
Mechanisms of Resistance to PI3K Inhibitors in Cancer: Adaptive Responses, Drug Tolerance and Cellular Plasticity


Abstract
Simple Summary
Abstract
1. Introduction
2. PI3K Pathway
3. Pharmacological Targeting of the PI3K Pathway
3.1. mTOR Inhibitors
3.1.1. Allosteric mTOR Inhibitors
3.1.2. Active Site or Catalytic mTOR Inhibitors
3.2. Dual mTOR and PI3K Inhibitors
3.3. Pan-Class I PI3K Inhibitors
3.4. Isoform-Selective PI3K Inhibitors
3.5. AKT Inhibitor
4. Reactivation of PI3K Signalling
4.1. Insulin Signalling and PI3K Reactivation
4.2. Receptor Tyrosine Kinase Reactivation
4.3. Endocrine-Mediated Resistance
4.4. Cellular Plasticity and PI3K Inhibitor Resistance
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cantley, L.C.; Whitman, M.; Chahwala, S.; Fleischman, L.; Kaplan, D.R.; Schaffhausen, B.S.; Roberts, T.M. Oncogenes and phosphatidylinositol turnover. Ann. N. Y. Acad. Sci. 1986, 488, 481–490. [Google Scholar] [CrossRef]
- Thorpe, L.M.; Yuzugullu, H.; Zhao, J.J. PI3K in cancer: Divergent roles of isoforms, modes of activation and therapeutic targeting. Nat. Rev. Cancer 2015, 15, 7–24. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Luo, J.; Cantley, L.C. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 2006, 7, 606–619. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.K.; Engelman, J.A.; Cantley, L.C. Targeting the PI3K signaling pathway in cancer. Curr. Opin. Genet. Dev. 2009, 20, 87–90. [Google Scholar] [CrossRef]
- Vanhaesebroeck, B.; Leevers, S.J.; Panayotou, G.; Waterfield, M.D. Phosphoinositide 3-kinases: A conserved family of signal transducers. Trends Biochem. Sci. 1997, 22, 267–272. [Google Scholar] [CrossRef]
- Yuan, T.L.; Cantley, L.C. PI3K pathway alterations in cancer: Variations on a theme. Oncogene 2008, 27, 5497–5510. [Google Scholar] [CrossRef] [PubMed]
- Vanhaesebroeck, B.; Guillermet-Guibert, J.; Graupera, M.; Bilanges, B. The emerging mechanisms of isoform-specific PI3K signalling. Nat. Rev. Mol. Cell Biol. 2010, 11, 329–341. [Google Scholar] [CrossRef]
- Auger, K.R.; Serunian, L.A.; Soltoff, S.P.; Libby, P.; Cantley, L.C. PDGF-dependent tyrosine phosphorylation stimulates production of novel polyphosphoinositides in intact cells. Cell 1989, 57, 167–175. [Google Scholar] [CrossRef]
- Ruderman, N.B.; Kapeller, R.; White, M.F.; Cantley, L.C. Activation of phosphatidylinositol 3-kinase by insulin. Proc. Natl. Acad. Sci. USA 1990, 87, 1411–1415. [Google Scholar] [CrossRef] [PubMed]
- Songyang, Z.; Shoelson, S.E.; Chaudhuri, M.; Gish, G.; Pawson, T.; Haser, W.G.; King, F.; Roberts, T.; Ratnofsky, S.; Lechleider, R.J.; et al. SH2 domains recognize specific phosphopeptide sequences. Cell 1993, 72, 767–778. [Google Scholar] [CrossRef] [PubMed]
- Dannemann, N.; Hart, J.R.; Ueno, L.; Vogt, P.K. Phosphatidylinositol 4,5-bisphosphate-specific AKT1 is oncogenic. Int. J. Cancer 2010, 127, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Miao, B.; Skidan, I.; Yang, J.; Lugovskoy, A.; Reibarkh, M.; Long, K.; Brazell, T.; Durugkar, K.A.; Maki, J.; Ramana, C.V.; et al. Small molecule inhibition of phosphatidylinositol-3,4,5-triphosphate (PIP3) binding to pleckstrin homology domains. Proc. Natl. Acad. Sci. USA 2010, 107, 20126–20131. [Google Scholar] [CrossRef] [PubMed]
- James, S.R.; Downes, C.P.; Gigg, R.; Grove, S.J.; Holmes, A.B.; Alessi, D.R. Specific binding of the Akt-1 protein kinase to phosphatidylinositol 3,4,5-trisphosphate without subsequent activation. Biochem. J. 1996, 315, 709–713. [Google Scholar] [CrossRef] [PubMed]
- Alessi, D.R.; Andjelkovic, M.; Caudwell, B.; Cron, P.; Morrice, N.; Cohen, P.; Hemmings, B.A. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996, 15, 6541–6551. [Google Scholar] [CrossRef] [PubMed]
- Alessi, D.R.; James, S.R.; Downes, C.P.; Holmes, A.B.; Gaffney, P.R.; Reese, C.B.; Cohen, P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr. Biol. Cb 1997, 7, 261–269. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Cron, P.; Good, V.M.; Thompson, V.; Hemmings, B.A.; Barford, D. Crystal structure of an activated Akt/protein kinase B ternary complex with GSK3-peptide and AMP-PNP. Nat. Struct. Biol. 2002, 9, 940–944. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yen, C.; Liaw, D.; Podsypanina, K.; Bose, S.; Wang, S.I.; Puc, J.; Miliaresis, C.; Rodgers, L.; McCombie, R.; et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997, 275, 1943–1947. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Shen, Y.; Wang, M.; Yang, J.; Lv, M.; Li, P.; Chen, Z.; Yang, J. Loss of PTEN expression in breast cancer: Association with clinicopathological characteristics and prognosis. Oncotarget 2017, 8, 32043–32054. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Sancak, Y.; Thoreen, C.C.; Peterson, T.R.; Lindquist, R.A.; Kang, S.A.; Spooner, E.; Carr, S.A.; Sabatini, D.M. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol. Cell 2007, 25, 903–915. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Harris, T.E.; Lawrence, J.C., Jr. Regulation of proline-rich Akt substrate of 40 kDa (PRAS40) function by mammalian target of rapamycin complex 1 (mTORC1)-mediated phosphorylation. J. Biol. Chem. 2008, 283, 15619–15627. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Sarbassov, D.D.; Ali, S.M.; King, J.E.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002, 110, 163–175. [Google Scholar] [CrossRef]
- Kim, D.H.; Sarbassov, D.D.; Ali, S.M.; Latek, R.R.; Guntur, K.V.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. GbetaL, a positive regulator of the rapamycin-sensitive pathway required for the nutrient-sensitive interaction between raptor and mTOR. Mol. Cell 2003, 11, 895–904. [Google Scholar] [CrossRef]
- Hara, K.; Maruki, Y.; Long, X.; Yoshino, K.; Oshiro, N.; Hidayat, S.; Tokunaga, C.; Avruch, J.; Yonezawa, K. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 2002, 110, 177–189. [Google Scholar] [CrossRef]
- Peterson, T.R.; Laplante, M.; Thoreen, C.C.; Sancak, Y.; Kang, S.A.; Kuehl, W.M.; Gray, N.S.; Sabatini, D.M. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell 2009, 137, 873–886. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Ali, S.M.; Kim, D.H.; Guertin, D.A.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. Cb 2004, 14, 1296–1302. [Google Scholar] [CrossRef] [PubMed]
- Frias, M.A.; Thoreen, C.C.; Jaffe, J.D.; Schroder, W.; Sculley, T.; Carr, S.A.; Sabatini, D.M. mSin1 is necessary for Akt/PKB phosphorylation, and its isoforms define three distinct mTORC2s. Curr. Biol. Cb 2006, 16, 1865–1870. [Google Scholar] [CrossRef]
- Ben-Sahra, I.; Manning, B.D. mTORC1 signaling and the metabolic control of cell growth. Curr. Opin. Cell Biol. 2017, 45, 72–82. [Google Scholar] [CrossRef]
- Hay, N.; Sonenberg, N. Upstream and downstream of mTOR. Genes Dev. 2004, 18, 1926–1945. [Google Scholar] [CrossRef] [PubMed]
- Meric-Bernstam, F.; Gonzalez-Angulo, A.M. Targeting the mTOR signaling network for cancer therapy. J. Clin. Oncol. 2009, 27, 2278–2287. [Google Scholar] [CrossRef] [PubMed]
- Braunstein, S.; Karpisheva, K.; Pola, C.; Goldberg, J.; Hochman, T.; Yee, H.; Cangiarella, J.; Arju, R.; Formenti, S.C.; Schneider, R.J. A hypoxia-controlled cap-dependent to cap-independent translation switch in breast cancer. Mol. Cell 2007, 28, 501–512. [Google Scholar] [CrossRef]
- Armengol, G.; Rojo, F.; Castellvi, J.; Iglesias, C.; Cuatrecasas, M.; Pons, B.; Baselga, J.; Ramon y Cajal, S. 4E-binding protein 1: A key molecular “funnel factor” in human cancer with clinical implications. Cancer Res. 2007, 67, 7551–7555. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Guo, J.; Li, H.; Wang, J. Meta-analysis of the prognostic value of p-4EBP1 in human malignancies. Oncotarget 2018, 9, 2761–2769. [Google Scholar] [CrossRef] [PubMed]
- Jastrzebski, K.; Hannan, K.M.; Tchoubrieva, E.B.; Hannan, R.D.; Pearson, R.B. Coordinate regulation of ribosome biogenesis and function by the ribosomal protein S6 kinase, a key mediator of mTOR function. Growth Factors 2007, 25, 209–226. [Google Scholar] [CrossRef] [PubMed]
- Holz, M.K. The role of S6K1 in ER-positive breast cancer. Cell Cycle 2012, 11, 3159–3165. [Google Scholar] [CrossRef][Green Version]
- Amaral, C.L.; Freitas, L.B.; Tamura, R.E.; Tavares, M.R.; Pavan, I.C.; Bajgelman, M.C.; Simabuco, F.M. S6Ks isoforms contribute to viability, migration, docetaxel resistance and tumor formation of prostate cancer cells. BMC Cancer 2016, 16, 602. [Google Scholar] [CrossRef]
- Sridharan, S.; Basu, A. Distinct Roles of mTOR Targets S6K1 and S6K2 in Breast Cancer. Int. J. Mol. Sci. 2020, 21, 1199. [Google Scholar] [CrossRef] [PubMed]
- Chauvin, C.; Koka, V.; Nouschi, A.; Mieulet, V.; Hoareau-Aveilla, C.; Dreazen, A.; Cagnard, N.; Carpentier, W.; Kiss, T.; Meyuhas, O.; et al. Ribosomal protein S6 kinase activity controls the ribosome biogenesis transcriptional program. Oncogene 2014, 33, 474–483. [Google Scholar] [CrossRef]
- Dufner, A.; Thomas, G. Ribosomal S6 kinase signaling and the control of translation. Exp. Cell Res. 1999, 253, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.; Wolgamott, L.; Yu, Y.; Blenis, J.; Yoon, S.O. Glycogen synthase kinase (GSK)-3 promotes p70 ribosomal protein S6 kinase (p70S6K) activity and cell proliferation. Proc. Natl. Acad. Sci. USA 2011, 108, E1204–E1213. [Google Scholar] [CrossRef]
- Zhou, H.; Huang, S. The complexes of mammalian target of rapamycin. Curr. Protein Pept. Sci. 2010, 11, 409–424. [Google Scholar] [CrossRef] [PubMed]
- Carracedo, A.; Ma, L.; Teruya-Feldstein, J.; Rojo, F.; Salmena, L.; Alimonti, A.; Egia, A.; Sasaki, A.T.; Thomas, G.; Kozma, S.C.; et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J. Clin. Investig. 2008, 118, 3065–3074. [Google Scholar] [CrossRef]
- McNamara, C.R.; Degterev, A. Small-molecule inhibitors of the PI3K signaling network. Future Med. Chem. 2011, 3, 549–565. [Google Scholar] [CrossRef] [PubMed]
- Arcaro, A.; Wymann, M.P. Wortmannin is a potent phosphatidylinositol 3-kinase inhibitor: The role of phosphatidylinositol 3,4,5-trisphosphate in neutrophil responses. Biochem. J. 1993, 296, 297–301. [Google Scholar] [CrossRef]
- Norman, B.H.; Shih, C.; Toth, J.E.; Ray, J.E.; Dodge, J.A.; Johnson, D.W.; Rutherford, P.G.; Schultz, R.M.; Worzalla, J.F.; Vlahos, C.J. Studies on the Mechanism of Phosphatidylinositol 3-Kinase Inhibition by Wortmannin and Related Analogs. J. Med. Chem. 1996, 39, 1106–1111. [Google Scholar] [CrossRef] [PubMed]
- Cleary, J.M.; Shapiro, G.I. Development of phosphoinositide-3 kinase pathway inhibitors for advanced cancer. Curr. Oncol. Rep. 2010, 12, 87–94. [Google Scholar] [CrossRef]
- Ihle, N.T.; Williams, R.; Chow, S.; Chew, W.; Berggren, M.I.; Paine-Murrieta, G.; Minion, D.J.; Halter, R.J.; Wipf, P.; Abraham, R.; et al. Molecular pharmacology and antitumor activity of PX-866, a novel inhibitor of phosphoinositide-3-kinase signaling. Mol. Cancer Ther. 2004, 3, 763–772. [Google Scholar] [PubMed]
- Gharbi, S.I.; Zvelebil, M.J.; Shuttleworth, S.J.; Hancox, T.; Saghir, N.; Timms, J.F.; Waterfield, M.D. Exploring the specificity of the PI3K family inhibitor LY294002. Biochem. J. 2007, 404, 15–21. [Google Scholar] [CrossRef]
- Vlahos, C.J.; Matter, W.F.; Hui, K.Y.; Brown, R.F. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). J. Biol. Chem. 1994, 269, 5241–5248. [Google Scholar] [CrossRef]
- Garlich, J.R.; De, P.; Dey, N.; Su, J.D.; Peng, X.; Miller, A.; Murali, R.; Lu, Y.; Mills, G.B.; Kundra, V.; et al. A vascular targeted pan phosphoinositide 3-kinase inhibitor prodrug, SF1126, with antitumor and antiangiogenic activity. Cancer Res. 2008, 68, 206–215. [Google Scholar] [CrossRef]
- Cafferkey, R.; Young, P.R.; McLaughlin, M.M.; Bergsma, D.J.; Koltin, Y.; Sathe, G.M.; Faucette, L.; Eng, W.K.; Johnson, R.K.; Livi, G.P. Dominant missense mutations in a novel yeast protein related to mammalian phosphatidylinositol 3-kinase and VPS34 abrogate rapamycin cytotoxicity. Mol. Cell. Biol. 1993, 13, 6012–6023. [Google Scholar] [CrossRef]
- Kunz, J.; Henriquez, R.; Schneider, U.; Deuter-Reinhard, M.; Movva, N.R.; Hall, M.N. Target of rapamycin in yeast, TOR2, is an essential phosphatidylinositol kinase homolog required for G1 progression. Cell 1993, 73, 585–596. [Google Scholar] [CrossRef]
- Zhang, M.; Jang, H.; Nussinov, R. PI3K inhibitors: Review and new strategies. Chem. Sci. 2020, 11, 5855–5865. [Google Scholar] [CrossRef] [PubMed]
- Janku, F.; Yap, T.A.; Meric-Bernstam, F. Targeting the PI3K pathway in cancer: Are we making headway? Nat. Rev. Clin. Oncol. 2018, 15, 273–291. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in cancer: Mechanisms and advances in clinical trials. Mol. Cancer 2019, 18, 26. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. mTOR signaling at a glance. J. Cell Sci. 2009, 122, 3589–3594. [Google Scholar] [CrossRef]
- Xie, J.; Wang, X.; Proud, C.G. mTOR inhibitors in cancer therapy. F1000Res 2016, 5. [Google Scholar] [CrossRef]
- Chan, S. Targeting the mammalian target of rapamycin (mTOR): A new approach to treating cancer. Br. J. Cancer 2004, 91, 1420–1424. [Google Scholar] [CrossRef] [PubMed]
- Agarwala, S.S.; Case, S. Everolimus (RAD001) in the treatment of advanced renal cell carcinoma: A review. Oncologist 2010, 15, 236–245. [Google Scholar] [CrossRef]
- Gnant, M.; Greil, R.; Hubalek, M.; Steger, G. Everolimus in postmenopausal, hormone receptor-positive advanced breast cancer: Summary and results of an austrian expert panel discussion. Breast Care 2013, 8, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.; Ito, T.; Jensen, R.T. Everolimus in the treatment of neuroendocrine tumors: Efficacy, side-effects, resistance, and factors affecting its place in the treatment sequence. Expert Opin. Pharm. 2018, 19, 909–928. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J.; Campone, M.; Piccart, M.; Burris, H.A.; Rugo, H.S.; Sahmoud, T.; Noguchi, S.; Gnant, M.; Pritchard, K.I.; Lebrun, F.; et al. Everolimus in Postmenopausal Hormone-Receptor-Positive Advanced Breast Cancer. N. Engl. J. Med. 2011. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Toral-Barza, L.; Shi, C.; Zhang, W.G.; Lucas, J.; Shor, B.; Kim, J.; Verheijen, J.; Curran, K.; Malwitz, D.J.; et al. Biochemical, cellular, and in vivo activity of novel ATP-competitive and selective inhibitors of the mammalian target of rapamycin. Cancer Res. 2009, 69, 6232–6240. [Google Scholar] [CrossRef] [PubMed]
- Hua, H.; Kong, Q.; Zhang, H.; Wang, J.; Luo, T.; Jiang, Y. Targeting mTOR for cancer therapy. J. Hematol. Oncol. 2019, 12, 71. [Google Scholar] [CrossRef] [PubMed]
- Brana, I.; LoRusso, P.; Baselga, J.; Heath, E.I.; Patnaik, A.; Gendreau, S.; Laird, A.; Papadopoulos, K. A phase I dose-escalation study of the safety, pharmacokinetics (PK), and pharmacodynamics of XL765 (SAR245409), a PI3K/TORC1/TORC2 inhibitor administered orally to patients (pts) with advanced malignancies. J. Clin. Oncol. 2010, 28, 3030. [Google Scholar] [CrossRef]
- Funakoshi, T.; Latif, A.; Galsky, M.D. Risk of hematologic toxicities in patients with solid tumors treated with everolimus: A systematic review and meta-analysis. Crit. Rev. Oncol. Hematol. 2013, 88, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Tabernero, J.; Rojo, F.; Calvo, E.; Burris, H.; Judson, I.; Hazell, K.; Martinelli, E.; Ramon y Cajal, S.; Jones, S.; Vidal, L.; et al. Dose- and schedule-dependent inhibition of the mammalian target of rapamycin pathway with everolimus: A phase I tumor pharmacodynamic study in patients with advanced solid tumors. J. Clin. Oncol. 2008, 26, 1603–1610. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Fort, M.K.; Case, E.C.; Rosen, A.C.; Cerci, F.B.; Wu, S.; Lacouture, M.E. Rash to the mTOR inhibitor everolimus: Systematic review and meta-analysis. Am. J. Clin. Oncol. 2014, 37, 266–271. [Google Scholar] [CrossRef]
- Walker, E.H.; Perisic, O.; Ried, C.; Stephens, L.; Williams, R.L. Structural insights into phosphoinositide 3-kinase catalysis and signalling. Nature 1999, 402, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Rudge, D.G.; Koos, J.D.; Vaidialingam, B.; Yang, H.J.; Pavletich, N.P. mTOR kinase structure, mechanism and regulation. Nature 2013, 497, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Hay, N. The Akt-mTOR tango and its relevance to cancer. Cancer Cell 2005, 8, 179–183. [Google Scholar] [CrossRef]
- O’Reilly, K.E.; Rojo, F.; She, Q.-B.; Solit, D.; Mills, G.B.; Smith, D.; Lane, H.; Hofmann, F.; Hicklin, D.J.; Ludwig, D.L.; et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006, 66, 1500–1508. [Google Scholar] [CrossRef]
- Maira, S.M.; Stauffer, F.; Brueggen, J.; Furet, P.; Schnell, C.; Fritsch, C.; Brachmann, S.; Chene, P.; De Pover, A.; Schoemaker, K.; et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol. Cancer Ther. 2008, 7, 1851–1863. [Google Scholar] [CrossRef]
- Serra, V.; Markman, B.; Scaltriti, M.; Eichhorn, P.J.A.; Valero, V.; Guzman, M.; Botero, M.L.; Llonch, E.; Atzori, F.; Di Cosimo, S.; et al. NVP-BEZ235, a dual PI3K/mTOR inhibitor, prevents PI3K signaling and inhibits the growth of cancer cells with activating PI3K mutations. Cancer Res. 2008, 68, 8022–8030. [Google Scholar] [CrossRef] [PubMed]
- Eichhorn, P.J.; Gili, M.; Scaltriti, M.; Serra, V.; Guzman, M.; Nijkamp, W.; Beijersbergen, R.L.; Valero, V.; Seoane, J.; Bernards, R.; et al. Phosphatidylinositol 3-kinase hyperactivation results in lapatinib resistance that is reversed by the mTOR/phosphatidylinositol 3-kinase inhibitor NVP-BEZ235. Cancer Res. 2008, 68, 9221–9230. [Google Scholar] [CrossRef] [PubMed]
- Rodon, J.; Pérez-Fidalgo, A.; Krop, I.E.; Burris, H.; Guerrero-Zotano, A.; Britten, C.D.; Becerra, C.; Schellens, J.; Richards, D.A.; Schuler, M.; et al. Phase 1/1b dose escalation and expansion study of BEZ235, a dual PI3K/mTOR inhibitor, in patients with advanced solid tumors including patients with advanced breast cancer. Cancer Chemother. Pharm. 2018, 82, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Pongas, G.; Fojo, T. BEZ235: When Promising Science Meets Clinical Reality. Oncologist 2016, 21, 1033–1034. [Google Scholar] [CrossRef]
- Wen, P.Y.; Cloughesy, T.F.; Olivero, A.G.; Morrissey, K.M.; Wilson, T.R.; Lu, X.; Mueller, L.U.; Coimbra, A.F.; Ellingson, B.M.; Gerstner, E.; et al. First-in-Human Phase I Study to Evaluate the Brain-Penetrant PI3K/mTOR Inhibitor GDC-0084 in Patients with Progressive or Recurrent High-Grade Glioma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2020, 26, 1820–1828. [Google Scholar] [CrossRef]
- Ippen, F.M.; Alvarez-Breckenridge, C.A.; Kuter, B.M.; Fink, A.L.; Bihun, I.V.; Lastrapes, M.; Penson, T.; Schmidt, S.P.; Wojtkiewicz, G.R.; Ning, J.; et al. The Dual PI3K/mTOR Pathway Inhibitor GDC-0084 Achieves Antitumor Activity in PIK3CA-Mutant Breast Cancer Brain Metastases. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 3374–3383. [Google Scholar] [CrossRef]
- Folkes, A.J.; Ahmadi, K.; Alderton, W.K.; Alix, S.; Baker, S.J.; Box, G.; Chuckowree, I.S.; Clarke, P.A.; Depledge, P.; Eccles, S.A.; et al. The Identification of 2-(1H-Indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-thieno[3,2-d]pyrimidine (GDC-0941) as a Potent, Selective, Orally Bioavailable Inhibitor of Class I PI3 Kinase for the Treatment of Cancer. J. Med. Chem. 2008, 51, 5522–5532. [Google Scholar] [CrossRef] [PubMed]
- Maira, S.M.; Pecchi, S.; Huang, A.; Burger, M.; Knapp, M.; Sterker, D.; Schnell, C.; Guthy, D.; Nagel, T.; Wiesmann, M.; et al. Identification and characterization of NVP-BKM120, an orally available pan-class I PI3-kinase inhibitor. Mol. Cancer Ther. 2012, 11, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Bendell, J.C.; Rodon, J.; Burris, H.A.; de Jonge, M.; Verweij, J.; Birle, D.; Demanse, D.; De Buck, S.S.; Ru, Q.C.; Peters, M.; et al. Phase I, Dose-Escalation Study of BKM120, an Oral Pan-Class I PI3K Inhibitor, in Patients With Advanced Solid Tumors. J. Clin. Oncol. 2011, 30, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, Y.H.; García-García, C.; Serra, V.; He, L.; Torres-Lockhart, K.; Prat, A.; Anton, P.; Cozar, P.; Guzmán, M.; Grueso, J.; et al. PI3K inhibition impairs BRCA1/2 expression and sensitizes BRCA-proficient triple-negative breast cancer to PARP inhibition. Cancer Discov. 2012, 2, 1036–1047. [Google Scholar] [CrossRef]
- Rodon, J.; Braña, I.; Siu, L.L.; De Jonge, M.J.; Homji, N.; Mills, D.; Di Tomaso, E.; Sarr, C.; Trandafir, L.; Massacesi, C.; et al. Phase I dose-escalation and -expansion study of buparlisib (BKM120), an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. Investig. New Drugs 2014, 32, 670–681. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ide, J.L.; Norton, I.; Marchionni, M.A.; Ebling, M.C.; Wang, L.Y.; Davis, E.; Sauvageot, C.M.; Kesari, S.; Kellersberger, K.A.; et al. Molecular imaging of drug transit through the blood-brain barrier with MALDI mass spectrometry imaging. Sci. Rep. 2013, 3, 2859. [Google Scholar] [CrossRef]
- Salphati, L.; Shahidi-Latham, S.; Quiason, C.; Barck, K.; Nishimura, M.; Alicke, B.; Pang, J.; Carano, R.A.; Olivero, A.G.; Phillips, H.S. Distribution of the phosphatidylinositol 3-kinase inhibitors Pictilisib (GDC-0941) and GNE-317 in U87 and GS2 intracranial glioblastoma models-assessment by matrix-assisted laser desorption ionization imaging. Drug Metab. Dispos. 2014, 42, 1110–1116. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A. Targeting PI3K signalling in cancer: Opportunities, challenges and limitations. Nat. Rev. Cancer 2009, 9, 550–562. [Google Scholar] [CrossRef] [PubMed]
- Sarker, D.; Ang, J.E.; Baird, R.; Kristeleit, R.; Shah, K.; Moreno, V.; Clarke, P.A.; Raynaud, F.I.; Levy, G.; Ware, J.A.; et al. First-in-human phase I study of pictilisib (GDC-0941), a potent pan-class I phosphatidylinositol-3-kinase (PI3K) inhibitor, in patients with advanced solid tumors. Clin. Cancer Res. 2015, 21, 77–86. [Google Scholar] [CrossRef]
- Markham, A. Copanlisib: First Global Approval. Drugs 2017, 77, 2057–2062. [Google Scholar] [CrossRef]
- Kim, R.D.; Alberts, S.R.; Peña, C.; Genvresse, I.; Ajavon-Hartmann, A.; Xia, C.; Kelly, A.; Grilley-Olson, J.E. Phase I dose-escalation study of copanlisib in combination with gemcitabine or cisplatin plus gemcitabine in patients with advanced cancer. Br. J. Cancer 2018, 118, 462–470. [Google Scholar] [CrossRef]
- Dong, S.; Guinn, D.; Dubovsky, J.A.; Zhong, Y.; Lehman, A.; Kutok, J.; Woyach, J.A.; Byrd, J.C.; Johnson, A.J. IPI-145 antagonizes intrinsic and extrinsic survival signals in chronic lymphocytic leukemia cells. Blood 2014, 124, 3583–3586. [Google Scholar] [CrossRef] [PubMed]
- Flinn, I.; Oki, Y.; Patel, M.; Horwitz, S.M.; Foss, F.M.; Sweeney, J.; Allen, K.; Douglas, M.; Steelman, L.; Dunbar, J.; et al. A Phase 1 Evaluation of Duvelisib (IPI-145), a PI3K-δ,γ Inhibitor, in Patients with Relapsed/Refractory iNHL. Blood 2014, 124, 802. [Google Scholar] [CrossRef]
- Flinn, I.W.; Hillmen, P.; Montillo, M.; Nagy, Z.; Illés, Á.; Etienne, G.; Delgado, J.; Kuss, B.J.; Tam, C.S.; Gasztonyi, Z.; et al. The phase 3 DUO trial: Duvelisib vs ofatumumab in relapsed and refractory CLL/SLL. Blood 2018, 132, 2446–2455. [Google Scholar] [CrossRef]
- Flinn, I.W.; Miller, C.B.; Ardeshna, K.M.; Tetreault, S.; Assouline, S.E.; Zinzani, P.L.; Mayer, J.; Merli, M.; Lunin, S.D.; Pettitt, A.R.; et al. Dynamo: A Phase 2 Study Demonstrating the Clinical Activity of Duvelisib in Patients with Relapsed Refractory Indolent Non-Hodgkin Lymphoma. Blood 2016, 128, 1218. [Google Scholar] [CrossRef]
- Rodrigues, D.A.; Sagrillo, F.S.; Fraga, C.A.M. Duvelisib: A 2018 Novel FDA-Approved Small Molecule Inhibiting Phosphoinositide 3-Kinases. Pharmaceuticals 2019, 12, 69. [Google Scholar] [CrossRef]
- Tamura, K.; Kodaira, M.; Shimizu, C.; Yonemori, K.; Yunokawa, M.; Shimomura, A.; Kobayashi, T.; Nakano, K.; Tomomatsu, J.; Ito, Y.; et al. Phase I study of taselisib in Japanese patients with advanced solid tumors or hormone receptor-positive advanced breast cancer. Cancer Sci. 2018, 109, 1592–1601. [Google Scholar] [CrossRef]
- Dent, S.; Cortes, J.; Im, Y.H.; Dieras, V.; Harbeck, N.; Krop, I.E.; Wilson, T.R.; Cui, N.; Schimmoller, F.; Hsu, J.Y.; et al. Phase III randomized study of taselisib or placebo with fulvestrant in estrogen receptor-positive, PIK3CA-mutant, HER2-negative, advanced breast cancer: The SANDPIPER trial. Ann. Oncol. 2021, 32, 197–207. [Google Scholar] [CrossRef]
- Foster, P.; Yamaguchi, K.; Hsu, P.P.; Qian, F.; Du, X.; Wu, J.; Won, K.-A.; Yu, P.; Jaeger, C.T.; Zhang, W.; et al. The Selective PI3K Inhibitor XL147 (SAR245408) Inhibits Tumor Growth and Survival and Potentiates the Activity of Chemotherapeutic Agents in Preclinical Tumor Models. Mol. Cancer Ther. 2015, 14, 931–940. [Google Scholar] [CrossRef]
- Matulonis, U.; Vergote, I.; Backes, F.; Martin, L.P.; McMeekin, S.; Birrer, M.; Campana, F.; Xu, Y.; Egile, C.; Ghamande, S. Phase II study of the PI3K inhibitor pilaralisib (SAR245408; XL147) in patients with advanced or recurrent endometrial carcinoma. Gynecol. Oncol. 2015, 136, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Herman, S.E.; Gordon, A.L.; Wagner, A.J.; Heerema, N.A.; Zhao, W.; Flynn, J.M.; Jones, J.; Andritsos, L.; Puri, K.D.; Lannutti, B.J.; et al. Phosphatidylinositol 3-kinase-δ inhibitor CAL-101 shows promising preclinical activity in chronic lymphocytic leukemia by antagonizing intrinsic and extrinsic cellular survival signals. Blood 2010, 116, 2078–2088. [Google Scholar] [CrossRef] [PubMed]
- Gopal, A.K.; Kahl, B.S.; de Vos, S.; Wagner-Johnston, N.D.; Schuster, S.J.; Jurczak, W.J.; Flinn, I.W.; Flowers, C.R.; Martin, P.; Viardot, A.; et al. PI3Kδ inhibition by idelalisib in patients with relapsed indolent lymphoma. N. Engl. J. Med. 2014, 370, 1008–1018. [Google Scholar] [CrossRef] [PubMed]
- Furman, R.R.; Sharman, J.P.; Coutre, S.E.; Cheson, B.D.; Pagel, J.M.; Hillmen, P.; Barrientos, J.C.; Zelenetz, A.D.; Kipps, T.J.; Flinn, I.; et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N. Engl. J. Med. 2014, 370, 997–1007. [Google Scholar] [CrossRef]
- Furet, P.; Guagnano, V.; Fairhurst, R.A.; Imbach-Weese, P.; Bruce, I.; Knapp, M.; Fritsch, C.; Blasco, F.; Blanz, J.; Aichholz, R.; et al. Discovery of NVP-BYL719 a potent and selective phosphatidylinositol-3 kinase alpha inhibitor selected for clinical evaluation. Bioorganic. Med. Chem. Lett. 2013, 23, 3741–3748. [Google Scholar] [CrossRef] [PubMed]
- Bosch, A.; Li, Z.; Bergamaschi, A.; Ellis, H.; Toska, E.; Prat, A.; Tao, J.J.; Spratt, D.E.; Viola-Villegas, N.T.; Castel, P.; et al. PI3K inhibition results in enhanced estrogen receptor function and dependence in hormone receptor-positive breast cancer. Sci. Transl. Med. 2015, 7, 283ra251. [Google Scholar] [CrossRef] [PubMed]
- Toska, E.; Osmanbeyoglu, H.U.; Castel, P.; Chan, C.; Hendrickson, R.C.; Elkabets, M.; Dickler, M.N.; Scaltriti, M.; Leslie, C.S.; Armstrong, S.A.; et al. PI3K pathway regulates ER-dependent transcription in breast cancer through the epigenetic regulator KMT2D. Science 2017, 355, 1324–1330. [Google Scholar] [CrossRef] [PubMed]
- Toska, E.; Castel, P.; Chhangawala, S.; Arruabarrena-Aristorena, A.; Chan, C.; Hristidis, V.C.; Cocco, E.; Sallaku, M.; Xu, G.; Park, J.; et al. PI3K Inhibition Activates SGK1 via a Feedback Loop to Promote Chromatin-Based Regulation of ER-Dependent Gene Expression. Cell Rep. 2019, 27, 294–306.e295. [Google Scholar] [CrossRef]
- Andre, F.; Ciruelos, E.; Rubovszky, G.; Campone, M.; Loibl, S.; Rugo, H.S.; Iwata, H.; Conte, P.; Mayer, I.A.; Kaufman, B.; et al. Alpelisib for PIK3CA-Mutated, Hormone Receptor-Positive Advanced Breast Cancer. N. Engl J. Med. 2019, 380, 1929–1940. [Google Scholar] [CrossRef] [PubMed]
- Mayer, I.A.; Abramson, V.G.; Formisano, L.; Balko, J.M.; Estrada, M.V.; Sanders, M.E.; Juric, D.; Solit, D.; Berger, M.F.; Won, H.H.; et al. A Phase Ib Study of Alpelisib (BYL719), a PI3Kα-Specific Inhibitor, with Letrozole in ER+/HER2− Metastatic Breast Cancer. Clin. Cancer Res. 2017, 23, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Yan, L. Abstract #DDT01-1: MK-2206: A potent oral allosteric AKT inhibitor. Cancer Res. 2009, 69, DDT01-01. [Google Scholar]
- Davies, B.R.; Greenwood, H.; Dudley, P.; Crafter, C.; Yu, D.-H.; Zhang, J.; Li, J.; Gao, B.; Ji, Q.; Maynard, J.; et al. Preclinical Pharmacology of AZD5363, an Inhibitor of AKT: Pharmacodynamics, Antitumor Activity, and Correlation of Monotherapy Activity with Genetic Background. Mol. Cancer Ther. 2012, 11, 873–887. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Serra, V.; Prudkin, L.; Scaltriti, M.; Murli, S.; Rodríguez, O.; Guzman, M.; Sampath, D.; Nannini, M.; Xiao, Y.; et al. Evaluation and Clinical Analyses of Downstream Targets of the Akt Inhibitor GDC-0068. Clin. Cancer Res. 2013, 19, 6976–6986. [Google Scholar] [CrossRef]
- Nitulescu, G.M.; Van De Venter, M.; Nitulescu, G.; Ungurianu, A.; Juzenas, P.; Peng, Q.; Olaru, O.T.; Gradinaru, D.; Tsatsakis, A.; Tsoukalas, D.; et al. The Akt pathway in oncology therapy and beyond (Review). Int. J. Oncol. 2018, 53, 2319–2331. [Google Scholar] [CrossRef]
- Fang, Z.; Simard, J.R.; Plenker, D.; Nguyen, H.D.; Phan, T.; Wolle, P.; Baumeister, S.; Rauh, D. Discovery of inter-domain stabilizers-a novel assay system for allosteric akt inhibitors. ACS Chem. Biol. 2015, 10, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Sangai, T.; Akcakanat, A.; Chen, H.; Tarco, E.; Wu, Y.; Do, K.A.; Miller, T.W.; Arteaga, C.L.; Mills, G.B.; Gonzalez-Angulo, A.M.; et al. Biomarkers of response to Akt inhibitor MK-2206 in breast cancer. Clin. Cancer Res. 2012, 18, 5816–5828. [Google Scholar] [CrossRef]
- Mimura, N.; Hideshima, T.; Shimomura, T.; Suzuki, R.; Ohguchi, H.; Rizq, O.; Kikuchi, S.; Yoshida, Y.; Cottini, F.; Jakubikova, J.; et al. Selective and potent Akt inhibition triggers anti-myeloma activities and enhances fatal endoplasmic reticulum stress induced by proteasome inhibition. Cancer Res. 2014, 74, 4458–4469. [Google Scholar] [CrossRef] [PubMed]
- Saura, C.; Roda, D.; Rosello, S.; Oliveira, M.; Macarulla, T.; Perez-Fidalgo, J.A.; Morales-Barrera, R.; Sanchis-Garcia, J.M.; Musib, L.; Budha, N.; et al. A First-in-Human Phase I Study of the ATP-Competitive AKT Inhibitor Ipatasertib Demonstrates Robust and Safe Targeting of AKT in Patients with Solid Tumors. Cancer Discov. 2017, 7, 102–113. [Google Scholar] [CrossRef]
- Bose, S.; Kalinsky, K. Durable Clinical Activity to the AKT Inhibitor Ipatasertib in a Heavily Pretreated Patient With an AKT1 E17K Mutant Metastatic Breast Cancer. Clin. Breast Cancer 2020. [Google Scholar] [CrossRef] [PubMed]
- Hyman, D.M.; Smyth, L.M.; Donoghue, M.T.A.; Westin, S.N.; Bedard, P.L.; Dean, E.J.; Bando, H.; El-Khoueiry, A.B.; Pérez-Fidalgo, J.A.; Mita, A.; et al. AKT Inhibition in Solid Tumors With AKT1 Mutations. J. Clin. Oncol. 2017, 35, 2251–2259. [Google Scholar] [CrossRef] [PubMed]
- Juric, D.; Castel, P.; Griffith, M.; Griffith, O.L.; Won, H.H.; Ellis, H.; Ebbesen, S.H.; Ainscough, B.J.; Ramu, A.; Iyer, G.; et al. Convergent loss of PTEN leads to clinical resistance to a PI(3)Kalpha inhibitor. Nature 2015, 518, 240–244. [Google Scholar] [CrossRef] [PubMed]
- Serra, V.; Eichhorn, P.J.; Garcia-Garcia, C.; Ibrahim, Y.H.; Prudkin, L.; Sanchez, G.; Rodriguez, O.; Anton, P.; Parra, J.L.; Marlow, S.; et al. RSK3/4 mediate resistance to PI3K pathway inhibitors in breast cancer. J. Clin. Investig. 2013, 123, 2551–2563. [Google Scholar] [CrossRef]
- Elkabets, M.; Pazarentzos, E.; Juric, D.; Sheng, Q.; Pelossof, R.A.; Brook, S.; Benzaken, A.O.; Rodon, J.; Morse, N.; Yan, J.J.; et al. AXL mediates resistance to PI3Kalpha inhibition by activating the EGFR/PKC/mTOR axis in head and neck and esophageal squamous cell carcinomas. Cancer Cell 2015, 27, 533–546. [Google Scholar] [CrossRef] [PubMed]
- Le, X.; Antony, R.; Razavi, P.; Treacy, D.J.; Luo, F.; Ghandi, M.; Castel, P.; Scaltriti, M.; Baselga, J.; Garraway, L.A. Systematic Functional Characterization of Resistance to PI3K Inhibition in Breast Cancer. Cancer Discov. 2016, 6, 1134–1147. [Google Scholar] [CrossRef]
- Ros, S.; Wright, A.J.; D’Santos, P.; Hu, D.E.; Hesketh, R.L.; Lubling, Y.; Georgopoulou, D.; Lerda, G.; Couturier, D.L.; Razavi, P.; et al. Metabolic Imaging Detects Resistance to PI3Kalpha Inhibition Mediated by Persistent FOXM1 Expression in ER(+) Breast Cancer. Cancer Cell 2020, 38, 516–533.e5. [Google Scholar] [CrossRef]
- Vora, S.R.; Juric, D.; Kim, N.; Mino-Kenudson, M.; Huynh, T.; Costa, C.; Lockerman, E.L.; Pollack, S.F.; Liu, M.; Li, X.; et al. CDK 4/6 inhibitors sensitize PIK3CA mutant breast cancer to PI3K inhibitors. Cancer Cell 2014, 26, 136–149. [Google Scholar] [CrossRef] [PubMed]
- Muellner, M.K.; Uras, I.Z.; Gapp, B.V.; Kerzendorfer, C.; Smida, M.; Lechtermann, H.; Craig-Mueller, N.; Colinge, J.; Duernberger, G.; Nijman, S.M. A chemical-genetic screen reveals a mechanism of resistance to PI3K inhibitors in cancer. Nat. Chem. Biol. 2011, 7, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Chen, L.; Tan, X.; Crosby, K.; Guimaraes, A.R.; Upadhyay, R.; Maira, M.; McNamara, K.; Perera, S.A.; Song, Y.; et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat. Med. 2008, 14, 1351–1356. [Google Scholar] [CrossRef] [PubMed]
- Castel, P.; Ellis, H.; Bago, R.; Toska, E.; Razavi, P.; Carmona, F.J.; Kannan, S.; Verma, C.S.; Dickler, M.; Chandarlapaty, S.; et al. PDK1-SGK1 Signaling Sustains AKT-Independent mTORC1 Activation and Confers Resistance to PI3Kalpha Inhibition. Cancer Cell 2016, 30, 229–242. [Google Scholar] [CrossRef] [PubMed]
- Gasser, J.A.; Inuzuka, H.; Lau, A.W.; Wei, W.; Beroukhim, R.; Toker, A. SGK3 mediates INPP4B-dependent PI3K signaling in breast cancer. Mol. Cell 2014, 56, 595–607. [Google Scholar] [CrossRef] [PubMed]
- Saei, A.; Eichhorn, P.J.A. Adaptive Responses as Mechanisms of Resistance to BRAF Inhibitors in Melanoma. Cancers 2019, 11, 1176. [Google Scholar] [CrossRef] [PubMed]
- Marine, J.C.; Dawson, S.J.; Dawson, M.A. Non-genetic mechanisms of therapeutic resistance in cancer. Nat. Rev. Cancer 2020, 20, 743–756. [Google Scholar] [CrossRef]
- Hoxhaj, G.; Manning, B.D. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef]
- Augustin, R. The protein family of glucose transport facilitators: It’s not only about glucose after all. Iubmb Life 2010, 62, 315–333. [Google Scholar] [CrossRef]
- Yuasa, T.; Uchiyama, K.; Ogura, Y.; Kimura, M.; Teshigawara, K.; Hosaka, T.; Tanaka, Y.; Obata, T.; Sano, H.; Kishi, K.; et al. The Rab GTPase-activating protein AS160 as a common regulator of insulin- and Galphaq-mediated intracellular GLUT4 vesicle distribution. Endocr. J. 2009, 56, 345–359. [Google Scholar] [CrossRef] [PubMed]
- Rathmell, J.C.; Fox, C.J.; Plas, D.R.; Hammerman, P.S.; Cinalli, R.M.; Thompson, C.B. Akt-directed glucose metabolism can prevent Bax conformation change and promote growth factor-independent survival. Mol. Cell. Biol. 2003, 23, 7315–7328. [Google Scholar] [CrossRef]
- Bentley, J.; Itchayanan, D.; Barnes, K.; McIntosh, E.; Tang, X.; Downes, C.P.; Holman, G.D.; Whetton, A.D.; Owen-Lynch, P.J.; Baldwin, S.A. Interleukin-3-mediated cell survival signals include phosphatidylinositol 3-kinase-dependent translocation of the glucose transporter GLUT1 to the cell surface. J. Biol. Chem. 2003, 278, 39337–39348. [Google Scholar] [CrossRef]
- Siska, P.J.; van der Windt, G.J.W.; Kishton, R.J.; Cohen, S.; Eisner, W.; MacIver, N.J.; Kater, A.P.; Weinberg, J.B.; Rathmell, J.C. Suppression of Glut1 and Glucose Metabolism by Decreased Akt/mTORC1 Signaling Drives T Cell Impairment in B Cell Leukemia. J. Immunol. 2016, 197, 2532–2540. [Google Scholar] [CrossRef]
- Wieman, H.L.; Wofford, J.A.; Rathmell, J.C. Cytokine stimulation promotes glucose uptake via phosphatidylinositol-3 kinase/Akt regulation of Glut1 activity and trafficking. Mol. Biol. Cell 2007, 18, 1437–1446. [Google Scholar] [CrossRef]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef]
- Osthus, R.C.; Shim, H.; Kim, S.; Li, Q.; Reddy, R.; Mukherjee, M.; Xu, Y.; Wonsey, D.; Lee, L.A.; Dang, C.V. Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J. Biol. Chem. 2000, 275, 21797–21800. [Google Scholar] [CrossRef] [PubMed]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef]
- Ando, Y.; Inada-Inoue, M.; Mitsuma, A.; Yoshino, T.; Ohtsu, A.; Suenaga, N.; Sato, M.; Kakizume, T.; Robson, M.; Quadt, C.; et al. Phase I dose-escalation study of buparlisib (BKM120), an oral pan-class I PI3K inhibitor, in Japanese patients with advanced solid tumors. Cancer Sci. 2014, 105, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Yan, L.; Patnaik, A.; Fearen, I.; Olmos, D.; Papadopoulos, K.; Baird, R.D.; Delgado, L.; Taylor, A.; Lupinacci, L.; et al. First-in-man clinical trial of the oral pan-AKT inhibitor MK-2206 in patients with advanced solid tumors. J. Clin. Oncol. 2011, 29, 4688–4695. [Google Scholar] [CrossRef]
- Khan, K.H.; Wong, M.; Rihawi, K.; Bodla, S.; Morganstein, D.; Banerji, U.; Molife, L.R. Hyperglycemia and Phosphatidylinositol 3-Kinase/Protein Kinase B/Mammalian Target of Rapamycin (PI3K/AKT/mTOR) Inhibitors in Phase I Trials: Incidence, Predictive Factors, and Management. Oncologist 2016, 21, 855–860. [Google Scholar] [CrossRef]
- Luo, J.; Field, S.J.; Lee, J.Y.; Engelman, J.A.; Cantley, L.C. The p85 regulatory subunit of phosphoinositide 3-kinase down-regulates IRS-1 signaling via the formation of a sequestration complex. J. Cell Biol 2005, 170, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Barbour, L.A.; Shao, J.; Qiao, L.; Leitner, W.; Anderson, M.; Friedman, J.E.; Draznin, B. Human placental growth hormone increases expression of the p85 regulatory unit of phosphatidylinositol 3-kinase and triggers severe insulin resistance in skeletal muscle. Endocrinology 2004, 145, 1144–1150. [Google Scholar] [CrossRef]
- Hopkins, B.D.; Pauli, C.; Du, X.; Wang, D.G.; Li, X.; Wu, D.; Amadiume, S.C.; Goncalves, M.D.; Hodakoski, C.; Lundquist, M.R.; et al. Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nature 2018, 560, 499–503. [Google Scholar] [CrossRef]
- Goncalves, M.D.; Hopkins, B.D.; Cantley, L.C. Phosphatidylinositol 3-Kinase, Growth Disorders, and Cancer. N. Engl. J. Med. 2018, 379, 2052–2062. [Google Scholar] [CrossRef] [PubMed]
- Hanker, A.B.; Kaklamani, V.; Arteaga, C.L. Challenges for the Clinical Development of PI3K Inhibitors: Strategies to Improve Their Impact in Solid Tumors. Cancer Discov. 2019, 9, 482. [Google Scholar] [CrossRef]
- Hillis, A.L.; Toker, A. Lactate Lights up PI3K Inhibitor Resistance in Breast Cancer. Cancer Cell 2020, 38, 441–443. [Google Scholar] [CrossRef]
- Haruta, T.; Uno, T.; Kawahara, J.; Takano, A.; Egawa, K.; Sharma, P.M.; Olefsky, J.M.; Kobayashi, M. A rapamycin-sensitive pathway down-regulates insulin signaling via phosphorylation and proteasomal degradation of insulin receptor substrate-1. Mol. Endocrinol. 2000, 14, 783–794. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, F.; Marette, A. Amino acid and insulin signaling via the mTOR/p70 S6 kinase pathway. A negative feedback mechanism leading to insulin resistance in skeletal muscle cells. J. Biol. Chem. 2001, 276, 38052–38060. [Google Scholar] [CrossRef] [PubMed]
- Berg, C.E.; Lavan, B.E.; Rondinone, C.M. Rapamycin partially prevents insulin resistance induced by chronic insulin treatment. Biochem. Biophys. Res. Commun. 2002, 293, 1021–1027. [Google Scholar] [CrossRef]
- Chandarlapaty, S.; Sawai, A.; Scaltriti, M.; Rodrik-Outmezguine, V.; Grbovic-Huezo, O.; Serra, V.; Majumder, P.K.; Baselga, J.; Rosen, N. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell 2011, 19, 58–71. [Google Scholar] [CrossRef]
- Serra, V.; Scaltriti, M.; Prudkin, L.; Eichhorn, P.J.; Ibrahim, Y.H.; Chandarlapaty, S.; Markman, B.; Rodriguez, O.; Guzman, M.; Rodriguez, S.; et al. PI3K inhibition results in enhanced HER signaling and acquired ERK dependency in HER2-overexpressing breast cancer. Oncogene 2011, 30, 2547–2557. [Google Scholar] [CrossRef]
- Chakrabarty, A.; Sánchez, V.; Kuba, M.G.; Rinehart, C.; Arteaga, C.L. Feedback upregulation of HER3 (ErbB3) expression and activity attenuates antitumor effect of PI3K inhibitors. Proc. Natl. Acad. Sci. USA 2012, 109, 2718–2723. [Google Scholar] [CrossRef]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef]
- Muranen, T.; Selfors, L.M.; Worster, D.T.; Iwanicki, M.P.; Song, L.; Morales, F.C.; Gao, S.; Mills, G.B.; Brugge, J.S. Inhibition of PI3K/mTOR leads to adaptive resistance in matrix-attached cancer cells. Cancer Cell 2012, 21, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.; Piao, H.L.; Zhuang, L.; dos Sarbassov, D.; Ma, L.; Gan, B. FoxO transcription factors promote AKT Ser473 phosphorylation and renal tumor growth in response to pharmacologic inhibition of the PI3K-AKT pathway. Cancer Res. 2014, 74, 1682–1693. [Google Scholar] [CrossRef]
- Jia, S.; Liu, Z.; Zhang, S.; Liu, P.; Zhang, L.; Lee, S.H.; Zhang, J.; Signoretti, S.; Loda, M.; Roberts, T.M.; et al. Essential roles of PI(3)K-p110beta in cell growth, metabolism and tumorigenesis. Nature 2008, 454, 776–779. [Google Scholar] [CrossRef]
- Costa, C.; Ebi, H.; Martini, M.; Beausoleil, S.A.; Faber, A.C.; Jakubik, C.T.; Huang, A.; Wang, Y.; Nishtala, M.; Hall, B.; et al. Measurement of PIP3 levels reveals an unexpected role for p110beta in early adaptive responses to p110alpha-specific inhibitors in luminal breast cancer. Cancer Cell 2015, 27, 97–108. [Google Scholar] [CrossRef]
- Schwartz, S.; Wongvipat, J.; Trigwell, C.B.; Hancox, U.; Carver, B.S.; Rodrik-Outmezguine, V.; Will, M.; Yellen, P.; de Stanchina, E.; Baselga, J.; et al. Feedback suppression of PI3Kalpha signaling in PTEN-mutated tumors is relieved by selective inhibition of PI3Kbeta. Cancer Cell 2015, 27, 109–122. [Google Scholar] [CrossRef]
- Mukherjee, R.; Vanaja, K.G.; Boyer, J.A.; Gadal, S.; Solomon, H.; Chandarlapaty, S.; Levchenko, A.; Rosen, N. Regulation of PTEN translation by PI3K signaling maintains pathway homeostasis. Mol. Cell 2021, 81, 708–723.e705. [Google Scholar] [CrossRef] [PubMed]
- Vasudevan, K.M.; Barbie, D.A.; Davies, M.A.; Rabinovsky, R.; McNear, C.J.; Kim, J.J.; Hennessy, B.T.; Tseng, H.; Pochanard, P.; Kim, S.Y.; et al. AKT-independent signaling downstream of oncogenic PIK3CA mutations in human cancer. Cancer Cell 2009, 16, 21–32. [Google Scholar] [CrossRef]
- Bago, R.; Sommer, E.; Castel, P.; Crafter, C.; Bailey, F.P.; Shpiro, N.; Baselga, J.; Cross, D.; Eyers, P.A.; Alessi, D.R. The hVps34-SGK3 pathway alleviates sustained PI3K/Akt inhibition by stimulating mTORC1 and tumour growth. Embo J. 2016, 35, 1902–1922. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhou, D.; Phung, S.; Masri, S.; Smith, D.; Chen, S. SGK3 is an estrogen-inducible kinase promoting estrogen-mediated survival of breast cancer cells. Mol. Endocrinol. 2011, 25, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Britschgi, A.; Andraos, R.; Brinkhaus, H.; Klebba, I.; Romanet, V.; Muller, U.; Murakami, M.; Radimerski, T.; Bentires-Alj, M. JAK2/STAT5 inhibition circumvents resistance to PI3K/mTOR blockade: A rationale for cotargeting these pathways in metastatic breast cancer. Cancer Cell 2012, 22, 796–811. [Google Scholar] [CrossRef] [PubMed]
- Clement, E.; Inuzuka, H.; Nihira, N.T.; Wei, W.; Toker, A. Skp2-dependent reactivation of AKT drives resistance to PI3K inhibitors. Sci. Signal. 2018, 11. [Google Scholar] [CrossRef]
- Chan, C.H.; Li, C.F.; Yang, W.L.; Gao, Y.; Lee, S.W.; Feng, Z.; Huang, H.Y.; Tsai, K.K.C.; Flores, L.G.; Shao, Y.; et al. The Skp2-SCF E3 Ligase Regulates Akt Ubiquitination, Glycolysis, Herceptin Sensitivity, and Tumorigenesis. Cell 2012, 151, 913–914. [Google Scholar] [CrossRef][Green Version]
- Rexer, B.N.; Chanthaphaychith, S.; Dahlman, K.; Arteaga, C.L. Direct inhibition of PI3K in combination with dual HER2 inhibitors is required for optimal antitumor activity in HER2+ breast cancer cells. Breast Cancer Res. 2014, 16, R9. [Google Scholar] [CrossRef] [PubMed]
- García-García, C.; Ibrahim, Y.H.; Serra, V.; Calvo, M.T.; Guzmán, M.; Grueso, J.; Aura, C.; Pérez, J.; Jessen, K.; Liu, Y.; et al. Dual mTORC1/2 and HER2 Blockade Results in Antitumor Activity in Preclinical Models of Breast Cancer Resistant to Anti-HER2 Therapy. Clin. Cancer Res. 2012, 18, 2603. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.H.; Xue, A.; Julovi, S.M.; Pavlakis, N.; Samra, J.S.; Hugh, T.J.; Gill, A.J.; Peters, L.; Baxter, R.C.; Smith, R.C. Cotargeting of epidermal growth factor receptor and PI3K overcomes PI3K-Akt oncogenic dependence in pancreatic ductal adenocarcinoma. Clin. Cancer Res. 2014, 20, 4047–4058. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.J.; Castel, P.; Radosevic-Robin, N.; Elkabets, M.; Auricchio, N.; Aceto, N.; Weitsman, G.; Barber, P.; Vojnovic, B.; Ellis, H.; et al. Antagonism of EGFR and HER3 enhances the response to inhibitors of the PI3K-Akt pathway in triple-negative breast cancer. Sci. Signal. 2014, 7, ra29. [Google Scholar] [CrossRef]
- Makki, J. Diversity of Breast Carcinoma: Histological Subtypes and Clinical Relevance. Clin. Med. Insights Pathol. 2015, 8, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Ciriello, G.; Gatza, M.L.; Beck, A.H.; Wilkerson, M.D.; Rhie, S.K.; Pastore, A.; Zhang, H.; McLellan, M.; Yau, C.; Kandoth, C.; et al. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell 2015, 163, 506–519. [Google Scholar] [CrossRef] [PubMed]
- Van Tine, B.A.; Crowder, R.J.; Ellis, M.J. ER and PI3K independently modulate endocrine resistance in ER-positive breast cancer. Cancer Discov. 2011, 1, 287–288. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Slingerland, J.M. Links between oestrogen receptor activation and proteolysis: Relevance to hormone-regulated cancer therapy. Nat. Rev. Cancer 2014, 14, 26–38. [Google Scholar] [CrossRef]
- Hata, A.N.; Niederst, M.J.; Archibald, H.L.; Gomez-Caraballo, M.; Siddiqui, F.M.; Mulvey, H.E.; Maruvka, Y.E.; Ji, F.; Bhang, H.E.; Krishnamurthy Radhakrishna, V.; et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat. Med. 2016, 22, 262–269. [Google Scholar] [CrossRef]
- Boumahdi, S.; de Sauvage, F.J. The great escape: Tumour cell plasticity in resistance to targeted therapy. Nat. Rev. Drug Discov. 2020, 19, 39–56. [Google Scholar] [CrossRef] [PubMed]
- Arozarena, I.; Wellbrock, C. Phenotype plasticity as enabler of melanoma progression and therapy resistance. Nat. Rev. Cancer 2019, 19, 377–391. [Google Scholar] [CrossRef]
- Rambow, F.; Rogiers, A.; Marin-Bejar, O.; Aibar, S.; Femel, J.; Dewaele, M.; Karras, P.; Brown, D.; Chang, Y.H.; Debiec-Rychter, M.; et al. Toward Minimal Residual Disease-Directed Therapy in Melanoma. Cell 2018, 174, 843–855.e819. [Google Scholar] [CrossRef]
- Vasan, N.; Baselga, J.; Hyman, D.M. A view on drug resistance in cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Settleman, J. EMT, cancer stem cells and drug resistance: An emerging axis of evil in the war on cancer. Oncogene 2010, 29, 4741–4751. [Google Scholar] [CrossRef] [PubMed]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef]
- Pei, Y.; Liu, K.W.; Wang, J.; Garancher, A.; Tao, R.; Esparza, L.A.; Maier, D.L.; Udaka, Y.T.; Murad, N.; Morrissy, S.; et al. HDAC and PI3K Antagonists Cooperate to Inhibit Growth of MYC-Driven Medulloblastoma. Cancer Cell 2016, 29, 311–323. [Google Scholar] [CrossRef]
- Xu, W.; Yang, Z.; Lu, N. A new role for the PI3K/Akt signaling pathway in the epithelial-mesenchymal transition. Cell Adh Migr 2015, 9, 317–324. [Google Scholar] [CrossRef]
- Hussein, A.M.; Wang, Y.; Mathieu, J.; Margaretha, L.; Song, C.; Jones, D.C.; Cavanaugh, C.; Miklas, J.W.; Mahen, E.; Showalter, M.R.; et al. Metabolic Control over mTOR-Dependent Diapause-like State. Dev. Cell 2020, 52, 236–250.e237. [Google Scholar] [CrossRef] [PubMed]
- Bulut-Karslioglu, A.; Biechele, S.; Jin, H.; Macrae, T.A.; Hejna, M.; Gertsenstein, M.; Song, J.S.; Ramalho-Santos, M. Inhibition of mTOR induces a paused pluripotent state. Nature 2016, 540, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Noda, T. Regulation of Autophagy through TORC1 and mTORC1. Biomolecules 2017, 7, 52. [Google Scholar] [CrossRef] [PubMed]
- Rehman, S.K.; Haynes, J.; Collignon, E.; Brown, K.R.; Wang, Y.; Nixon, A.M.L.; Bruce, J.P.; Wintersinger, J.A.; Singh Mer, A.; Lo, E.B.L.; et al. Colorectal Cancer Cells Enter a Diapause-like DTP State to Survive Chemotherapy. Cell 2021, 184, 226–242.e221. [Google Scholar] [CrossRef] [PubMed]
- Pascual-Garcia, M.; Bonfill-Teixidor, E.; Planas-Rigol, E.; Rubio-Perez, C.; Iurlaro, R.; Arias, A.; Cuartas, I.; Sala-Hojman, A.; Escudero, L.; Martinez-Ricarte, F.; et al. LIF regulates CXCL9 in tumor-associated macrophages and prevents CD8(+) T cell tumor-infiltration impairing anti-PD1 therapy. Nat. Commun. 2019, 10, 2416. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
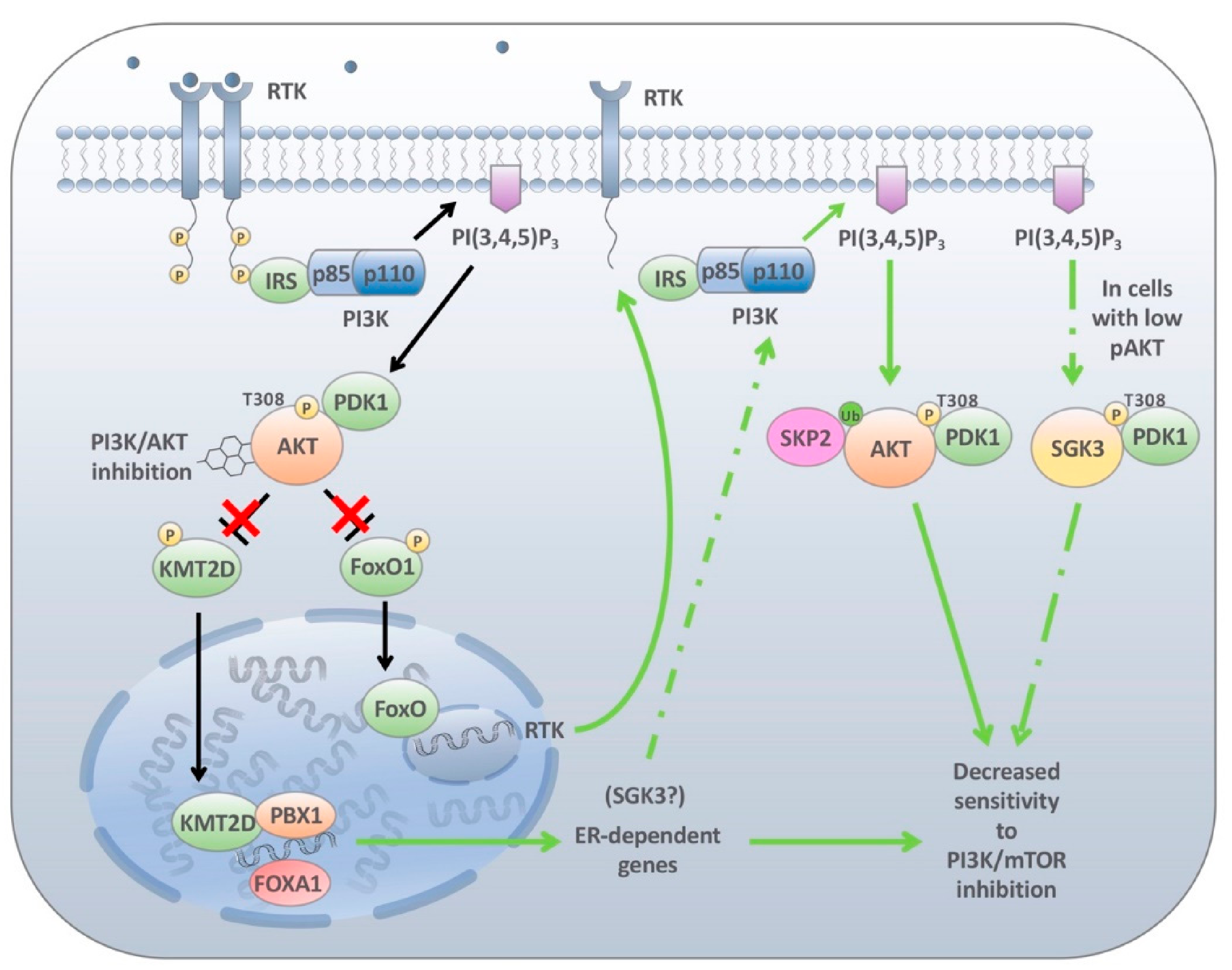{kind=link}
| Target | Drug | Cancer Targets | FDA Status |
|---|---|---|---|
| Pan-PI3K Inhibitors | Copanlisib (BAY 80-6946) | Refractory follicular lymphoma (FL) | Approved |
| Duvelisib (IPI-145) | Refractory follicular lymphoma (FL); refractory chronic lymphocytic leukaemia (CLL); small lymphocytic lymphoma (SLL); refractory follicular B-cell non-Hodgkin lymphoma (NHL) | Approved | |
| Buparlisib (BKM120) | Discontinued | ||
| Pictilisib (GDC-0941) | Under clinical development in breast cancer | Under clinical development | |
| Isoform-Specific PI3K Inhibitors | Alpelisib (BYL719) | Hormone receptor-positive/HER2-negative (HR+/HER2-) PIK3CA mutant breast cancer in combination with fulvestrant | Approved |
| Idelalisib (CAL101) | Second-line treatment for patients with Chronic lymphocytic leukaemia (CLL) in combination with rituximab; follicular B-cell non-Hodgkin lymphoma (FL) and relapsed small lymphocytic lymphoma (SLL), both in patients who have received at least two prior systemic therapies. | Approved | |
| Serabelisib (INK1117/TAK-117) | Under clinical development for various tumours including breast, and endometrial cancer. | Active but not recruiting | |
| AKT Inhibitors | MK2206 | Under clinical development for PIK3CA and/or PTEN mutant breast cancer; non-small cell lung cancer, and ovarian cancers. | Under clinical development |
| TAS-117 | Under clinical development | ||
| Capivasertib (AZD5363) | Under clinical development for patients with AKT E17K mutations | Under Clinical development | |
| mTOR Inhibitors | Everolimus (RAD001) | Advanced renal cell carcinoma; hormone receptor-positive/HER2-negative (HR+/HER2-) breast cancer; gastrointestinal/lung neuroendocrine tumours (NET) | Approved |
| Temsirolimus (CCI-779) | Advanced-stage renal cell carcinoma | Approved | |
| Torkinib (PP242) | Discontinued | ||
| Sapanisertib (MLN0128) | Under clinical development for multiple solid tumours | Under clinical development | |
| Vistusertib (AZD2014) | Under clinical development for multiple solid tumours | Under clinical development | |
| Nab-sirolimus (ABI-009) | Perivascular epithelioid cell neoplasms (PEComa) | Approved | |
| Dual PI3K/mTOR Inhibitors | Dactolisib (BEZ235) | Discontinued | |
| Apitolisib (GDC-0980) | Under clinical development for prostate cancer. | Under clinical development | |
| Vortalisib (XL765) | Discontinued | ||
| Gedatolisib (PF-05212384) | Under clinical development for breast cancer | Under clinical development |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wright, S.C.E.; Vasilevski, N.; Serra, V.; Rodon, J.; Eichhorn, P.J.A. Mechanisms of Resistance to PI3K Inhibitors in Cancer: Adaptive Responses, Drug Tolerance and Cellular Plasticity. Cancers 2021, 13, 1538. https://doi.org/10.3390/cancers13071538
Wright SCE, Vasilevski N, Serra V, Rodon J, Eichhorn PJA. Mechanisms of Resistance to PI3K Inhibitors in Cancer: Adaptive Responses, Drug Tolerance and Cellular Plasticity. Cancers. 2021; 13(7):1538. https://doi.org/10.3390/cancers13071538
Chicago/Turabian StyleWright, Sarah Christine Elisabeth, Natali Vasilevski, Violeta Serra, Jordi Rodon, and Pieter Johan Adam Eichhorn. 2021. "Mechanisms of Resistance to PI3K Inhibitors in Cancer: Adaptive Responses, Drug Tolerance and Cellular Plasticity" Cancers 13, no. 7: 1538. https://doi.org/10.3390/cancers13071538
APA StyleWright, S. C. E., Vasilevski, N., Serra, V., Rodon, J., & Eichhorn, P. J. A. (2021). Mechanisms of Resistance to PI3K Inhibitors in Cancer: Adaptive Responses, Drug Tolerance and Cellular Plasticity. Cancers, 13(7), 1538. https://doi.org/10.3390/cancers13071538