
Immune Milieu Established by Postpartum Liver Involution Promotes Breast Cancer Liver Metastasis


Abstract
Simple Summary
Abstract
1. Introduction
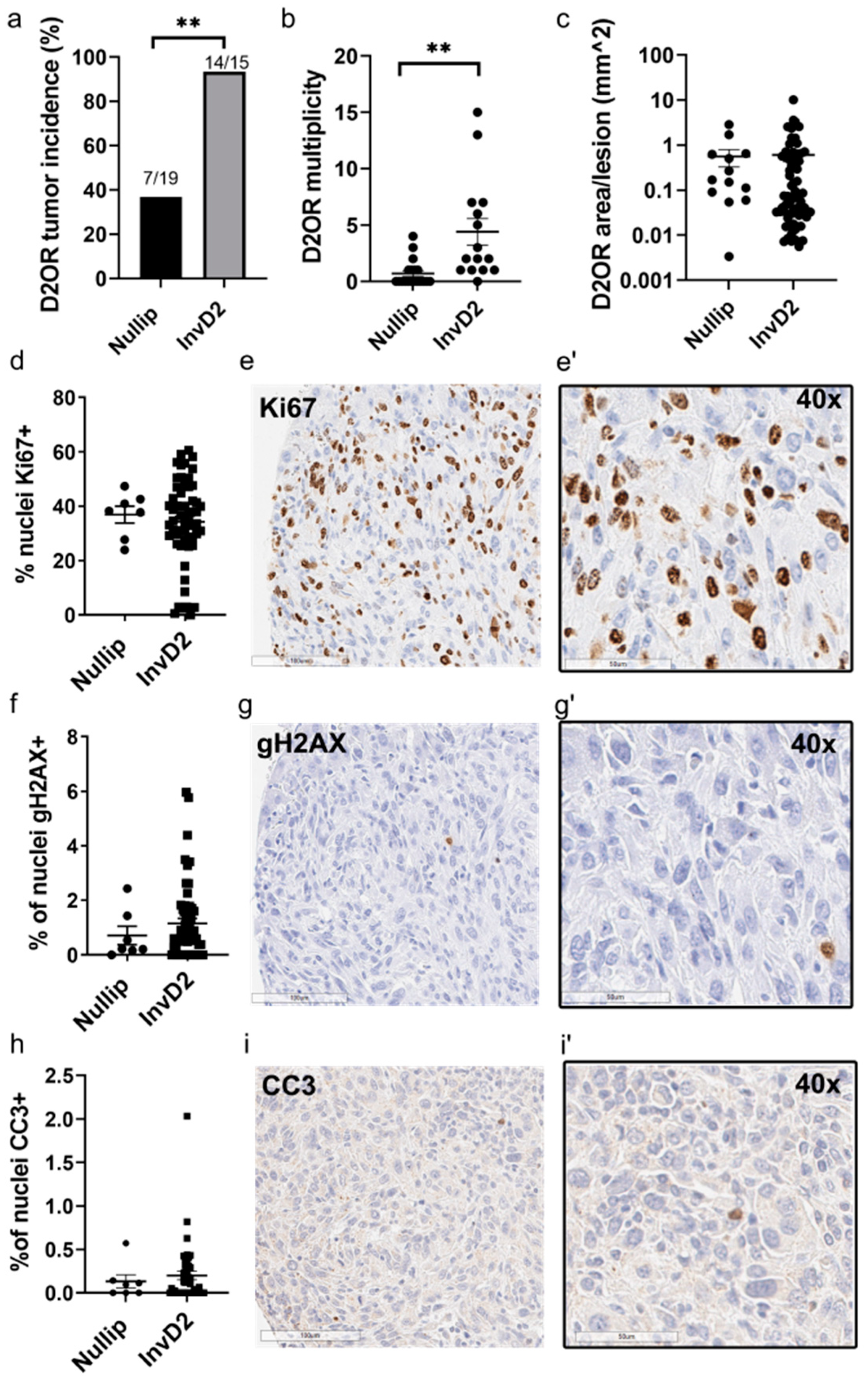
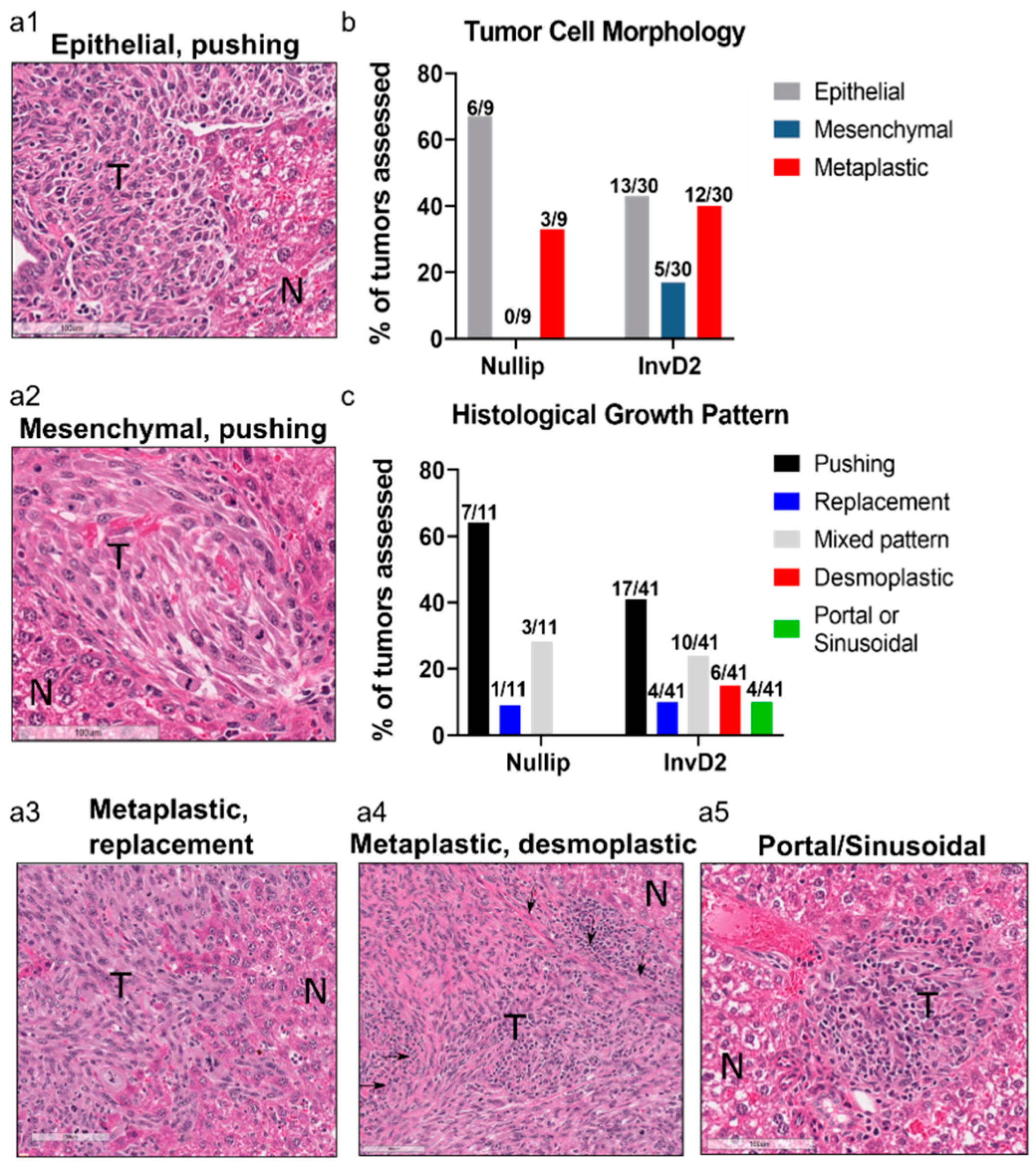
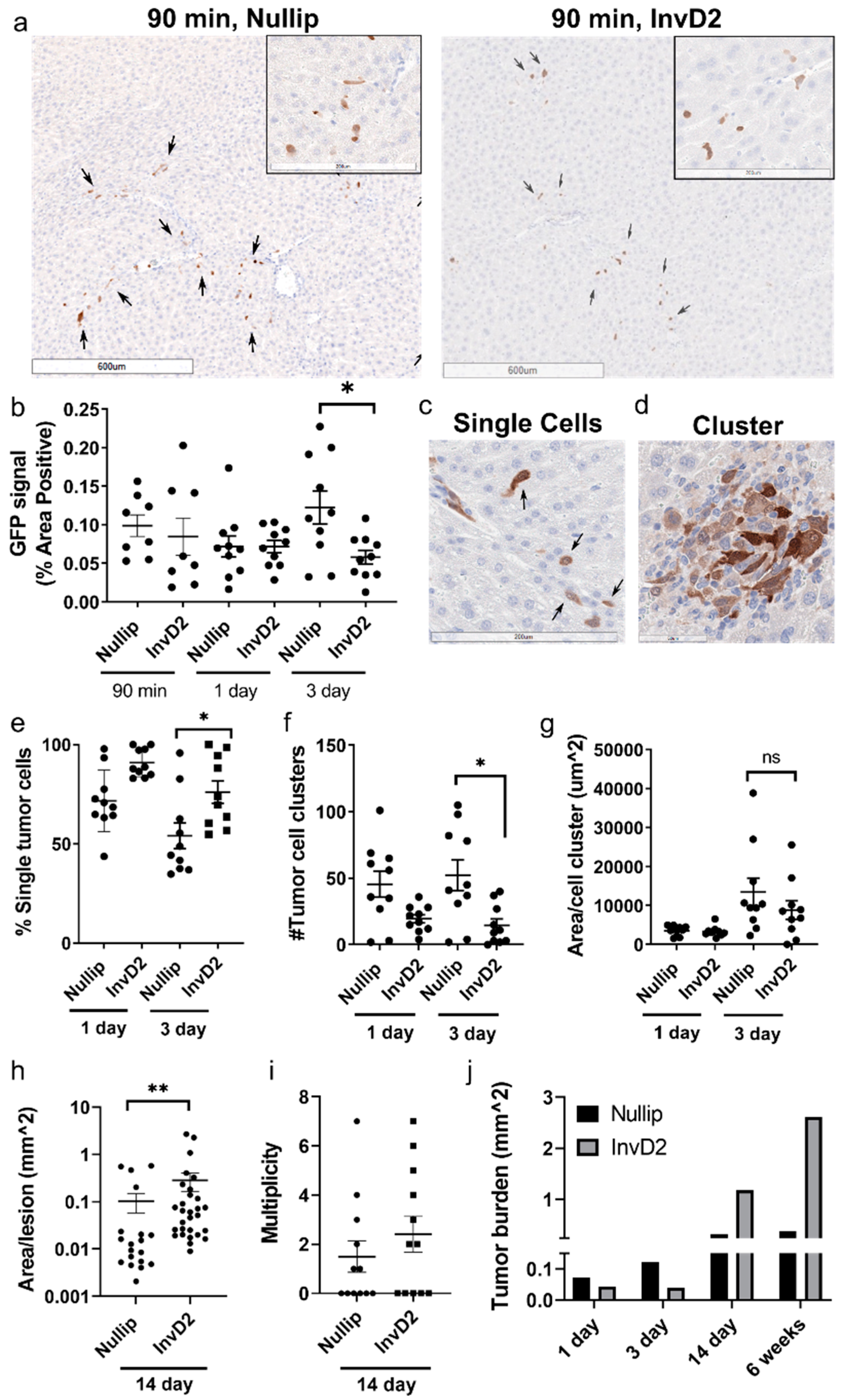
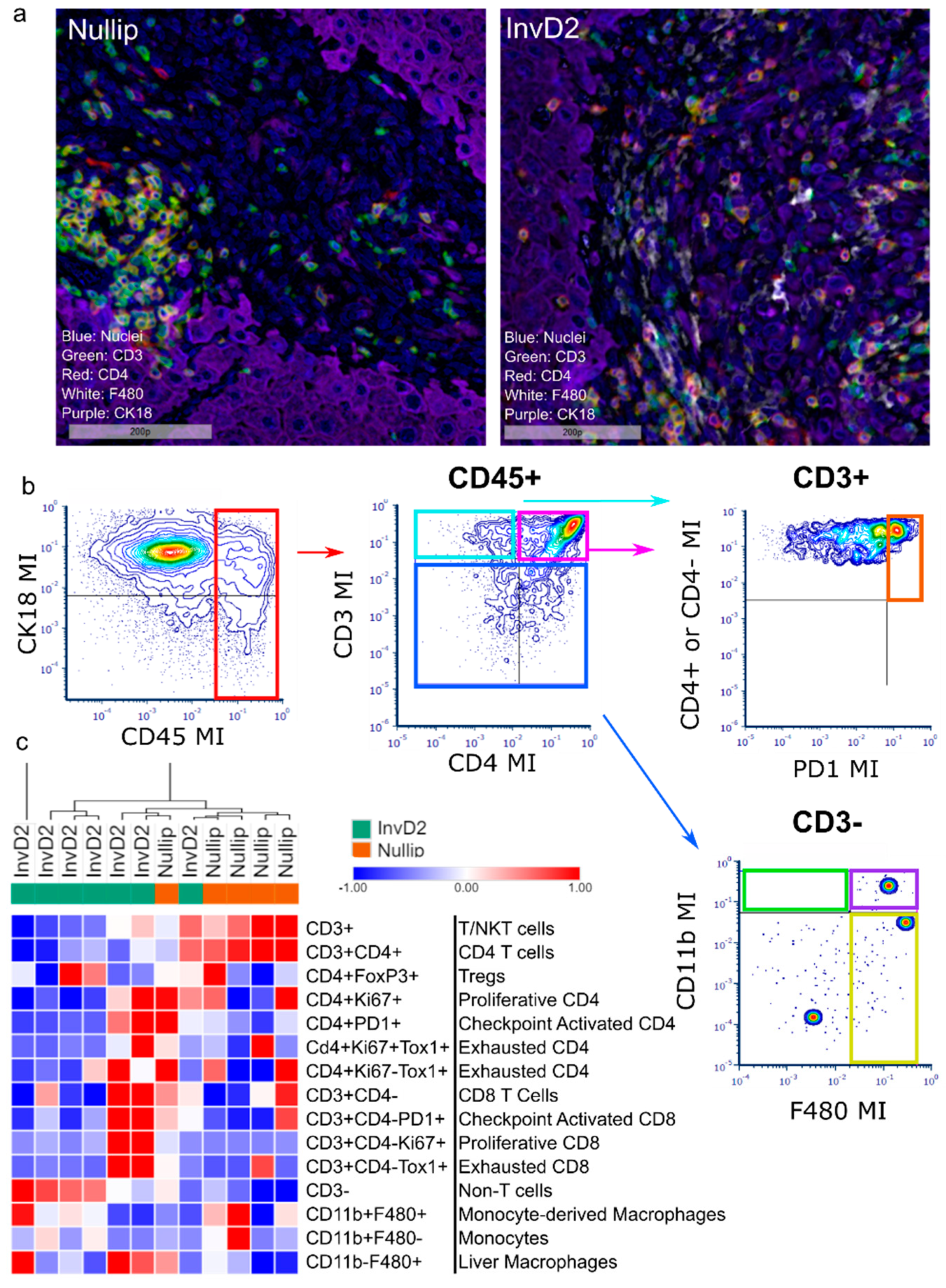
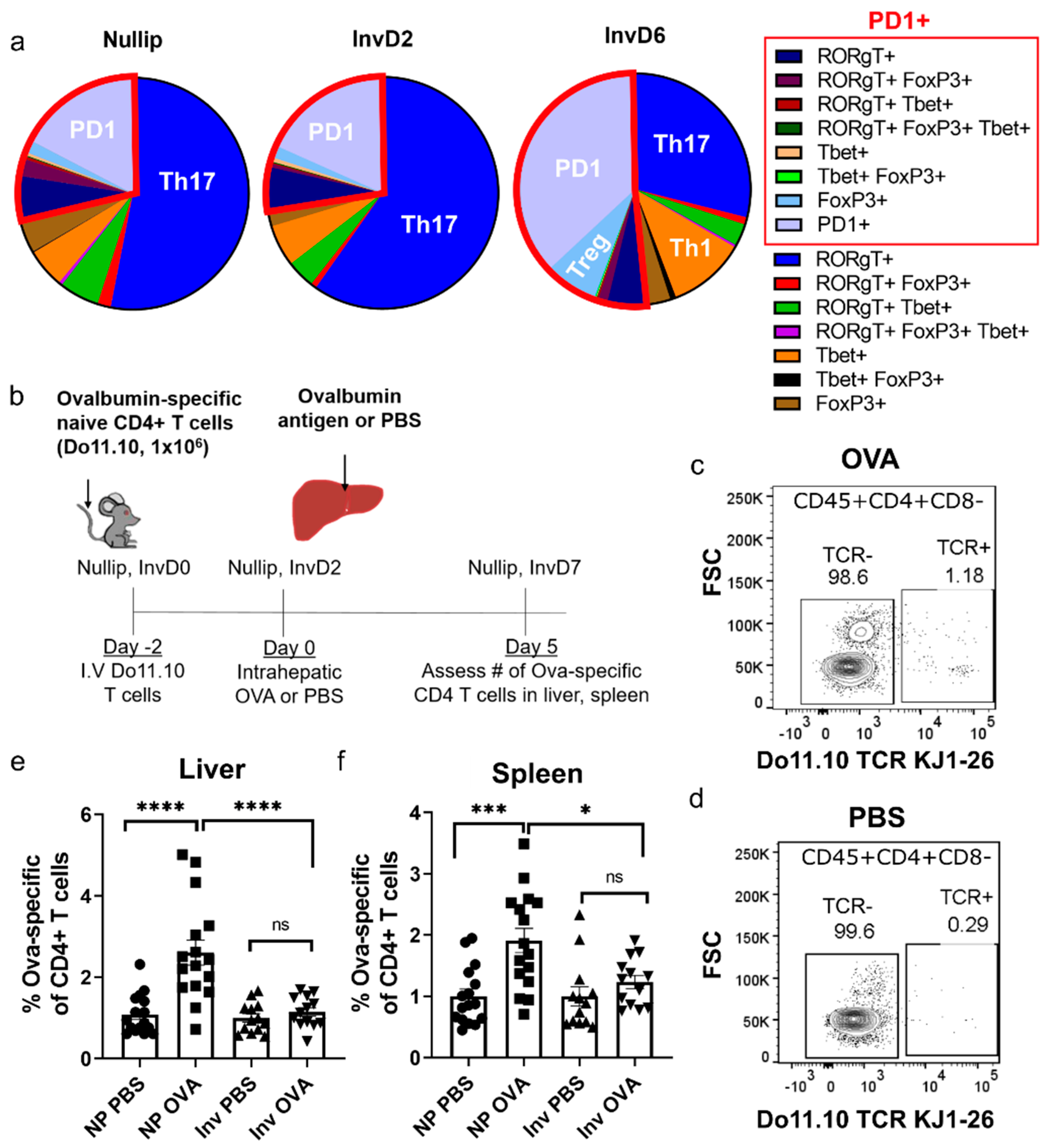
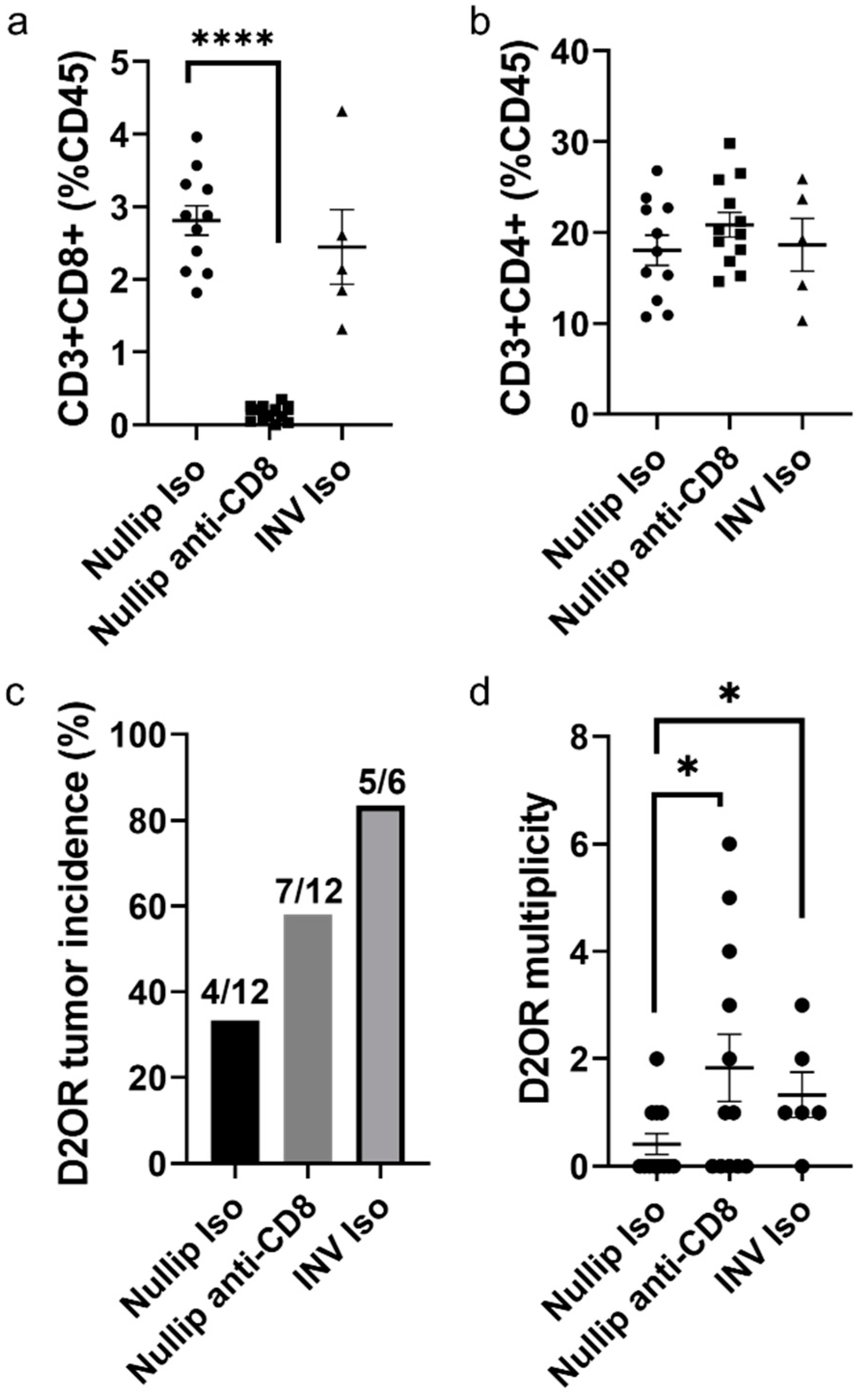
2. Results
3. Discussion
4. Materials and Methods
4.1. Animal Husbandry
4.2. Cell Culture
4.3. Liver Metastases Studies
4.4. CD8 Depletion Experiment
4.5. Histological Analyses
4.6. Immunohistochemistry
4.7. Multiplex IHC, Image Processing and Data Analysis
4.8. Flow Cytometry
4.9. Adoptive Transfer In Vivo T Cell Activation Assay
4.10. Statistical Analysis and Hierarchical Clustering
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Anders, C.K.; Johnson, R.; Litton, J.; Phillips, M.; Bleyer, A. Breast cancer before age 40 years. Semin. Oncol. 2009, 36, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Gnerlich, J.L.; Deshpande, A.D.; Jeffe, D.B.; Sweet, A.; White, N.; Margenthaler, J.A. Elevated breast cancer mortality in women younger than age 40 years compared with older women is attributed to poorer survival in early-stage disease. J. Am. Coll. Surg. 2009, 208, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Anders, C.K.; Hsu, D.S.; Broadwater, G.; Acharya, C.R.; Foekens, J.A.; Zhang, Y.; Wang, Y.; Marcom, P.K.; Marks, J.R.; Febbo, P.G.; et al. Young age at diagnosis correlates with worse prognosis and defines a subset of breast cancers with shared patterns of gene expression. J. Clin. Oncol. 2008, 26, 3324–3330. [Google Scholar] [CrossRef]
- Partridge, A.H.; Hughes, M.E.; Warner, E.T.; Ottesen, R.A.; Wong, Y.N.; Edge, S.B.; Theriault, R.L.; Blayney, D.W.; Niland, J.C.; Winer, E.P.; et al. Subtype-Dependent Relationship Between Young Age at Diagnosis and Breast Cancer Survival. J. Clin. Oncol. 2016, 34, 3308–3314. [Google Scholar] [CrossRef] [PubMed]
- Ursini-Siegel, J.; Siegel, P.M. The influence of the pre-metastatic niche on breast cancer metastasis. Cancer Lett. 2016, 380, 281–288. [Google Scholar] [CrossRef]
- Kaplan, R.N.; Riba, R.D.; Zacharoulis, S.; Bramley, A.H.; Vincent, L.; Costa, C.; MacDonald, D.D.; Jin, D.K.; Shido, K.; Kerns, S.A.; et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 2005, 438, 820–827. [Google Scholar] [CrossRef]
- Peinado, H.; Zhang, H.; Matei, I.R.; Costa-Silva, B.; Hoshino, A.; Rodrigues, G.; Psaila, B.; Kaplan, R.N.; Bromberg, J.F.; Kang, Y.; et al. Pre-metastatic niches: Organ-specific homes for metastases. Nat. Rev. Cancer 2017, 17, 302–317. [Google Scholar] [CrossRef]
- Psaila, B.; Lyden, D. The metastatic niche: Adapting the foreign soil. Nat. Rev. Cancer 2009, 9, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Fidler, I.J. The pathogenesis of cancer metastasis: The ’seed and soil’ hypothesis revisited. Nat. Rev. Cancer 2003, 3, 453–458. [Google Scholar] [CrossRef]
- Valastyan, S.; Weinberg, R.A. Tumor metastasis: Molecular insights and evolving paradigms. Cell 2011, 147, 275–292. [Google Scholar] [CrossRef]
- Costa-Silva, B.; Aiello, N.M.; Ocean, A.J.; Singh, S.; Zhang, H.; Thakur, B.K.; Becker, A.; Hoshino, A.; Mark, M.T.; Molina, H.; et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat. Cell Biol. 2015, 17, 816–826. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.R.; Quaranta, V.; Linford, A.; Emeagi, P.; Rainer, C.; Santos, A.; Ireland, L.; Sakai, T.; Sakai, K.; Kim, Y.S.; et al. Macrophage-secreted granulin supports pancreatic cancer metastasis by inducing liver fibrosis. Nat. Cell Biol. 2016, 18, 549–560. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Stone, M.L.; Porrett, P.M.; Thomas, S.K.; Komar, C.A.; Li, J.H.; Delman, D.; Graham, K.; Gladney, W.L.; Hua, X.; et al. Hepatocytes direct the formation of a pro-metastatic niche in the liver. Nature 2019, 567, 249–252. [Google Scholar] [CrossRef]
- Kok, S.Y.; Oshima, H.; Takahashi, K.; Nakayama, M.; Murakami, K.; Ueda, H.R.; Miyazono, K.; Oshima, M. Malignant subclone drives metastasis of genetically and phenotypically heterogenous cell clusters through fibrotic niche generation. Nat. Commun. 2021, 12, 863. [Google Scholar] [CrossRef]
- Dai, G.; Bustamante, J.J.; Zou, Y.; Myronovych, A.; Bao, Q.; Kumar, S.; Soares, M.J. Maternal hepatic growth response to pregnancy in the mouse. Exp. Biol. Med. (Maywood) 2011, 236, 1322–1332. [Google Scholar] [CrossRef] [PubMed]
- Hollister, A.; Okubara, P.; Watson, J.G.; Chaykin, S. Reproduction in mice: Liver enlargement in mice during pregnancy and lactation. Life Sci. 1987, 40, 11–18. [Google Scholar] [CrossRef]
- Hyatt, H.W.; Zhang, Y.; Hood, W.R.; Kavazis, A.N. Changes in Metabolism, Mitochondrial Function, and Oxidative Stress Between Female Rats Under Nonreproductive and 3 Reproductive Conditions. Reprod. Sci. 2019, 26, 114–127. [Google Scholar] [CrossRef]
- Goddard, E.T.; Hill, R.C.; Nemkov, T.; D’Alessandro, A.; Hansen, K.C.; Maller, O.; Mongoue-Tchokote, S.; Mori, M.; Partridge, A.H.; Borges, V.F.; et al. The Rodent Liver Undergoes Weaning-Induced Involution and Supports Breast Cancer Metastasis. Cancer Discov. 2017, 7, 177–187. [Google Scholar] [CrossRef]
- Cummings, M.C.; Simpson, P.T.; Reid, L.E.; Jayanthan, J.; Skerman, J.; Song, S.; McCart Reed, A.E.; Kutasovic, J.R.; Morey, A.L.; Marquart, L.; et al. Metastatic progression of breast cancer: Insights from 50 years of autopsies. J. Pathol. 2014, 232, 23–31. [Google Scholar] [CrossRef]
- Goddard, E.T.; Bassale, S.; Schedin, T.; Jindal, S.; Johnston, J.; Cabral, E.; Latour, E.; Lyons, T.R.; Mori, M.; Schedin, P.J.; et al. Association Between Postpartum Breast Cancer Diagnosis and Metastasis and the Clinical Features Underlying Risk. JAMA Netw. Open 2019, 2, e186997. [Google Scholar] [CrossRef] [PubMed]
- Morris, V.L.; Tuck, A.B.; Wilson, S.M.; Percy, D.; Chambers, A.F. Tumor progression and metastasis in murine D2 hyperplastic alveolar nodule mammary tumor cell lines. Clin. Exp. Metastasis 1993, 11, 103–112. [Google Scholar] [CrossRef]
- Morris, V.L.; Koop, S.; MacDonald, I.C.; Schmidt, E.E.; Grattan, M.; Percy, D.; Chambers, A.F.; Groom, A.C. Mammary carcinoma cell lines of high and low metastatic potential differ not in extravasation but in subsequent migration and growth. Clin. Exp. Metastasis 1994, 12, 357–367. [Google Scholar] [CrossRef]
- Goddard, E.T.; Fischer, J.; Schedin, P. A Portal Vein Injection Model to Study Liver Metastasis of Breast Cancer. J. Vis. Exp. 2016. [Google Scholar] [CrossRef]
- Van Dam, P.J.; Van der Stok, E.P.; Teuwen, L.A.; Van den Eynden, G.G.; Illemann, M.; Frentzas, S.; Majeed, A.W.; Eefsen, R.L.; Coebergh van den Braak, R.R.J.; Lazaris, A.; et al. International consensus guidelines for scoring the histopathological growth patterns of liver metastasis. Br. J. Cancer 2017, 117, 1427–1441. [Google Scholar] [CrossRef]
- Day, C.P.; Carter, J.; Weaver Ohler, Z.; Bonomi, C.; El Meskini, R.; Martin, P.; Graff-Cherry, C.; Feigenbaum, L.; Tuting, T.; Van Dyke, T.; et al. “Glowing head” mice: A genetic tool enabling reliable preclinical image-based evaluation of cancers in immunocompetent allografts. PLoS ONE 2014, 9, e0109956. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Kaech, S.M.; Wherry, E.J.; Ahmed, R. Effector and memory T-cell differentiation: Implications for vaccine development. Nat. Rev. Immunol. 2002, 2, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Blank, C.; Mackensen, A. Contribution of the PD-L1/PD-1 pathway to T-cell exhaustion: An update on implications for chronic infections and tumor evasion. Cancer Immunol. Immunother. 2007, 56, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Khan, O.; Giles, J.R.; McDonald, S.; Manne, S.; Ngiow, S.F.; Patel, K.P.; Werner, M.T.; Huang, A.C.; Alexander, K.A.; Wu, J.E.; et al. TOX transcriptionally and epigenetically programs CD8(+) T cell exhaustion. Nature 2019, 571, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Borst, J.; Ahrends, T.; Babala, N.; Melief, C.J.M.; Kastenmuller, W. CD4(+) T cell help in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2018, 18, 635–647. [Google Scholar] [CrossRef] [PubMed]
- Sheu, B.C.; Hsu, S.M.; Ho, H.N.; Lin, R.H.; Torng, P.L.; Huang, S.C. Reversed CD4/CD8 ratios of tumor-infiltrating lymphocytes are correlated with the progression of human cervical carcinoma. Cancer 1999, 86, 1537–1543. [Google Scholar] [CrossRef]
- Shah, W.; Yan, X.; Jing, L.; Zhou, Y.; Chen, H.; Wang, Y. A reversed CD4/CD8 ratio of tumor-infiltrating lymphocytes and a high percentage of CD4(+)FOXP3(+) regulatory T cells are significantly associated with clinical outcome in squamous cell carcinoma of the cervix. Cell. Mol. Immunol. 2011, 8, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Steinman, R.M.; Turley, S.; Mellman, I.; Inaba, K. The induction of tolerance by dendritic cells that have captured apoptotic cells. J. Exp. Med. 2000, 191, 411–416. [Google Scholar] [CrossRef] [PubMed]
- Erwig, L.P.; Henson, P.M. Immunological consequences of apoptotic cell phagocytosis. Am. J. Pathol. 2007, 171, 2–8. [Google Scholar] [CrossRef]
- Jacob, L.S.; Vanharanta, S.; Obenauf, A.C.; Pirun, M.; Viale, A.; Socci, N.D.; Massague, J. Metastatic Competence Can Emerge with Selection of Preexisting Oncogenic Alleles without a Need of New Mutations. Cancer Res. 2015, 75, 3713–3719. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.X.; Massague, J. Genetic determinants of cancer metastasis. Nat. Rev. Genet. 2007, 8, 341–352. [Google Scholar] [CrossRef]
- Pereira, B.; Chin, S.F.; Rueda, O.M.; Vollan, H.K.; Provenzano, E.; Bardwell, H.A.; Pugh, M.; Jones, L.; Russell, R.; Sammut, S.J.; et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat. Commun. 2016, 7, 11479. [Google Scholar] [CrossRef]
- Ursini-Siegel, J.; Schade, B.; Cardiff, R.D.; Muller, W.J. Insights from transgenic mouse models of ERBB2-induced breast cancer. Nat. Rev. Cancer 2007, 7, 389–397. [Google Scholar] [CrossRef]
- Kruger, A. Premetastatic niche formation in the liver: Emerging mechanisms and mouse models. J. Mol. Med. 2015, 93, 1193–1201. [Google Scholar] [CrossRef]
- Van den Eynden, G.G.; Majeed, A.W.; Illemann, M.; Vermeulen, P.B.; Bird, N.C.; Hoyer-Hansen, G.; Eefsen, R.L.; Reynolds, A.R.; Brodt, P. The multifaceted role of the microenvironment in liver metastasis: Biology and clinical implications. Cancer Res. 2013, 73, 2031–2043. [Google Scholar] [CrossRef]
- Lee, J.W.; Beatty, G.L. Inflammatory networks cultivate cancer cell metastasis to the liver. Cell Cycle 2020, 19, 642–651. [Google Scholar] [CrossRef] [PubMed]
- Eefsen, R.L.; Vermeulen, P.B.; Christensen, I.J.; Laerum, O.D.; Mogensen, M.B.; Rolff, H.C.; Van den Eynden, G.G.; Hoyer-Hansen, G.; Osterlind, K.; Vainer, B.; et al. Growth pattern of colorectal liver metastasis as a marker of recurrence risk. Clin. Exp. Metastasis 2015, 32, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Bohlok, A.; Botzenhart, L.; Lucidi, V.; Noel, J.C.; Demetter, P.; Larsimont, D.; Donckier, V.; Vermeulen, P.B. Histological growth pattern as a potential prognostic factor in patients operated for breast cancer liver metastases. J. Clin. Oncol. 2019, 37, e12576. [Google Scholar] [CrossRef]
- Wu, P.H.; Phillip, J.M.; Khatau, S.B.; Chen, W.C.; Stirman, J.; Rosseel, S.; Tschudi, K.; Van Patten, J.; Wong, M.; Gupta, S.; et al. Evolution of cellular morpho-phenotypes in cancer metastasis. Sci. Rep. 2015, 5, 18437. [Google Scholar] [CrossRef] [PubMed]
- Lyons, S.M.; Alizadeh, E.; Mannheimer, J.; Schuamberg, K.; Castle, J.; Schroder, B.; Turk, P.; Thamm, D.; Prasad, A. Changes in cell shape are correlated with metastatic potential in murine and human osteosarcomas. Biol. Open 2016, 5, 289–299. [Google Scholar] [CrossRef]
- Anderson, A.R.; Weaver, A.M.; Cummings, P.T.; Quaranta, V. Tumor morphology and phenotypic evolution driven by selective pressure from the microenvironment. Cell 2006, 127, 905–915. [Google Scholar] [CrossRef]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef]
- Nagaharu, K.; Zhang, X.; Yoshida, T.; Katoh, D.; Hanamura, N.; Kozuka, Y.; Ogawa, T.; Shiraishi, T.; Imanaka-Yoshida, K. Tenascin C induces epithelial-mesenchymal transition-like change accompanied by SRC activation and focal adhesion kinase phosphorylation in human breast cancer cells. Am. J. Pathol. 2011, 178, 754–763. [Google Scholar] [CrossRef]
- Park, J.; Schwarzbauer, J.E. Mammary epithelial cell interactions with fibronectin stimulate epithelial-mesenchymal transition. Oncogene 2014, 33, 1649–1657. [Google Scholar] [CrossRef]
- Polyak, K.; Weinberg, R.A. Transitions between epithelial and mesenchymal states: Acquisition of malignant and stem cell traits. Nat. Rev. Cancer 2009, 9, 265–273. [Google Scholar] [CrossRef]
- Akalay, I.; Janji, B.; Hasmim, M.; Noman, M.Z.; Thiery, J.P.; Mami-Chouaib, F.; Chouaib, S. EMT impairs breast carcinoma cell susceptibility to CTL-mediated lysis through autophagy induction. Autophagy 2013, 9, 1104–1106. [Google Scholar] [CrossRef]
- Kudo-Saito, C.; Shirako, H.; Takeuchi, T.; Kawakami, Y. Cancer metastasis is accelerated through immunosuppression during Snail-induced EMT of cancer cells. Cancer Cell 2009, 15, 195–206. [Google Scholar] [CrossRef]
- Soundararajan, R.; Fradette, J.J.; Konen, J.M.; Moulder, S.; Zhang, X.; Gibbons, D.L.; Varadarajan, N.; Wistuba, I.I.; Tripathy, D.; Bernatchez, C.; et al. Targeting the Interplay between Epithelial-to-Mesenchymal-Transition and the Immune System for Effective Immunotherapy. Cancers 2019, 11, 714. [Google Scholar] [CrossRef]
- Dongre, A.; Rashidian, M.; Eaton, E.N.; Reinhardt, F.; Thiru, P.; Zagorulya, M.; Nepal, S.; Banaz, T.; Martner, A.; Spranger, S.; et al. Direct and Indirect Regulators of Epithelial-Mesenchymal Transition (EMT)-mediated Immunosuppression in Breast Carcinomas. Cancer Discov. 2020. [Google Scholar] [CrossRef]
- Kienast, Y.; Von Baumgarten, L.; Fuhrmann, M.; Klinkert, W.E.; Goldbrunner, R.; Herms, J.; Winkler, F. Real-time imaging reveals the single steps of brain metastasis formation. Nat. Med. 2010, 16, 116–122. [Google Scholar] [CrossRef]
- Luzzi, K.J.; MacDonald, I.C.; Schmidt, E.E.; Kerkvliet, N.; Morris, V.L.; Chambers, A.F.; Groom, A.C. Multistep nature of metastatic inefficiency: Dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. Am. J. Pathol. 1998, 153, 865–873. [Google Scholar] [CrossRef]
- Goddard, E.T.; Hill, R.C.; Barrett, A.; Betts, C.; Guo, Q.; Maller, O.; Borges, V.F.; Hansen, K.C.; Schedin, P. Quantitative extracellular matrix proteomics to study mammary and liver tissue microenvironments. Int. J. Biochem. Cell Biol. 2016, 81, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Barkan, D.; El Touny, L.H.; Michalowski, A.M.; Smith, J.A.; Chu, I.; Davis, A.S.; Webster, J.D.; Hoover, S.; Simpson, R.M.; Gauldie, J.; et al. Metastatic growth from dormant cells induced by a col-I-enriched fibrotic environment. Cancer Res. 2010, 70, 5706–5716. [Google Scholar] [CrossRef] [PubMed]
- Hoye, A.M.; Erler, J.T. Structural ECM components in the premetastatic and metastatic niche. Am. J. Physiol. Cell Physiol. 2016, 310, C955–C967. [Google Scholar] [CrossRef]
- Kai, F.; Drain, A.P.; Weaver, V.M. The Extracellular Matrix Modulates the Metastatic Journey. Dev. Cell 2019, 49, 332–346. [Google Scholar] [CrossRef]
- Oskarsson, T.; Acharyya, S.; Zhang, X.H.; Vanharanta, S.; Tavazoie, S.F.; Morris, P.G.; Downey, R.J.; Manova-Todorova, K.; Brogi, E.; Massague, J. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat. Med. 2011, 17, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Winkler, J.; Abisoye-Ogunniyan, A.; Metcalf, K.J.; Werb, Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat. Commun. 2020, 11, 5120. [Google Scholar] [CrossRef] [PubMed]
- Brodt, P. Role of the Microenvironment in Liver Metastasis: From Pre- to Prometastatic Niches. Clin. Cancer Res. 2016, 22, 5971–5982. [Google Scholar] [CrossRef] [PubMed]
- Coffelt, S.B.; Kersten, K.; Doornebal, C.W.; Weiden, J.; Vrijland, K.; Hau, C.S.; Verstegen, N.J.M.; Ciampricotti, M.; Hawinkels, L.; Jonkers, J.; et al. IL-17-producing gammadelta T cells and neutrophils conspire to promote breast cancer metastasis. Nature 2015, 522, 345–348. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.C.; Mehdizadeh, S.; Smith, J.; Young, A.; Mufazalov, I.A.; Mowery, C.T.; Daud, A.; Bluestone, J.A. Regulatory T cell control of systemic immunity and immunotherapy response in liver metastasis. Sci. Immunol. 2020, 5, eaba0759. [Google Scholar] [CrossRef]
- Kitamura, T.; Qian, B.Z.; Pollard, J.W. Immune cell promotion of metastasis. Nat. Rev. Immunol. 2015, 15, 73–86. [Google Scholar] [CrossRef] [PubMed]
- Betts, C.B.; Pennock, N.D.; Caruso, B.P.; Ruffell, B.; Borges, V.F.; Schedin, P. Mucosal Immunity in the Female Murine Mammary Gland. J. Immunol. 2018, 201, 734–746. [Google Scholar] [CrossRef]
- Noelia, A.G.; Bensinger, S.J.; Hong, C.; Beceiro, S.; Bradley, M.N.; Zelcer, N.; Deniz, J.; Ramirez, C.; Diaz, M.; Gallardo, G.; et al. Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity 2009, 31, 245–258. [Google Scholar] [CrossRef]
- Gordon-Weeks, A.; Yuzhalin, A.E. Cancer Extracellular Matrix Proteins Regulate Tumour Immunity. Cancers 2020, 12, 3331. [Google Scholar] [CrossRef]
- Jachetti, E.; Caputo, S.; Mazzoleni, S.; Brambillasca, C.S.; Parigi, S.M.; Grioni, M.; Piras, I.S.; Restuccia, U.; Calcinotto, A.; Freschi, M.; et al. Tenascin-C Protects Cancer Stem-like Cells from Immune Surveillance by Arresting T-cell Activation. Cancer Res. 2015, 75, 2095–2108. [Google Scholar] [CrossRef]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pages, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef]
- Zhang, L.; Conejo-Garcia, J.R.; Katsaros, D.; Gimotty, P.A.; Massobrio, M.; Regnani, G.; Makrigiannakis, A.; Gray, H.; Schlienger, K.; Liebman, M.N.; et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N. Engl. J. Med. 2003, 348, 203–213. [Google Scholar] [CrossRef]
- Callihan, E.B.; Gao, D.; Jindal, S.; Lyons, T.R.; Manthey, E.; Edgerton, S.; Urquhart, A.; Schedin, P.; Borges, V.F. Postpartum diagnosis demonstrates a high risk for metastasis and merits an expanded definition of pregnancy-associated breast cancer. Breast. Cancer Res. Treat. 2013, 138, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Azim, H.A., Jr.; Santoro, L.; Russell-Edu, W.; Pentheroudakis, G.; Pavlidis, N.; Peccatori, F.A. Prognosis of pregnancy-associated breast cancer: A meta-analysis of 30 studies. Cancer Treat. Rev. 2012, 38, 834–842. [Google Scholar] [CrossRef]
- Shao, C.; Yu, Z.; Xiao, J.; Liu, L.; Hong, F.; Zhang, Y.; Jia, H. Prognosis of pregnancy-associated breast cancer: A meta-analysis. BMC Cancer 2020, 20, 746. [Google Scholar] [CrossRef]
- Bartlett, A.Q.; Vesco, K.K.; Purnell, J.Q.; Francisco, M.; Goddard, E.; DeBarber, A.; Leo, M.C.; Baetscher, E.; Rooney, W.; Naugler, W.; et al. Pregnancy and weaning regulate human maternal liver size and function. bioRxiv 2021. [Google Scholar] [CrossRef]
- Martinson, H.A.; Jindal, S.; Durand-Rougely, C.; Borges, V.F.; Schedin, P. Wound healing-like immune program facilitates postpartum mammary gland involution and tumor progression. Int. J. Cancer 2015, 136, 1803–1813. [Google Scholar] [CrossRef] [PubMed]
- Lyons, T.R.; O’Brien, J.; Borges, V.F.; Conklin, M.W.; Keely, P.J.; Eliceiri, K.W.; Marusyk, A.; Tan, A.C.; Schedin, P. Postpartum mammary gland involution drives progression of ductal carcinoma in situ through collagen and COX-2. Nat. Med. 2011, 17, 1109–1115. [Google Scholar] [CrossRef] [PubMed]
- Jindal, S.; Narasimhan, J.; Borges, V.F.; Schedin, P. Characterization of weaning-induced breast involution in women: Implications for young women’s breast cancer. NPJ Breast. Cancer 2020, 6, 55. [Google Scholar] [CrossRef] [PubMed]
- Tsujikawa, T.; Kumar, S.; Borkar, R.N.; Azimi, V.; Thibault, G.; Chang, Y.H.; Balter, A.; Kawashima, R.; Choe, G.; Sauer, D.; et al. Quantitative Multiplex Immunohistochemistry Reveals Myeloid-Inflamed Tumor-Immune Complexity Associated with Poor Prognosis. Cell Rep. 2017, 19, 203–217. [Google Scholar] [CrossRef] [PubMed]
- Pennock, N.D.; Martinson, H.A.; Guo, Q.; Betts, C.B.; Jindal, S.; Tsujikawa, T.; Coussens, L.M.; Borges, V.F.; Schedin, P. Ibuprofen supports macrophage differentiation, T cell recruitment, and tumor suppression in a model of postpartum breast cancer. J. Immunother. Cancer 2018, 6, 98. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primary Antibody | Manufacturer | Catalog # | Lot# | Concentration | Incubation | Secondary Antibody |
|---|---|---|---|---|---|---|
| CD4 | Cell Signaling | ab25229 | Lot:4 | 1:50 | O/N 4 °C | anti-Rb |
| CD45 | BDPharminigen | 550539 | Lot: 4141820 & 9301732 | 1:50 | 60 min | anti-Rt |
| Ki67 | Cell Signaling | 12202 | Lot: 6 (11/20) | 1:800 | 60 min | anti-Rb |
| FoxP3 | eBioscience | 14-5773-82 | Lot:E023634 + 2172602 | 1:100 | 60 min | anti-Rt |
| CD3 | Abcam | ab16669 | Lot: GR291605-1 | 1:100 | 60 min | Anti-Rb |
| Tox1 | Abcam | ab237009 | Lot: GR3241900-3 | 1:300 | 60 min | anti-Rt |
| PD1 | Cell Signaling | 84651 | Lot: 4 (11/20) | 1:200 | 60 min | anti-Rb |
| CD11b | Abcam | ab133357 | EPR1344 | 1:30k | 60 min | anti-Rb |
| F480 | Cell Signaling | 70076S | Lot: | 1:500 | 60 min | anti-Rb |
| CK18 | Abcam | ab181597 | Lot: GR321105-11 | 1:1000 | 60 min | anti-Rb |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bartlett, A.Q.; Pennock, N.D.; Klug, A.; Schedin, P. Immune Milieu Established by Postpartum Liver Involution Promotes Breast Cancer Liver Metastasis. Cancers 2021, 13, 1698. https://doi.org/10.3390/cancers13071698
Bartlett AQ, Pennock ND, Klug A, Schedin P. Immune Milieu Established by Postpartum Liver Involution Promotes Breast Cancer Liver Metastasis. Cancers. 2021; 13(7):1698. https://doi.org/10.3390/cancers13071698
Chicago/Turabian StyleBartlett, Alexandra Q., Nathan D. Pennock, Alex Klug, and Pepper Schedin. 2021. "Immune Milieu Established by Postpartum Liver Involution Promotes Breast Cancer Liver Metastasis" Cancers 13, no. 7: 1698. https://doi.org/10.3390/cancers13071698
APA StyleBartlett, A. Q., Pennock, N. D., Klug, A., & Schedin, P. (2021). Immune Milieu Established by Postpartum Liver Involution Promotes Breast Cancer Liver Metastasis. Cancers, 13(7), 1698. https://doi.org/10.3390/cancers13071698