
Hot and Cold Tumors: Is Endoglin (CD105) a Potential Target for Vessel Normalization?


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
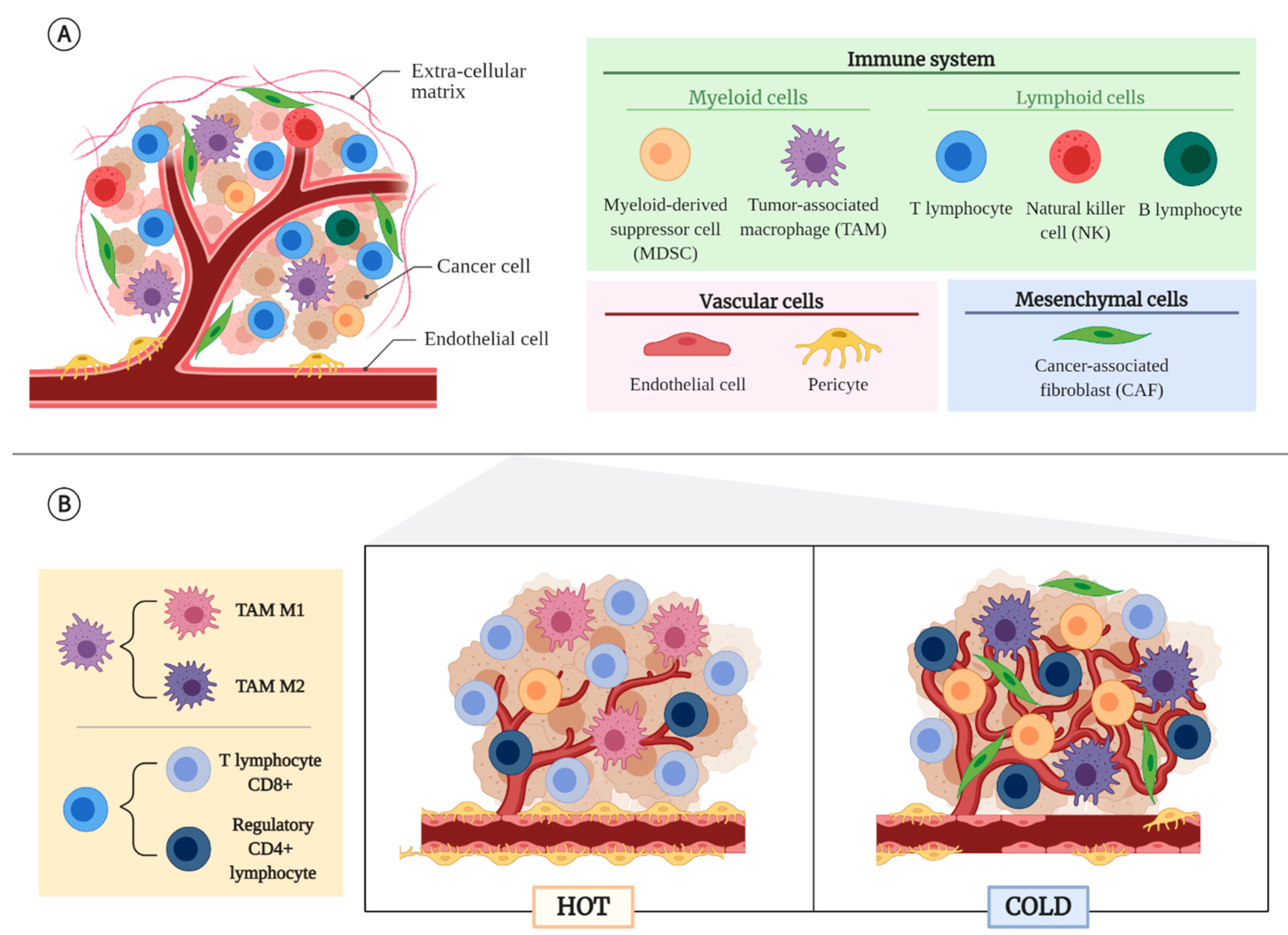
2. Tumor Microenvironment (TME)
2.1. Non-Cancerous Cellular Component of the TME
2.2. Tumor Types According to Their TME
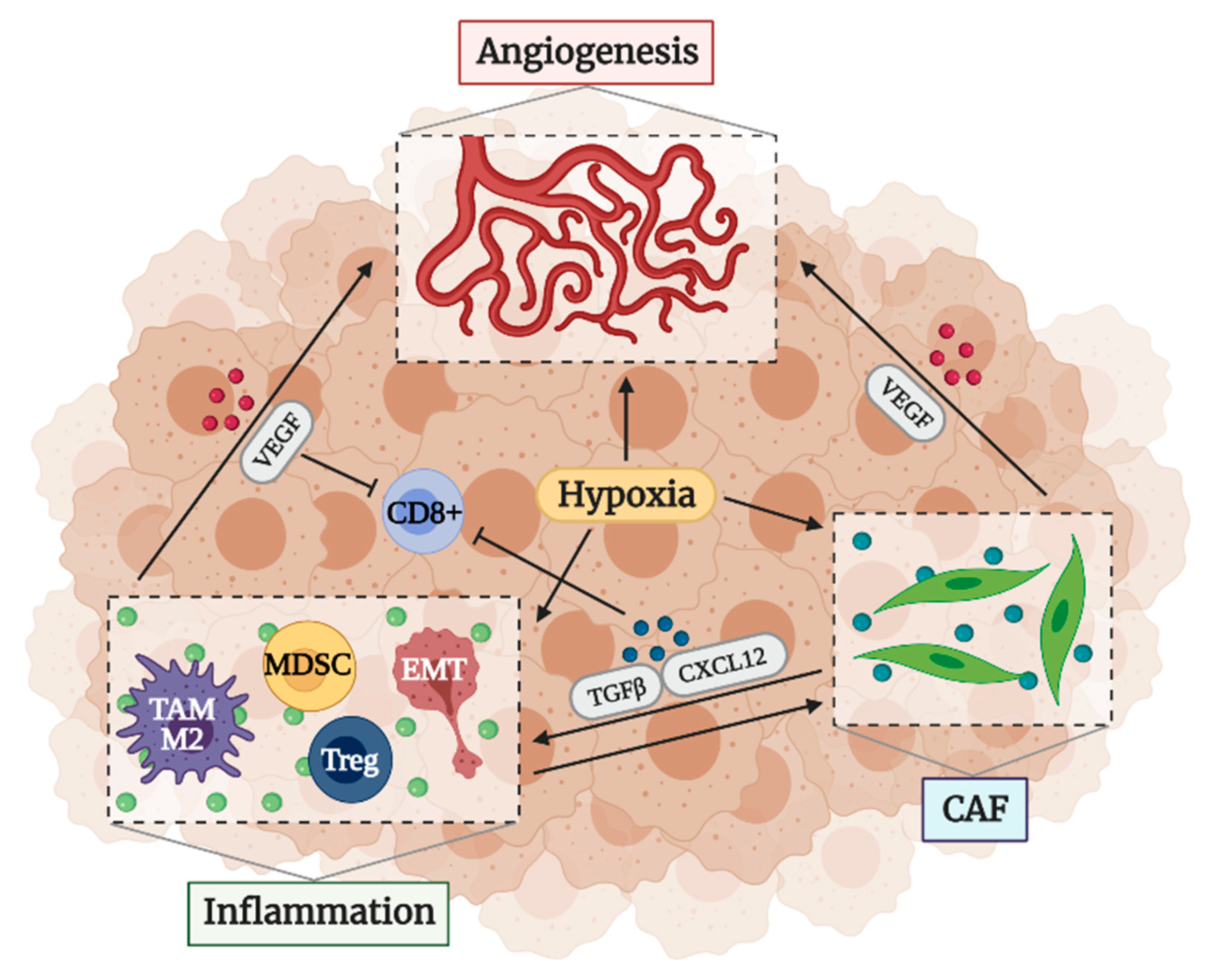
2.3. Factors That Determine an Immunosuppressive TME
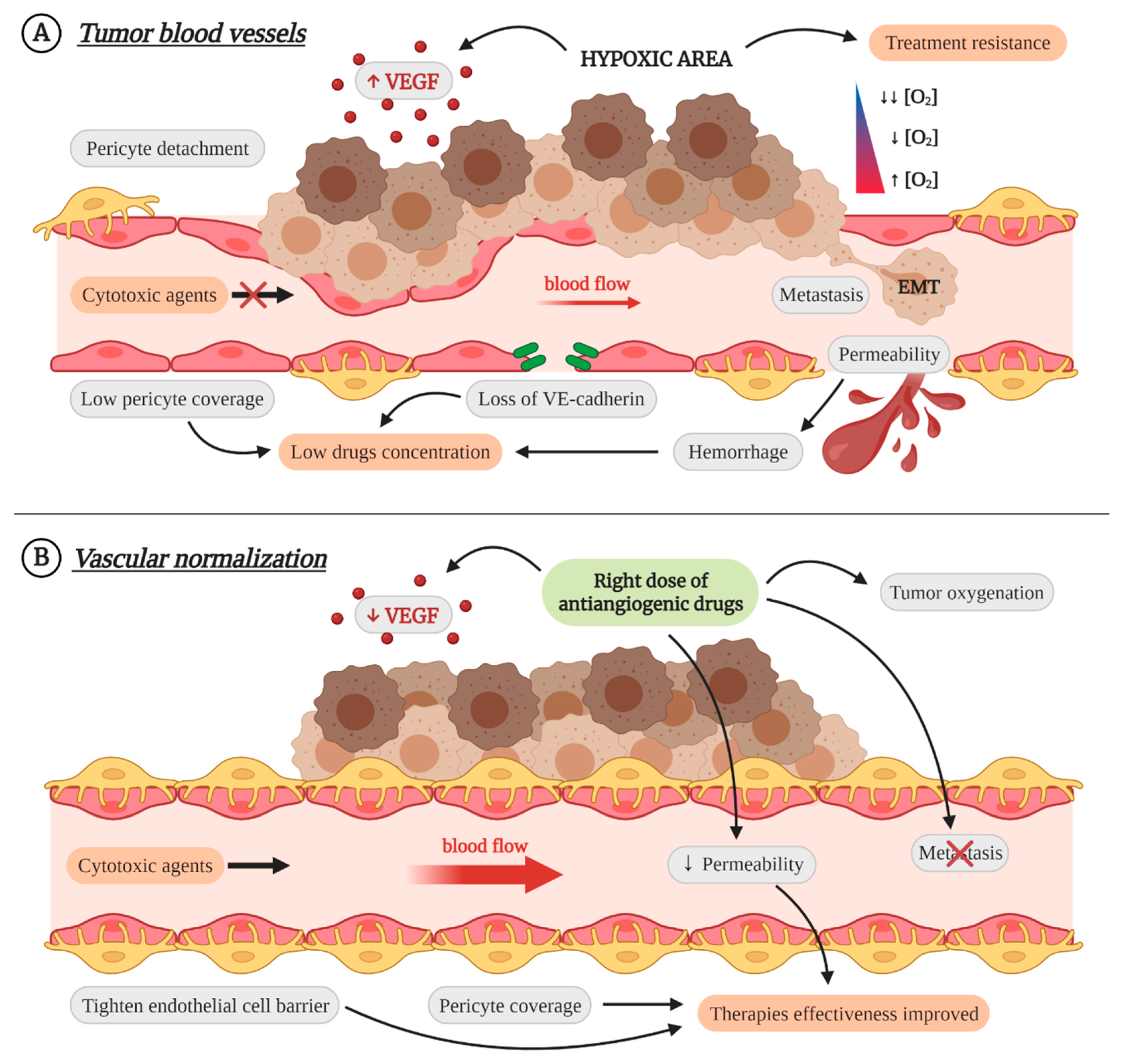
3. Vascular Normalization as Therapeutic Strategy
4. Endoglin
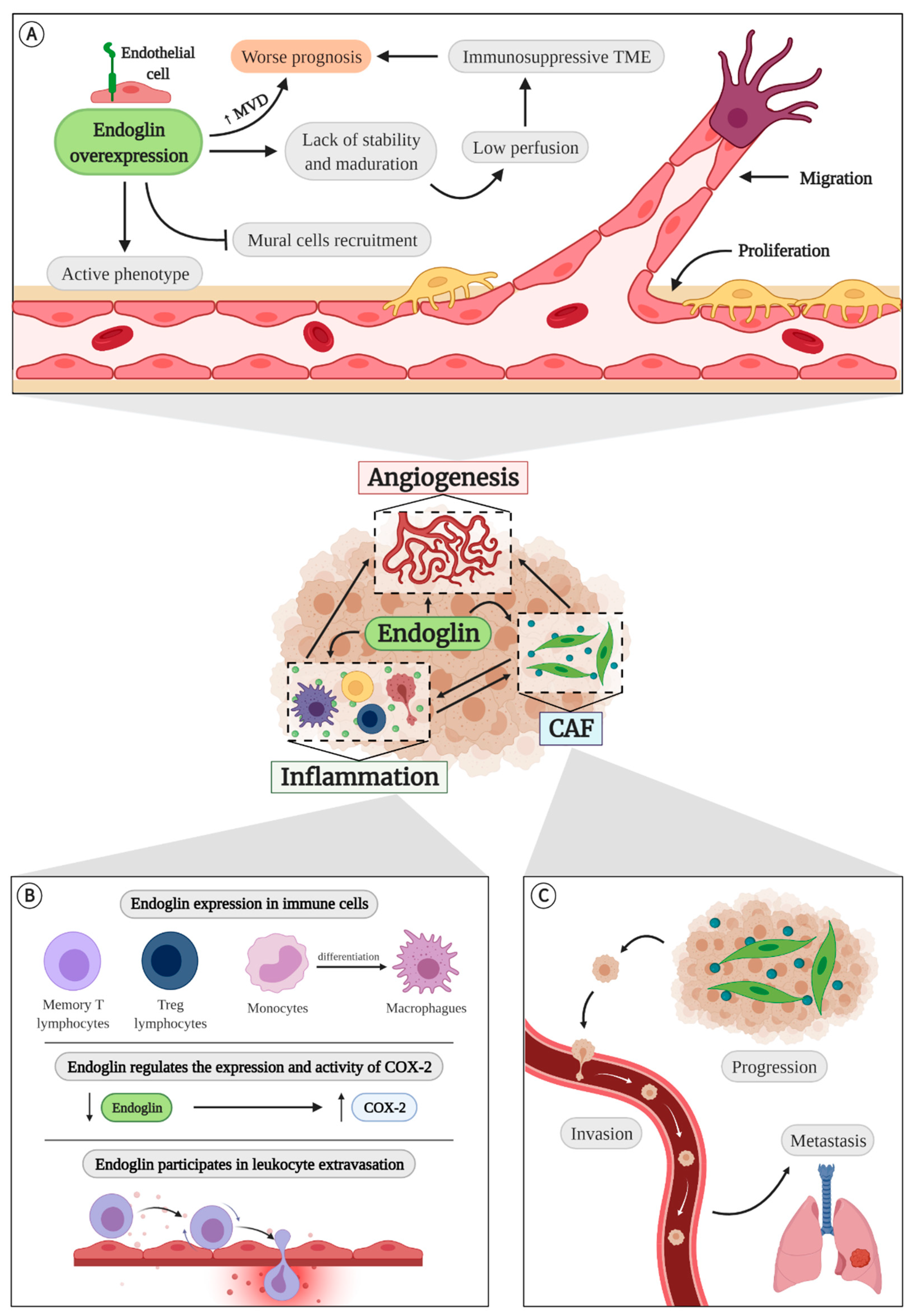
4.1. Endoglin in Angiogenesis
4.2. Endoglin in Inflammation
4.3. Endoglin and Cancer-Associated Fibroblasts (CAFs)
5. Anti-Endoglin Therapies
6. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ADT | androgen signaling deprivation therapy |
| ATP | adenosine triphosphate |
| ATX | autotaxin |
| AVM | arteriovenous malformation |
| BOEC | blood outgrowth endothelial cell |
| CAF | cancer-associated fibroblast |
| COX-2 | cyclooxygenase-2 |
| DNA | deoxyribonucleic acid |
| EC | endothelial cell |
| ECM | extracellular matrix |
| EMT | epithelium-mesenchyme transition |
| EPC | endothelial progenitor cell |
| FAP | fibroblast activation protein |
| GET | gene electrotransfer |
| HHT | hereditary hemorrhagic telangiectasia |
| HUVEC | human umbilical vein endothelial cell |
| IFN | interferon |
| IL | interleukin |
| LPA | Lysophosphatide |
| MDSC | myeloid-derived suppressor cell |
| MEEC | murine embryonic endothelial cell |
| miRNA | micro ribonucleic acid |
| mRNA | messenger ribonucleic acid |
| MSC | mesenchymal stromal cell |
| NK | natural killer |
| OIR | oxygen-induced ischemic retinopathy |
| PDGF | platelet-derived growth factor |
| PDT | photodynamic therapy |
| PET | positron emission tomography |
| PGE2 | prostaglandin E2 |
| ROS | reactive oxygen species |
| shRNA | short hairpin ribonucleic acid |
| siRNA | small interfering ribonucleic acid |
| S1P | sphingosine-1-phosphate |
| TβRI | TGF-β receptor I kinase |
| TβRII | TGF-β receptor II kinase |
| TAM | tumor-associated macrophage |
| TGF-β | transforming growth factor β |
| TME | tumor microenvironment |
| VEGF | vascular endothelial growth factor |
| VEGFR | VEGF-receptor |
| WT | wild type |
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hanahan, D.; Coussens, L.M. Accessories to the Crime: Functions of Cells Recruited to the Tumor Microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The tumor microenvironment at a glance. J. Cell Sci. 2012, 125, 5591–5596. [Google Scholar] [CrossRef]
- Najafi, M.; Goradel, N.H.; Farhood, B.; Salehi, E.; Solhjoo, S.; Toolee, H.; Kharazinejad, E.; Mortezaee, K. Tumor microenvironment: Interactions and therapy. J. Cell. Physiol. 2019, 234, 5700–5721. [Google Scholar] [CrossRef]
- Duan, Q.; Zhang, H.; Zheng, J.; Zhang, L. Turning Cold into Hot: Firing up the Tumor Microenvironment. Trends Cancer 2020, 6, 605–618. [Google Scholar] [CrossRef]
- Jarosz-Biej, M.; Kamińska, N.; Matuszczak, S.; Cichoń, T.; Pamuła-Piłat, J.; Czapla, J.; Smolarczyk, R.; Skwarzyńska, D.; Kulik, K.; Szala, S. M1-like macrophages change tumor blood vessels and microenvironment in murine melanoma. PLoS ONE 2018, 13, e0191012. [Google Scholar] [CrossRef]
- Leibovici, J.; Itzhaki, O.; Huszar, M.; Sinai, J. The tumor microenvironment: Part 1. Immunotherapy 2011, 3, 1367–1384. [Google Scholar] [CrossRef] [PubMed]
- Shiao, S.L.; Preethi Ganesan, A.; Rugo, H.S.; Coussens, L.M. Immune microenvironments in solid tumors: New targets for therapy. Genes Dev. 2011, 25, 2559–2572. [Google Scholar] [CrossRef] [PubMed]
- Datta, M.; Coussens, L.M.; Nishikawa, H.; Hodi, F.S.; Jain, R.K. Reprogramming the Tumor Microenvironment to Improve Immunotherapy: Emerging Strategies and Combination Therapies. Am. Soc. Clin. Oncol. Educ. B 2019, 39, 165–174. [Google Scholar] [CrossRef]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Casazza, A.; Di Conza, G.; Wenes, M.; Finisguerra, V.; Deschoemaeker, S.; Mazzone, M. Tumor stroma: A complexity dictated by the hypoxic tumor microenvironment. Oncogene 2014, 33, 1743–1754. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Goel, S.; Duda, D.G.; Fukumura, D.; Jain, R.K. Vascular normalization as an emerging strategy to enhance cancer immunotherapy. Cancer Res. 2013, 73, 2943–2948. [Google Scholar] [CrossRef]
- Biffi, G.; Tuveson, D.A. Diversity and Biology of Cancer-Associated Fibroblasts. Physiol. Rev. 2021, 101, 147–176. [Google Scholar] [CrossRef]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef]
- Fearon, D.T. The carcinoma-associated fibroblast expressing fibroblast activation protein and escape from immune surveillance. Cancer Immunol. Res. 2014, 2, 187–193. [Google Scholar] [CrossRef]
- Monteran, L.; Erez, N. The dark side of fibroblasts: Cancer-associated fibroblasts as mediators of immunosuppression in the tumor microenvironment. Front. Immunol. 2019, 10, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Barbazán, J.; Matic Vignjevic, D. Cancer associated fibroblasts: Is the force the path to the dark side? Curr. Opin. Cell Biol. 2019, 56, 71–79. [Google Scholar] [CrossRef]
- De Guillebon, E.; Dardenne, A.; Saldmann, A.; Séguier, S.; Tran, T.; Paolini, L.; Lebbe, C.; Tartour, E. Beyond the concept of cold and hot tumors for the development of novel predictive biomarkers and the rational design of immunotherapy combination. Int. J. Cancer 2020, 147, 1509–1518. [Google Scholar] [CrossRef] [PubMed]
- Gajewski, T.F.; Corrales, L.; Williams, J.; Horton, B.; Sivan, A.; Spranger, S. Cancer immunotherapy targets based on understanding the t cell-inflamed versus non-t cell-inflamed tumor microenvironment. In Advances in Experimental Medicine and Biology; Springer: Berlin/Heidelberg, Germany, 2017. [Google Scholar]
- Trujillo, J.A.; Sweis, R.F.; Bao, R.; Luke, J.J. T cell–inflamed versus Non-T cell–inflamed tumors: A conceptual framework for cancer immunotherapy drug development and combination therapy selection. Cancer Immunol. Res. 2018, 6, 990–1000. [Google Scholar] [CrossRef]
- Luo, J.L.; Tan, W.; Ricono, J.M.; Korchynskyi, O.; Zhang, M.; Gonias, S.L.; Cheresh, D.A.; Karin, M. Nuclear cytokine-activated IKKα controls prostate cancer metastasis by repressing Maspin. Nature 2007, 446, 690–694. [Google Scholar] [CrossRef] [PubMed]
- Benesch, M.G.K.; Ko, Y.M.; McMullen, T.P.W.; Brindley, D.N. Autotaxin in the crosshairs: Taking aim at cancer and other inflammatory conditions. FEBS Lett. 2014, 588, 2712–2727. [Google Scholar] [CrossRef]
- Riboni, L.; Abdel Hadi, L.; Navone, S.E.; Guarnaccia, L.; Campanella, R.; Marfia, G. Sphingosine-1-Phosphate in the Tumor Microenvironment: A Signaling Hub Regulating Cancer Hallmarks. Cells 2020, 9, 337. [Google Scholar] [CrossRef]
- Wang, P.; Yuan, Y.; Lin, W.; Zhong, H.; Xu, K.; Qi, X. Roles of sphingosine-1-phosphate signaling in cancer. Cancer Cell Int. 2019, 19, 295–312. [Google Scholar] [CrossRef]
- Balaji Ragunathrao, V.A.; Anwar, M.; Akhter, M.Z.; Chavez, A.; Mao, D.Y.; Natarajan, V.; Lakshmikanthan, S.; Chrzanowska-Wodnicka, M.; Dudek, A.Z.; Claesson-Welsh, L.; et al. Sphingosine-1-Phosphate Receptor 1 Activity Promotes Tumor Growth by Amplifying VEGF-VEGFR2 Angiogenic Signaling. Cell Rep. 2019, 29, 3472–3487.e4. [Google Scholar] [CrossRef]
- Gaengel, K.; Niaudet, C.; Hagikura, K.; Siemsen, B.L.; Muhl, L.; Hofmann, J.J.; Ebarasi, L.; Nyström, S.; Rymo, S.; Chen, L.L.; et al. The Sphingosine-1-Phosphate Receptor S1PR1 Restricts Sprouting Angiogenesis by Regulating the Interplay between VE-Cadherin and VEGFR2. Dev. Cell 2012, 23, 587–599. [Google Scholar] [CrossRef]
- Zhuang, J.; Lu, Q.; Shen, B.; Huang, X.; Shen, L.; Zheng, X.; Huang, R.; Yan, J.; Guo, H. TGFβ1 secreted by cancer-associated fibroblasts induces epithelial-mesenchymal transition of bladder cancer cells through lncRNA-ZEB2NAT. Sci. Rep. 2015, 5, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ansems, M.; Span, P.N. The tumor microenvironment and radiotherapy response; a central role for cancer-associated fibroblasts. Clin. Transl. Radiat. Oncol. 2020, 22, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Nieman, K.M.; Kenny, H.A.; Penicka, C.V.; Ladanyi, A.; Buell-Gutbrod, R.; Zillhardt, M.R.; Romero, I.L.; Carey, M.S.; Mills, G.B.; Hotamisligil, G.S.; et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 2011, 17, 1498–1503. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Tumor angiogenesis: Therapeutic implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar]
- Núñez-Gómez, E.; Pericacho, M.; Ollauri-Ibáñez, C.; Bernabéu, C.; López-Novoa, J.M. The role of endoglin in post-ischemic revascularization. Angiogenesis 2017, 20, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Ollauri-Ibáñez, C.; Núñez-Gómez, E.; Egido-Turrión, C.; Silva-Sousa, L.; Díaz-Rodríguez, E.; Rodríguez-Barbero, A.; López-Novoa, J.M.; Pericacho, M. Continuous endoglin (CD105) overexpression disrupts angiogenesis and facilitates tumor cell metastasis. Angiogenesis 2020, 23, 231–247. [Google Scholar] [CrossRef]
- Ollauri-Ibáñez, C.; López-Novoa, J.M.; Pericacho, M. Endoglin-based biological therapy in the treatment of angiogenesis-dependent pathologies. Expert Opin. Biol. Ther. 2017, 17, 1053–1063. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yuan, J.; Righi, E.; Kamoun, W.S.; Ancukiewicz, M.; Nezivar, J.; Santosuosso, M.; Martin, J.D.; Martin, M.R.; Vianello, F.; et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc. Natl. Acad. Sci. USA 2012, 109, 17561–17566. [Google Scholar] [CrossRef]
- Alipour, M.; Majidi, A.; Molaabasi, F.; Sheikhnejad, R.; Hosseinkhani, S. In vivo tumor gene delivery using novel peptideticles: PH-responsive and ligand targeted core–shell nanoassembly. Int. J. Cancer 2018, 143, 2017–2028. [Google Scholar] [CrossRef]
- Jain, R.K.; Martin, J.D.; Stylianopoulos, T. The role of mechanical forces in tumor growth and therapy. Annu. Rev. Biomed. Eng. 2014, 16, 321–346. [Google Scholar] [CrossRef]
- Reymond, N.; D’Água, B.B.; Ridley, A.J. Crossing the endothelial barrier during metastasis. Nat. Rev. Cancer 2013, 13, 858–870. [Google Scholar] [CrossRef]
- Liu, Y.; Zhen, W.; Jin, L.; Zhang, S.; Sun, G.; Zhang, T.; Xu, X.; Song, S.; Wang, Y.; Liu, J.; et al. All-in-One Theranostic Nanoagent with Enhanced Reactive Oxygen Species Generation and Modulating Tumor Microenvironment Ability for Effective Tumor Eradication. ACS Nano 2018, 12, 4886–4893. [Google Scholar] [CrossRef]
- Huang, Y.; Chen, X.; Dikov, M.M.; Novitskiy, S.V.; Mosse, C.A.; Yang, L.; Carbone, D.P. Distinct roles of VEGFR-1 and VEGFR-2 in the aberrant hematopoiesis associated with elevated levels of VEGF. Blood 2007, 110, 624–631. [Google Scholar] [CrossRef] [PubMed]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.B.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef] [PubMed]
- Lakins, M.A.; Ghorani, E.; Munir, H.; Martins, C.P.; Shields, J.D. Cancer-associated fibroblasts induce antigen-specific deletion of CD8 + T Cells to protect tumour cells. Nat. Commun. 2018, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Donthireddy, L.; Marvel, D.; Condamine, T.; Wang, F.; Lavilla-Alonso, S.; Hashimoto, A.; Vonteddu, P.; Behera, R.; Goins, M.A.; et al. Cancer-Associated Fibroblasts Neutralize the Anti-tumor Effect of CSF1 Receptor Blockade by Inducing PMN-MDSC Infiltration of Tumors. Cancer Cell 2017, 32, 654–668.e5. [Google Scholar] [CrossRef]
- Albrengues, J.; Bertero, T.; Grasset, E.; Bonan, S.; Maiel, M.; Bourget, I.; Philippe, C.; Herraiz Serrano, C.; Benamar, S.; Croce, O.; et al. Epigenetic switch drives the conversion of fibroblasts into proinvasive cancer-associated fibroblasts. Nat. Commun. 2015, 6, 10204. [Google Scholar] [CrossRef]
- Albrengues, J.; Bourget, I.; Pons, C.; Butet, V.; Hofman, P.; Tartare-Deckert, S.; Feral, C.C.; Meneguzzi, G.; Gaggioli, C. LIF mediates proinvasive activation of stromal fibroblasts in cancer. Cell Rep. 2014, 7, 1664–1678. [Google Scholar] [CrossRef]
- Avery, D.; Govindaraju, P.; Jacob, M.; Todd, L.; Monslow, J.; Puré, E. Extracellular matrix directs phenotypic heterogeneity of activated fibroblasts. Matrix Biol. 2018, 67, 90–106. [Google Scholar] [CrossRef]
- Comito, G.; Giannoni, E.; Segura, C.P.; Barcellos-De-Souza, P.; Raspollini, M.R.; Baroni, G.; Lanciotti, M.; Serni, S.; Chiarugi, P. Cancer-associated fibroblasts and M2-polarized macrophages synergize during prostate carcinoma progression. Oncogene 2014, 33, 2423–2431. [Google Scholar] [CrossRef]
- Mathew, E.; Brannon, A.L.; Del Vecchio, A.C.; Garcia, P.E.; Penny, M.K.; Kane, K.T.; Vinta, A.; Buckanovich, R.J.; Di Magliano, M.P. Mesenchymal Stem Cells Promote Pancreatic Tumor Growth by Inducing Alternative Polarization of Macrophages. Neoplasia 2016, 18, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J. Putting the Immunologic Brakes on Cancer. Cell 2018, 175, 1452–1454. [Google Scholar] [CrossRef]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef] [PubMed]
- Petrillo, M.; Scambia, G.; Ferrandina, G. Novel targets for VEGF-independent anti-angiogenic drugs. Expert Opin. Investig. Drugs 2012, 21, 451–472. [Google Scholar] [CrossRef]
- Zhao, Y.; Adjei, A.A. New Drug Development and Clinical Pharmacology Targeting Angiogenesis in Cancer Therapy: Moving Beyond Vascular Endothelial Growth Factor. Oncologist 2015, 20, 660–673. [Google Scholar] [CrossRef]
- Ollauri-Ibáñez, C.; Astigarraga, I. Use of Antiangiogenic Therapies in Pediatric Solid Tumors. Cancers 2021, 13, 253. [Google Scholar] [CrossRef]
- Ramjiawan, R.R.; Griffioen, A.W.; Duda, D.G. Anti-angiogenesis for cancer revisited: Is there a role for combinations with immunotherapy? Angiogenesis 2017, 20, 185–204. [Google Scholar] [CrossRef]
- van Beijnum, J.R.; Nowak-Sliwinska, P.; Huijbers, E.J.M.; Thijssen, V.L.; Griffioen, A.W. The great escape; the hallmarks of resistance to antiangiogenic therapy. Pharmacol. Rev. 2015, 67, 441–461. [Google Scholar] [CrossRef] [PubMed]
- Haibe, Y.; Kreidieh, M.; El Hajj, H.; Khalifeh, I.; Mukherji, D.; Temraz, S.; Shamseddine, A. Resistance Mechanisms to Anti-angiogenic Therapies in Cancer. Front. Oncol. 2020, 10, 221. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Normalization of tumor vasculature: An emerging concept in antiangiogenic therapy. Science 2005, 307, 58–62. [Google Scholar] [CrossRef]
- Goel, S.; Duda, D.G.; Xu, L.; Munn, L.L.; Boucher, Y.; Fukumura, D.; Jain, R.K. Normalization of the vasculature for treatment of cancer and other diseases. Physiol. Rev. 2011, 91, 1071–1121. [Google Scholar] [CrossRef]
- Wikström, P.; Lissbrant, I.F.; Stattin, P.; Egevad, L.; Bergh, A. Endoglin (CDI05) is expressed on immature blood vessels and is a marker for survival in prostate cancer. Prostate 2002, 51, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Paauwe, M.; Schoonderwoerd, M.J.A.; Helderman, R.F.C.P.; Harryvan, T.J.; Groenewoud, A.; Van Pelt, G.W.; Bor, R.; Hemmer, D.M.; Versteeg, H.H.; Ewa Snaar-Jagalska, B.; et al. Endoglin expression on cancer-associated fibroblasts regulates invasion and stimulates colorectal cancer metastasis. Clin. Cancer Res. 2018, 24, 6331–6344. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Placencio-Hickok, V.R.; Madhav, A.; Haldar, S.; Tripathi, M.; Billet, S.; Mishra, R.; Smith, B.; Rohena-Rivera, K.; Agarwal, P.; et al. Heterogeneous cancer-associated fibroblast population potentiates neuroendocrine differentiation and castrate resistance in a CD105-dependent manner. Oncogene 2019, 38, 716–730. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.S.; Xin, Z.C.; Dai, J.; Lue, T.F. Commonly used mesenchymal stem cell markers and tracking labels: Limitations and challenges. Histol. Histopathol. 2013, 28, 1109–1116. [Google Scholar]
- Zhang, J.; Yuan, B.; Zhang, H.; Li, H. Human epithelial ovarian cancer cells expressing cd105, cd44 and cd106 surface markers exhibit increased invasive capacity and drug resistance. Oncol. Lett. 2019, 17, 5351–5360. [Google Scholar] [CrossRef]
- Kassouf, W.; Ismail, H.R.A.; Aprikian, A.G.; Chevalier, S. Whole-mount prostate sections reveal differential endoglin expression in stromal, epithelial, and endothelial cells with the development of prostate cancer. Prostate Cancer Prostatic Dis. 2004, 7, 105–110. [Google Scholar] [CrossRef][Green Version]
- Grange, C.; Tapparo, M.; Collino, F.; Vitillo, L.; Damasco, C.; Deregibus, M.C.; Tetta, C.; Bussolati, B.; Camussi, G. Microvesicles released from human renal cancer stem cells stimulate angiogenesis and formation of lung premetastatic niche. Cancer Res. 2011, 71, 5346–5356. [Google Scholar] [CrossRef]
- Meurer, S.K.; Weiskirchen, R. Endoglin: An ‘accessory’ receptor regulating blood cell development and inflammation. Int. J. Mol. Sci. 2020, 21, 9247. [Google Scholar] [CrossRef]
- Aristorena, M.; Gallardo-Vara, E.; Vicen, M.; Casas-Engel, M.D.L.; Ojeda-Fernandez, L.; Nieto, C.; Blanco, F.J.; Valbuena-Diez, A.C.; Botella, L.M.; Nachtigal, P.; et al. MMP-12, secreted by pro-inflammatory macrophages, targets endoglin in human macrophages and endothelial cells. Int. J. Mol. Sci. 2019, 20, 3107. [Google Scholar] [CrossRef]
- Ojeda-Fernández, L.; Recio-Poveda, L.; Aristorena, M.; Lastres, P.; Blanco, F.J.; Sanz-Rodríguez, F.; Gallardo-Vara, E.; De las Casas-Engel, M.; Corbí, Á.; Arthur, H.M.; et al. Mice Lacking Endoglin in Macrophages Show an Impaired Immune Response. PLoS Genet. 2016, 12, e1005935. [Google Scholar] [CrossRef] [PubMed]
- Nowaczyk, R.M.; Jursza-Piotrowska, E.; Gram, A.; Siemieniuch, M.J.; Boos, A.; Kowalewski, M.P. Cells expressing CD4, CD8, MHCII and endoglin in the canine corpus luteum of pregnancy, and prepartum activation of the luteal TNFα system. Theriogenology 2017, 98, 123–132. [Google Scholar] [CrossRef]
- Schmidt-Weber, C.B.; Letarte, M.; Kunzmann, S.; Rückert, B.; Bernabéu, C.; Blaser, K. TGF-{beta} signaling of human T cells is modulated by the ancillary TGF-{beta} receptor endoglin. Int. Immunol. 2005, 17, 921–930. [Google Scholar] [CrossRef][Green Version]
- Schoonderwoerd, M.J.A.; Koops, M.F.M.; Angela, R.A.; Koolmoes, B.; Toitou, M.; Paauwe, M.; Barnhoorn, M.C.; Liu, Y.; Sier, C.F.M.; Hardwick, J.C.H.; et al. Targeting Endoglin-Expressing Regulatory T Cells in the Tumor Microenvironment Enhances the Effect of PD1 Checkpoint Inhibitor Immunotherapy. Clin. Cancer Res. 2020, 26, 3831–3842. [Google Scholar] [CrossRef] [PubMed]
- Schoonderwoerd, M.J.A.; Goumans, M.J.T.H.; Hawinkels, L.J.A.C. Endoglin: Beyond the endothelium. Biomolecules 2020, 10, 289. [Google Scholar] [CrossRef]
- Lastres, P.; Bellon, T.; Cabañas, C.; Sanchez-Madrid, F.; Acevedo, A.; Gougos, A.; Letarte, M.; Bernabeu, C. Regulated expression on human macrophages of endoglin, an Arg-Gly-Asp-containing surface antigen. Eur. J. Immunol. 1992, 22, 393–397. [Google Scholar] [CrossRef]
- O’Connell, P.; McKenzie, A.; Fisicaro, N.; Rockman, S.; Pearse, M.; D’Apice, A. Endoglin: A 180-kD endothelial cell and macrophage restricted differentiation molecule. Clin. Exp. Immunol. 1992, 90, 154–159. [Google Scholar] [CrossRef]
- Gougos, A.; Letarte, M. Primary structure of endoglin, an RGD-containing glycoprotein of human endothelial cells. J. Biol. Chem. 1990, 265, 8361–8364. [Google Scholar] [CrossRef]
- Velasco, S.; Alvarez-Munoz, P.; Pericacho, M.; Dijke, P.T.; Bernabeu, C.; Lopez-Novoa, J.M.; Rodriguez-Barbero, A. L- and S-endoglin differentially modulate TGF 1 signaling mediated by ALK1 and ALK5 in L6E9 myoblasts. J. Cell Sci. 2008, 121, 913–919. [Google Scholar] [CrossRef] [PubMed]
- Blanco, F.J.; Bernabeu, C. Alternative splicing factor or splicing factor-2 plays a key role in intron retention of the endoglin gene during endothelial senescence. Aging Cell 2011, 10, 896–907. [Google Scholar] [CrossRef]
- Blanco, F.J.; Grande, M.T.; Langa, C.; Oujo, B.; Velasco, S.; Rodriguez-Barbero, A.; Perez-Gomez, E.; Quintanilla, M.; López-Novoa, J.M.; Bernabeu, C. S-endoglin expression is induced in senescent endothelial cells and contributes to vascular pathology. Circ. Res. 2008, 103, 1383–1392. [Google Scholar] [CrossRef]
- Aristorena, M.; Blanco, F.J.; De Las Casas-Engel, M.; Ojeda-Fernandez, L.; Gallardo-Vara, E.; Corbi, A.; Botella, L.M.; Bernabeu, C. Expression of endoglin isoforms in the myeloid lineage and their role during aging and macrophage polarization. J. Cell Sci. 2014, 127, 2723–2735. [Google Scholar] [CrossRef]
- Hawinkels, L.J.A.C.; Kuiper, P.; Wiercinska, E.; Verspaget, H.W.; Liu, Z.; Pardali, E.; Sier, C.F.M.; Ten Dijke, P. Matrix metalloproteinase-14 (MT1-MMP)-mediated endoglin shedding inhibits tumor angiogenesis. Cancer Res. 2010, 70, 4141–4150. [Google Scholar] [CrossRef]
- Li, C.G.; Wilson, P.B.; Bernabeu, C.; Raab, U.; Wang, J.M.; Kumar, S. Immunodetection and characterisation of soluble CD105-TGFβ complexes. J. Immunol. Methods 1998, 218, 85–93. [Google Scholar] [CrossRef]
- Castonguay, R.; Werner, E.D.; Matthews, R.G.; Presman, E.; Mulivor, A.W.; Solban, N.; Sako, D.; Pearsall, R.S.; Underwood, K.W.; Seehra, J.; et al. Soluble endoglin specifically binds bone morphogenetic proteins 9 and 10 via its orphan domain, inhibits blood vessel formation, and suppresses tumor growth. J. Biol. Chem. 2011, 286, 30034–30046. [Google Scholar] [CrossRef]
- Gallardo-Vara, E.; Ruiz-Llorente, L.; Casado-Vela, J.; Ruiz-Rodríguez, M.J.; López-Andrés, N.; Pattnaik, A.K.; Quintanilla, M.; Bernabeu, C. Endoglin Protein Interactome Profiling Identifies TRIM21 and Galectin-3 as New Binding Partners. Cells 2019, 8, 1082. [Google Scholar] [CrossRef] [PubMed]
- Lawera, A.; Tong, Z.; Thorikay, M.; Redgrave, R.E.; Cai, J.; Van Dinther, M.; Morrell, N.W.; Afink, G.B.; Charnock-Jones, D.S.; Arthur, H.M.; et al. Role of soluble endoglin in BMP9 signaling. Proc. Natl. Acad. Sci. USA 2019, 116, 17800–17808. [Google Scholar] [CrossRef] [PubMed]
- Venkatesha, S.; Toporsian, M.; Lam, C.; Hanai, J.I.; Mammoto, T.; Kim, Y.M.; Bdolah, Y.; Lim, K.H.; Yuan, H.T.; Libermann, T.A.; et al. Soluble endoglin contributes to the pathogenesis of preeclampsia. Nat. Med. 2006, 12, 642–649. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Roque, L.; Núñez-Gómez, E.; Rodríguez-Barbero, A.; Bernabéu, C.; López-Novoa, J.M.; Pericacho, M. Pregnancy-Induced High Plasma Levels of Soluble Endoglin in Mice Lead to Preeclampsia Symptoms and Placental Abnormalities. Int. J. Mol. Sci. 2020, 22, 165. [Google Scholar] [CrossRef]
- Li, C.; Guo, B.; Wilson, P.B.; Stewart, A.; Byrne, G.; Bundred, N.; Kumar, S. Plasma levels of soluble CD105 correlate with metastasis in patients with breast cancer. Int. J. Cancer 2000, 89, 122–126. [Google Scholar] [CrossRef]
- Barnett, J.M.; Suarez, S.; McCollum, G.W.; Penn, J.S. Endoglin promotes angiogenesis in cell- and animal- based models of retinal neovascularization. Investig. Ophthalmol. Vis. Sci. 2014, 55, 6490–6498. [Google Scholar] [CrossRef]
- Jin, Y.; Muhl, L.; Burmakin, M.; Wang, Y.; Duchez, A.C.; Betsholtz, C.; Arthur, H.M.; Jakobsson, L. Endoglin prevents vascular malformation by regulating flow-induced cell migration and specification through VEGFR2 signalling. Nat. Cell Biol. 2017, 19, 639–652. [Google Scholar] [CrossRef]
- Van Laake, L.W.; Van Den Driesche, S.; Post, S.; Feijen, A.; Jansen, M.A.; Driessens, M.H.; Mager, J.J.; Snijder, R.J.; Westermann, C.J.J.; Doevendans, P.A.; et al. Endoglin has a crucial role in blood cell-mediated vascular repair. Circulation 2006, 114, 2288–2297. [Google Scholar] [CrossRef] [PubMed]
- Munoz, R.; Arias, Y.; Miguel Ferreras, J.; Jimenez, P.; Angeles Rojo, M.; Bernabeu, C.; Cordoba-Diaz, D.; Girbes, T. Transient injury-dependent up-regulation of CD105 and its specific targeting with an anti-vascular anti-mouse endoglin-nigrin b immunotoxin. Med. Chem. 2012, 8, 996–1002. [Google Scholar] [PubMed]
- Park, S.; DiMaio, T.A.; Liu, W.; Wang, S.; Sorenson, C.M.; Sheibani, N. Endoglin regulates the activation and quiescence of endothelium by participating in canonical and non-canonical TGF- signaling pathways. J. Cell Sci. 2013, 126, 1392–1405. [Google Scholar] [CrossRef] [PubMed]
- Ardelean, D.S.; Yin, M.; Jerkic, M.; Peter, M.; Ngan, B.; Kerbel, R.S.; Foster, F.S.; Letarte, M. Anti-VEGF therapy reduces intestinal inflammation in Endoglin heterozygous mice subjected to experimental colitis. Angiogenesis 2014, 17, 641–659. [Google Scholar] [CrossRef]
- Paauwe, M.; Ten Dijke, P.; Hawinkels, L.J.A.C. Endoglin for tumor imaging and targeted cancer therapy. Expert Opin. Ther. Targets 2013, 17, 421–435. [Google Scholar] [CrossRef]
- Arthur, H.M.; Ure, J.; Smith, A.J.H.; Renforth, G.; Wilson, D.I.; Torsney, E.; Charlton, R.; Parums, D.V.; Jowett, T.; Marchuk, D.A.; et al. Endoglin, an ancillary TGFβ receptor, is required for extraembryonic angiogenesis and plays a key role in heart development. Dev. Biol. 2000, 217, 42–53. [Google Scholar] [CrossRef]
- Goumans, M.J.; Ten Dijke, P. TGF-β signaling in control of cardiovascular function. Cold Spring Harb. Perspect. Biol. 2018, 10, a022210. [Google Scholar] [CrossRef] [PubMed]
- Li, D.Y.; Sorensen, L.K.; Brooke, B.S.; Urness, L.D.; Davis, E.C.; Taylor, D.G.; Boak, B.B.; Wendel, D.P. Defective angiogenesis in mice lacking endoglin. Science 1999, 284, 1534–1537. [Google Scholar] [CrossRef] [PubMed]
- Sugden, W.W.; Meissner, R.; Aegerter-Wilmsen, T.; Tsaryk, R.; Leonard, E.V.; Bussmann, J.; Hamm, M.J.; Herzog, W.; Jin, Y.; Jakobsson, L.; et al. Endoglin controls blood vessel diameter through endothelial cell shape changes in response to haemodynamic cues. Nat. Cell Biol. 2017, 19, 653–665. [Google Scholar] [CrossRef]
- McDonald, J.; Wooderchak-Donahue, W.; VanSant Webb, C.; Whitehead, K.; Stevenson, D.A.; Bayrak-Toydemir, P. Hereditary hemorrhagic telangiectasia: Genetics and molecular diagnostics in a new era. Front. Genet. 2015, 6, 1. [Google Scholar] [CrossRef]
- Porteous, M.E.; Burn, J.; Proctor, S.J. Hereditary haemorrhagic telangiectasia: A clinical analysis. J. Med. Genet. 1992, 29, 527–530. [Google Scholar] [CrossRef]
- Shovlin, C.L.; Guttmacher, A.E.; Buscarini, E.; Faughnan, M.E.; Hyland, R.H.; Westermann, C.J.J.; Kjeldsen, A.D.; Plauchu, H. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome). Am. J. Med. Genet. 2000, 91, 66–67. [Google Scholar] [CrossRef]
- Bernabeu, C.; Bayrak-Toydemir, P.; McDonald, J.; Letarte, M. Potential Second-Hits in Hereditary Hemorrhagic Telangiectasia. J. Clin. Med. 2020, 9, 3571. [Google Scholar] [CrossRef]
- Jerkic, M.; Rodríguez-Barbero, A.; Prieto, M.; Toporsian, M.; Pericacho, M.; Rivas-Elena, J.V.; Obreo, J.; Wang, A.; Pérez-Barriocanal, F.; Arévalo, M.; et al. Reduced angiogenic responses in adult endoglin heterozygous mice. Cardiovasc. Res. 2006, 69, 845–854. [Google Scholar] [CrossRef]
- Seghers, L.; De Vries, M.R.; Pardali, E.; Hoefer, I.E.; Hierck, B.P.; Ten Dijke, P.; Goumans, M.J.; Quax, P.H.A. Shear induced collateral artery growth modulated by endoglin but not by ALK1. J. Cell. Mol. Med. 2012, 16, 2440–2450. [Google Scholar] [CrossRef]
- Lebrin, F.; Srun, S.; Raymond, K.; Martin, S.; Van Den Brink, S.; Freitas, C.; Bréant, C.; Mathivet, T.; Larrivée, B.; Thomas, J.L.; et al. Thalidomide stimulates vessel maturation and reduces epistaxis in individuals with hereditary hemorrhagic telangiectasia. Nat. Med. 2010, 16, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, M.; Allinson, K.R.; Zhai, Z.; Oakenfull, R.; Ghandi, P.; Adams, R.H.; Fruttiger, M.; Arthur, H.M. Pathogenesis of arteriovenous malformations in the absence of endoglin. Circ. Res. 2010, 106, 1425–1433. [Google Scholar] [CrossRef] [PubMed]
- Tual-Chalot, S.; Mahmoud, M.; Allinson, K.R.; Redgrave, R.E.; Zhai, Z.; Oh, S.P.; Fruttiger, M.; Arthur, H.M. Endothelial depletion of Acvrl1 in mice leads to arteriovenous malformations associated with reduced endoglin expression. PLoS ONE 2014, 9, e98646. [Google Scholar] [CrossRef]
- Singh, E.; Redgrave, R.E.; Phillips, H.M.; Arthur, H.M. Arterial endoglin does not protect against arteriovenous malformations. Angiogenesis 2020, 23, 559–566. [Google Scholar] [CrossRef]
- Rossi, E.; Smadja, D.M.; Boscolo, E.; Langa, C.; Arevalo, M.A.; Pericacho, M.; Gamella-Pozuelo, L.; Kauskot, A.; Botella, L.M.; Gaussem, P.; et al. Endoglin regulates mural cell adhesion in the circulatory system. Cell. Mol. Life Sci. 2016, 73, 1715–1739. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.C.; Kumar, S.; Shah, N.; Hoyt, D.G.; Hawinkels, L.J.A.C.; Mythreye, K.; Lee, N.Y. Src-mediated post-translational regulation of endoglin stability and function is critical for angiogenesis. J. Biol. Chem. 2014, 289, 25486–25496. [Google Scholar] [CrossRef]
- Pan, C.C.; Bloodworth, J.C.; Mythreye, K.; Lee, N.Y. Endoglin inhibits ERK-induced c-Myc and cyclin D1 expression to impede endothelial cell proliferation. Biochem. Biophys. Res. Commun. 2012, 424, 620–623. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pece-Barbara, N.; Vera, S.; Kathirkamathamby, K.; Liebner, S.; Di Guglielmo, G.M.; Dejana, E.; Wrana, J.L.; Letarte, M. Endoglin null endothelial cells proliferate faster and are more responsive to transforming growth factor β1 with higher affinity receptors and an activated Alk1 pathway. J. Biol. Chem. 2005, 280, 27800–27808. [Google Scholar] [CrossRef] [PubMed]
- Bernabeu, C.; Lopez-Novoa, J.M.; Quintanilla, M. The emerging role of TGF-β superfamily coreceptors in cancer. Biochim. Biophys. Acta Mol. Basis Dis. 2009, 1792, 954–973. [Google Scholar] [CrossRef]
- Liu, Y.Q.; Jovanovic, B.; Pins, M.; Lee, C.; Bergan, R.C. Over expression of endoglin in human prostate cancer suppresses cell detachment, migration and invasion. Oncogene 2002, 21, 8272–8281. [Google Scholar] [CrossRef][Green Version]
- Conley, B.A.; Koleva, R.; Smith, J.D.; Kacer, D.; Zhang, D.; Bernabéu, C.; Vary, C.P.H. Endoglin controls cell migration and composition of focal adhesions: Function of the cytosolic domain. J. Biol. Chem. 2004, 279, 27440–27449. [Google Scholar] [CrossRef]
- Sanz-Rodriguez, F.; Guerrero-Esteo, M.; Botella, L.M.; Banville, D.; Vary, C.P.H.; Bernabéu, C. Endoglin regulates cytoskeletal organization through binding to ZRP-1, a member of the LIM family of proteins. J. Biol. Chem. 2004, 279, 32858–32868. [Google Scholar] [CrossRef]
- Fernandez-Lopez, A.; Garrido-Martin, E.M.; Sanz-Rodriguez, F.; Pericacho, M.; Rodriguez-Barbero, A.; Eleno, N.; Lopez-Novoa, J.M.; Düwell, A.; Vega, M.A.; Bernabeu, C.; et al. Gene expression fingerprinting for human hereditary hemorrhagic telangiectasia. Hum. Mol. Genet. 2007, 16, 1515–1533. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-L, A.; Sanz-Rodriguez, F.; Zarrabeitia, R.; Pérez-Molino, A.; Hebbel, R.P.; Nguyen, J.; Bernabéu, C.; Botella, L.M. Blood outgrowth endothelial cells from Hereditary Haemorrhagic Telangiectasia patients reveal abnormalities compatible with vascular lesions. Cardiovasc. Res. 2005, 68, 235–248. [Google Scholar] [CrossRef]
- Zucco, L.; Zhang, Q.; Kuliszewski, M.A.; Kandic, I.; Faughnan, M.E.; Stewart, D.J.; Kutryk, M.J. Circulating angiogenic cell dysfunction in patients with hereditary hemorrhagic telangiectasia. PLoS ONE 2014, 9, e89927. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Tian, H.; Blobe, G.C.; Theuer, C.P.; Hurwitz, H.I.; Nixon, A.B. Effects of the combination of TRC105 and bevacizumab on endothelial cell biology. Investig. New Drugs 2014, 32, 851–859. [Google Scholar] [CrossRef] [PubMed]
- Goumans, M.J.; Valdimarsdottir, G.; Itoh, S.; Rosendahl, A.; Sideras, P.; Ten Dijke, P. Balancing the activation state of the endothelium via two distinct TGF-β type I receptors. EMBO J. 2002, 21, 1743–1753. [Google Scholar] [CrossRef]
- Ray, B.N.; Lee, N.Y.; How, T.; Blobe, G.C. ALK5 phosphorylation of the endoglin cytoplasmic domain regulates Smad1/5/8 signaling and endothelial cell migration. Carcinogenesis 2010, 31, 435–441. [Google Scholar] [CrossRef]
- Anderberg, C.; Cunha, S.I.; Zhai, Z.; Cortez, E.; Pardali, E.; Johnson, J.R.; Franco, M.; Páez-Ribes, M.; Cordiner, R.; Fuxe, J.; et al. Deficiency for endoglin in tumor vasculature weakens the endothelial barrier to metastatic dissemination. J. Exp. Med. 2013, 210, 563–579. [Google Scholar] [CrossRef]
- Jerkic, M.; Peter, M.; Ardelean, D.; Fine, M.; Konerding, M.A.; Letarte, M. Dextran sulfate sodium leads to chronic colitis and pathological angiogenesis in endoglin heterozygous mice. Inflamm. Bowel Dis. 2010, 16, 1859–1870. [Google Scholar] [CrossRef] [PubMed]
- Jerkic, M.; Letarte, M. Increased endothelial cell permeability in endoglin-deficient cells. FASEB J. 2015, 29, 3678–3688. [Google Scholar] [CrossRef]
- Mancini, M.L.; Terzic, A.; Conley, B.A.; Oxburgh, L.H.; Nicola, T.; Vary, C.P.H. Endoglin plays distinct roles in vascular smooth muscle cell recruitment and regulation of arteriovenous identity during angiogenesis. Dev. Dyn. 2009, 238, 2479–2493. [Google Scholar] [CrossRef]
- Orlova, V.V.; Liu, Z.; Goumans, M.J.; Ten Dijke, P. Controlling angiogenesis by two unique TGF-β type I receptor signaling pathways. Histol. Histopathol. 2011, 26, 1219–1230. [Google Scholar] [PubMed]
- Fonsatti, E.; Del Vecchio, L.; Altomonte, M.; Sigalotti, L.; Nicotra, M.R.; Coral, S.; Natali, P.G.; Maio, M. Endoglin: An accessory component of the TGF-β-binding receptor-complex with diagnostic, prognostic, and bioimmunotherapeutic potential in human malignancies. J. Cell. Physiol. 2001, 188, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Huang, C.C.; Ou, Y.C.; Huang, E.Y.; Changchien, C.C.; Tseng, C.W.; Fu, H.C.; Wu, C.H.; Li, C.J.; Ma, Y.Y. High immunohistochemical expression of TGF-β1 predicts a poor prognosis in cervical cancer patients who harbor enriched endoglin microvessel density. Int. J. Gynecol. Pathol. 2012, 31, 482–489. [Google Scholar] [CrossRef]
- Behrem, S.; Zarkovic, K.; Eskinja, N.; Jonjic, N. Endoglin is a better marker than CD31 in evaluation of angiogenesis in glioblastoma. Croat. Med. J. 2005, 46, 417–422. [Google Scholar] [PubMed]
- Rosen, L.S.; Gordon, M.S.; Robert, F.; Matei, D.E. Endoglin for targeted cancer treatment. Curr. Oncol. Rep. 2014, 16, 365. [Google Scholar] [CrossRef]
- Yao, Y.; Kubota, T.; Takeuchi, H.; Sato, K. Prognostic significance of microvessel density determined by an anti-CD105/endoglin monoclonal antibody in astrocytic tumors: Comparison with an anti-CD31 monoclonal antibody. Neuropathology 2005, 25, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Paolo, V.D.; Russo, I.; Boldrini, R.; Ravà, L.; Pezzullo, M.; Benedetti, M.C.; Galardi, A.; Colletti, M.; Rota, R.; Orlando, D.; et al. Evaluation of endoglin (CD105) expression in pediatric rhabdomyosarcoma. BMC Cancer 2018, 18, 31. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, F.; Zhang, Z.; Wang, D.; Cui, B.; Zeng, F.; Huang, L.; Zhang, Q.; Sun, Q. High expression levels of Cyr61 and VEGF are associated with poor prognosis in osteosarcoma. Pathol. Res. Pract. 2017, 213, 895–899. [Google Scholar] [CrossRef]
- Düwel, A.; Eleno, N.; Jerkic, M.; Arevalo, M.; Bolaños, J.P.; Bernabeu, C.; López-Novoa, J.M. Reduced tumor growth and angiogenesis in endoglin-haploinsufficient mice. Tumor Biol. 2006, 28, 1–8. [Google Scholar]
- Zhang, L.; Yang, B.; Li, X.; Zhang, Y.; Zhao, J.; Wang, W.; Yu, X.; Zhai, Z.; Sun, H. The targeting of endoglin on vascular endothelial cells affects the infiltration of M2 macrophages into the breast cancer microenvironment by modulating the interleukin-6 (IL-6) level. Transl. Cancer Res. 2018, 7, 4. [Google Scholar] [CrossRef]
- Pérez-Gómez, E.; Villa-Morales, M.; Santos, J.; Fernández-Piqueras, J.; Gamallo, C.; Dotor, J.; Bernabéu, C.; Quintanilla, M. A role for endoglin as a suppressor of malignancy during mouse skin carcinogenesis. Cancer Res. 2007, 67, 10268–10277. [Google Scholar] [CrossRef]
- Romero, D.; O’Neill, C.; Terzic, A.; Contois, L.; Young, K.; Conley, B.A.; Bergan, R.C.; Brooks, P.C.; Vary, C.P.H. Endoglin regulates cancer-stromal cell interactions in prostate tumors. Cancer Res. 2011, 71, 3482–3493. [Google Scholar] [CrossRef]
- Duarte, C.W.; Black, A.W.; Lucas, F.; Calvin, L.; Vary, P.H. Cancer incidence in patients with hereditary hemorrhagic telangiectasia. J. Cancer Res. Clin. Oncol. 2017, 143, 209–214. [Google Scholar] [CrossRef]
- Hosman, A.E.; Devlin, H.L.; Silva, B.M.; Shovlin, C.L. Specific cancer rates may differ in patients with hereditary haemorrhagic telangiectasia compared to controls. Orphanet J. Rare Dis. 2013, 8, 195. [Google Scholar] [CrossRef]
- Duarte, C.W.; Murray, K.; Lucas, F.L.; Fairfield, K.; Miller, H.; Brooks, P.; Vary, C.P.H. Improved survival outcomes in cancer patients with hereditary hemorrhagic telangiectasia. Cancer Epidemiol. Biomarkers Prev. 2014, 23, 117–125. [Google Scholar] [CrossRef]
- Guilhem, A.; Malcus, C.; Clarivet, B.; Plauchu, H.; Dupuis-Girod, S. Immunological abnormalities associated with hereditary haemorrhagic telangiectasia. J. Intern. Med. 2013, 274, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Cirulli, A.; Loria, M.; Dambra, P.; Di Serio, F.; Ventura, M.; Amati, L.; Jirillo, E.; Sabba, C. Patients with Hereditary Hemorrhagic Telangectasia (HHT) Exhibit a Deficit of Polymorphonuclear Cell and Monocyte Oxidative Burst and Phagocytosis: A Possible Correlation with Altered Adaptive Immune Responsiveness in HHT. Curr. Pharm. Des. 2006, 12, 1209–1215. [Google Scholar] [CrossRef] [PubMed]
- Dingenouts, C.K.E.; Goumans, M.J.; Bakker, W. Mononuclear cells and vascular repair in HHT. Front. Genet. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Quackenbush, E.J.; Letarte, M. Identification of several cell surface proteins of non-T, non-B acute lymphoblastic leukemia by using monoclonal antibodies. J. Immunol. 1985, 134, 1276–1285. [Google Scholar] [PubMed]
- Lastres, P.; Letamendía, A.; Zhang, H.; Rius, C.; Almendro, N.; Raab, U.; López, L.A.; Langa, C.; Fabra, A.; Letarte, M.; et al. Endoglin modulates cellular responses to TGF-β1. J. Cell Biol. 1996, 133, 1109–1121. [Google Scholar] [CrossRef]
- Sundström, C.; Nilsson, K. Establishment and characterization of a human histiocytic lymphoma cell line (U-937). Int. J. Cancer 1976. [Google Scholar] [CrossRef]
- Scharpfenecker, M.; Floot, B.; Russell, N.S.; Stewart, F.A. The TGF-β co-receptor endoglin regulates macrophage infiltration and cytokine production in the irradiated mouse kidney. Radiother. Oncol. 2012, 105, 313–320. [Google Scholar] [CrossRef]
- Han, Z.; Shaligram, S.; Faughnan, M.E.; Clark, D.; Sun, Z.; Su, H. Reduction of endoglin receptor impairs mononuclear cell-migration. Explor. Med. 2020, 1, 136–148. [Google Scholar] [CrossRef]
- Jerkic, M.; Rivas-Elena, J.V.; Santibanez, J.F.; Prieto, M.; Rodríguez-Barbero, A.; Perez-Barriocanal, F.; Pericacho, M.; Arévalo, M.; Vary, C.P.H.; Letarte, M.; et al. Endoglin regulates cyclooxygenase-2 expression and activity. Circ. Res. 2006, 99, 248–256. [Google Scholar] [CrossRef]
- Hashemi Goradel, N.; Najafi, M.; Salehi, E.; Farhood, B.; Mortezaee, K. Cyclooxygenase-2 in cancer: A review. J. Cell. Physiol. 2019, 234, 5683–5699. [Google Scholar] [CrossRef] [PubMed]
- Rossi, E.; Sanz-Rodriguez, F.; Eleno, N.; Düwell, A.; Blanco, F.J.; Langa, C.; Botella, L.M.; Cabañas, C.; Lopez-Novoa, J.M.; Bernabeu, C. Endothelial endoglin is involved in inflammation: Role in leukocyte adhesion and transmigration. Blood 2013, 121, 403–415. [Google Scholar] [CrossRef] [PubMed]
- Numakura, S.; Uozaki, H.; Kikuchi, Y.; Watabe, S.; Togashi, A.; Watanabe, M. Mesenchymal Stem Cell Marker Expression in Gastric Cancer Stroma. Anticancer Res. 2019, 39, 387–393. [Google Scholar] [CrossRef]
- Wang, X.; Liu, Y.; Zhou, K.; Zhang, G.; Wang, F.; Ren, J. Isolation and characterization of CD105+/CD90+ subpopulation in breast cancer MDA-MB-231 cell line. Int. J. Clin. Exp. Pathol. 2015, 8, 5105–5112. [Google Scholar] [PubMed]
- Nolan-Stevaux, O.; Zhong, W.; Culp, S.; Shaffer, K.; Hoover, J.; Wickramasinghe, D.; Ruefli-Brasse, A. Endoglin Requirement for BMP9 Signaling in Endothelial Cells Reveals New Mechanism of Action for Selective Anti-Endoglin Antibodies. PLoS ONE 2012, 7, e50920. [Google Scholar] [CrossRef]
- Apolo, A.B.; Karzai, F.H.; Trepel, J.B.; Alarcon, S.; Lee, S.; Lee, M.-J.; Tomita, Y.; Cao, L.; Yu, Y.; Merino, M.J.; et al. A Phase II Clinical Trial of TRC105 (Anti-Endoglin Antibody) in Adults with Advanced/Metastatic Urothelial Carcinoma. Clin. Genitourin. Cancer 2017, 15, 77–85. [Google Scholar] [CrossRef]
- Wu, H.W.; Sheard, M.A.; Malvar, J.; Fernandez, G.E.; DeClerck, Y.A.; Blavier, L.; Shimada, H.; Theuer, C.P.; Sposto, R.; Seeger, R.C. Anti-CD105 antibody eliminates tumor microenvironment cells and enhances Anti-GD2 antibody immunotherapy of neuroblastoma with activated natural killer cells. Clin. Cancer Res. 2019, 25, 4761–4774. [Google Scholar] [CrossRef]
- Paauwe, M.; Heijkants, R.C.; Oudt, C.H.; Van Pelt, G.W.; Cui, C.; Theuer, C.P.; Hardwick, J.C.H.; Sier, C.F.M.; Hawinkels, L.J.A.C. Endoglin targeting inhibits tumor angiogenesis and metastatic spread in breast cancer. Oncogene 2016, 35, 4069–4079. [Google Scholar] [CrossRef]
- Karzai, F.H.; Apolo, A.B.; Cao, L.; Madan, R.A.; Adelberg, D.E.; Parnes, H.; McLeod, D.G.; Harold, N.; Peer, C.; Yu, Y.; et al. A phase i study of TRC105 anti-endoglin (CD105) antibody in metastatic castration-resistant prostate cancer. BJU Int. 2015, 116, 546–555. [Google Scholar] [CrossRef] [PubMed]
- Duffy, A.G.; Ma, C.; Ulahannan, S.V.; Rahma, O.E.; Makarova-Rusher, O.; Cao, L.; Yu, Y.; Kleiner, D.E.; Trepel, J.; Lee, M.J.; et al. Phase I and preliminary phase II study of TRC105 in combination with sorafenib in hepatocellular carcinoma. Clin. Cancer Res. 2017, 23, 4633–4641. [Google Scholar] [CrossRef]
- Dorff, T.B.; Longmate, J.A.; Pal, S.K.; Stadler, W.M.; Fishman, M.N.; Vaishampayan, U.N.; Rao, A.; Pinksi, J.K.; Hu, J.S.; Quinn, D.I.; et al. Bevacizumab alone or in combination with TRC105 for patients with refractory metastatic renal cell cancer. Cancer 2017, 123, 4566–4573. [Google Scholar] [CrossRef]
- Rosen, L.S.; Hurwitz, H.I.; Wong, M.K.; Goldman, J.; Mendelson, D.S.; Figg, W.D.; Spencer, S.; Adams, B.J.; Alvarez, D.; Seon, B.K.; et al. A phase I first-in-human study of TRC105 (anti-endoglin antibody) in patients with advanced cancer. Clin. Cancer Res. 2012, 18, 4820–4829. [Google Scholar] [CrossRef] [PubMed]
- Tansi, F.L.; Rüger, R.; Kollmeier, A.M.; Rabenhold, M.; Steiniger, F.; Kontermann, R.E.; Teichgräber, U.K.; Fahr, A.; Hilger, I. Targeting the Tumor Microenvironment with Fluorescence-Activatable Bispecific Endoglin/Fibroblast Activation Protein Targeting Liposomes. Pharmaceutics 2020, 12, 370. [Google Scholar] [CrossRef] [PubMed]
- Rabenhold, M.; Steiniger, F.; Fahr, A.; Kontermann, R.E.; Rüger, R. Bispecific single-chain diabody-immunoliposomes targeting endoglin (CD105) and fibroblast activation protein (FAP) simultaneously. J. Control. Release 2015, 201, 56–67. [Google Scholar] [CrossRef]
- Dolinsek, T.; Markelc, B.; Sersa, G.; Coer, A.; Stimac, M.; Lavrencak, J.; Brozic, A.; Kranjc, S.; Cemazar, M. Multiple Delivery of siRNA against Endoglin into Murine Mammary Adenocarcinoma Prevents Angiogenesis and Delays Tumor Growth. PLoS ONE 2013, 8, e58723. [Google Scholar] [CrossRef] [PubMed]
- Stimac, M.; Dolinsek, T.; Lampreht, U.; Cemazar, M.; Sersa, G. Gene electrotransfer of plasmid with tissue specific promoter encoding shRNA against endoglin exerts antitumor efficacy against murine TS/A tumors by vascular targeted effects. PLoS ONE 2015, 10, e0124913. [Google Scholar] [CrossRef]
- Tesic, N.; Kamensek, U.; Sersa, G.; Kranjc, S.; Stimac, M.; Lampreht, U.; Preat, V.; Vandermeulen, G.; Butinar, M.; Turk, B.; et al. Endoglin (CD105) Silencing Mediated by shRNA Under the Control of Endothelin-1 Promoter for Targeted Gene Therapy of Melanoma. Mol. Ther. Nucleic Acids 2015, 4, e239. [Google Scholar] [CrossRef]
- Dolinsek, T.; Markelc, B.; Bosnjak, M.; Blagus, T.; Prosen, L.; Kranjc, S.; Stimac, M.; Lampreht, U.; Sersa, G.; Cemazar, M. Endoglin silencing has significant antitumor effect on murine mammary adenocarcinoma mediated by vascular targeted effect. Curr. Gene Ther. 2015, 15, 228–244. [Google Scholar] [CrossRef]
- Stimac, M.; Kamensek, U.; Cemazar, M.; Kranjc, S.; Coer, A.; Sersa, G. Tumor radiosensitization by gene therapy against endoglin. Cancer Gene Ther. 2016, 23, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Jarosz, M.; Jazowiecka-Rakus, J.; Cichoń, T.; Głowala-Kosińska, M.; Smolarczyk, R.; Smagur, A.; Malina, S.; Sochanik, A.; Szala, S. Therapeutic antitumor potential of endoglin-based DNA vaccine combined with immunomodulatory agents. Gene Ther. 2013, 20, 262–273. [Google Scholar] [CrossRef]
- Zhang, F.-P.; Huang, Y.-P.; Luo, W.-X.; Deng, W.-Y.; Liu, C.-Q.; Xu, L.-B.; Liu, C. Construction of a risk score prognosis model based on hepatocellular carcinoma microenvironment. World J. Gastroenterol. 2020, 26, 134–153. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ollauri-Ibáñez, C.; Ayuso-Íñigo, B.; Pericacho, M. Hot and Cold Tumors: Is Endoglin (CD105) a Potential Target for Vessel Normalization? Cancers 2021, 13, 1552. https://doi.org/10.3390/cancers13071552
Ollauri-Ibáñez C, Ayuso-Íñigo B, Pericacho M. Hot and Cold Tumors: Is Endoglin (CD105) a Potential Target for Vessel Normalization? Cancers. 2021; 13(7):1552. https://doi.org/10.3390/cancers13071552
Chicago/Turabian StyleOllauri-Ibáñez, Claudia, Blanca Ayuso-Íñigo, and Miguel Pericacho. 2021. "Hot and Cold Tumors: Is Endoglin (CD105) a Potential Target for Vessel Normalization?" Cancers 13, no. 7: 1552. https://doi.org/10.3390/cancers13071552
APA StyleOllauri-Ibáñez, C., Ayuso-Íñigo, B., & Pericacho, M. (2021). Hot and Cold Tumors: Is Endoglin (CD105) a Potential Target for Vessel Normalization? Cancers, 13(7), 1552. https://doi.org/10.3390/cancers13071552