C.; Gradilone, A.; Giannini, G.; De Falco, E.; Chimenti, I.; Varriale, E.; Hauch, S.; Plappert, L.; Cortesi, E.; et al. Circulating Tumor Cells: Exploring Intratumor Heterogeneity of Colorectal Cancer. Cancer Biol. Ther. 2014, 15, 496–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berg, K.C.G.; Eide, P.W.; Eilertsen, I.A.; Johannessen, B.; Bruun, J.; Danielsen, S.A.; Bjørnslett, M.; Meza-Zepeda, L.A.; Eknæs, M.; Lind, G.E.; et al. Multi-Omics of 34 Colorectal Cancer Cell Lines—A Resource for Biomedical Studies. Mol. Cancer 2017, 16, 116. [Google Scholar] [CrossRef]
- Sahin, I.H.; Akce, M.; Alese, O.; Shaib, W.; Lesinski, G.B.; El-Rayes, B.; Wu, C. Immune Checkpoint Inhibitors for the Treatment of MSI-H/MMR-D Colorectal Cancer and a Perspective on Resistance Mechanisms. Br. J. Cancer 2019, 121, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Sinha, S.; Paul, R.N. The Impact of Microsatellite Stability Status in Colorectal Cancer. Curr. Probl. Cancer 2018, 42, 548–559. [Google Scholar] [CrossRef]
- Brandi, J.; Manfredi, M.; Speziali, G.; Gosetti, F.; Marengo, E.; Cecconi, D. Proteomic Approaches to Decipher Cancer Cell Secretome. Semin. Cell Dev. Biol. 2018, 78, 93–101. [Google Scholar] [CrossRef]
- Mohebbi, B.; Ashtibaghaei, K.; Hashemi, M.; Hashemi, M.; Asadzadeh Aghdaei, H.; Zali, M.R. Conditioned Medium from Cultured Colorectal Cancer Cells Affects Peripheral Blood Mononuclear Cells Inflammatory Phenotype in Vitro. Iran. J. Med. Sci. 2019, 44, 334–341. [Google Scholar] [CrossRef]
- Lee, C.; Jeong, H.; Bae, Y.; Shin, K.; Kang, S.; Kim, H.; Oh, J.; Bae, H. Targeting of M2-like Tumor-Associated Macrophages with a Melittin-Based pro-Apoptotic Peptide. J. Immunother. Cancer 2019, 7, 147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Mitri, D.; Mirenda, M.; Vasilevska, J.; Calcinotto, A.; Delaleu, N.; Revandkar, A.; Gil, V.; Boysen, G.; Losa, M.; Mosole, S.; et al. Re-Education of Tumor-Associated Macrophages by CXCR2 Blockade Drives Senescence and Tumor Inhibition in Advanced Prostate Cancer. Cell Rep. 2019, 28, 2156–2168.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andón, F.T.; Digifico, E.; Maeda, A.; Erreni, M.; Mantovani, A.; Alonso, M.J.; Allavena, P. Targeting Tumor Associated Macrophages: The New Challenge for Nanomedicine. Semin. Immunol. 2017, 34, 103–113. [Google Scholar] [CrossRef]
- Grugan, K.D.; McCabe, F.L.; Kinder, M.; Greenplate, A.R.; Harman, B.C.; Ekert, J.E.; van Rooijen, N.; Anderson, G.M.; Nemeth, J.A.; Strohl, W.R.; et al. Tumor-Associated Macrophages Promote Invasion While Retaining Fc-Dependent Anti-Tumor Function. J. Immunol. 2012, 189, 5457–5466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solinas, G.; Schiarea, S.; Liguori, M.; Fabbri, M.; Pesce, S.; Zammataro, L.; Pasqualini, F.; Nebuloni, M.; Chiabrando, C.; Mantovani, A.; et al. Tumor-Conditioned Macrophages Secrete Migration-Stimulating Factor: A New Marker for M2-Polarization, Influencing Tumor Cell Motility. J. Immunol. 2010, 185, 642–652. [Google Scholar] [CrossRef] [Green Version]
- Benner, B.; Scarberry, L.; Suarez-Kelly, L.P.; Duggan, M.C.; Campbell, A.R.; Smith, E.; Lapurga, G.; Jiang, K.; Butchar, J.P.; Tridandapani, S.; et al. Generation of Monocyte-Derived Tumor-Associated Macrophages Using Tumor-Conditioned Media Provides a Novel Method to Study Tumor-Associated Macrophages in Vitro. J. Immunother. Cancer 2019, 7, 140. [Google Scholar] [CrossRef] [Green Version]
- Dong, H.; Yang, Y.; Gao, C.; Sun, H.; Wang, H.; Hong, C.; Wang, J.; Gong, F.; Gao, X. Lactoferrin-Containing Immunocomplex Mediates Antitumor Effects by Resetting Tumor-Associated Macrophages to M1 Phenotype. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef]
- Sawa-Wejksza, K.; Dudek, A.; Lemieszek, M.; Kaławaj, K.; Kandefer-Szerszeń, M. Colon Cancer-Derived Conditioned Medium Induces Differentiation of THP-1 Monocytes into a Mixed Population of M1/M2 Cells. Tumour Biol. 2018, 40, 1010428318797880. [Google Scholar] [CrossRef] [Green Version]
- Adil, A.A.M.; Vallinayagam, L.; Chitra, K.; Jamal, S.; Pandurangan, A.K.; Ahmed, N. Increased Expression of TGF-β and IFN-γ in Peripheral Blood Mononuclear Cells (PBMCs) Cultured in Conditioned Medium (CM) of K562 Cell Culture. J. Environ. Pathol. Toxicol. Oncol. 2019, 38, 173–183. [Google Scholar] [CrossRef]
- Teng, L.; Liu, L.; Su, Y.; Yuan, X.; Li, J.; Fu, Q.; Chen, S.; Wang, C. Suppression of Alloimmunity in Mice by Regulatory T Cells Converted with Conditioned Media. J. Surg. Res. 2011, 171, 797–806. [Google Scholar] [CrossRef]
- Wang, D.; Yang, L.; Yu, W.; Wu, Q.; Lian, J.; Li, F.; Liu, S.; Li, A.; He, Z.; Liu, J.; et al. Colorectal Cancer Cell-Derived CCL20 Recruits Regulatory T Cells to Promote Chemoresistance via FOXO1/CEBPB/NF-ΚB Signaling. J. Immunother. Cancer 2019, 7, 215. [Google Scholar] [CrossRef] [Green Version]
- Heeran, A.B.; Dunne, M.R.; Morrissey, M.E.; Buckley, C.E.; Clarke, N.; Cannon, A.; Donlon, N.E.; Nugent, T.S.; Durand, M.; Dunne, C.; et al. The Protein Secretome Is Altered in Rectal Cancer Tissue Compared to Normal Rectal Tissue, and Alterations in the Secretome Induce Enhanced Innate Immune Responses. Cancers 2021, 13, 571. [Google Scholar] [CrossRef]
- Madden, E.C.; Gorman, A.M.; Logue, S.E.; Samali, A. Tumour Cell Secretome in Chemoresistance and Tumour Recurrence. Trends Cancer 2020, 6, 489–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kather, J.N.; Halama, N.; Jaeger, D. Genomics and Emerging Biomarkers for Immunotherapy of Colorectal Cancer. Semin. Cancer Biol. 2018, 52, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Slaney, C.Y.; Kershaw, M.H.; Darcy, P.K. Trafficking of T Cells into Tumors. Cancer Res. 2014, 74, 7168–7174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kmiecik, J.; Poli, A.; Brons, N.H.C.; Waha, A.; Eide, G.E.; Enger, P.Ø.; Zimmer, J.; Chekenya, M. Elevated CD3+ and CD8+ Tumor-Infiltrating Immune Cells Correlate with Prolonged Survival in Glioblastoma Patients despite Integrated Immunosuppressive Mechanisms in the Tumor Microenvironment and at the Systemic Level. J. Neuroimmunol. 2013, 264, 71–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyrer, P.C.; Bean, E.G.; Ruth Foxwell, A.; Pavli, P. Effects of Bacterial Products on Enterocyte-Macrophage Interactions in Vitro. Biochem. Biophys. Res. Commun. 2011, 413, 336–341. [Google Scholar] [CrossRef]
- Nagata, M.; Yamamoto, H.; Tabe, K.; Sakamoto, Y. Eosinophil Transmigration across VCAM-1-Expressing Endothelial Cells Is Upregulated by Antigen-Stimulated Mononuclear Cells. Int. Arch. Allergy Immunol. 2001, 125 (Suppl. S1), 7–11. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, H.; He, H.; Ai, K.; Yu, W.; Xiao, X.; Qin, Y.; Zhang, L.; Xiong, H.; Zhou, G. LRCH1 Suppresses Migration of CD4+ T Cells and Refers to Disease Activity in Ulcerative Colitis. Int. J. Med. Sci. 2020, 17, 599–608. [Google Scholar] [CrossRef] [Green Version]
- Huang, R.-P. An Array of Possibilities in Cancer Research Using Cytokine Antibody Arrays. Expert Rev. Proteom. 2007, 4, 299–308. [Google Scholar] [CrossRef]
- Zhang, Y.; Du, W.; Chen, Z.; Xiang, C. Upregulation of PD-L1 by SPP1 Mediates Macrophage Polarization and Facilitates Immune Escape in Lung Adenocarcinoma. Exp. Cell Res. 2017, 359, 449–457. [Google Scholar] [CrossRef]
- Wang, C.-J.; Zhu, C.-C.; Xu, J.; Wang, M.; Zhao, W.-Y.; Liu, Q.; Zhao, G.; Zhang, Z.-Z. The LncRNA UCA1 Promotes Proliferation, Migration, Immune Escape and Inhibits Apoptosis in Gastric Cancer by Sponging Anti-Tumor MiRNAs. Mol. Cancer 2019, 18, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harlin, H.; Meng, Y.; Peterson, A.C.; Zha, Y.; Tretiakova, M.; Slingluff, C.; McKee, M.; Gajewski, T.F. Chemokine Expression in Melanoma Metastases Associated with CD8+ T-Cell Recruitment. Cancer Res. 2009, 69, 3077–3085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.-Q.; Zhou, W.-J.; Luo, X.-Z.; Tao, Y.; Li, D.-J. Synergistic Effect of Regulatory T Cells and Proinflammatory Cytokines in Angiogenesis in the Endometriotic Milieu. Hum. Reprod. 2017, 32, 1304–1317. [Google Scholar] [CrossRef]
- Hennel, R.; Brix, N.; Seidl, K.; Ernst, A.; Scheithauer, H.; Belka, C.; Lauber, K. Release of Monocyte Migration Signals by Breast Cancer Cell Lines after Ablative and Fractionated γ-Irradiation. Radiat. Oncol. 2014, 9, 85. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Blokhuis, B.; Derks, Y.; Kumari, S.; Garssen, J.; Redegeld, F. Human Mast Cells Promote Colon Cancer Growth via Bidirectional Crosstalk: Studies in 2D and 3D Coculture Models. Oncoimmunology 2018, 7, e1504729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Liu, R.; Wang, B.; Lian, J.; Yao, Y.; Sun, H.; Zhang, C.; Fang, L.; Guan, X.; Shi, J.; et al. Blocking IL-17A Enhances Tumor Response to Anti-PD-1 Immunotherapy in Microsatellite Stable Colorectal Cancer. J. Immunother. Cancer 2021, 9. [Google Scholar] [CrossRef]
- Grützkau, A.; Radbruch, A. Small but Mighty: How the MACS-Technology Based on Nanosized Superparamagnetic Particles Has Helped to Analyze the Immune System within the Last 20 Years. Cytom. A 2010, 77, 643–647. [Google Scholar] [CrossRef] [PubMed]
- Rios, F.J.; Touyz, R.M.; Montezano, A.C. Isolation and Differentiation of Human Macrophages. Methods Mol. Biol. 2017, 1527, 311–320. [Google Scholar] [CrossRef]
- Trickett, A.; Kwan, Y.L. T Cell Stimulation and Expansion Using Anti-CD3/CD28 Beads. J. Immunol. Methods 2003, 275, 251–255. [Google Scholar] [CrossRef]
- Tomchuck, S.L.; Leung, W.H.; Dallas, M.H. Enhanced Cytotoxic Function of Natural Killer and CD3+CD56+ Cells in Cord Blood after Culture. Biol. Blood Marrow Transplant. 2015, 21, 39–49. [Google Scholar] [CrossRef] [Green Version]
- Vogel, D.Y.S.; Glim, J.E.; Stavenuiter, A.W.D.; Breur, M.; Heijnen, P.; Amor, S.; Dijkstra, C.D.; Beelen, R.H.J. Human Macrophage Polarization in Vitro: Maturation and Activation Methods Compared. Immunobiology 2014, 219, 695–703. [Google Scholar] [CrossRef]
- Melief, J.; Wickström, S.; Kiessling, R.; Pico de Coaña, Y. Assessment of Antitumor T-Cell Responses by Flow Cytometry After Coculture of Tumor Cells with Autologous Tumor-Infiltrating Lymphocytes. Methods Mol. Biol. 2019, 1913, 133–140. [Google Scholar] [CrossRef]
- Minute, L.; Teijeira, A.; Sanchez-Paulete, A.R.; Ochoa, M.C.; Alvarez, M.; Otano, I.; Etxeberrria, I.; Bolaños, E.; Azpilikueta, A.; Garasa, S.; et al. Cellular Cytotoxicity Is a Form of Immunogenic Cell Death. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef]
- Yu, W.; Wang, Y.; Guo, P. Notch Signaling Pathway Dampens Tumor-Infiltrating CD8+ T Cells Activity in Patients with Colorectal Carcinoma. Biomed. Pharmacother. Biomed. Pharmacother. 2018, 97, 535–542. [Google Scholar] [CrossRef]
- He, W.; Zhang, H.; Han, F.; Chen, X.; Lin, R.; Wang, W.; Qiu, H.; Zhuang, Z.; Liao, Q.; Zhang, W.; et al. CD155T/TIGIT Signaling Regulates CD8+ T-Cell Metabolism and Promotes Tumor Progression in Human Gastric Cancer. Cancer Res. 2017, 77, 6375–6388. [Google Scholar] [CrossRef] [Green Version]
- Zaidi, N.E.; Shazali, N.A.H.; Chor, A.L.T.; Osman, M.A.; Ibrahim, K.; Jaoi-Edward, M.; Afizan Nik Abd Rahman, N.M. Time-Lapse 2D Imaging of Phagocytic Activity in M1 Macrophage-4T1 Mouse Mammary Carcinoma Cells in Co-Cultures. J. Vis. Exp. JoVE 2019, 154, e60281. [Google Scholar] [CrossRef] [PubMed]
- Souza, A.G.; Silva, I.B.B.; Campos-Fernandez, E.; Barcelos, L.S.; Souza, J.B.; Marangoni, K.; Goulart, L.R.; Alonso-Goulart, V. Comparative Assay of 2D and 3D Cell Culture Models: Proliferation, Gene Expression and Anticancer Drug Response. Curr. Pharm. Des. 2018, 24, 1689–1694. [Google Scholar] [CrossRef] [PubMed]
- Tung, Y.-C.; Hsiao, A.Y.; Allen, S.G.; Torisawa, Y.; Ho, M.; Takayama, S. High-Throughput 3D Spheroid Culture and Drug Testing Using a 384 Hanging Drop Array. Analyst 2011, 136, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Breslin, S.; O’Driscoll, L. Three-Dimensional Cell Culture: The Missing Link in Drug Discovery. Drug Discov. Today 2013, 18, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Silva-Almeida, C.; Ewart, M.-A.; Wilde, C. 3D Gastrointestinal Models and Organoids to Study Metabolism in Human Colon Cancer. Semin. Cell Dev. Biol. 2020, 98, 98–104. [Google Scholar] [CrossRef]
- Vinci, M.; Gowan, S.; Boxall, F.; Patterson, L.; Zimmermann, M.; Court, W.; Lomas, C.; Mendiola, M.; Hardisson, D.; Eccles, S.A. Advances in Establishment and Analysis of Three-Dimensional Tumor Spheroid-Based Functional Assays for Target Validation and Drug Evaluation. BMC Biol. 2012, 10, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.B. Three-Dimensional Tissue Culture Models in Cancer Biology. Semin. Cancer Biol. 2005, 15, 365–377. [Google Scholar] [CrossRef]
- Foty, R. A Simple Hanging Drop Cell Culture Protocol for Generation of 3D Spheroids. J. Vis. Exp. JoVE 2011. [Google Scholar] [CrossRef]
- Weiswald, L.-B.; Bellet, D.; Dangles-Marie, V. Spherical Cancer Models in Tumor Biology. Neoplasia 2015, 17, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelm, J.M.; Timmins, N.E.; Brown, C.J.; Fussenegger, M.; Nielsen, L.K. Method for Generation of Homogeneous Multicellular Tumor Spheroids Applicable to a Wide Variety of Cell Types. Biotechnol. Bioeng. 2003, 83, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, J.; Ebner, R.; Kunz-Schughart, L.A. Experimental Anti-Tumor Therapy in 3-D: Spheroids--Old Hat or New Challenge? Int. J. Radiat. Biol. 2007, 83, 849–871. [Google Scholar] [CrossRef]
- Hamilton, G.; Rath, B. Applicability of Tumor Spheroids for in Vitro Chemosensitivity Assays. Expert Opin. Drug Metab. Toxicol. 2019, 15, 15–23. [Google Scholar] [CrossRef]
- Grimes, D.R.; Kelly, C.; Bloch, K.; Partridge, M. A Method for Estimating the Oxygen Consumption Rate in Multicellular Tumour Spheroids. J. R. Soc. Interface 2014, 11, 20131124. [Google Scholar] [CrossRef] [Green Version]
- Gong, X.; Lin, C.; Cheng, J.; Su, J.; Zhao, H.; Liu, T.; Wen, X.; Zhao, P. Generation of Multicellular Tumor Spheroids with Microwell-Based Agarose Scaffolds for Drug Testing. PLoS ONE 2015, 10, e0130348. [Google Scholar] [CrossRef] [Green Version]
- Dubessy, C.; Merlin, J.M.; Marchal, C.; Guillemin, F. Spheroids in Radiobiology and Photodynamic Therapy. Crit. Rev. Oncol. Hematol. 2000, 36, 179–192. [Google Scholar] [CrossRef]
- Mó, I.; Alves, C.G.; de Melo-Diogo, D.; Lima-Sousa, R.; Correia, I.J. Assessing the Combinatorial Chemo-Photothermal Therapy Mediated by Sulfobetaine Methacrylate-Functionalized Nanoparticles in 2D and 3D In Vitro Cancer Models. Biotechnol. J. 2020, 15, e2000219. [Google Scholar] [CrossRef] [PubMed]
- Imamura, Y.; Mukohara, T.; Shimono, Y.; Funakoshi, Y.; Chayahara, N.; Toyoda, M.; Kiyota, N.; Takao, S.; Kono, S.; Nakatsura, T.; et al. Comparison of 2D- and 3D-Culture Models as Drug-Testing Platforms in Breast Cancer. Oncol. Rep. 2015, 33, 1837–1843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunes, A.S.; Costa, E.C.; Barros, A.S.; de Melo-Diogo, D.; Correia, I.J. Establishment of 2D Cell Cultures Derived From 3D MCF-7 Spheroids Displaying a Doxorubicin Resistant Profile. Biotechnol. J. 2019, 14, e1800268. [Google Scholar] [CrossRef]
- Azharuddin, M.; Roberg, K.; Dhara, A.K.; Jain, M.V.; Darcy, P.; Hinkula, J.; Slater, N.K.H.; Patra, H.K. Dissecting Multi Drug Resistance in Head and Neck Cancer Cells Using Multicellular Tumor Spheroids. Sci. Rep. 2019, 9, 20066. [Google Scholar] [CrossRef] [PubMed]
- Minchinton, A.I.; Tannock, I.F. Drug Penetration in Solid Tumours. Nat. Rev. Cancer 2006, 6, 583–592. [Google Scholar] [CrossRef]
- Rebelo, S.P.; Pinto, C.; Martins, T.R.; Harrer, N.; Estrada, M.F.; Loza-Alvarez, P.; Cabeçadas, J.; Alves, P.M.; Gualda, E.J.; Sommergruber, W.; et al. 3D-3-Culture: A Tool to Unveil Macrophage Plasticity in the Tumour Microenvironment. Biomaterials 2018, 163, 185–197. [Google Scholar] [CrossRef]
- Herter, S.; Morra, L.; Schlenker, R.; Sulcova, J.; Fahrni, L.; Waldhauer, I.; Lehmann, S.; Reisländer, T.; Agarkova, I.; Kelm, J.M.; et al. A Novel Three-Dimensional Heterotypic Spheroid Model for the Assessment of the Activity of Cancer Immunotherapy Agents. Cancer Immunol. Immunother. 2017, 66, 129–140. [Google Scholar] [CrossRef]
- Alonso-Nocelo, M.; Abuín, C.; López-López, R.; de la Fuente, M. Development and Characterization of a Three-Dimensional Co-Culture Model of Tumor T Cell Infiltration. Biofabrication 2016, 8, 025002. [Google Scholar] [CrossRef]
- Courau, T.; Bonnereau, J.; Chicoteau, J.; Bottois, H.; Remark, R.; Assante Miranda, L.; Toubert, A.; Blery, M.; Aparicio, T.; Allez, M.; et al. Cocultures of Human Colorectal Tumor Spheroids with Immune Cells Reveal the Therapeutic Potential of MICA/B and NKG2A Targeting for Cancer Treatment. J. Immunother. Cancer 2019, 7, 74. [Google Scholar] [CrossRef] [Green Version]
- Hickman, J.A.; Graeser, R.; de Hoogt, R.; Vidic, S.; Brito, C.; Gutekunst, M.; van der Kuip, H.; IMI PREDECT Consortium. Three-Dimensional Models of Cancer for Pharmacology and Cancer Cell Biology: Capturing Tumor Complexity in Vitro/Ex Vivo. Biotechnol. J. 2014, 9, 1115–1128. [Google Scholar] [CrossRef]
- Ji, D.-B.; Wu, A.-W. Organoid in Colorectal Cancer: Progress and Challenges. Chin. Med. J. 2020, 133, 1971–1977. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Larsson, P.; Ljuslinder, I.; Öhlund, D.; Myte, R.; Löfgren-Burström, A.; Zingmark, C.; Ling, A.; Edin, S.; Palmqvist, R. Ex Vivo Organoid Cultures Reveal the Importance of the Tumor Microenvironment for Maintenance of Colorectal Cancer Stem Cells. Cancers 2020, 12, 923. [Google Scholar] [CrossRef] [PubMed]
- Van de Wetering, M.; Francies, H.E.; Francis, J.M.; Bounova, G.; Iorio, F.; Pronk, A.; van Houdt, W.; van Gorp, J.; Taylor-Weiner, A.; Kester, L.; et al. Prospective Derivation of a Living Organoid Biobank of Colorectal Cancer Patients. Cell 2015, 161, 933–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, N.; Clevers, H. Studying Cellular Heterogeneity and Drug Sensitivity in Colorectal Cancer Using Organoid Technology. Curr. Opin. Genet. Dev. 2018, 52, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Roerink, S.F.; Sasaki, N.; Lee-Six, H.; Young, M.D.; Alexandrov, L.B.; Behjati, S.; Mitchell, T.J.; Grossmann, S.; Lightfoot, H.; Egan, D.A.; et al. Intra-Tumour Diversification in Colorectal Cancer at the Single-Cell Level. Nature 2018, 556, 457–462. [Google Scholar] [CrossRef]
- Riedhammer, C.; Halbritter, D.; Weissert, R. Peripheral Blood Mononuclear Cells: Isolation, Freezing, Thawing, and Culture. Methods Mol. Biol. 2016, 1304, 53–61. [Google Scholar] [CrossRef]
- Bar-Ephraim, Y.E.; Kretzschmar, K.; Clevers, H. Organoids in Immunological Research. Nat. Rev. Immunol. 2020, 20, 279–293. [Google Scholar] [CrossRef]
- Gonzalez-Exposito, R.; Semiannikova, M.; Griffiths, B.; Khan, K.; Barber, L.J.; Woolston, A.; Spain, G.; von Loga, K.; Challoner, B.; Patel, R.; et al. CEA Expression Heterogeneity and Plasticity Confer Resistance to the CEA-Targeting Bispecific Immunotherapy Antibody Cibisatamab (CEA-TCB) in Patient-Derived Colorectal Cancer Organoids. J. Immunother. Cancer 2019, 7, 101. [Google Scholar] [CrossRef] [Green Version]
- Dijkstra, K.K.; Cattaneo, C.M.; Weeber, F.; Chalabi, M.; van de Haar, J.; Fanchi, L.F.; Slagter, M.; van der Velden, D.L.; Kaing, S.; Kelderman, S.; et al. Generation of Tumor-Reactive T Cells by Co-Culture of Peripheral Blood Lymphocytes and Tumor Organoids. Cell 2018, 174, 1586–1598.e12. [Google Scholar] [CrossRef] [Green Version]
- Usui, T.; Sakurai, M.; Umata, K.; Yamawaki, H.; Ohama, T.; Sato, K. Preparation of Human Primary Colon Tissue-Derived Organoid Using Air Liquid Interface Culture. Curr. Protoc. Toxicol. 2018, 75, 22.6.1–22.6.7. [Google Scholar] [CrossRef]
- Li, X.; Ootani, A.; Kuo, C. An Air-Liquid Interface Culture System for 3D Organoid Culture of Diverse Primary Gastrointestinal Tissues. Methods Mol. Biol. 2016, 1422, 33–40. [Google Scholar] [CrossRef]
- Finnberg, N.K.; Gokare, P.; Lev, A.; Grivennikov, S.I.; MacFarlane, A.W.; Campbell, K.S.; Winters, R.M.; Kaputa, K.; Farma, J.M.; Abbas, A.E.-S.; et al. Application of 3D Tumoroid Systems to Define Immune and Cytotoxic Therapeutic Responses Based on Tumoroid and Tissue Slice Culture Molecular Signatures. Oncotarget 2017, 8, 66747–66757. [Google Scholar] [CrossRef] [Green Version]
- Neal, J.T.; Li, X.; Zhu, J.; Giangarra, V.; Grzeskowiak, C.L.; Ju, J.; Liu, I.H.; Chiou, S.-H.; Salahudeen, A.A.; Smith, A.R.; et al. Organoid Modeling of the Tumor Immune Microenvironment. Cell 2018, 175, 1972–1988.e16. [Google Scholar] [CrossRef] [Green Version]
- You, D.; Hillerman, S.; Locke, G.; Chaudhry, C.; Stromko, C.; Murtaza, A.; Fan, Y.; Koenitzer, J.; Chen, Y.; Briceno, S.; et al. Enhanced Antitumor Immunity by a Novel Small Molecule HPK1 Inhibitor. J. Immunother. Cancer 2021, 9. [Google Scholar] [CrossRef]
- Selby, M.J.; Engelhardt, J.J.; Johnston, R.J.; Lu, L.-S.; Han, M.; Thudium, K.; Yao, D.; Quigley, M.; Valle, J.; Wang, C.; et al. Preclinical Development of Ipilimumab and Nivolumab Combination Immunotherapy: Mouse Tumor Models, In Vitro Functional Studies, and Cynomolgus Macaque Toxicology. PLoS ONE 2016, 11, e0161779. [Google Scholar] [CrossRef] [Green Version]
- Goggi, J.L.; Tan, Y.X.; Hartimath, S.V.; Jieu, B.; Hwang, Y.Y.; Jiang, L.; Boominathan, R.; Cheng, P.; Yuen, T.Y.; Chin, H.X.; et al. Granzyme B PET Imaging of Immune Checkpoint Inhibitor Combinations in Colon Cancer Phenotypes. Mol. Imaging Biol. 2020, 22, 1392–1402. [Google Scholar] [CrossRef]
- Um, W.; Ko, H.; You, D.G.; Lim, S.; Kwak, G.; Shim, M.K.; Yang, S.; Lee, J.; Song, Y.; Kim, K.; et al. Necroptosis-Inducible Polymeric Nanobubbles for Enhanced Cancer Sonoimmunotherapy. Adv. Mater. 2020, 32, e1907953. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, L.K.; Fröhlich, C.; Christensen, C.; Melander, M.C.; Poulsen, T.T.; Galler, G.R.; Lantto, J.; Horak, I.D.; Kragh, M.; Nielsen, C.H.; et al. CD4+ and CD8a+ PET Imaging Predicts Response to Novel PD-1 Checkpoint Inhibitor: Studies of Sym021 in Syngeneic Mouse Cancer Models. Theranostics 2019, 9, 8221–8238. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, S.; Matrone, N.; Muddassir, A.L.; Martini, G.; Sorokin, A.; De Falco, V.; Giunta, E.F.; Ciardiello, D.; Martinelli, E.; Belli, V.; et al. Triple Blockade of EGFR, MEK and PD-L1 Has Antitumor Activity in Colorectal Cancer Models with Constitutive Activation of MAPK Signaling and PD-L1 Overexpression. J. Exp. Clin. Cancer Res. 2019, 38, 492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schweickert, P.G.; Yang, Y.; White, E.E.; Cresswell, G.M.; Elzey, B.D.; Ratliff, T.L.; Arumugam, P.; Antoniak, S.; Mackman, N.; Flick, M.J.; et al. Thrombin-PAR1 Signaling in Pancreatic Cancer Promotes an Immunosuppressive Microenvironment. J. Thromb. Haemost. 2021, 19, 161–172. [Google Scholar] [CrossRef]
- Kim, J.S.; Kim, E.J.; Lee, S.; Tan, X.; Liu, X.; Park, S.; Kang, K.; Yoon, J.-S.; Ko, Y.H.; Kurie, J.M.; et al. MiR-34a and MiR-34b/c Have Distinct Effects on the Suppression of Lung Adenocarcinomas. Exp. Mol. Med. 2019, 51, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Juneja, V.R.; McGuire, K.A.; Manguso, R.T.; LaFleur, M.W.; Collins, N.; Haining, W.N.; Freeman, G.J.; Sharpe, A.H. PD-L1 on Tumor Cells Is Sufficient for Immune Evasion in Immunogenic Tumors and Inhibits CD8 T Cell Cytotoxicity. J. Exp. Med. 2017, 214, 895–904. [Google Scholar] [CrossRef] [PubMed]
- Vandeveer, A.J.; Fallon, J.K.; Tighe, R.; Sabzevari, H.; Schlom, J.; Greiner, J.W. Systemic Immunotherapy of Non-Muscle Invasive Mouse Bladder Cancer with Avelumab, an Anti-PD-L1 Immune Checkpoint Inhibitor. Cancer Immunol. Res. 2016, 4, 452–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rigo, V.; Emionite, L.; Daga, A.; Astigiano, S.; Corrias, M.V.; Quintarelli, C.; Locatelli, F.; Ferrini, S.; Croce, M. Combined Immunotherapy with Anti-PDL-1/PD-1 and Anti-CD4 Antibodies Cures Syngeneic Disseminated Neuroblastoma. Sci. Rep. 2017, 7, 14049. [Google Scholar] [CrossRef] [Green Version]
- Yakkundi, P.; Gonsalves, E.; Galou-Lameyer, M.; Selby, M.J.; Chan, W.K. Aryl Hydrocarbon Receptor Acts as a Tumor Suppressor in a Syngeneic MC38 Colon Carcinoma Tumor Model. Hypoxia 2019, 7, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Zhong, W.; Myers, J.S.; Wang, F.; Wang, K.; Lucas, J.; Rosfjord, E.; Lucas, J.; Hooper, A.T.; Yang, S.; Lemon, L.A.; et al. Comparison of the Molecular and Cellular Phenotypes of Common Mouse Syngeneic Models with Human Tumors. BMC Genom. 2020, 21, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castle, J.C.; Loewer, M.; Boegel, S.; de Graaf, J.; Bender, C.; Tadmor, A.D.; Boisguerin, V.; Bukur, T.; Sorn, P.; Paret, C.; et al. Immunomic, Genomic and Transcriptomic Characterization of CT26 Colorectal Carcinoma. BMC Genom. 2014, 15, 190. [Google Scholar] [CrossRef] [Green Version]
- Gentles, A.J.; Newman, A.M.; Liu, C.L.; Bratman, S.V.; Feng, W.; Kim, D.; Nair, V.S.; Xu, Y.; Khuong, A.; Hoang, C.D.; et al. The Prognostic Landscape of Genes and Infiltrating Immune Cells across Human Cancers. Nat. Med. 2015, 21, 938–945. [Google Scholar] [CrossRef]
- Mosely, S.I.S.; Prime, J.E.; Sainson, R.C.A.; Koopmann, J.-O.; Wang, D.Y.Q.; Greenawalt, D.M.; Ahdesmaki, M.J.; Leyland, R.; Mullins, S.; Pacelli, L.; et al. Rational Selection of Syngeneic Preclinical Tumor Models for Immunotherapeutic Drug Discovery. Cancer Immunol. Res. 2017, 5, 29–41. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Zhang, X.; Du, L.; Wang, Y.; Liu, X.; Tian, H.; Wang, L.; Li, P.; Zhao, Y.; Duan, W.; et al. Exosome-Transmitted MiR-128-3p Increase Chemosensitivity of Oxaliplatin-Resistant Colorectal Cancer. Mol. Cancer 2019, 18, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, H.-H.; Chen, M.-C.; Baskaran, R.; Lin, Y.-M.; Day, C.H.; Lin, Y.-J.; Tu, C.-C.; Vijaya Padma, V.; Kuo, W.-W.; Huang, C.-Y. Oxaliplatin Resistance in Colorectal Cancer Cells Is Mediated via Activation of ABCG2 to Alleviate ER Stress Induced Apoptosis. J. Cell. Physiol. 2018, 233, 5458–5467. [Google Scholar] [CrossRef] [PubMed]
- Tesniere, A.; Schlemmer, F.; Boige, V.; Kepp, O.; Martins, I.; Ghiringhelli, F.; Aymeric, L.; Michaud, M.; Apetoh, L.; Barault, L.; et al. Immunogenic Death of Colon Cancer Cells Treated with Oxaliplatin. Oncogene 2010, 29, 482–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taniura, T.; Iida, Y.; Kotani, H.; Ishitobi, K.; Tajima, Y.; Harada, M. Immunogenic Chemotherapy in Two Mouse Colon Cancer Models. Cancer Sci. 2020, 111, 3527–3539. [Google Scholar] [CrossRef] [PubMed]
- Combès, E.; Andrade, A.F.; Tosi, D.; Michaud, H.-A.; Coquel, F.; Garambois, V.; Desigaud, D.; Jarlier, M.; Coquelle, A.; Pasero, P.; et al. Inhibition of Ataxia-Telangiectasia Mutated and RAD3-Related (ATR) Overcomes Oxaliplatin Resistance and Promotes Antitumor Immunity in Colorectal Cancer. Cancer Res. 2019, 79, 2933–2946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golchin, S.; Alimohammadi, R.; Rostami Nejad, M.; Jalali, S.A. Synergistic Antitumor Effect of Anti-PD-L1 Combined with Oxaliplatin on a Mouse Tumor Model. J. Cell. Physiol. 2019, 234, 19866–19874. [Google Scholar] [CrossRef]
- Eng, C.; Kim, T.W.; Bendell, J.; Argilés, G.; Tebbutt, N.C.; Di Bartolomeo, M.; Falcone, A.; Fakih, M.; Kozloff, M.; Segal, N.H.; et al. Atezolizumab with or without Cobimetinib versus Regorafenib in Previously Treated Metastatic Colorectal Cancer (IMblaze370): A Multicentre, Open-Label, Phase 3, Randomised, Controlled Trial. Lancet Oncol. 2019, 20, 849–861. [Google Scholar] [CrossRef]
- Yu, J.; Green, M.D.; Li, S.; Sun, Y.; Journey, S.N.; Choi, J.E.; Rizvi, S.M.; Qin, A.; Waninger, J.J.; Lang, X.; et al. Liver Metastasis Restrains Immunotherapy Efficacy via Macrophage-Mediated T Cell Elimination. Nat. Med. 2021, 27, 152–164. [Google Scholar] [CrossRef]
- Pietrocola, F.; Pol, J.; Vacchelli, E.; Rao, S.; Enot, D.P.; Baracco, E.E.; Levesque, S.; Castoldi, F.; Jacquelot, N.; Yamazaki, T.; et al. Caloric Restriction Mimetics Enhance Anticancer Immunosurveillance. Cancer Cell 2016, 30, 147–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Pu, Y.; Cron, K.; Deng, L.; Kline, J.; Frazier, W.A.; Xu, H.; Peng, H.; Fu, Y.-X.; Xu, M.M. CD47 Blockade Triggers T Cell-Mediated Destruction of Immunogenic Tumors. Nat. Med. 2015, 21, 1209–1215. [Google Scholar] [CrossRef] [Green Version]
- Lu, W.; Yu, W.; He, J.; Liu, W.; Yang, J.; Lin, X.; Zhang, Y.; Wang, X.; Jiang, W.; Luo, J.; et al. Reprogramming Immunosuppressive Myeloid Cells Facilitates Immunotherapy for Colorectal Cancer. EMBO Mol. Med. 2020, e12798. [Google Scholar] [CrossRef]
- Mestas, J.; Hughes, C.C.W. Of Mice and Not Men: Differences between Mouse and Human Immunology. J. Immunol. 2004, 172, 2731–2738. [Google Scholar] [CrossRef] [Green Version]
- Shultz, L.D.; Brehm, M.A.; Garcia-Martinez, J.V.; Greiner, D.L. Humanized Mice for Immune System Investigation: Progress, Promise and Challenges. Nat. Rev. Immunol. 2012, 12, 786–798. [Google Scholar] [CrossRef]
- Huntington, N.D.; Di Santo, J.P. Humanized Immune System (HIS) Mice as a Tool to Study Human NK Cell Development. Curr. Top. Microbiol. Immunol. 2008, 324, 109–124. [Google Scholar] [CrossRef]
- Wege, A.K. Humanized Mouse Models for the Preclinical Assessment of Cancer Immunotherapy. BioDrugs Clin. Immunother. Biopharm. Gene Ther. 2018, 32, 245–266. [Google Scholar] [CrossRef] [PubMed]
- Okada, S.; Vaeteewoottacharn, K.; Kariya, R. Establishment of a Patient-Derived Tumor Xenograft Model and Application for Precision Cancer Medicine. Chem. Pharm. Bull. 2018, 66, 225–230. [Google Scholar] [CrossRef] [Green Version]
- Walsh, N.C.; Kenney, L.L.; Jangalwe, S.; Aryee, K.-E.; Greiner, D.L.; Brehm, M.A.; Shultz, L.D. Humanized Mouse Models of Clinical Disease. Annu. Rev. Pathol. 2017, 12, 187–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, N.; Flutter, B.; Sanchez Rodriguez, R.; Sharif-Paghaleh, E.; Barber, L.D.; Lombardi, G.; Nestle, F.O. Xenogeneic Graft-versus-Host-Disease in NOD-Scid IL-2Rγnull Mice Display a T-Effector Memory Phenotype. PLoS ONE 2012, 7, e44219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, R.; Takahashi, T.; Ito, M. Humanized Mouse Models: Application to Human Diseases. J. Cell. Physiol. 2018, 233, 3723–3728. [Google Scholar] [CrossRef] [PubMed]
- De La Rochere, P.; Guil-Luna, S.; Decaudin, D.; Azar, G.; Sidhu, S.S.; Piaggio, E. Humanized Mice for the Study of Immuno-Oncology. Trends Immunol. 2018, 39, 748–763. [Google Scholar] [CrossRef]
- Allen, T.M.; Brehm, M.A.; Bridges, S.; Ferguson, S.; Kumar, P.; Mirochnitchenko, O.; Palucka, K.; Pelanda, R.; Sanders-Beer, B.; Shultz, L.D.; et al. Humanized Immune System Mouse Models: Progress, Challenges and Opportunities. Nat. Immunol. 2019, 20, 770–774. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Yao, L.-C.; Cheng, M.; Cai, D.; Martinek, J.; Pan, C.-X.; Shi, W.; Ma, A.-H.; De Vere White, R.W.; Airhart, S.; et al. Humanized Mice in Studying Efficacy and Mechanisms of PD-1-Targeted Cancer Immunotherapy. FASEB J. 2018, 32, 1537–1549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klichinsky, M.; Ruella, M.; Shestova, O.; Lu, X.M.; Best, A.; Zeeman, M.; Schmierer, M.; Gabrusiewicz, K.; Anderson, N.R.; Petty, N.E.; et al. Human Chimeric Antigen Receptor Macrophages for Cancer Immunotherapy. Nat. Biotechnol. 2020, 38, 947–953. [Google Scholar] [CrossRef]
- Yao, L.-C.; Aryee, K.-E.; Cheng, M.; Kaur, P.; Keck, J.G.; Brehm, M.A. Creation of PDX-Bearing Humanized Mice to Study Immuno-Oncology. Methods Mol. Biol. 2019, 1953, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Lazzari, L.; Corti, G.; Picco, G.; Isella, C.; Montone, M.; Arcella, P.; Durinikova, E.; Zanella, E.R.; Novara, L.; Barbosa, F.; et al. Patient-Derived Xenografts and Matched Cell Lines Identify Pharmacogenomic Vulnerabilities in Colorectal Cancer. Clin. Cancer Res. 2019, 25, 6243–6259. [Google Scholar] [CrossRef] [Green Version]
- Inoue, A.; Deem, A.K.; Kopetz, S.; Heffernan, T.P.; Draetta, G.F.; Carugo, A. Current and Future Horizons of Patient-Derived Xenograft Models in Colorectal Cancer Translational Research. Cancers 2019, 11, 1321. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Lee, S.H.; Wang, C.; Gao, Y.; Li, J.; Xu, W. Establishing Metastatic Patient-Derived Xenograft Model for Colorectal Cancer. Jpn. J. Clin. Oncol. 2020, 50, 1108–1116. [Google Scholar] [CrossRef]
- Hidalgo, M.; Amant, F.; Biankin, A.V.; Budinská, E.; Byrne, A.T.; Caldas, C.; Clarke, R.B.; de Jong, S.; Jonkers, J.; Mælandsmo, G.M.; et al. Patient-Derived Xenograft Models: An Emerging Platform for Translational Cancer Research. Cancer Discov. 2014, 4, 998–1013. [Google Scholar] [CrossRef] [Green Version]
- Gitto, S.B.; Kim, H.; Rafail, S.; Omran, D.K.; Medvedev, S.; Kinose, Y.; Rodriguez-Garcia, A.; Flowers, A.J.; Xu, H.; Schwartz, L.E.; et al. An Autologous Humanized Patient-Derived-Xenograft Platform to Evaluate Immunotherapy in Ovarian Cancer. Gynecol. Oncol. 2020, 156, 222–232. [Google Scholar] [CrossRef] [Green Version]
- Capasso, A.; Lang, J.; Pitts, T.M.; Jordan, K.R.; Lieu, C.H.; Davis, S.L.; Diamond, J.R.; Kopetz, S.; Barbee, J.; Peterson, J.; et al. Characterization of Immune Responses to Anti-PD-1 Mono and Combination Immunotherapy in Hematopoietic Humanized Mice Implanted with Tumor Xenografts. J. Immunother. Cancer 2019, 7, 37. [Google Scholar] [CrossRef]
- Chan, T.; Wiltrout, R.H.; Weiss, J.M. Immunotherapeutic Modulation of the Suppressive Liver and Tumor Microenvironments. Int. Immunopharmacol. 2011, 11, 879–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halama, N.; Spille, A.; Lerchl, T.; Brand, K.; Herpel, E.; Welte, S.; Keim, S.; Lahrmann, B.; Klupp, F.; Kahlert, C.; et al. Hepatic Metastases of Colorectal Cancer Are Rather Homogeneous but Differ from Primary Lesions in Terms of Immune Cell Infiltration. Oncoimmunology 2013, 2, e24116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skardal, A.; Shupe, T.; Atala, A. Organoid-on-a-Chip and Body-on-a-Chip Systems for Drug Screening and Disease Modeling. Drug Discov. Today 2016, 21, 1399–1411. [Google Scholar] [CrossRef]
- Duzagac, F.; Saorin, G.; Memeo, L.; Canzonieri, V.; Rizzolio, F. Microfluidic Organoids-on-a-Chip: Quantum Leap in Cancer Research. Cancers 2021, 13, 737. [Google Scholar] [CrossRef]
- Skardal, A.; Devarasetty, M.; Forsythe, S.; Atala, A.; Soker, S. A Reductionist Metastasis-on-a-Chip Platform for in Vitro Tumor Progression Modeling and Drug Screening. Biotechnol. Bioeng. 2016, 113, 2020–2032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parlato, S.; De Ninno, A.; Molfetta, R.; Toschi, E.; Salerno, D.; Mencattini, A.; Romagnoli, G.; Fragale, A.; Roccazzello, L.; Buoncervello, M.; et al. 3D Microfluidic Model for Evaluating Immunotherapy Efficacy by Tracking Dendritic Cell Behaviour toward Tumor Cells. Sci. Rep. 2017, 7, 1093. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.P.; Shin, W.; Ahn, E.H.; Kim, H.J.; Kim, D.-H. Engineering Microphysiological Immune System Responses on Chips. Trends Biotechnol. 2020, 38, 857–872. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| CMS | Type | Colorectal Cell Lines |
|---|---|---|
| CMS1 | MSI, Immune | Co115, DLD-1, HCT15, KM12, LoVo, SW48, Colo205, HCC2998 |
| CMS2 | Canonical | EB, FRI, IS3, LS1034, NCI-H508, SW116, SW1463, SW403, V9P |
| CMS3 | Metabolic | CL-34, LS174T, CL-40, HT29, SW948, WiDr |
| CMS4 | Mesenchymal | HCT116, RKO, TC71, CaCo2, CL-11, Colo678, IS1, SW480, SW837 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pramil, E.; Dillard, C.; Escargueil, A.E. Colorectal Cancer and Immunity: From the Wet Lab to Individuals. Cancers 2021, 13, 1713. https://doi.org/10.3390/cancers13071713
Pramil E, Dillard C, Escargueil AE. Colorectal Cancer and Immunity: From the Wet Lab to Individuals. Cancers. 2021; 13(7):1713. https://doi.org/10.3390/cancers13071713
Chicago/Turabian StylePramil, Elodie, Clémentine Dillard, and Alexandre E. Escargueil. 2021. "Colorectal Cancer and Immunity: From the Wet Lab to Individuals" Cancers 13, no. 7: 1713. https://doi.org/10.3390/cancers13071713
APA StylePramil, E., Dillard, C., & Escargueil, A. E. (2021). Colorectal Cancer and Immunity: From the Wet Lab to Individuals. Cancers, 13(7), 1713. https://doi.org/10.3390/cancers13071713