
Death Receptor 5 (TNFRSF10B) Is Upregulated and TRAIL Resistance Is Reversed in Hypoxia and Normoxia in Colorectal Cancer Cell Lines after Treatment with Skyrin, the Active Metabolite of Hypericum spp.

 ,
,  , , , , and
, , , , and
Simple Summary
Abstract
1. Introduction
2. Results
2.1. SKR Selectively Decreases the Metabolic Activity of Cancer Cells and Their Clonogenic Ability in Hypoxia
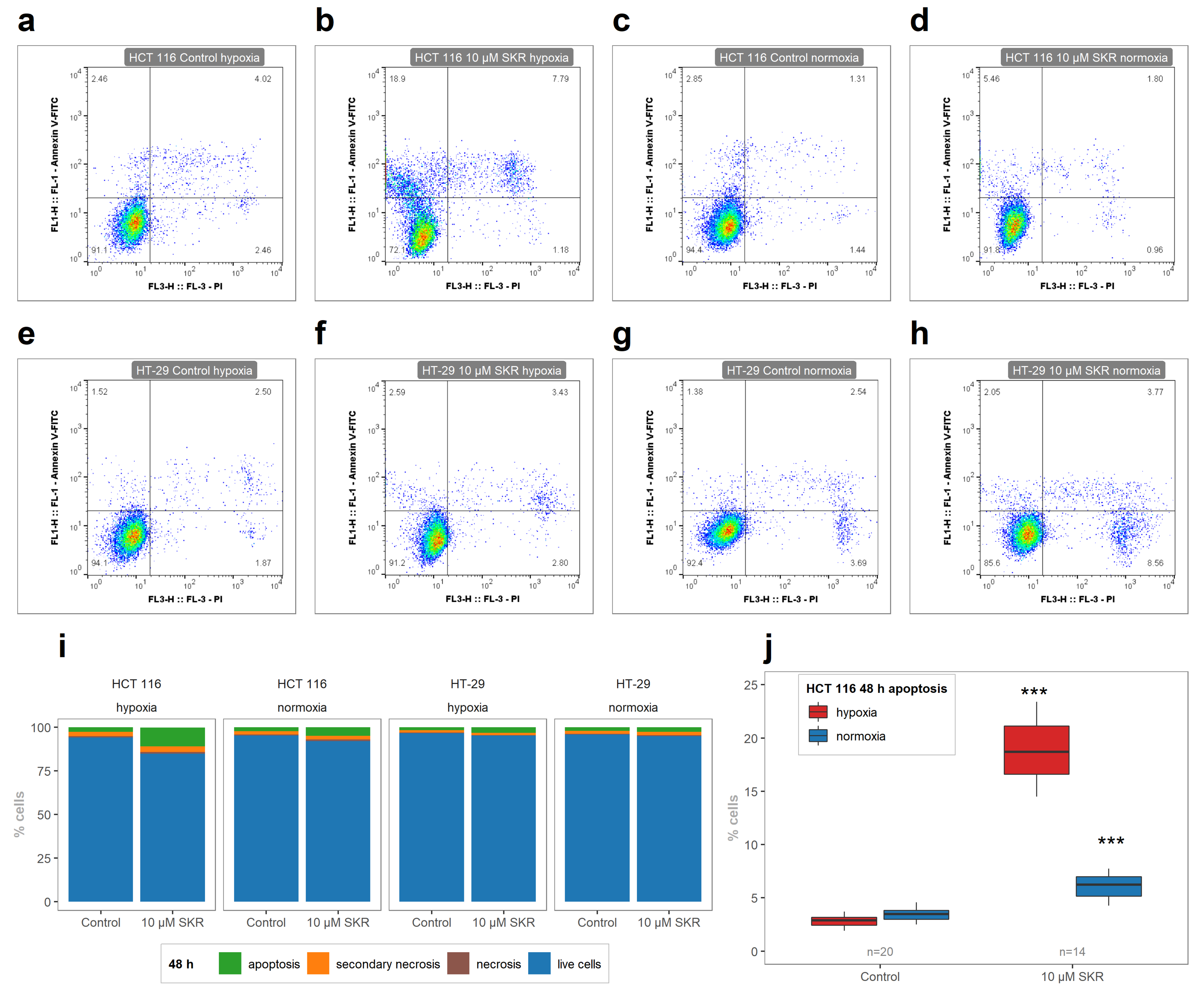
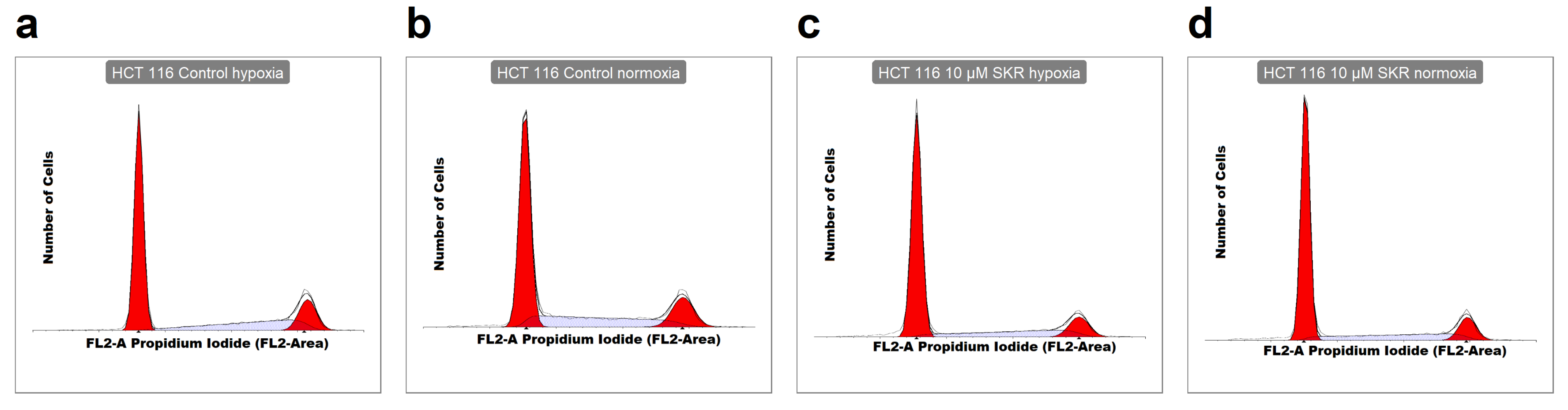
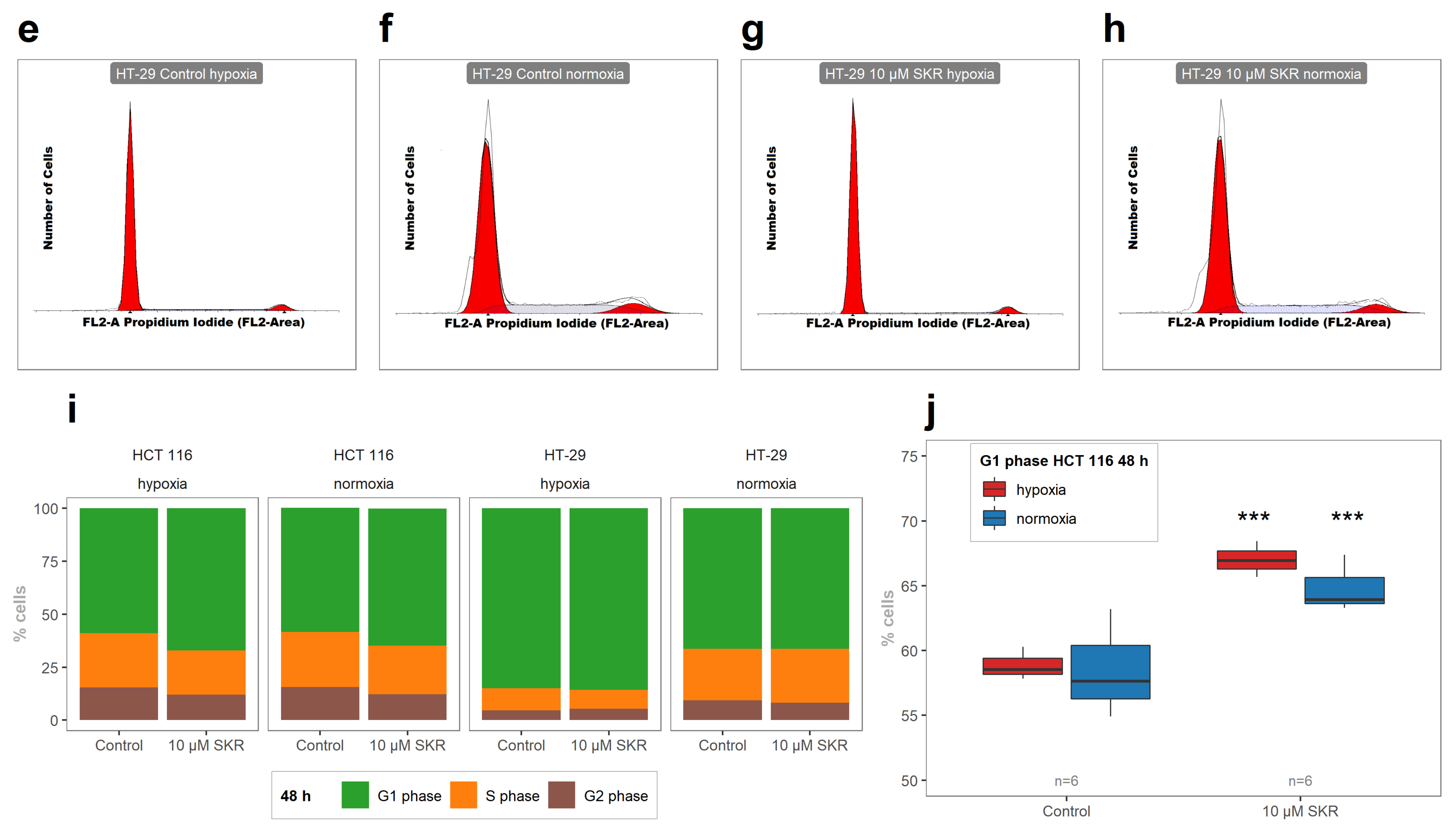
2.2. SKR Increases the Accumulation of Cells in G1 Phase and Induces Apoptosis
2.3. SKR Upregulates Pathways Associated with Mitophagy and TRAIL Signalization
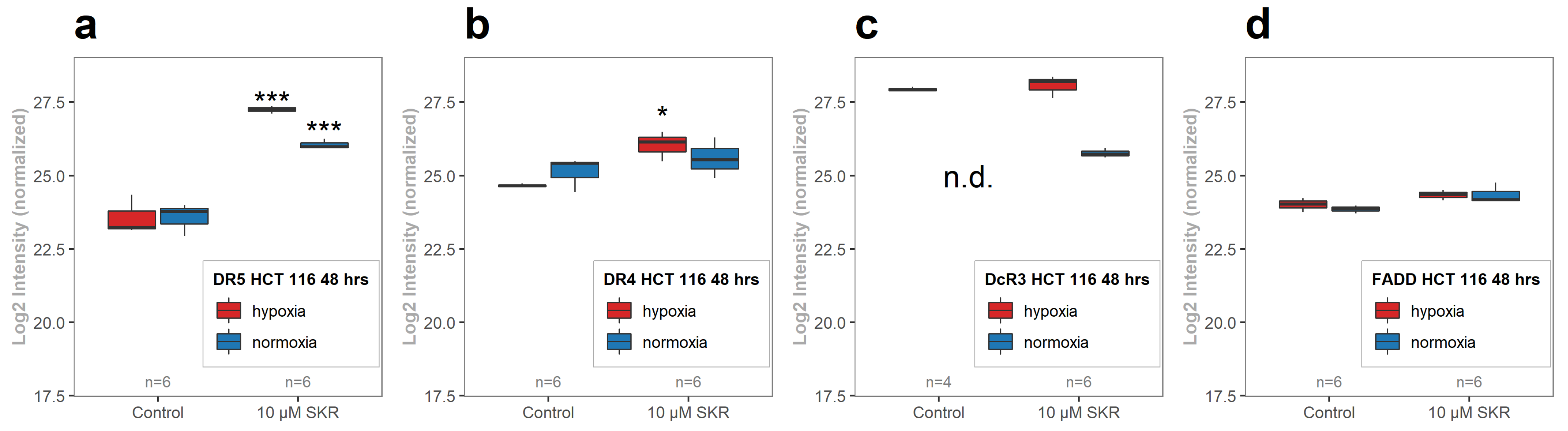
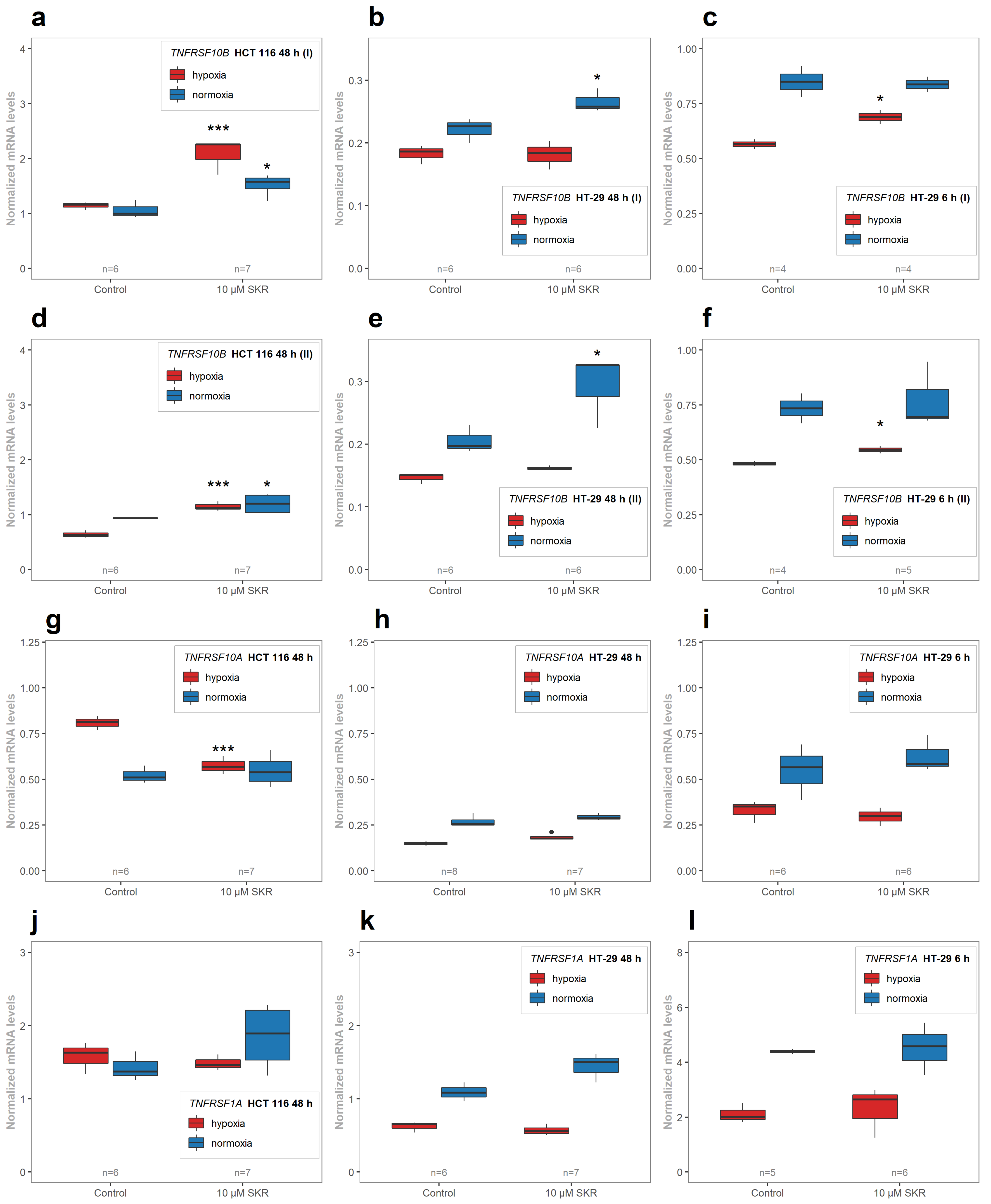
2.4. SKR Significantly Upregulates Death Receptor 5 in HCT 116 Cells in Hypoxia and Normoxia
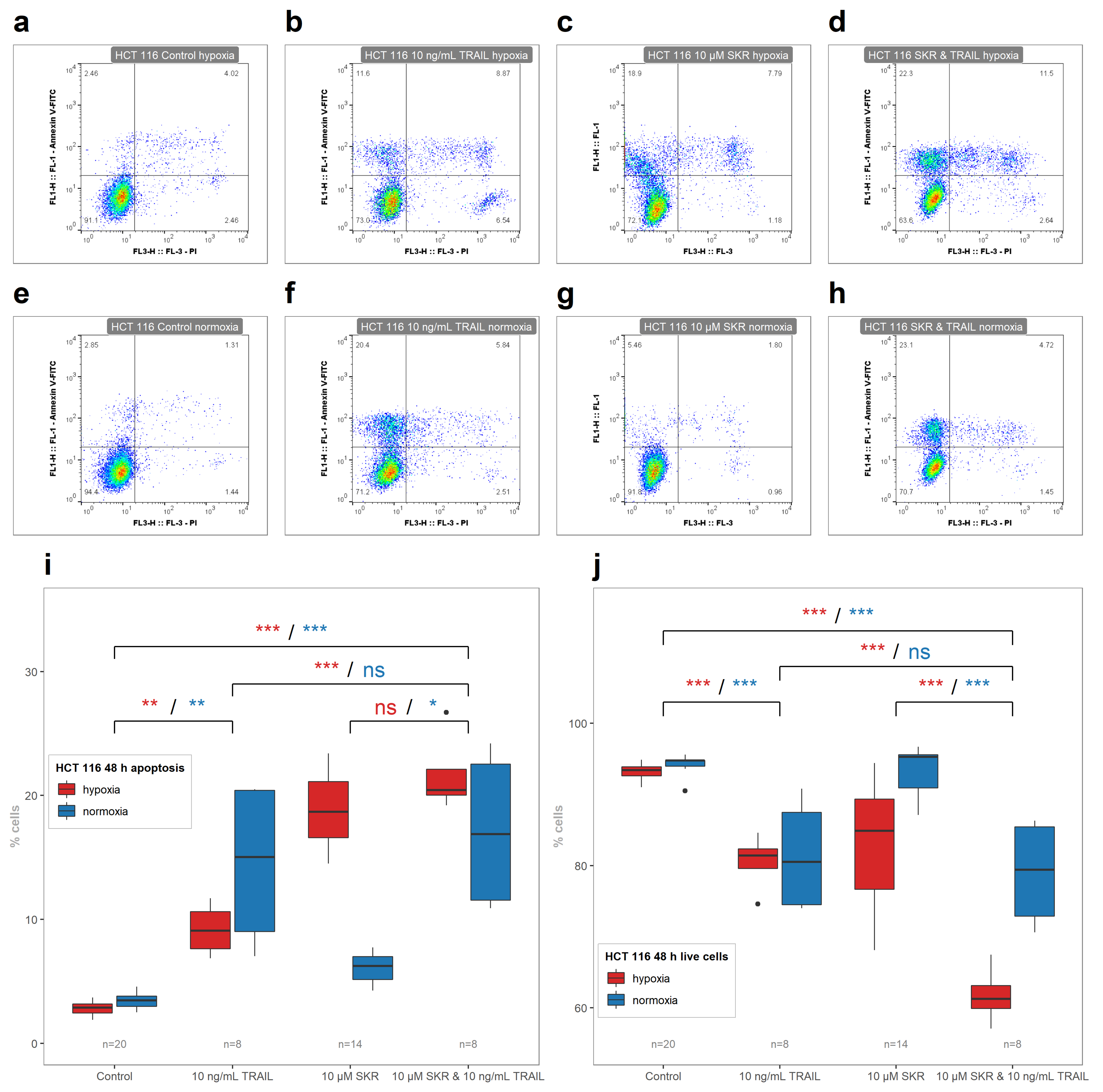
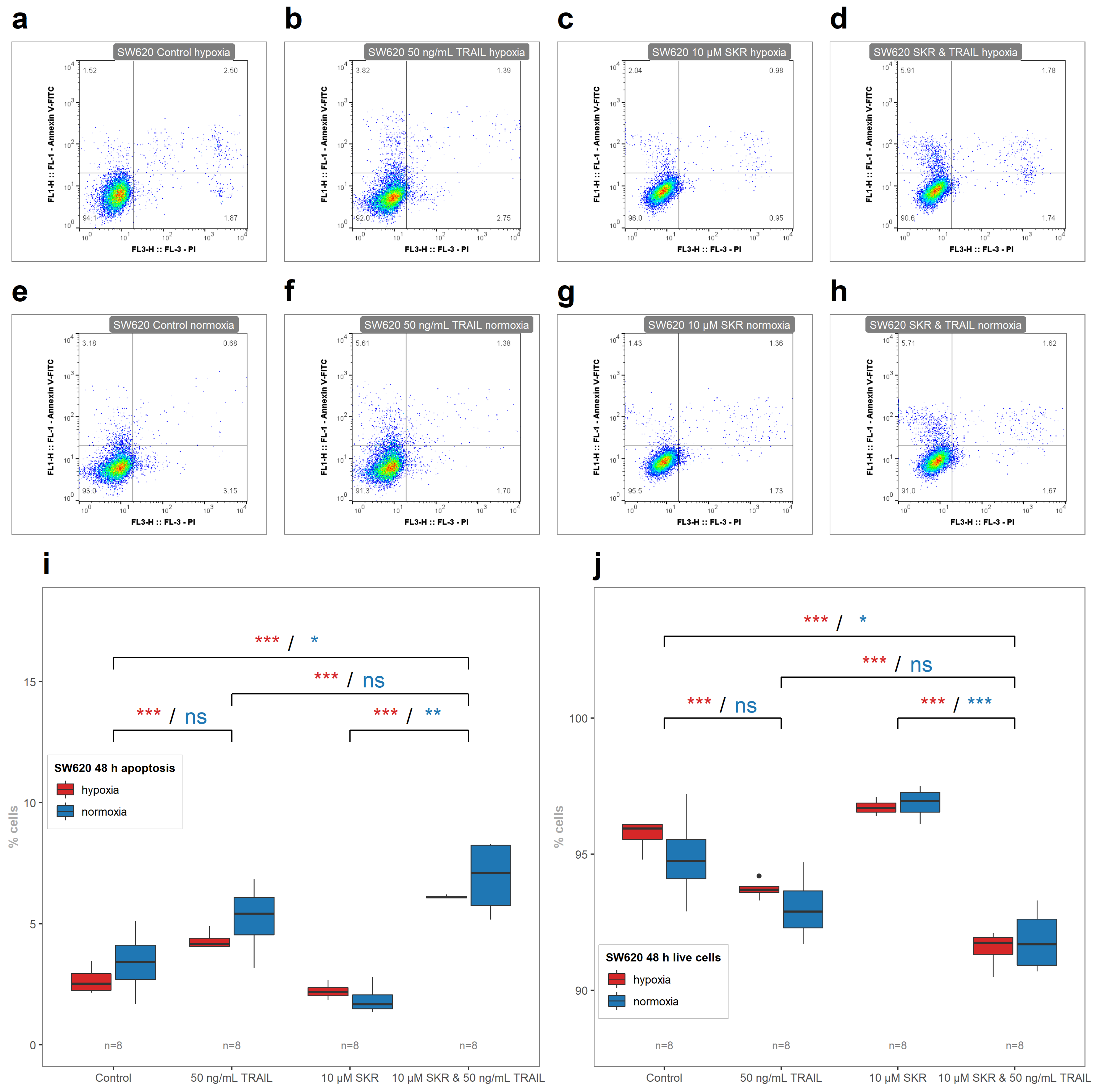
2.5. SKR Reverse TRAIL Resistance in p53 Mutated Cancer Cell Line Even in Hypoxia
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Cultures
4.3. Cultivation Conditions and Experimental Design
4.4. Metabolic Activity Assay
4.5. Analysis of Cellularity and Cell Size
4.6. Phosphatidylserine Externalization Analysis
4.7. Analysis of Cell Cycle Distribution
4.8. Colony Forming Assay
4.9. Mass Spectrometry
4.9.1. Sample Preparation
4.9.2. MS Measurement and Analysis
4.10. RT-qPCR Analysis
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ABCG2 | ATP-binding cassette super-family G member 2 |
| DMSO | Dimethylsulfoxid |
| DR5 | Death receptor 5; known as TNFRSF10B (Tumor necrosis factor receptor superfamily 10B) |
| DTT | Dithiothreitol |
| FACS | Fluorescence-activated cell sorting |
| FDR | False discovery rate |
| GN | gene name |
| IC50 | Half maximal inhibitory concentration |
| LC-MS | Liquid chromatography–mass spectrometry |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| PBS | Phosphate-buffered saline |
| PTEN | Phosphatase and tensin homolog |
| RT-qPCR | Reverse transcription quantitative polymerase chain reaction |
| SDS | Sodium dodecyl sulfate |
| SKR | Skyrin |
| TNF | Tumor necrosis factor |
| TRAIL | TNF-related apoptosis-inducing ligand |
References
- Jahn, L.; Schafhauser, T.; Wibberg, D.; Rückert, C.; Winkler, A.; Kulik, A.; Weber, T.; Flor, L.; van Pée, K.H.; Kalinowski, J.; et al. Linking secondary metabolites to biosynthesis genes in the fungal endophyte Cyanodermella asteris: The anti-cancer bisanthraquinone skyrin. J. Biotechnol. 2017, 257, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Kimáková, K.; Kimáková, A.; Idkowiak, J.; Stobiecki, M.; Rodziewicz, P.; Marczak, Ł.; Čellárová, E. Phenotyping the genus Hypericum by secondary metabolite profiling: Emodin vs. skyrin, two possible key intermediates in hypericin biosynthesis. Anal. Bioanal. Chem. 2018, 410, 7689–7699. [Google Scholar] [CrossRef]
- Rizzo, P.; Altschmied, L.; Stark, P.; Rutten, T.; Gündel, A.; Scharfenberg, S.; Franke, K.; Bäumlein, H.; Wessjohann, L.; Koch, M.; et al. Discovery of key regulators of dark gland development and hypericin biosynthesis in St. John’s Wort (Hypericum perforatum). Plant Biotechnol. J. 2019, 17, 2299–2312. [Google Scholar] [CrossRef] [PubMed]
- Howard, B.H.; Raistrick, H. Studies in the biochemistry of micro-organisms. 91. The colouring matters of Penicillium islandicum Sopp. Part 3. Skyrin and flavoskyrin. Biochem. J. 1954, 56, 56–65. [Google Scholar] [CrossRef]
- Shibata, S.; Shoji, J.; Ohta, A.; Watanabe, M. Metabolic products of fungi. XI. Some observation on the occurrence of skyrin and rugulosin in mold metabolites, with a reference to structural relationship between penicilliopsin and skyrin. Pharm. Bull. 1957, 5, 380–382. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yanagi, Y.; Nakata, M.; Suzuki, N. Selective Inhibition of Viral RNA Transcription by Skyrin. J. Pestic. Sci. 1976, 1, 107–114. [Google Scholar] [CrossRef][Green Version]
- Kiyoshi, K.; Taketoshi, K.; Hideki, M.; Jiro, K.; Yoshinori, N. A comparative study on cytotoxicities and biochemical properties of anthraquinone mycotoxins emodin and skyrin from Penicillium islandicum sopp. Toxicol. Lett. 1984, 20, 155–160. [Google Scholar] [CrossRef]
- Ueno, Y.; Umemori, K.; Niimi, E.C.C.; Tanuma, S.I.I.; Nagata, S.; Sugamata, M.; Ihara, T.; Sekijima, M.; Kawai, K.I.I.; Ueno, I.; et al. Induction of apoptosis by T-2 toxin and other natural toxins in HL-60 human promyelotic leukemia cells. Nat. Toxins 1995, 3, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Brady, S.F.; Singh, M.P.; Janso, J.E.; Clardy, J. Cytoskyrins A and B, new BIA active bisanthraquinones isolated from an endophytic fungus. Org. Lett. 2000, 2, 4047–4049. [Google Scholar] [CrossRef]
- Parker, J.C.; McPherson, R.K.; Andrews, K.M.; Levy, C.B.; Dubins, J.S.; Chin, J.E.; Perry, P.V.; Hulin, B.; Perry, D.A.; Inagaki, T.; et al. Effects of skyrin, a receptor-selective glucagon antagonist, in rat and human hepatocytes. Diabetes 2000, 49, 2079–2086. [Google Scholar] [CrossRef][Green Version]
- Lin, L.C.; Chou, C.J.; Kuo, Y.C. Cytotoxic principles from Ventilago leiocarpa. J. Nat. Prod. 2001, 64, 674–676. [Google Scholar] [CrossRef] [PubMed]
- Watts, P.; Kittakoop, P.; Veeranondha, S.; Wanasith, S.; Thongwichian, R.; Saisaha, P.; Intamas, S.; Hywel-Jones, N.L. Cytotoxicity against insect cells of entomopathogenic fungi of the genera Hypocrella (anamorph Aschersonia): Possible agents for biological control. Mycol. Res. 2003, 107, 581–586. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, K.C.; Papageorgiou, C.D.; Piper, J.L.; Chadha, R.K. The cytoskyrin cascade: A facile entry into cytoskyrin A, deoxyrubroskyrin, rugulin, skyrin, and flavoskyrin model systems. Angew. Chemie Int. Ed. 2005, 44, 5846–5851. [Google Scholar] [CrossRef] [PubMed]
- Vargas, F.; Rivas, C.; Zoltan, T.; Lopez, V.; Ortega, J.; Izzo, C.; Pineda, M.; Medina, J.; Medina, E.; Rosales, L. Antioxydant and scavenging activity of skyrin on free radical and some reactive oxygen species. Av. Quim. 2008, 3, 7–14. [Google Scholar]
- Bräse, S.; Gläser, F.; Kramer, C.; Lindner, S.; Linsenmeier, A.M.; Masters, K.S.; Meister, A.C.; Ruff, B.M.; Zhong, S. The Chemistry of Mycotoxins; Progress in the Chemistry of Organic Natural Products; Springer: Vienna, Austria, 2013; Volume 97, pp. 139–151. [Google Scholar] [CrossRef]
- Koul, M.; Meena, S.; Kumar, A.; Sharma, P.R.; Singamaneni, V.; Riyaz-Ul-Hassan, S.; Hamid, A.; Chaubey, A.; Prabhakar, A.; Gupta, P.; et al. Secondary Metabolites from Endophytic Fungus Penicillium pinophilum Induce ROS-Mediated Apoptosis through Mitochondrial Pathway in Pancreatic Cancer Cells. Planta Med. 2016, 82, 344–355. [Google Scholar] [CrossRef] [PubMed]
- Revuru, B.; Bálintová, M.; Henzelyová, J.; Čellárová, E.; Kusari, S. MALDI-HRMS Imaging Maps the Localization of Skyrin, the Precursor of Hypericin, and Pathway Intermediates in Leaves of Hypericum Species. Molecules 2020, 25. [Google Scholar] [CrossRef]
- Hatfield, G.; Slagle, D. Isolation of Skyrin from Hypomyces lactifluorum. Lloydia 1973, 36, 354–356. [Google Scholar]
- Bara, R.A. Natural Products from Endophytic Fungus Talaromyces Wortmannii: Their Structure Elucidation and Mechanism of Actions. Ph.D. Thesis, Heinrich-Heine Universität Düsseldorf, Düsseldorf, Germany, 2012. [Google Scholar]
- Bara, R.; Aly, A.H.; Pretsch, A.; Wray, V.; Wang, B.; Proksch, P.; Debbab, A. Antibiotically active metabolites from Talaromyces wortmannii, an endophyte of Aloe vera. J. Antibiot. 2013, 66, 491–493. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Jin, Q.; Yang, S.; Zhang, D.; Wang, Q.; Li, J.; Song, S.; Sun, Z.; Ni, Y.; Zhang, J.; et al. Synthesis and Evaluation of 131I-Skyrin as a Necrosis Avid Agent for Potential Targeted Radionuclide Therapy of Solid Tumors. Mol. Pharm. 2016, 13, 180–189. [Google Scholar] [CrossRef]
- Zaman, A. Docking studies and network analyses reveal capacity of compounds from Kandelia rheedii to strengthen cellular immunity by interacting with host proteins during tuberculosis infection. Bioinformation 2012, 8, 1012–1020. [Google Scholar] [CrossRef]
- Jendželovská, Z.; Jendželovský, R.; Hilovská, L.; Koval, J.; Mikeš, J.; Fedoročko, P. Single pre-treatment with hypericin, a St. John’s wort secondary metabolite, attenuates cisplatin- and mitoxantrone-induced cell death in A2780, A2780cis and HL-60 cells. Toxicol. Vitr. 2014, 28, 1259–1273. [Google Scholar] [CrossRef]
- Jendzelovská, Z.; Jendželovský, R.; Kuchárová, B.; Fedoročko, P. Hypericin in the Light and in the Dark: Two Sides of the Same Coin. Front. Plant Sci. 2016, 7, 1–20. [Google Scholar] [CrossRef]
- Hockel, M.; Vaupel, P. Tumor Hypoxia: Definitions and Current Clinical, Biologic, and Molecular Aspects. JNCI J. Natl. Cancer Inst. 2001, 93, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression angiogenesis metastasis and resistane to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V.; Semenza, G.L. Oncogenic alterations of metabolism. Trends Biochem. Sci. 1999, 24, 68–72. [Google Scholar] [CrossRef]
- Challapalli, A.; Carroll, L.; Aboagye, E.O. Molecular mechanisms of hypoxia in cancer. Clin. Transl. Imaging 2017, 5, 225–253. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef]
- Wang, G.L.; Semenza, G.L. Purification and Characterization of Hypoxia-inducible Factor 1. J. Biol. Chem. 1995, 270, 1230–1237. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef]
- Rademakers, S.E.; Span, P.N.; Kaanders, J.H.A.M.; Sweep, F.C.G.J.; van der Kogel, A.J.; Bussink, J. Molecular aspects of tumour hypoxia. Mol. Oncol. 2008, 2, 41–53. [Google Scholar] [CrossRef]
- Vaupel, P. The Role of Hypoxia-Induced Factors in Tumor Progression. Oncologist 2004, 9, 10–17. [Google Scholar] [CrossRef]
- Abraham, J.; Salama, N.N.; Azab, A.K. The role of P-glycoprotein in drug resistance in multiple myeloma Leuk. Lymphoma 2015, 56, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Das, B.; Tsuchida, R.; Malkin, D.; Koren, G.; Baruchel, S.; Yeger, H. Hypoxia Enhances Tumor Stemness by Increasing the Invasive and Tumorigenic Side Population Fraction. Stem Cells 2008, 26, 1818–1830. [Google Scholar] [CrossRef] [PubMed]
- Duiker, E.W.; Mom, C.H.; de Jong, S.; Willemse, P.H.; Gietema, J.A.; van der Zee, A.G.; de Vries, E.G. The clinical trail of TRAIL. Eur. J. Cancer 2006, 42, 2233–2240. [Google Scholar] [CrossRef] [PubMed]
- Stolfi, C.; Pallone, F.; Monteleone, G. Molecular Targets of TRAIL-Sensitizing Agents in Colorectal Cancer. Int. J. Mol. Sci. 2012, 13, 7886–7901. [Google Scholar] [CrossRef]
- Szliszka, E.; Zydowicz, G.; Mizgala, E.; Krol, W. Artepillin C (3,5-diprenyl-4-hydroxycinnamic acid) sensitizes LNCaP prostate cancer cells to TRAIL-induced apoptosis. Int. J. Oncol. 2012, 41, 818–828. [Google Scholar] [CrossRef] [PubMed]
- Cormier, Z. Small-molecule drug drives cancer cells to suicide. Nature 2013. [Google Scholar] [CrossRef]
- Mahajan, S.; Dammai, V.; Hsu, T.; Kraft, A. Hypoxia-inducible factor-2α regulates the expression of TRAIL receptor DR5 in renal cancer cells. Carcinogenesis 2008, 29, 1734–1741. [Google Scholar] [CrossRef]
- Kim, M.; Park, S.Y.; Pai, H.S.; Kim, T.H.; Billiar, T.R.; Seol, D.W. Hypoxia inhibits tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis by blocking Bax translocation. Cancer Res. 2004, 64, 4078–4081. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, N.S.; Vigneswaran, N.; Zacharias, W. Hypoxia inhibits TRAIL-induced tumor cell apoptosis: Involvement of lysosomal cathepsins. Apoptosis 2007, 12, 125–139. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Fan, L.; Ren, J.; Pang, Z.; Ren, Y.; Li, J.; Wen, Z.; Jiang, X. A novel combination of TRAIL and doxorubicin enhances antitumor effect based on passive tumor-targeting of liposomes. Nanotechnology 2011, 22, 265105. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.A.; Choi, B.H.; Kwak, M.K. The c-MET/PI3K signaling is associated with cancer resistance to doxorubicin and photodynamic therapy by elevating BCRP/ABCG2 expression. Mol. Pharmacol. 2015, 87, 465–476. [Google Scholar] [CrossRef]
- Subramaniam, A.; Loo, S.Y.; Rajendran, P.; Manu, K.A.; Perumal, E.; Li, F.; Shanmugam, M.K.; Siveen, K.S.; Park, J.I.; Ahn, K.S.; et al. An anthraquinone derivative, emodin sensitizes hepatocellular carcinoma cells to TRAIL induced apoptosis through the induction of death receptors and downregulation of cell survival proteins. Apoptosis 2013, 18, 1175–1187. [Google Scholar] [CrossRef] [PubMed]
- Todo, M.; Horinaka, M.; Tomosugi, M.; Tanaka, R.; Ikawa, H.; Sowa, Y.; Ishikawa, H.; Fujiwara, H.; Otsuji, E.; Sakai, T. Ibuprofen enhances TRAIL-induced apoptosis through DR5 upregulation. Oncol. Rep. 2013, 30, 2379–2384. [Google Scholar] [CrossRef] [PubMed]
- Senbabaoglu, F.; Cingoz, A.; Kaya, E.; Kazancioglu, S.; Lack, N.A.; Acilan, C.; Bagci-Onder, T. Identification of Mitoxantrone as a TRAIL-sensitizing agent for Glioblastoma Multiforme. Cancer Biol. Ther. 2016, 17, 546–557. [Google Scholar] [CrossRef] [PubMed]
- Petrova, V.; Annicchiarico-Petruzzelli, M.; Melino, G.; Amelio, I. The hypoxic tumour microenvironment. Oncogenesis 2018, 7. [Google Scholar] [CrossRef]
- Majerník, M.; Jendželovský, R.; Babinčák, M.; Košuth, J.; Ševc, J.; Tonelli Gombalová, Z.; Jendželovská, Z.; Buríková, M.; Fedoročko, P. Novel Insights into the Effect of Hyperforin and Photodynamic Therapy with Hypericin on Chosen Angiogenic Factors in Colorectal Micro-Tumors Created on Chorioallantoic Membrane. Int. J. Mol. Sci. 2019, 20, 3004. [Google Scholar] [CrossRef]
- Xu, K.; Zhan, Y.; Yuan, Z.; Qiu, Y.; Wang, H.; Fan, G.; Wang, J.; Li, W.; Cao, Y.; Shen, X.; et al. Hypoxia Induces Drug Resistance in Colorectal Cancer through the HIF-1α/miR-338-5p/IL-6 Feedback Loop. Mol. Ther. 2019, 27, 1810–1824. [Google Scholar] [CrossRef]
- Knoll, G.; Bittner, S.; Kurz, M.; Jantsch, J.; Ehrenschwender, M. Hypoxia regulates TRAIL sensitivity of colorectal cancer cells through mitochondrial autophagy. Oncotarget 2016, 7, 41488–41504. [Google Scholar] [CrossRef]
- Yao, K.; Gietema, J.A.; Shida, S.; Selvakumaran, M.; Fonrose, X.; Haas, N.B.; Testa, J.; O’Dwyer, P.J. In vitro hypoxia-conditioned colon cancer cell lines derived from HCT116 and HT29 exhibit altered apoptosis susceptibility and a more angiogenic profile in vivo. Br. J. Cancer 2005, 93, 1356–1363. [Google Scholar] [CrossRef]
- Bunz, F.; Hwang, P.M.; Torrance, C.; Waldman, T.; Zhang, Y.; Dillehay, L.; Williams, J.; Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Disruption of p53 in human cancer cells alters the responses to therapeutic agents. J. Clin. Investig. 1999, 104, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yu, J.; Park, B.H.; Kinzler, K.W.; Vogelstein, B. Role of BAX in the apoptotic response to anticancer agents. Science 2000, 290, 989–992. [Google Scholar] [CrossRef] [PubMed]
- Aires, V.; Colin, D.J.; Doreau, A.; Di Pietro, A.; Heydel, J.M.; Artur, Y.; Latruffe, N.; Delmas, D. P-Glycoprotein 1 Affects Chemoactivities of Resveratrol against Human Colorectal Cancer Cells. Nutrients 2019, 11, 2098. [Google Scholar] [CrossRef] [PubMed]
- Jendželovský, R.; Mikeš, J.; Koval’, J.; Souček, K.; Procházková, J.; Kello, M.; Sačková, V.; Hofmanová, J.; Kozubík, A.; Fedoročko, P. Drug efflux transporters, MRP1 and BCRP, affect the outcome of hypericin-mediated photodynamic therapy in HT-29 adenocarcinoma cells. Photochem. Photobiol. Sci. 2009, 8, 1716–1723. [Google Scholar] [CrossRef]
- Jendželovský, R.; Jendželovská, Z.; Kuchárová, B.; Fedoročko, P. Breast cancer resistance protein is the enemy of hypericin accumulation and toxicity of hypericin-mediated photodynamic therapy. Biomed. Pharmacother. 2019, 109, 2173–2181. [Google Scholar] [CrossRef]
- Vargová, J.; Mikeš, J.; Jendželovský, R.; Mikešová, L.; Kuchárová, B.; Čulka, L.; Fedr, R.; Remšík, J.; Souček, K.; Kozubík, A.; et al. Hypericin affects cancer side populations via competitive inhibition of BCRP. Biomed. Pharmacother. 2018, 99, 511–522. [Google Scholar] [CrossRef]
- Doyle, L.A.; Ross, D.D. Multidrug resistance mediated by the breast cancer resistance protein BCRP (ABCG2). Oncogene 2003, 22, 7340–7358. [Google Scholar] [CrossRef]
- Rosenberg, M.F.; Bikadi, Z.; Chan, J.; Liu, X.; Ni, Z.; Cai, X.; Ford, R.C.; Mao, Q. The Human Breast Cancer Resistance Protein (BCRP/ABCG2) Shows Conformational Changes with Mitoxantrone. Structure 2010, 18, 482–493. [Google Scholar] [CrossRef]
- Fetsch, P.A.; Abati, A.; Litman, T.; Morisaki, K.; Honjo, Y.; Mittal, K.; Bates, S.E. Localization of the ABCG2 mitoxantrone resistance-associated protein in normal tissues. Cancer Lett. 2006, 235, 84–92. [Google Scholar] [CrossRef]
- Hilovska, L.; Jendželovský, R.; Jendželovská, Z.; Koval’, J.; Fedoročko, P. Downregulation of BCRP and anti-apoptotic proteins by proadifen (SKF-525A) is responsible for the enhanced mitoxantrone accumulation and toxicity in mitoxantrone-resistant human promyelocytic leukemia cells. Int. J. Oncol. 2015, 47, 1572–1584. [Google Scholar] [CrossRef][Green Version]
- He, X.; Wang, J.; Wei, W.; Shi, M.; Xin, B.; Zhang, T.; Shen, X. Hypoxia regulates ABCG2 activity through the activivation of ERK1/2/HIF-1α and contributes to chemoresistance in pancreatic cancer cells. Cancer Biol. Ther. 2016, 17, 188–198. [Google Scholar] [CrossRef]
- Martin, C.M.; Ferdous, A.; Gallardo, T.; Humphries, C.; Sadek, H.; Caprioli, A.; Garcia, J.A.; Szweda, L.I.; Garry, M.G.; Garry, D.J. Hypoxia-Inducible Factor-2α Transactivates Abcg2 and Promotes Cytoprotection in Cardiac Side Population Cells. Circ. Res. 2008, 102, 1075–1081. [Google Scholar] [CrossRef]
- Jing, X.; Yang, F.; Shao, C.; Wei, K.; Xie, M.; Shen, H.; Shu, Y. Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol. Cancer 2019, 18, 1–15. [Google Scholar] [CrossRef]
- Triner, D.; Shah, Y.M. Hypoxia-inducible factors: A central link between inflammation and cancer. J. Clin. Investig. 2016, 126, 3689–3698. [Google Scholar] [CrossRef]
- Seo, S.B.; Hur, J.G.; Kim, M.J.; Lee, J.W.; Kim, H.B.; Bae, J.H.; Kim, D.W.; Kang, C.D.; Kim, S.H. TRAIL sensitize MDR cells to MDR-related drugs by down-regulation of P-glycoprotein through inhibition of DNA-PKcs/Akt/GSK-3β pathway and activation of caspases. Mol. Cancer 2010, 9, 199. [Google Scholar] [CrossRef]
- Sadarangani, A.; Kato, S.; Espinoza, N.; Lange, S.; Llados, C.; Espinosa, M.; Villalón, M.; Lipkowitz, S.; Cuello, M.; Owen, G.I. TRAIL mediates apoptosis in cancerous but not normal primary cultured cells of the human reproductive tract. Apoptosis 2007, 12, 73–85. [Google Scholar] [CrossRef]
- Van Dijk, M.; Halpin-McCormick, A.; Sessler, T.; Samali, A.; Szegezdi, E. Resistance to TRAIL in non-transformed cells is due to multiple redundant pathways. Cell Death Dis. 2013, 4, e702. [Google Scholar] [CrossRef]
- Huang, Y.; Yang, X.; Xu, T.; Kong, Q.; Zhang, Y.; Shen, Y.; Wei, Y.; Wang, G.; Chang, K.J. Overcoming resistance to TRAIL-induced apoptosis in solid tumor cells by simultaneously targeting death receptors, c-FLIP and IAPs. Int. J. Oncol. 2016, 49, 153–163. [Google Scholar] [CrossRef]
- Kretz, A.L.; Trauzold, A.; Hillenbrand, A.; Knippschild, U.; Henne-Bruns, D.; von Karstedt, S.; Lemke, J. Trailblazing strategies for cancer treatment. Cancers 2019, 11, 456. [Google Scholar] [CrossRef]
- Zhang, B.; Liu, B.; Chen, D.; Setroikromo, R.; Haisma, H.J.; Quax, W.J. Histone Deacetylase Inhibitors Sensitize TRAIL-Induced Apoptosis in Colon Cancer Cells. Cancers 2019, 11, 645. [Google Scholar] [CrossRef] [PubMed]
- Sophonnithiprasert, T.; Nilwarangkoon, S.; Nakamura, Y.; Watanapokasin, R. Goniothalamin enhances TRAIL-induced apoptosis in colorectal cancer cells through DR5 upregulation and cFLIP downregulation. Int. J. Oncol. 2015, 47, 2188–2196. [Google Scholar] [CrossRef]
- Chen, M.; Wang, X.; Zha, D.; Cai, F.; Zhang, W.; He, Y.; Huang, Q.; Zhuang, H.; Hua, Z.C. Apigenin potentiates TRAIL therapy of non-small cell lung cancer via upregulating DR4/DR5 expression in a p53-dependent manner. Sci. Rep. 2016, 6, 35468. [Google Scholar] [CrossRef]
- Yao, Z.; Chen, A.; Li, X.; Zhu, Z.; Jiang, X. Hsp90 inhibitor sensitizes TRAIL-mediated apoptosis via chop-dependent DR5 upregulation in colon cancer cells. Am. J. Transl. Res. 2017, 9, 4945. [Google Scholar] [PubMed]
- Rasheduzzaman, M.; Jeong, J.K.; Park, S.Y. Resveratrol sensitizes lung cancer cell to TRAIL by p53 independent and suppression of Akt/NF-κB signaling. Life Sci. 2018, 208, 208–220. [Google Scholar] [CrossRef] [PubMed]
- Dilshara, M.G.; Molagoda, I.M.N.; Jayasooriya, R.G.P.T.; Choi, Y.H.; Park, C.; Lee, K.T.; Lee, S.; Kim, G.Y. P53-mediated oxidative stress enhances indirubin-3 -monoxime-induced apoptosis in HCT116 colon cancer cells by upregulating death receptor 5 and TNF-related apoptosis-inducing ligand expression. Antioxidants 2019, 8, 423. [Google Scholar] [CrossRef]
- Na, Y.J.; Lee, D.H.; Kim, J.L.; Kim, B.R.; Park, S.H.; Jo, M.J.; Jeong, S.; Kim, H.J.; young Lee, S.; Jeong, Y.A.; et al. Cyclopamine sensitizes TRAIL-resistant gastric cancer cells to TRAIL-induced apoptosis via endoplasmic reticulum stress-mediated increase of death receptor 5 and survivin degradation. Int. J. Biochem. Cell Biol. 2017, 89, 147–156. [Google Scholar] [CrossRef]
- Schempp, C.M.; Simon-Haarhaus, B.; Termeer, C.C.; Simon, J.C. Hypericin photo-induced apoptosis involves the tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and activation of caspase-8. FEBS Lett. 2001, 493, 26–30. [Google Scholar] [CrossRef]
- Zhang, P.; Wang, H.; Chen, Y.; Lodhi, A.F.; Sun, C.; Sun, F.; Yan, L.; Deng, Y.; Ma, H. DR5 related autophagy can promote apoptosis in gliomas after irradiation. Biochem. Biophys. Res. Commun. 2020, 522, 910–916. [Google Scholar] [CrossRef]
- Fagerberg, L.; Hallstrom, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteomics 2014, 13, 397–406. [Google Scholar] [CrossRef]
- Hellwig, C.T.; Rehm, M. TRAIL signaling and synergy mechanisms used in TRAIL-based combination therapies. Mol. Cancer Ther. 2012, 11, 3–13. [Google Scholar] [CrossRef]
- Falschlehner, C.; Emmerich, C.H.; Gerlach, B.; Walczak, H. TRAIL signalling: Decisions between life and death. Int. J. Biochem. Cell Biol. 2007, 39, 1462–1475. [Google Scholar] [CrossRef] [PubMed]
- Mills, K.R.; Reginato, M.; Debnath, J.; Queenan, B.; Brugge, J.S. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is required for induction of autophagy during lumen formation in vitro. Proc. Natl. Acad. Sci. USA 2004, 101, 3438–3443. [Google Scholar] [CrossRef] [PubMed]
- Pespeni, M.H.; Hodnett, M.; Abayasiriwardana, K.S.; Roux, J.; Howard, M.; Broaddus, V.C.; Pittet, J.F. Sensitization of mesothelioma cells to tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis by heat stress via the inhibition of the 3-phosphoinositide-dependent kinase 1/Akt pathway. Cancer Res. 2007, 67, 2865–2871. [Google Scholar] [CrossRef]
- Li, L.; Wen, X.Z.; Bu, Z.D.; Cheng, X.J.; Xing, X.F.; Wang, X.H.; Zhang, L.H.; Guo, T.; Du, H.; Hu, Y.; et al. Paclitaxel enhances tumoricidal potential of TRAIL via inhibition of MAPK in resistant gastric cancer cells. Oncol. Rep. 2016, 35, 3009–3017. [Google Scholar] [CrossRef]
- Fei, P.; Wang, W.; Kim, S.H.; Wang, S.; Burns, T.F.; Sax, J.K.; Buzzai, M.; Dicker, D.T.; McKenna, W.G.; Bernhard, E.J.; et al. Bnip3L is induced by p53 under hypoxia, and its knockdown promotes tumor growth. Cancer Cell 2004, 6, 597–609. [Google Scholar] [CrossRef]
- Shao, C.; Li, Z.; Ahmad, N.; Liu, X. Regulation of PTEN degradation and NEDD4–1 E3 ligase activity by Numb. Cell Cycle 2017, 16, 957–967. [Google Scholar] [CrossRef]
- Jouan-Lanhouet, S.; Arshad, M.I.; Piquet-Pellorce, C.; Martin-Chouly, C.; Le Moigne-Muller, G.; Van Herreweghe, F.; Takahashi, N.; Sergent, O.; Lagadic-Gossmann, D.; Vandenabeele, P.; et al. TRAIL induces necroptosis involving RIPK1/RIPK3-dependent PARP-1 activation. Cell Death Differ. 2012, 19, 2003–2014. [Google Scholar] [CrossRef]
- Luebke, T.; Schwarz, L.; Beer, Y.Y.; Schumann, S.; Misterek, M.; Sander, F.E.; Plaza-Sirvent, C.; Schmitz, I. c-FLIP and CD95 signaling are essential for survival of renal cell carcinoma. Cell Death Dis. 2019, 10, 1–12. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Cox, J.; Neuhauser, N.; Michalski, A.; Scheltema, R.A.; Olsen, J.V.; Mann, M. Andromeda: A Peptide Search Engine Integrated into the MaxQuant Environment. J. Proteome Res. 2011, 10, 1794–1805. [Google Scholar] [CrossRef]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef]
- Okonechnikov, K.; Golosova, O.; Fursov, M.; Varlamov, A.; Vaskin, Y.; Efremov, I.; German Grehov, O.G.; Kandrov, D.; Rasputin, K.; Syabro, M.; et al. Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Venables, W.N.; Ripley, B.D. Modern Applied Statistics with S, 4th ed.; Springer: New York, NY, USA, 2002. [Google Scholar]
- Wickham, H. Reshaping Data with the {reshape} Package. J. Stat. Softw. 2007, 21, 1–20. [Google Scholar] [CrossRef]
- Sarkar, D. Lattice: Multivariate Data Visualization with R; Springer: New York, NY, USA, 2008. [Google Scholar]
- Clark, N.; Hafner, M.; Kouril, M.; Muhlich, J.; Niepel, M.; Williams, E.; Sorger, P.; Medvedovic, M. GRcalculator: An online tool for calculating and mining drug response data. BMC Cancer 2017. [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Auguie, B. Miscellaneous Functions for “Grid” Graphics; R Foundation for Statistical Computing: Vienna, Austria, 2017. [Google Scholar]
- Sievert, C. Plotly for R; R Foundation for Statistical Computing: Vienna, Austria, 2018. [Google Scholar]
- Xiao, N. ggsci: Scientific Journal and Sci-Fi Themed Color Palettes for ‘ggplot2’; R Foundation for Statistical Computing: Vienna, Austria, 2018. [Google Scholar]
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.D.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the {tidyverse}. J. Open Source Softw. 2019, 4, 1686. [Google Scholar] [CrossRef]
- Wickham, H.; François, R.; Henry, L.; Müller, K. Dplyr: A Grammar of Data Manipulation; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- R Core Team. Foreign: Read Data Stored by ‘Minitab’, ‘S’, ‘SAS’, ‘SPSS’, ‘Stata’, ‘Systat’, ‘Weka’, ‘dBase’,...; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Wickham, H.; Henry, L. Tidyr: Tidy Messy Data; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Wickham, H. Forcats: Tools for Working with Categorical Variables (Factors); R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 24 h Normoxia | 24 h Hypoxia | 48 h Normoxia | 48 h Hypoxia | |
|---|---|---|---|---|
| HCT 116 | 17.88 ± 1.56 | 21.96 ± 2.19 | 9.50 ± 0.80 | 6.70 ± 0.34 |
| HT-29 | 25.67 ± 2.80 | 16.45 ± 1.17 | 15.83 ± 1.15 | 11.36 ± 0.65 |
| Accession | Description of Protein Group (Gene Name) | Log2FC (H) | adj.p Value (H) | Log2FC (N) | adj.p Value (N) | Hypoxia | Normoxia |
|---|---|---|---|---|---|---|---|
| O14763 | Tumor necrosis factor receptor superfamily member 10B (GN = TNFRSF10B) | 3.66 | 0 | 2.48 | 0.01 | ↑ | ↑ |
| Q6IQ22 | Ras-related protein Rab-12 (GN = RAB12) | 2.39 | 0.05 | 2.33 | 0.01 | ↑ | ↑ |
| Q9Y2H2 | Phosphatidylinositide phosphatase SAC2 (GN = INPP5F) | 3.65 | 0.01 | 1.05 | 0.03 | ↑ | |
| P15407 | Fos-related antigen 1 (GN = FOSL1) | 1.52 | 0 | 2.91 | 0 | ↑ | ↑ |
| Q9GZQ8 | Microtubule-associated proteins 1A/1B light chain 3B (GN = MAP1LC3B) | 2.54 | 0 | 1.78 | 0 | ↑ | ↑ |
| Q9HBI6 | Phylloquinone omega-hydroxylase CYP4F11 (GN = CYP4F11) | 1.36 | 0.04 | 2.89 | 0.01 | ↑ | |
| Q03405 | Urokinase plasminogen activator surface receptor (GN = PLAUR) | 1.95 | 0 | 2.22 | 0 | ↑ | ↑ |
| Q7Z4Q2 | HEAT repeat-containing protein 3 (GN = HEATR3) | 2.09 | 0 | 1.82 | 0 | ↑ | ↑ |
| Q53EU6 | Glycerol-3-phosphate acyltransferase 3 (GN = GPAT3) | 1.83 | 0.05 | 1.77 | 0 | ↑ | ↑ |
| Q9BSH5 | Haloacid dehalogenase-like hydrolase domain-containing protein 3 (GN = HDHD3) | 3.01 | 0.01 | 0.44 | 0.02 | ↑ | |
| P08243 | Asparagine synthetase [glutamine-hydrolyzing] (GN = ASNS) | 2.06 | 0 | 1.37 | 0 | ↑ | |
| Q16850 | Lanosterol 14-alpha demethylase (GN = CYP51A1) | 1.84 | 0 | 1.33 | 0 | ↑ | |
| Q9NUM4 | Transmembrane protein 106B (GN = TMEM106B) | 1.34 | 0.01 | 1.66 | 0.01 | ↑ | |
| H3BU86 | STX16-NPEPL1 readthrough (NMD candidate) (GN = STX16-NPEPL1) | 1.8 | 0.02 | 1.18 | 0.05 | ↑ | |
| Q8ND82 | Zinc finger protein 280C (GN = ZNF280C) | 1.86 | 0 | 1.06 | 0.01 | ↑ | |
| Q9H6H4 | Receptor expression-enhancing protein 4 (GN = REEP4) | 1.84 | 0.01 | 0.98 | 0.04 | ↑ | |
| Q6WCQ1 | Myosin phosphatase Rho-interacting protein (GN = MPRIP) | 0.92 | 0.04 | 1.78 | 0 | ↑ | |
| O95352 | Ubiquitin-like modifier-activating enzyme ATG7 (GN = ATG7) | 1.56 | 0.01 | 1.11 | 0.02 | ↑ | |
| Q8IVF2 | Protein AHNAK2 (GN = AHNAK2) | 1.93 | 0.02 | 0.7 | 0.05 | ↑ | |
| O15231 | Zinc finger protein 185 (GN = ZNF185) | 1.51 | 0 | 1.09 | 0.05 | ↑ | |
| Q9NQW6 | Anillin (GN = ANLN) | 0.52 | 0.04 | 1.84 | 0 | ↑ | |
| Q9Y617 | Phosphoserine aminotransferase (GN = PSAT1) | 1.5 | 0 | 0.83 | 0 | ↑ | |
| Q9NR12 | PDZ and LIM domain protein 7 (GN = PDLIM7) | 1.51 | 0 | 0.72 | 0.01 | ↑ | |
| Q9NP97 | Dynein light chain roadblock-type 1 (GN = DYNLRB1) | −1.22 | 0.04 | 2.37 | 0 | ↑ | |
| Q8N5M9 | Protein jagunal homolog 1 (GN = JAGN1) | −2.59 | 0.01 | 3.24 | 0 | ↓ | ↑ |
| Q9NUQ3 | Gamma-taxilin (GN = TXLNG) | −0.91 | 0.02 | 1.52 | 0.01 | ↑ | |
| P15153 | Ras-related C3 botulinum toxin substrate 2 (GN = RAC2) | 1.57 | 0.02 | −1.14 | 0.01 | ↑ | |
| P14635 | G2/mitotic-specific cyclin-B1 (GN = CCNB1) | 1.86 | 0 | −1.45 | 0.04 | ↑ | |
| Q9H2C0 | Gigaxonin (GN = GAN) | −2.48 | 0.02 | 2.43 | 0.04 | ↓ | ↑ |
| Q9UL03 | Integrator complex subunit 6 (GN = INTS6) | −1.77 | 0 | 1.5 | 0 | ↓ | |
| Q14BN4 | Sarcolemmal membrane-associated protein (GN = SLMAP) | 4.26 | 0.04 | −4.77 | 0 | ↑ | ↓ |
| Q9HCS7 | Pre-mRNA-splicing factor SYF1 (GN = XAB2) | −1.79 | 0.02 | 0.65 | 0.04 | ↓ | |
| P31483 | Nucleolysin TIA-1 isoform p40 (GN = TIA1) | 0.65 | 0.03 | −1.83 | 0.02 | ↓ | |
| Q16555 | Dihydropyrimidinase-related protein 2 (GN = DPYSL2) | −0.46 | 0.03 | −1.53 | 0 | ↓ | |
| P82664 | 28S ribosomal protein S10, mitochondrial (GN = MRPS10) | −1.65 | 0 | −0.45 | 0.03 | ↓ | |
| P55011 | Solute carrier family 12 member 2 (GN = SLC12A2) | −1.72 | 0.01 | −0.45 | 0.04 | ↓ | |
| Q9H8V3 | Protein ECT2 (GN = ECT2) | −1.83 | 0.01 | −0.42 | 0.04 | ↓ | |
| Q9P2R3 | Rabankyrin-5 (GN = ANKFY1) | −1.67 | 0.01 | −0.88 | 0.01 | ↓ | |
| P00395 | Cytochrome c oxidase subunit 1 (GN = MT-CO1) | −0.91 | 0.03 | −1.66 | 0 | ↓ | |
| Q92466 | DNA damage-binding protein 2 (GN = DDB2) | −1.64 | 0.04 | −1.15 | 0.03 | ↓ | |
| Q9UHL4 | Dipeptidyl peptidase 2 (GN = DPP7) | −1.68 | 0.03 | −1.12 | 0.03 | ↓ | |
| Q9BRK5 | 45 kDa calcium-binding protein (GN = SDF4) | −2.02 | 0.01 | −0.81 | 0 | ↓ | |
| Q6SPF0 | Atherin (GN = SAMD1) | −1.49 | 0.03 | −1.59 | 0.01 | ↓ | |
| Q9NRX4 | 14 kDa phosphohistidine phosphatase (GN = PHPT1) | −1.76 | 0 | −1.32 | 0.01 | ↓ | |
| P14406 | Cytochrome c oxidase subunit 7A2, mitochondrial (GN = COX7A2) | −2.14 | 0 | −1.01 | 0 | ↓ | |
| P35611 | Alpha-adducin (GN=ADD1) | −1.61 | 0.01 | −1.56 | 0 | ↓ | ↓ |
| Q52LW3 | Rho GTPase-activating protein 29 (GN = ARHGAP29) | −2.1 | 0 | −1.09 | 0 | ↓ | |
| Q6UX53 | Methyltransferase-like protein 7B (GN = METTL7B) | −1.73 | 0 | −1.69 | 0 | ↓ | ↓ |
| P22830 | Ferrochelatase, mitochondrial (GN = FECH) | −1.81 | 0.01 | −1.91 | 0.01 | ↓ | ↓ |
| O94832 | Unconventional myosin-Id (GN = MYO1D) | −2.52 | 0.01 | −1.23 | 0.01 | ↓ | |
| P33552 | Cyclin-dependent kinases regulatory subunit 2 (GN = CKS2) | −1.77 | 0.01 | −2.01 | 0 | ↓ | ↓ |
| Q92817 | Envoplakin (GN = EVPL) | −2.76 | 0.01 | −1.54 | 0.01 | ↓ | ↓ |
| A6NJ78 | Probable methyltransferase-like protein 15 (GN = METTL15) | −2.16 | 0.02 | −3.27 | 0 | ↓ | ↓ |
| P50748 | Kinetochore-associated protein 1 (GN = KNTC1) | −3.94 | 0 | −2.16 | 0.02 | ↓ | ↓ |
| Q14517 | Protocadherin Fat 1 (GN = FAT1) | −5.73 | 0 | −0.49 | 0.05 | ↓ | |
| P46934 | E3 ubiquitin-protein ligase NEDD4 (GN = NEDD4) | −3.7 | 0.01 | −3.94 | 0 | ↓ | ↓ |
| Pathway Name | Entities Found | Entities Total | Entities’ p-Value | Entities’ FDR |
|---|---|---|---|---|
| Rab regulation of trafficking | 2 | 123 | 0.00236 | 0.0398 |
| Attachment of GPI anchor to uPAR | 1 | 7 | 0.00429 | 0.0398 |
| TRAIL signaling | 1 | 8 | 0.0049 | 0.0398 |
| Regulation by c-FLIP | 1 | 11 | 0.00674 | 0.0398 |
| Dimerization of procaspase-8 | 1 | 11 | 0.00674 | 0.0398 |
| Receptor-mediated mitophagy | 1 | 11 | 0.00674 | 0.0398 |
| Pexophagy | 1 | 11 | 0.00674 | 0.0398 |
| TP53 regulates transcription of death receptors and ligands | 1 | 12 | 0.00735 | 0.0398 |
| Dissolution of Fibrin Clot | 1 | 13 | 0.00796 | 0.0398 |
| CASP8 activity is inhibited | 1 | 14 | 0.00857 | 0.0421 |
| Caspase activation via death receptors in the presence of ligand | 1 | 19 | 0.0116 | 0.0421 |
| Regulation of necroptotic cell death | 1 | 21 | 0.0128 | 0.0421 |
| Pink/Parkin mediated mitophagy | 1 | 22 | 0.0134 | 0.0421 |
| RIPK1-mediated regulated necrosis | 1 | 23 | 0.014 | 0.0421 |
| Regulated necrosis | 1 | 23 | 0.014 | 0.0421 |
| Mitophagy | 1 | 29 | 0.0177 | 0.0474 |
| Caspase activation via extrinsic apoptotic signaling pathway | 1 | 29 | 0.0177 | 0.0474 |
| Synthesis of PA | 1 | 39 | 0.0237 | 0.0474 |
| NGF-stimulated transcription | 1 | 39 | 0.0237 | 0.0474 |
| Pathway Name | Entities Found | Entities Total | Entities’ p-Value | Entities’ FDR |
|---|---|---|---|---|
| Regulation of PTEN localization | 1 | 9 | 0.0063 | 0.126 |
| Downregulation of ERBB4 signaling | 1 | 11 | 0.0077 | 0.126 |
| Caspase-mediated cleavage of cytoskeletal proteins | 1 | 12 | 0.00839 | 0.126 |
| Heme biosynthesis | 1 | 15 | 0.0105 | 0.126 |
| Miscellaneous transport and binding events | 1 | 26 | 0.0181 | 0.126 |
| Metabolism of porphyrins | 1 | 28 | 0.0195 | 0.126 |
| Apoptotic cleavage of cellular proteins | 1 | 38 | 0.0264 | 0.126 |
| XBP1(S) activates chaperone genes | 1 | 48 | 0.0332 | 0.126 |
| IRE1alpha activates chaperones | 1 | 50 | 0.0346 | 0.126 |
| Apoptotic execution phase | 1 | 52 | 0.0359 | 0.126 |
| Signaling by ERBB4 | 1 | 62 | 0.0427 | 0.126 |
| Regulation of PTEN stability and activity | 1 | 69 | 0.0474 | 0.126 |
| Gene (NCBI Gene ID) | GenBank Ref.seq | Primer Name | Sequence | Ta and Product Length |
|---|---|---|---|---|
| TNFRSF10B | NM_003842 | DR5_for | GTGATTCAGGTGAAGTGGAGC | 60 |
| (8795) | NM_147187 | DR5_rev | CGACCTTGACCATCCCTCTG | 147 bp |
| TNFRSF10B | NM_003842 | DR5.v1_for | ACTCCTGCCTCTCCCTGTTC | 60 |
| (8795) | DR5.v1_rev | AGGTCGTTGTGAGCTTCTGTC | 186 bp | |
| TNFRSF10B | NM_147187 | DR5.v2_for | CTAAGTCCCTGCACCACGAC | 60 |
| (8795) | DR5.v2_rev | TGACTCCTATGATGATGCCTGATT | 193 bp | |
| TNFRSF10A | NM_003844 | DR4_for | GTTGGTGGCTGTGCTGATTG | 60 |
| (8797) | DR4_rev | TGCGTTGCTCAGAATCTCGT | 151 bp | |
| TNF (TNF alpha) | NM_000594 | TNFa_for | TGTCCACACGATCCCAACAC | 60 |
| (7124) | TNFa_rev | GGCTGTCACACCCACAATCA | 152 bp | |
| TNFRSF1A | NM_001065 | TNFRSF1A_for | CGTGATCTCTATGCCCGAGT | 60 |
| (7132) | TNFRSF1A_rev | GACCAGTCCAATAACCCCTGA | 244 bp | |
| TNFRSF1B | NM_001066 | TNFRSF1B_for | TGTCCACACGATCCCAACAC | 60 |
| (7133) | TNFRSF1B_rev | GGCTGTCACACCCACAATCA | 152 bp | |
| TNFRSF6B | NM_003823 | DcR3_for | CTCTTCCTCCCATGACACCCT | 60 |
| (8771) | DcR3_rev | ATGGAGATGTCCTGGAAAGCC | 123 bp | |
| PMM1 | NM_002676 | PMM1_for | GCTCGCCAGAAAATTGACCCT | 61 |
| (5372) | PMM1_rev | ATACTGCACCGTCCCGTTCT | 177 bp |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Babinčák, M.; Jendželovský, R.; Košuth, J.; Majerník, M.; Vargová, J.; Mikulášek, K.; Zdráhal, Z.; Fedoročko, P. Death Receptor 5 (TNFRSF10B) Is Upregulated and TRAIL Resistance Is Reversed in Hypoxia and Normoxia in Colorectal Cancer Cell Lines after Treatment with Skyrin, the Active Metabolite of Hypericum spp. Cancers 2021, 13, 1646. https://doi.org/10.3390/cancers13071646
Babinčák M, Jendželovský R, Košuth J, Majerník M, Vargová J, Mikulášek K, Zdráhal Z, Fedoročko P. Death Receptor 5 (TNFRSF10B) Is Upregulated and TRAIL Resistance Is Reversed in Hypoxia and Normoxia in Colorectal Cancer Cell Lines after Treatment with Skyrin, the Active Metabolite of Hypericum spp. Cancers. 2021; 13(7):1646. https://doi.org/10.3390/cancers13071646
Chicago/Turabian StyleBabinčák, Marián, Rastislav Jendželovský, Ján Košuth, Martin Majerník, Jana Vargová, Kamil Mikulášek, Zbyněk Zdráhal, and Peter Fedoročko. 2021. "Death Receptor 5 (TNFRSF10B) Is Upregulated and TRAIL Resistance Is Reversed in Hypoxia and Normoxia in Colorectal Cancer Cell Lines after Treatment with Skyrin, the Active Metabolite of Hypericum spp." Cancers 13, no. 7: 1646. https://doi.org/10.3390/cancers13071646
APA StyleBabinčák, M., Jendželovský, R., Košuth, J., Majerník, M., Vargová, J., Mikulášek, K., Zdráhal, Z., & Fedoročko, P. (2021). Death Receptor 5 (TNFRSF10B) Is Upregulated and TRAIL Resistance Is Reversed in Hypoxia and Normoxia in Colorectal Cancer Cell Lines after Treatment with Skyrin, the Active Metabolite of Hypericum spp. Cancers, 13(7), 1646. https://doi.org/10.3390/cancers13071646