
Between a Rock and a Hard Place: An Epigenetic-Centric View of Testicular Germ Cell Tumors



Abstract
Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
1. Introduction
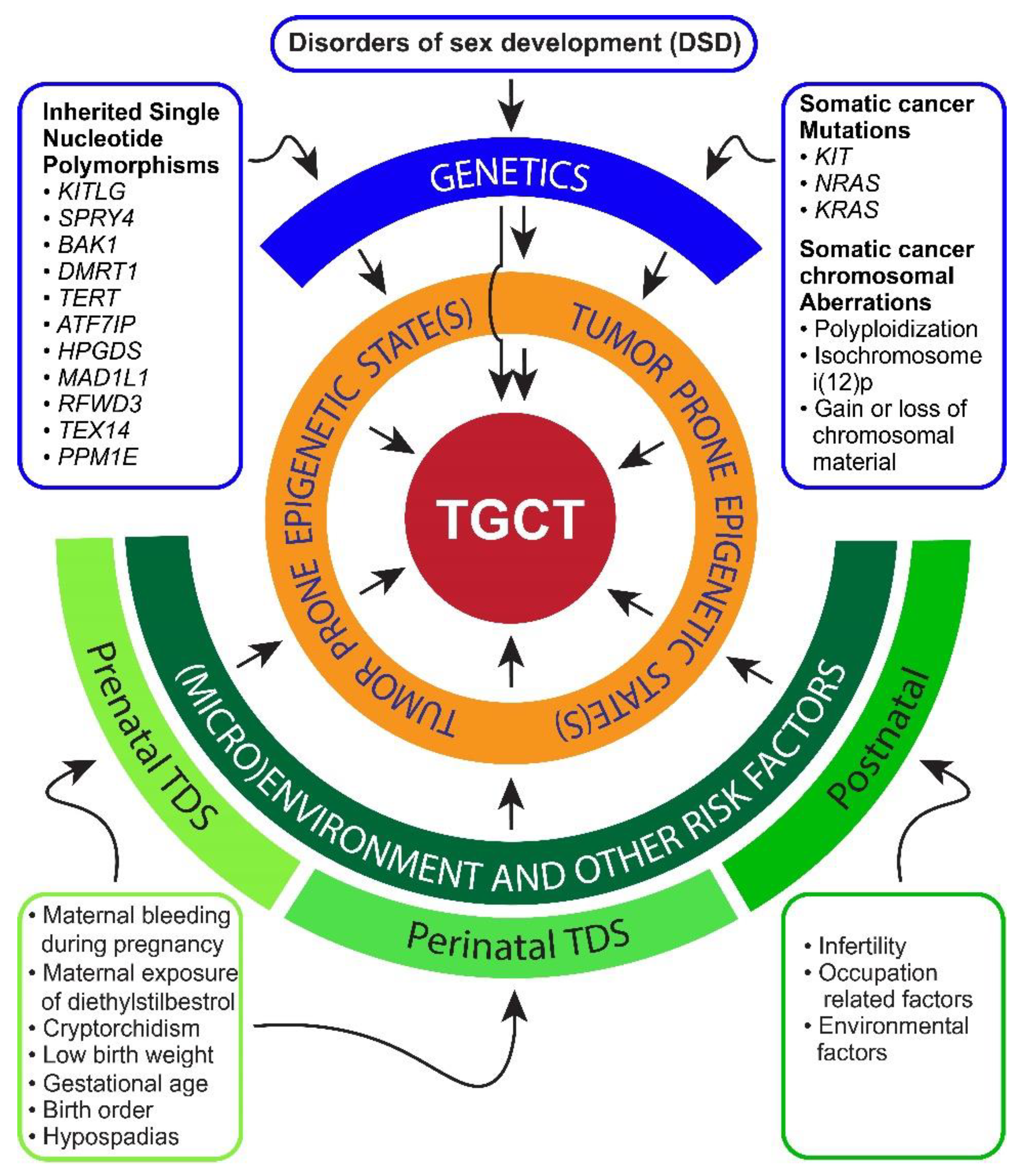
2. Risk Factors and Pathogenesis of TGCTs
3. Mechanisms of Chemotherapy Sensitivity and Resistance in TGCTs
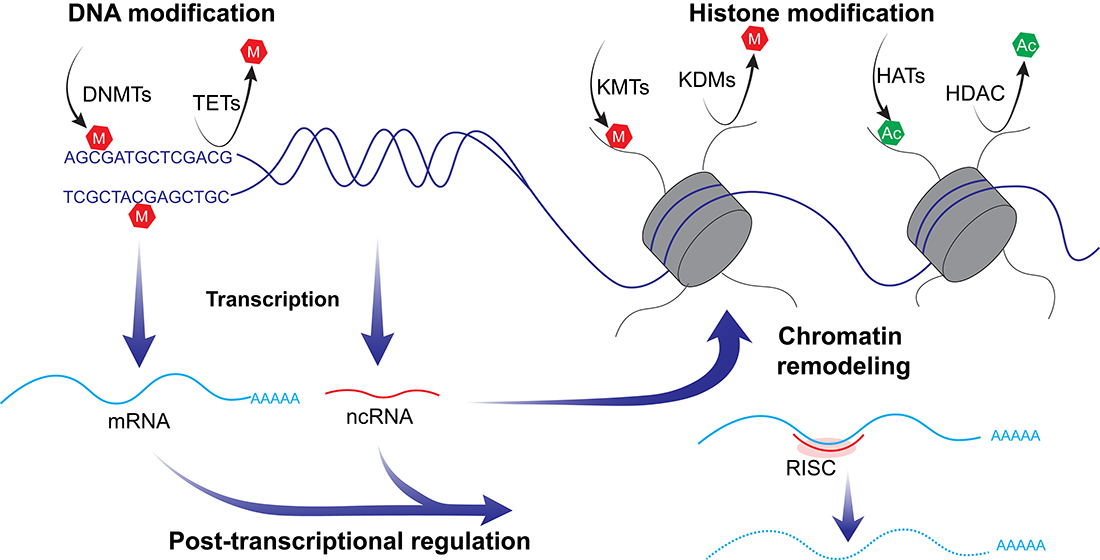
4. Epigenetic States in Testicular Germ Cell Tumors Associated with Tumorigenicity and Chemosensitivity
4.1. Differentiation
4.2. DNA Methylation
4.3. Histone Modifications
4.4. Non-Coding RNA
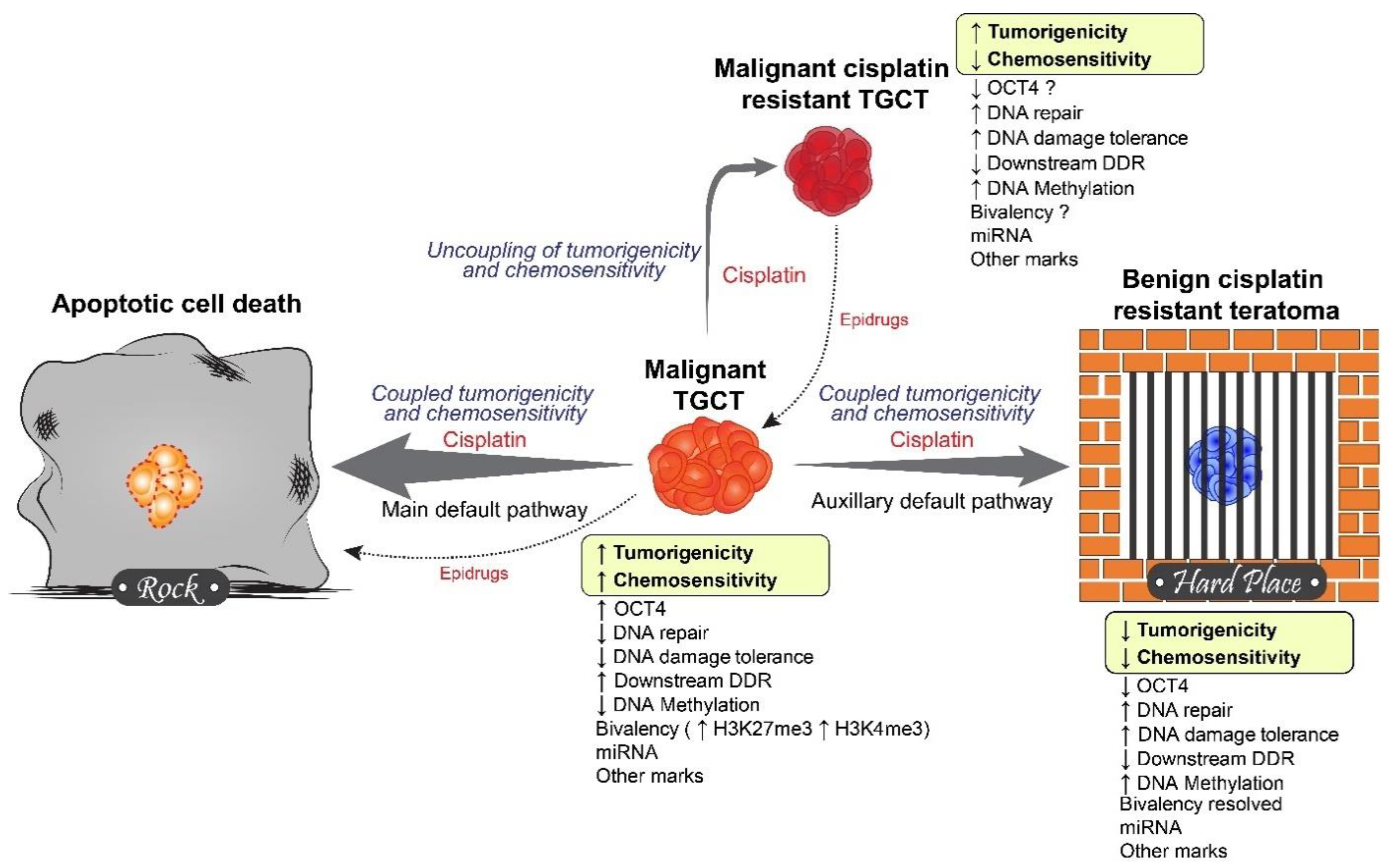
5. Rock and a Hard Place Model
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Batool, A.; Karimi, N.; Wu, X.N.; Chen, S.R.; Liu, Y.X. Testicular germ cell tumor: A comprehensive review. Cell. Mol. Life Sci. 2019, 76, 1713–1727. [Google Scholar] [CrossRef]
- Lobo, J.; Gillis, A.J.M.; Jerónimo, C.; Henrique, R.; Looijenga, L.H.J. Human germ cell tumors are developmental cancers: Impact of epigenetics on pathobiology and clinic. Int. J. Mol. Sci. 2019, 20, 258. [Google Scholar] [CrossRef] [PubMed]
- Williamson, S.R.; Delahunt, B.; Magi-Galluzzi, C.; Algaba, F.; Egevad, L.; Ulbright, T.M.; Tickoo, S.K.; Srigley, J.R.; Epstein, J.I.; Berney, D.M. The World Health Organization 2016 classification of testicular germ cell tumours: A review and update from the International Society of Urological Pathology Testis Consultation Panel. Histopathology 2017, 70, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Berney, D.M.; Looijenga, L.H.; Idrees, M.; Oosterhuis, J.W.; Rajpert-De Meyts, E.; Ulbright, T.M.; Skakkebaek, N.E. Germ cell neoplasia in situ (GCNIS): Evolution of the current nomenclature for testicular pre-invasive germ cell malignancy. Histopathology 2016, 69, 7–10. [Google Scholar] [CrossRef]
- Oosterhuis, J.W.; Looijenga, L.H. Testicular germ-cell tumours in a broader perspective. Nat. Rev. Cancer 2005, 5, 210–222. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Woldu, S.L.; Bagrodia, A. Update on epidemiologic considerations and treatment trends in testicular cancer. Curr. Opin. Urol. 2018, 28, 440–447. [Google Scholar] [CrossRef]
- Cai, Q.; Chen, Y.; Zhang, D.; Pan, J.; Xie, Z.; Xu, C.; Li, S.; Zhang, X.; Gao, Y.; Hou, J.; et al. Estimates of over-time trends in incidence and mortality of testicular cancer from 1990 to 2030. Transl. Androl. Urol. 2020, 9, 182–195. [Google Scholar] [CrossRef]
- Xing, J.S.; Bai, Z.M. Is testicular dysgenesis syndrome a genetic, endocrine, or environmental disease, or an unexplained reproductive disorder? Life Sci. 2018, 194, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Adra, N.; Einhorn, L.H. Testicular cancer update. Clin. Adv. Hematol. Oncol. 2017, 15, 386–396. [Google Scholar] [PubMed]
- Saju, S.V.; Radhakrishnan, V.; Ganesan, T.S.; Dhanushkodi, M.; Raja, A.; Selvaluxmy, G.; Sagar, T.G. Factors that impact the outcomes in testicular germ cell tumors in low-middle-income countries. Med. Oncol. 2019, 36, 28. [Google Scholar] [CrossRef]
- Giuliano, C.J.; Freemantle, S.J.; Spinella, M.J. Testicular germ cell tumors: A paradigm for the successful treatment of solid tumor stem cells. Curr. Cancer Ther. Rev. 2006, 2, 255–270. [Google Scholar] [CrossRef]
- Alsdorf, W.; Seidel, C.; Bokemeyer, C.; Oing, C. Current pharmacotherapy for testicular germ cell cancer. Expert Opin. Pharmacother. 2019, 20, 837–850. [Google Scholar] [CrossRef] [PubMed]
- Feldman, D.R.; Patil, S.; Trinos, M.J.; Carousso, M.; Ginsberg, M.S.; Sheinfeld, J.; Bajorin, D.F.; Bosl, G.J.; Motzer, R.J. Progression-free and overall survival in patients with relapsed/refractory germ cell tumors treated with single-agent chemotherapy: Endpoints for clinical trial design. Cancer 2012, 118, 981–986. [Google Scholar] [CrossRef]
- Porcu, P.; Bhatia, S.; Sharma, M.; Einhorn, L.H. Results of treatment after relapse from high-dose chemotherapy in germ cell tumors. J. Clin. Oncol. 2000, 18, 1181–1186. [Google Scholar] [CrossRef] [PubMed]
- Fung, C.; Dinh, P., Jr.; Ardeshir-Rouhani-Fard, S.; Schaffer, K.; Fossa, S.D.; Travis, L.B. Toxicities associated with cisplatin-based chemotherapy and radiotherapy in long-term testicular cancer survivors. Adv. Urol. 2018, 2018, 8671832. [Google Scholar] [CrossRef]
- Christensen, J.F.; Bandak, M.; Campbell, A.; Jones, L.W.; Højman, P. Treatment-related cardiovascular late effects and exercise training countermeasures in testicular germ cell cancer survivorship. Acta Oncol. 2015, 54, 592–599. [Google Scholar] [CrossRef]
- Curreri, S.A.; Fung, C.; Beard, C.J. Secondary malignant neoplasms in testicular cancer survivors. Urol. Oncol. 2015, 33, 392–398. [Google Scholar] [CrossRef]
- Fung, C.; Sesso, H.D.; Williams, A.M.; Kerns, S.L.; Monahan, P.; Abu Zaid, M.; Feldman, D.R.; Hamilton, R.J.; Vaughn, D.J.; Beard, C.J.; et al. Multi-institutional assessment of adverse health outcomes among north american testicular cancer survivors after modern cisplatin-based chemotherapy. J. Clin. Oncol. 2017, 35, 1211–1222. [Google Scholar] [CrossRef] [PubMed]
- Oosterhuis, J.W.; Looijenga, L.H.J. Human germ cell tumours from a developmental perspective. Nat. Rev. Cancer 2019, 19, 522–537. [Google Scholar] [CrossRef] [PubMed]
- Singla, N.; Lafin, J.T.; Ghandour, R.A.; Kaffenberger, S.; Amatruda, J.F.; Bagrodia, A. Genetics of testicular germ cell tumors. Curr. Opin. Urol. 2019, 29, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Elzinga-Tinke, J.E.; Dohle, G.R.; Looijenga, L.H. Etiology and early pathogenesis of malignant testicular germ cell tumors: Towards possibilities for preinvasive diagnosis. Asian J. Androl. 2015, 17, 381–393. [Google Scholar] [CrossRef] [PubMed]
- Baroni, T.; Arato, I.; Mancuso, F.; Calafiore, R.; Luca, G. On the origin of testicular germ cell tumors: From gonocytes to testicular cancer. Front. Endocrinol. 2019, 10, 343. [Google Scholar] [CrossRef]
- Litchfield, K.; Summersgill, B.; Yost, S.; Sultana, R.; Labreche, K.; Dudakia, D.; Renwick, A.; Seal, S.; Al-Saadi, R.; Broderick, P.; et al. Whole-exome sequencing reveals the mutational spectrum of testicular germ cell tumours. Nat. Commun. 2015, 6, 5973. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Shih, J.; Hollern, D.P.; Wang, L.; Bowlby, R.; Tickoo, S.K.; Thorsson, V.; Mungall, A.J.; Newton, Y.; Hegde, A.M.; et al. Integrated molecular characterization of testicular germ cell tumors. Cell Rep. 2018, 23, 3392–3406. [Google Scholar] [CrossRef]
- Boccellino, M.; Vanacore, D.; Zappavigna, S.; Cavaliere, C.; Rossetti, S.; D’Aniello, C.; Chieffi, P.; Amler, E.; Buonerba, C.; Di Lorenzo, G.; et al. Testicular cancer from diagnosis to epigenetic factors. Oncotarget 2017, 8, 104654–104663. [Google Scholar] [CrossRef]
- Ilijazi, D.; Shariat, S.F.; Hassler, M.R.; Lemberger, U.; Ertl, I.E. Epigenetic alterations of testicular germ cell tumours. Curr. Opin. Urol. 2020, 30, 264–270. [Google Scholar] [CrossRef]
- Buljubašić, R.; Buljubašić, M.; Bojanac, A.K.; Ulamec, M.; Vlahović, M.; Ježek, D.; Bulić-Jakuš, F.; Sinčić, N. Epigenetics and testicular germ cell tumors. Gene 2018, 661, 22–33. [Google Scholar] [CrossRef]
- Singh, R.; Fazal, Z.; Freemantle, S.J.; Spinella, M.J. Mechanisms of cisplatin sensitivity and resistance in testicular germ cell tumors. Cancer Drug Resist. 2019, 2, 580–594. [Google Scholar] [CrossRef]
- Oing, C.; Skowron, M.A.; Bokemeyer, C.; Nettersheim, D. Epigenetic treatment combinations to effectively target cisplatin-resistant germ cell tumors: Past, present, and future considerations. Andrology 2019, 7, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, A.R.; Lobo, J.; Miranda-Gonçalves, V.; Henrique, R.; Jerónimo, C. Epigenetic alterations as therapeutic targets in Testicular Germ Cell Tumours: Current and future application of ‘epidrugs’. Epigenetics 2020, 1–20. [Google Scholar] [CrossRef]
- Jostes, S.; Nettersheim, D.; Schorle, H. Epigenetic drugs and their molecular targets in testicular germ cell tumours. Nat. Rev. Urol. 2019, 16, 245–259. [Google Scholar] [CrossRef]
- Kratz, C.P.; Mai, P.L.; Greene, M.H. Familial testicular germ cell tumours. Best Pract. Res. Clin. Endocrinol. Metab. 2010, 24, 503–513. [Google Scholar] [CrossRef]
- Kanetsky, P.A.; Mitra, N.; Vardhanabhuti, S.; Li, M.; Vaughn, D.J.; Letrero, R.; Ciosek, S.L.; Doody, D.R.; Smith, L.M.; Weaver, J.; et al. Common variation in KITLG and at 5q31.3 predisposes to testicular germ cell cancer. Nat. Genet. 2009, 41, 811–815. [Google Scholar] [CrossRef]
- Rapley, E.A.; Turnbull, C.; Al Olama, A.A.; Dermitzakis, E.T.; Linger, R.; Huddart, R.A.; Renwick, A.; Hughes, D.; Hines, S.; Seal, S.; et al. A genome-wide association study of testicular germ cell tumor. Nat. Genet. 2009, 41, 807–810. [Google Scholar] [CrossRef]
- Turnbull, C.; Rapley, E.A.; Seal, S.; Pernet, D.; Renwick, A.; Hughes, D.; Ricketts, M.; Linger, R.; Nsengimana, J.; Deloukas, P.; et al. Variants near DMRT1, TERT and ATF7IP are associated with testicular germ cell cancer. Nat. Genet. 2010, 42, 604–607. [Google Scholar] [CrossRef]
- Kanetsky, P.A.; Mitra, N.; Vardhanabhuti, S.; Vaughn, D.J.; Li, M.; Ciosek, S.L.; Letrero, R.; D’Andrea, K.; Vaddi, M.; Doody, D.R.; et al. A second independent locus within DMRT1 is associated with testicular germ cell tumor susceptibility. Hum. Mol. Genet. 2011, 20, 3109–3117. [Google Scholar] [CrossRef]
- Chung, C.C.; Kanetsky, P.A.; Wang, Z.; Hildebrandt, M.A.; Koster, R.; Skotheim, R.I.; Kratz, C.P.; Turnbull, C.; Cortessis, V.K.; Bakken, A.C.; et al. Meta-analysis identifies four new loci associated with testicular germ cell tumor. Nat. Genet. 2013, 45, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Ruark, E.; Seal, S.; McDonald, H.; Zhang, F.; Elliot, A.; Lau, K.; Perdeaux, E.; Rapley, E.; Eeles, R.; Peto, J.; et al. Identification of nine new susceptibility loci for testicular cancer, including variants near DAZL and PRDM14. Nat. Genet. 2013, 45, 686–689. [Google Scholar] [CrossRef]
- Schumacher, F.R.; Wang, Z.; Skotheim, R.I.; Koster, R.; Chung, C.C.; Hildebrandt, M.A.; Kratz, C.P.; Bakken, A.C.; Bishop, D.T.; Cook, M.B.; et al. Testicular germ cell tumor susceptibility associated with the UCK2 locus on chromosome 1q23. Hum. Mol. Genet. 2013, 22, 2748–2753. [Google Scholar] [CrossRef]
- Litchfield, K.; Holroyd, A.; Lloyd, A.; Broderick, P.; Nsengimana, J.; Eeles, R.; Easton, D.F.; Dudakia, D.; Bishop, D.T.; Reid, A.; et al. Identification of four new susceptibility loci for testicular germ cell tumour. Nat. Commun. 2015, 6, 8690. [Google Scholar] [CrossRef]
- Litchfield, K.; Sultana, R.; Renwick, A.; Dudakia, D.; Seal, S.; Ramsay, E.; Powell, S.; Elliott, A.; Warren-Perry, M.; Eeles, R.; et al. Multi-stage genome-wide association study identifies new susceptibility locus for testicular germ cell tumour on chromosome 3q25. Hum. Mol. Genet. 2015, 24, 1169–1176. [Google Scholar] [CrossRef]
- Wang, Z.; McGlynn, K.A.; Rajpert-De Meyts, E.; Bishop, D.T.; Chung, C.C.; Dalgaard, M.D.; Greene, M.H.; Gupta, R.; Grotmol, T.; Haugen, T.B.; et al. Meta-analysis of five genome-wide association studies identifies multiple new loci associated with testicular germ cell tumor. Nat. Genet. 2017, 49, 1141–1147. [Google Scholar] [CrossRef] [PubMed]
- Litchfield, K.; Loveday, C.; Levy, M.; Dudakia, D.; Rapley, E.; Nsengimana, J.; Bishop, D.T.; Reid, A.; Huddart, R.; Broderick, P.; et al. Large-scale sequencing of testicular germ cell tumour (tgct) cases excludes major TGCT predisposition gene. Eur. Urol. 2018, 73, 828–831. [Google Scholar] [CrossRef] [PubMed]
- Loveday, C.; Law, P.; Litchfield, K.; Levy, M.; Holroyd, A.; Broderick, P.; Kote-Jarai, Z.; Dunning, A.M.; Muir, K.; Peto, J.; et al. Large-scale analysis demonstrates familial testicular cancer to have polygenic aetiology. Eur. Urol. 2018, 74, 248–252. [Google Scholar] [CrossRef]
- Dieckmann, K.P.; Rube, C.; Henke, R.P. Association of Down’s syndrome and testicular cancer. J. Urol. 1997, 157, 1701–1704. [Google Scholar] [CrossRef]
- Moller, H.; Skakkebaek, N.E. Risk of testicular cancer in subfertile men: Case-control study. BMJ 1999, 318, 559–562. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, R.; Bostofte, E.; Engholm, G.; Hansen, J.; Olsen, J.H.; Skakkebaek, N.E.; Moller, H. Risk of testicular cancer in men with abnormal semen characteristics: Cohort study. BMJ 2000, 321, 789–792. [Google Scholar] [CrossRef]
- Fossa, S.D.; Chen, J.; Schonfeld, S.J.; McGlynn, K.A.; McMaster, M.L.; Gail, M.H.; Travis, L.B. Risk of contralateral testicular cancer: A population-based study of 29,515 U.S. men. J. Natl. Cancer Inst. 2005, 97, 1056–1066. [Google Scholar] [CrossRef]
- Cook, M.B.; Akre, O.; Forman, D.; Madigan, M.P.; Richiardi, L.; McGlynn, K.A. A systematic review and meta-analysis of perinatal variables in relation to the risk of testicular cancer—Experiences of the mother. Int. J. Epidemiol. 2009, 38, 1532–1542. [Google Scholar] [CrossRef]
- Cook, M.B.; Akre, O.; Forman, D.; Madigan, M.P.; Richiardi, L.; McGlynn, K.A. A systematic review and meta-analysis of perinatal variables in relation to the risk of testicular cancer—Experiences of the son. Int. J. Epidemiol. 2010, 39, 1605–1618. [Google Scholar] [CrossRef] [PubMed]
- Maule, M.; Malavassi, J.L.; Richiardi, L. Age at puberty and risk of testicular cancer: A meta-analysis. Int. J. Androl. 2012, 35, 828–834. [Google Scholar] [CrossRef]
- Trabert, B.; Zugna, D.; Richiardi, L.; McGlynn, K.A.; Akre, O. Congenital malformations and testicular germ cell tumors. Int. J. Cancer 2013, 133, 1900–1904. [Google Scholar] [CrossRef]
- Hanson, H.A.; Anderson, R.E.; Aston, K.I.; Carrell, D.T.; Smith, K.R.; Hotaling, J.M. Subfertility increases risk of testicular cancer: Evidence from population-based semen samples. Fertil. Steril. 2016, 105, 322–328.e321. [Google Scholar] [CrossRef] [PubMed]
- Skakkebaek, N.E.; Rajpert-De Meyts, E.; Buck Louis, G.M.; Toppari, J.; Andersson, A.M.; Eisenberg, M.L.; Jensen, T.K.; Jørgensen, N.; Swan, S.H.; Sapra, K.J.; et al. Male reproductive disorders and fertility trends: Influences of environment and genetic susceptibility. Physiol. Rev. 2016, 96, 55–97. [Google Scholar] [CrossRef] [PubMed]
- Piltoft, J.S.; Larsen, S.B.; Dalton, S.O.; Johansen, C.; Baker, J.L.; Cederkvist, L.; Andersen, I. Early life risk factors for testicular cancer: A case-cohort study based on the Copenhagen School Health Records Register. Acta Oncol. 2017, 56, 220–224. [Google Scholar] [CrossRef] [PubMed]
- Skakkebaek, N.E.; Rajpert-De Meyts, E.; Main, K.M. Testicular dysgenesis syndrome: An increasingly common developmental disorder with environmental aspects. Hum. Reprod. 2001, 16, 972–978. [Google Scholar] [CrossRef]
- Selvi, I.; Ozturk, E.; Yikilmaz, T.N.; Sarikaya, S.; Basar, H. Effects of testicular dysgenesis syndrome components on testicular germ cell tumor prognosis and oncological outcomes. Int. Braz. J. Urol. 2020, 46, 725–740. [Google Scholar] [CrossRef]
- Sharma, A.; Mollier, J.; Brocklesby, R.W.K.; Caves, C.; Jayasena, C.N.; Minhas, S. Endocrine-disrupting chemicals and male reproductive health. Reprod. Med. Biol. 2020, 19, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Fénichel, P.; Chevalier, N. Is testicular germ cell cancer estrogen dependent? The Role of Endocrine Disrupting Chemicals. Endocrinology 2019, 160, 2981–2989. [Google Scholar] [CrossRef]
- Bhartiya, D.; Kaushik, A. Testicular stem cell dysfunction due to environmental insults could be responsible for deteriorating reproductive health of men. Reprod. Sci. 2021, 28, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Hersmus, R.; van Bever, Y.; Wolffenbuttel, K.P.; Biermann, K.; Cools, M.; Looijenga, L.H. The biology of germ cell tumors in disorders of sex development. Clin. Genet. 2017, 91, 292–301. [Google Scholar] [CrossRef] [PubMed]
- Looijenga, L.H.J.; Kao, C.S.; Idrees, M.T. Predicting gonadal germ cell cancer in people with disorders of sex development; insights from developmental biology. Int. J. Mol. Sci. 2019, 20, 5017. [Google Scholar] [CrossRef]
- Looijenga, L.H.; Van Agthoven, T.; Biermann, K. Development of malignant germ cells-the genvironmental hypothesis. Int. J. Dev. Biol. 2013, 57, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Rajpert-De Meyts, E.; Skakkebaek, N.E.; Toppari, J. Testicular Cancer Pathogenesis, Diagnosis and Endocrine Aspects. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dungan, K., Grossman, A., Hershman, J.M., Hofland, J., Kaltsas, G., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2018. [Google Scholar]
- Nicholls, P.K.; Schorle, H.; Naqvi, S.; Hu, Y.C.; Fan, Y.; Carmell, M.A.; Dobrinski, I.; Watson, A.L.; Carlson, D.F.; Fahrenkrug, S.C.; et al. Mammalian germ cells are determined after PGC colonization of the nascent gonad. Proc. Natl. Acad. Sci. USA 2019, 116, 25677–25687. [Google Scholar] [CrossRef]
- Rajpert-De Meyts, E. Developmental model for the pathogenesis of testicular carcinoma in situ: Genetic and environmental aspects. Hum. Reprod. Update 2006, 12, 303–323. [Google Scholar] [CrossRef] [PubMed]
- Rajpert-De Meyts, E.; Nielsen, J.E.; Skakkebaek, N.E.; Almstrup, K. Diagnostic markers for germ cell neoplasms: From placental-like alkaline phosphatase to micro-RNAs. Folia Histochem. Cytobiol. 2015, 53, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Lafin, J.T.; Bagrodia, A.; Woldu, S.; Amatruda, J.F. New insights into germ cell tumor genomics. Andrology 2019, 7, 507–515. [Google Scholar] [CrossRef]
- Baker, D.E.; Harrison, N.J.; Maltby, E.; Smith, K.; Moore, H.D.; Shaw, P.J.; Heath, P.R.; Holden, H.; Andrews, P.W. Adaptation to culture of human embryonic stem cells and oncogenesis in vivo. Nat. Biotechnol. 2007, 25, 207–215. [Google Scholar] [CrossRef]
- Loveday, C.; Litchfield, K.; Proszek, P.Z.; Cornish, A.J.; Santo, F.; Levy, M.; Macintyre, G.; Holryod, A.; Broderick, P.; Dudakia, D.; et al. Genomic landscape of platinum resistant and sensitive testicular cancers. Nat. Commun. 2020, 11, 2189. [Google Scholar] [CrossRef]
- Pathak, A.; Stewart, D.R.; Faucz, F.R.; Xekouki, P.; Bass, S.; Vogt, A.; Zhang, X.; Boland, J.; Yeager, M.; Loud, J.T.; et al. Rare inactivating PDE11A variants associated with testicular germ cell tumors. Endocr. Relat. Cancer 2015, 22, 909–917. [Google Scholar] [CrossRef] [PubMed]
- Dorssers, L.C.J.; Gillis, A.J.M.; Stoop, H.; van Marion, R.; Nieboer, M.M.; van Riet, J.; van de Werken, H.J.G.; Oosterhuis, J.W.; de Ridder, J.; Looijenga, L.H.J. Molecular heterogeneity and early metastatic clone selection in testicular germ cell cancer development. Br. J. Cancer 2019, 120, 444–452. [Google Scholar] [CrossRef]
- McGlynn, K.A.; Trabert, B. Adolescent and adult risk factors for testicular cancer. Nat. Rev. Urol. 2012, 9, 339–349. [Google Scholar] [CrossRef]
- Soteriades, E.S.; Kim, J.; Christophi, C.A.; Kales, S.N. Cancer incidence and mortality in firefighters: A state-of-the-art review and meta-analysis. Asian Pac. J. Cancer Prev. 2019, 20, 3221–3231. [Google Scholar] [CrossRef]
- Romerius, P.; Giwercman, A.; Moell, C.; Relander, T.; Cavallin-Stahl, E.; Wiebe, T.; Hallden, C.; Giwercman, Y.L. Estrogen receptor alpha single nucleotide polymorphism modifies the risk of azoospermia in childhood cancer survivors. Pharm. Genom. 2011, 21, 263–269. [Google Scholar] [CrossRef]
- Richiardi, L.; Bellocco, R.; Adami, H.O.; Torrang, A.; Barlow, L.; Hakulinen, T.; Rahu, M.; Stengrevics, A.; Storm, H.; Tretli, S.; et al. Testicular cancer incidence in eight northern European countries: Secular and recent trends. Cancer Epidemiol. Biomark. Prev. 2004, 13, 2157–2166. [Google Scholar]
- Figueroa, J.D.; Sakoda, L.C.; Graubard, B.I.; Chanock, S.; Rubertone, M.V.; Erickson, R.L.; McGlynn, K.A. Genetic variation in hormone metabolizing genes and risk of testicular germ cell tumors. Cancer Causes Control 2008, 19, 917–929. [Google Scholar] [CrossRef]
- Kristiansen, W.; Haugen, T.B.; Witczak, O.; Andersen, J.M.; Fossa, S.D.; Aschim, E.L. CYP1A1, CYP3A5 and CYP3A7 polymorphisms and testicular cancer susceptibility. Int. J. Androl. 2011, 34, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Lobo, J.; Jerónimo, C.; Henrique, R. Cisplatin resistance in testicular germ cell tumors: Current challenges from various perspectives. Cancers 2020, 12, 1601. [Google Scholar] [CrossRef] [PubMed]
- Schmidtova, S.; Kalavska, K.; Kucerova, L. Molecular mechanisms of cisplatin chemoresistance and its circumventing in testicular germ cell tumors. Curr. Oncol. Rep. 2018, 20, 88. [Google Scholar] [CrossRef] [PubMed]
- Kalavska, K.; Conteduca, V.; De Giorgi, U.; Mego, M. Molecular mechanisms of resistance in testicular germ cell tumors-clinical implications. Curr. Cancer Drug Targets 2018, 18, 967–978. [Google Scholar] [CrossRef]
- Barrett, M.T.; Lenkiewicz, E.; Malasi, S.; Stanton, M.; Slack, J.; Andrews, P.; Pagliaro, L.; Bryce, A.H. Clonal analyses of refractory testicular germ cell tumors. PLoS ONE 2019, 14, e0213815. [Google Scholar] [CrossRef]
- Taylor-Weiner, A.; Zack, T.; O’Donnell, E.; Guerriero, J.L.; Bernard, B.; Reddy, A.; Han, G.C.; AlDubayan, S.; Amin-Mansour, A.; Schumacher, S.E.; et al. Genomic evolution and chemoresistance in germ-cell tumours. Nature 2016, 540, 114–118. [Google Scholar] [CrossRef]
- Bilen, M.A.; Hess, K.R.; Campbell, M.T.; Wang, J.; Broaddus, R.R.; Karam, J.A.; Ward, J.F.; Wood, C.G.; Choi, S.L.; Rao, P.; et al. Intratumoral heterogeneity and chemoresistance in nonseminomatous germ cell tumor of the testis. Oncotarget 2016, 7, 86280–86289. [Google Scholar] [CrossRef]
- Bagrodia, A.; Lee, B.H.; Lee, W.; Cha, E.K.; Sfakianos, J.P.; Iyer, G.; Pietzak, E.J.; Gao, S.P.; Zabor, E.C.; Ostrovnaya, I.; et al. Genetic determinants of cisplatin resistance in patients with advanced germ cell tumors. J. Clin. Oncol. 2016, 34, 4000–4007. [Google Scholar] [CrossRef]
- Bloom, J.C.; Loehr, A.R.; Schimenti, J.C.; Weiss, R.S. Germline genome protection: Implications for gamete quality and germ cell tumorigenesis. Andrology 2019, 7, 516–526. [Google Scholar] [CrossRef]
- Cierna, Z.; Miskovska, V.; Roska, J.; Jurkovicova, D.; Pulzova, L.B.; Sestakova, Z.; Hurbanova, L.; Machalekova, K.; Chovanec, M.; Rejlekova, K.; et al. Increased levels of XPA might be the basis of cisplatin resistance in germ cell tumours. BMC Cancer 2020, 20, 17. [Google Scholar] [CrossRef] [PubMed]
- Awuah, S.G.; Riddell, I.A.; Lippard, S.J. Repair shielding of platinum-DNA lesions in testicular germ cell tumors by high-mobility group box protein 4 imparts cisplatin hypersensitivity. Proc. Natl. Acad. Sci. USA 2017, 114, 950–955. [Google Scholar] [CrossRef]
- Mendoza, J.; Martínez, J.; Hernández, C.; Pérez-Montiel, D.; Castro, C.; Fabián-Morales, E.; Santibáñez, M.; González-Barrios, R.; Díaz-Chávez, J.; Andonegui, M.A.; et al. Association between ERCC1 and XPA expression and polymorphisms and the response to cisplatin in testicular germ cell tumours. Br. J. Cancer 2013, 109, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Usanova, S.; Piée-Staffa, A.; Sied, U.; Thomale, J.; Schneider, A.; Kaina, B.; Köberle, B. Cisplatin sensitivity of testis tumour cells is due to deficiency in interstrand-crosslink repair and low ERCC1-XPF expression. Mol. Cancer 2010, 9, 248. [Google Scholar] [CrossRef] [PubMed]
- Honecker, F.; Wermann, H.; Mayer, F.; Gillis, A.J.; Stoop, H.; van Gurp, R.J.; Oechsle, K.; Steyerberg, E.; Hartmann, J.T.; Dinjens, W.N.; et al. Microsatellite instability, mismatch repair deficiency, and BRAF mutation in treatment-resistant germ cell tumors. J. Clin. Oncol. 2009, 27, 2129–2136. [Google Scholar] [CrossRef]
- Caggiano, C.; Cavallo, F.; Giannattasio, T.; Cappelletti, G.; Rossi, P.; Grimaldi, P.; Feldman, D.R.; Jasin, M.; Barchi, M. Testicular germ cell tumors acquire cisplatin resistance by rebalancing the usage of DNA repair pathways. Cancers 2021, 13, 787. [Google Scholar] [CrossRef]
- Sakurai, Y.; Ichinoe, M.; Yoshida, K.; Nakazato, Y.; Saito, S.; Satoh, M.; Nakada, N.; Sanoyama, I.; Umezawa, A.; Numata, Y.; et al. Inactivation of REV7 enhances chemosensitivity and overcomes acquired chemoresistance in testicular germ cell tumors. Cancer Lett. 2020, 489, 100–110. [Google Scholar] [CrossRef]
- Lobo, J.; Constâncio, V.; Guimarães-Teixeira, C.; Leite-Silva, P.; Miranda-Gonçalves, V.; Sequeira, J.P.; Pistoni, L.; Guimarães, R.; Cantante, M.; Braga, I.; et al. Promoter methylation of DNA homologous recombination genes is predictive of the responsiveness to PARP inhibitor treatment in testicular germ cell tumors. Mol. Oncol. 2021. [Google Scholar] [CrossRef]
- Cavallo, F.; Graziani, G.; Antinozzi, C.; Feldman, D.R.; Houldsworth, J.; Bosl, G.J.; Chaganti, R.S.; Moynahan, M.E.; Jasin, M.; Barchi, M. Reduced proficiency in homologous recombination underlies the high sensitivity of embryonal carcinoma testicular germ cell tumors to cisplatin and poly (adp-ribose) polymerase inhibition. PLoS ONE 2012, 7, e51563. [Google Scholar] [CrossRef] [PubMed]
- Olasz, J.; Mándoky, L.; Géczi, L.; Bodrogi, I.; Csuka, O.; Bak, M. Influence of hMLH1 methylation, mismatch repair deficiency and microsatellite instability on chemoresistance of testicular germ-cell tumors. Anticancer Res. 2005, 25, 4319–4324. [Google Scholar] [PubMed]
- Kurimoto, K.; Saitou, M. Epigenome regulation during germ cell specification and development from pluripotent stem cells. Curr. Opin. Genet. Dev. 2018, 52, 57–64. [Google Scholar] [CrossRef]
- Gainetdinov, I.V.; Skvortsova, Y.V.; Kondratieva, S.A.; Klimov, A.; Tryakin, A.A.; Azhikina, T.L. Assessment of piRNA biogenesis and function in testicular germ cell tumors and their precursor germ cell neoplasia in situ. BMC Cancer 2018, 18, 20. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, H.J.; Heyn, H.; Garcia del Muro, X.; Vidal, A.; Larriba, S.; Muñoz, C.; Villanueva, A.; Esteller, M. Epigenetic loss of the PIWI/piRNA machinery in human testicular tumorigenesis. Epigenetics 2014, 9, 113–118. [Google Scholar] [CrossRef]
- Killian, J.K.; Dorssers, L.C.; Trabert, B.; Gillis, A.J.; Cook, M.B.; Wang, Y.; Waterfall, J.J.; Stevenson, H.; Smith, W.I., Jr.; Noyes, N.; et al. Imprints and DPPA3 are bypassed during pluripotency- and differentiation-coupled methylation reprogramming in testicular germ cell tumors. Genome Res. 2016, 26, 1490–1504. [Google Scholar] [CrossRef]
- Rijlaarsdam, M.A.; Tax, D.M.; Gillis, A.J.; Dorssers, L.C.; Koestler, D.C.; de Ridder, J.; Looijenga, L.H. Genome wide DNA methylation profiles provide clues to the origin and pathogenesis of germ cell tumors. PLoS ONE 2015, 10, e0122146. [Google Scholar] [CrossRef]
- Ghodoussipour, S.; Daneshmand, S. Postchemotherapy resection of residual mass in nonseminomatous germ cell tumor. Urol. Clin. N. Am. 2019, 46, 389–398. [Google Scholar] [CrossRef]
- Pierpont, T.M.; Lyndaker, A.M.; Anderson, C.M.; Jin, Q.; Moore, E.S.; Roden, J.L.; Braxton, A.; Bagepalli, L.; Kataria, N.; Hu, H.Z.; et al. Chemotherapy-induced depletion of oct4-positive cancer stem cells in a mouse model of malignant testicular cancer. Cell Rep. 2017, 21, 1896–1909. [Google Scholar] [CrossRef]
- Mueller, T.; Mueller, L.P.; Luetzkendorf, J.; Voigt, W.; Simon, H.; Schmoll, H.J. Loss of Oct-3/4 expression in embryonal carcinoma cells is associated with induction of cisplatin resistance. Tumor Biol. 2006, 27, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Mueller, T.; Mueller, L.P.; Holzhausen, H.J.; Witthuhn, R.; Albers, P.; Schmoll, H.J. Histological evidence for the existence of germ cell tumor cells showing embryonal carcinoma morphology but lacking OCT4 expression and cisplatin sensitivity. Histochem. Cell. Biol. 2010, 134, 197–204. [Google Scholar] [CrossRef]
- Gutekunst, M.; Mueller, T.; Weilbacher, A.; Dengler, M.A.; Bedke, J.; Kruck, S.; Oren, M.; Aulitzky, W.E.; van der Kuip, H. Cisplatin hypersensitivity of testicular germ cell tumors is determined by high constitutive Noxa levels mediated by Oct-4. Cancer Res. 2013, 73, 1460–1469. [Google Scholar] [CrossRef]
- Koster, R.; di Pietro, A.; Timmer-Bosscha, H.; Gibcus, J.H.; van den Berg, A.; Suurmeijer, A.J.; Bischoff, R.; Gietema, J.A.; de Jong, S. Cytoplasmic p21 expression levels determine cisplatin resistance in human testicular cancer. J. Clin. Investig. 2010, 120, 3594–3605. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.C.; Ling, T.Y.; Lu, S.H.; Kuo, H.C.; Ho, H.N.; Yeh, S.D.; Shen, C.N.; Huang, Y.H. Chemotherapeutic sensitivity of testicular germ cell tumors under hypoxic conditions is negatively regulated by SENP1-controlled sumoylation of OCT4. Cancer Res. 2012, 72, 4963–4973. [Google Scholar] [CrossRef]
- Gutekunst, M.; Oren, M.; Weilbacher, A.; Dengler, M.A.; Markwardt, C.; Thomale, J.; Aulitzky, W.E.; van der Kuip, H. p53 hypersensitivity is the predominant mechanism of the unique responsiveness of testicular germ cell tumor (TGCT) cells to cisplatin. PLoS ONE 2011, 6, e19198. [Google Scholar] [CrossRef] [PubMed]
- Nettersheim, D.; Heimsoeth, A.; Jostes, S.; Schneider, S.; Fellermeyer, M.; Hofmann, A.; Schorle, H. SOX2 is essential for in vivo reprogramming of seminoma-like TCam-2 cells to an embryonal carcinoma-like fate. Oncotarget 2016, 7, 47095–47110. [Google Scholar] [CrossRef]
- Abad, E.; Graifer, D.; Lyakhovich, A. DNA damage response and resistance of cancer stem cells. Cancer Lett. 2020, 474, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Fazal, Z.; Singh, R.; Fang, F.; Bikorimana, E.; Baldwin, H.; Corbet, A.; Tomlin, M.; Yerby, C.; Adra, N.; Albany, C.; et al. Hypermethylation and global remodelling of DNA methylation is associated with acquired cisplatin resistance in testicular germ cell tumours. Epigenetics 2020, 1–14. [Google Scholar] [CrossRef]
- Koul, S.; McKiernan, J.M.; Narayan, G.; Houldsworth, J.; Bacik, J.; Dobrzynski, D.L.; Assaad, A.M.; Mansukhani, M.; Reuter, V.E.; Bosl, G.J.; et al. Role of promoter hypermethylation in Cisplatin treatment response of male germ cell tumors. Mol. Cancer 2004, 3, 16. [Google Scholar] [CrossRef][Green Version]
- Wermann, H.; Stoop, H.; Gillis, A.J.; Honecker, F.; van Gurp, R.J.; Ammerpohl, O.; Richter, J.; Oosterhuis, J.W.; Bokemeyer, C.; Looijenga, L.H. Global DNA methylation in fetal human germ cells and germ cell tumours: Association with differentiation and cisplatin resistance. J. Pathol. 2010, 221, 433–442. [Google Scholar] [CrossRef]
- Martinelli, C.; Lengert, A.V.H.; Cárcano, F.M.; Silva, E.C.A.; Brait, M.; Lopes, L.F.; Vidal, D.O. MGMT and CALCA promoter methylation are associated with poor prognosis in testicular germ cell tumor patients. Oncotarget 2017, 8, 50608–50617. [Google Scholar] [CrossRef] [PubMed]
- Lobo, J.; Nunes, S.P.; Gillis, A.J.M.; Barros-Silva, D.; Miranda-Gonçalves, V.; Berg, A.V.D.; Cantante, M.; Guimarães, R.; Henrique, R.; Jerónimo, C.; et al. XIST-promoter demethylation as tissue biomarker for testicular germ cell tumors and spermatogenesis quality. Cancers 2019, 11, 1385. [Google Scholar] [CrossRef]
- Bo, H.; Cao, K.; Tang, R.; Zhang, H.; Gong, Z.; Liu, Z.; Liu, J.; Li, J.; Fan, L. A network-based approach to identify DNA methylation and its involved molecular pathways in testicular germ cell tumors. J. Cancer 2019, 10, 893–902. [Google Scholar] [CrossRef] [PubMed]
- Lobo, J.; Guimarães, R.; Miranda-Gonçalves, V.; Monteiro-Reis, S.; Cantante, M.; Antunes, L.; Braga, I.; Maurício, J.; Looijenga, L.H.; Jerónimo, C.; et al. Differential expression of DNA methyltransferases and demethylases among the various testicular germ cell tumor subtypes. Epigenomics 2020, 12, 1579–1592. [Google Scholar] [CrossRef]
- Lobo, J.; Henrique, R.; Jerónimo, C. The role of DNA/histone modifying enzymes and chromatin remodeling complexes in testicular germ cell tumors. Cancers 2018, 11, 6. [Google Scholar] [CrossRef]
- Albany, C.; Hever-Jardine, M.P.; von Herrmann, K.M.; Yim, C.Y.; Tam, J.; Warzecha, J.M.; Shin, L.; Bock, S.E.; Curran, B.S.; Chaudhry, A.S.; et al. Refractory testicular germ cell tumors are highly sensitive to the second generation DNA methylation inhibitor guadecitabine. Oncotarget 2017, 8, 2949–2959. [Google Scholar] [CrossRef]
- Biswal, B.K.; Beyrouthy, M.J.; Hever-Jardine, M.P.; Armstrong, D.; Tomlinson, C.R.; Christensen, B.C.; Marsit, C.J.; Spinella, M.J. Acute hypersensitivity of pluripotent testicular cancer-derived embryonal carcinoma to low-dose 5-aza deoxycytidine is associated with global DNA Damage-associated p53 activation, anti-pluripotency and DNA demethylation. PLoS ONE 2012, 7, e53003. [Google Scholar] [CrossRef] [PubMed]
- Beyrouthy, M.J.; Garner, K.M.; Hever, M.P.; Freemantle, S.J.; Eastman, A.; Dmitrovsky, E.; Spinella, M.J. High DNA methyltransferase 3B expression mediates 5-aza-deoxycytidine hypersensitivity in testicular germ cell tumors. Cancer Res. 2009, 69, 9360–9366. [Google Scholar] [CrossRef]
- Oing, C.; Verem, I.; Mansour, W.Y.; Bokemeyer, C.; Dyshlovoy, S.; Honecker, F. 5-Azacitidine exerts prolonged pro-apoptotic effects and overcomes cisplatin-resistance in non-seminomatous germ cell tumor cells. Int. J. Mol. Sci. 2018, 20, 21. [Google Scholar] [CrossRef] [PubMed]
- Wongtrakoongate, P.; Li, J.; Andrews, P.W. Aza-deoxycytidine induces apoptosis or differentiation via DNMT3B and targets embryonal carcinoma cells but not their differentiated derivatives. Br. J. Cancer 2014, 110, 2131–2138. [Google Scholar] [CrossRef]
- Lobo, J.; Cardoso, A.R.; Miranda-Gonçalves, V.; Looijenga, L.H.J.; Lopez, M.; Arimondo, P.B.; Henrique, R.; Jerónimo, C. Targeting germ cell tumors with the newly synthesized flavanone-derived compound mlo1302 efficiently reduces tumor cell viability and induces apoptosis and cell cycle arrest. Pharmaceutics 2021, 13, 73. [Google Scholar] [CrossRef]
- Albany, C.; Fazal, Z.; Singh, R.; Bikorimana, E.; Adra, N.; Hanna, N.H.; Einhorn, L.H.; Perkins, S.M.; Sandusky, G.E.; Christensen, B.C.; et al. A phase 1 study of combined guadecitabine and cisplatin in platinum refractory germ cell cancer. Cancer Med. 2020. [Google Scholar] [CrossRef]
- Dhar, S.S.; Lee, S.H.; Chen, K.; Zhu, G.; Oh, W.; Allton, K.; Gafni, O.; Kim, Y.Z.; Tomoiga, A.S.; Barton, M.C.; et al. An essential role for UTX in resolution and activation of bivalent promoters. Nucleic Acids Res. 2016, 44, 3659–3674. [Google Scholar] [CrossRef]
- Li, F.; Wan, M.; Zhang, B.; Peng, Y.; Zhou, Y.; Pi, C.; Xu, X.; Ye, L.; Zhou, X.; Zheng, L. Bivalent Histone Modifications and Development. Curr. Stem Cell Res. Ther. 2018, 13, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Lobo, J.; Barros-Silva, D.; Henrique, R.; Jerónimo, C. The emerging role of epitranscriptomics in cancer: Focus on urological tumors. Genes 2018, 9, 552. [Google Scholar] [CrossRef] [PubMed]
- Lobo, J.; Guimarães-Teixeira, C.; Barros-Silva, D.; Miranda-Gonçalves, V.; Camilo, V.; Guimarães, R.; Cantante, M.; Braga, I.; Maurício, J.; Oing, C.; et al. Efficacy of HDAC inhibitors belinostat and panobinostat against cisplatin-sensitive and cisplatin-resistant testicular germ cell tumors. Cancers 2020, 12, 2903. [Google Scholar] [CrossRef]
- Wang, D.; Li, W.; Zhao, R.; Chen, L.; Liu, N.; Tian, Y.; Zhao, H.; Xie, M.; Lu, F.; Fang, Q.; et al. Stabilized peptide HDAC inhibitors derived from HDAC1 substrate H3K56 for the treatment of cancer stem-like cells in vivo. Cancer Res. 2019, 79, 1769–1783. [Google Scholar] [CrossRef]
- Steinemann, G.; Dittmer, A.; Kuzyniak, W.; Hoffmann, B.; Schrader, M.; Schobert, R.; Biersack, B.; Nitzsche, B.; Höpfner, M. Animacroxam, a novel dual-mode compound targeting histone deacetylases and cytoskeletal integrity of testicular germ cell cancer cells. Mol. Cancer Ther. 2017, 16, 2364–2374. [Google Scholar] [CrossRef] [PubMed]
- Nettersheim, D.; Berger, D.; Jostes, S.; Skowron, M.; Schorle, H. Deciphering the molecular effects of romidepsin on germ cell tumours: DHRS2 is involved in cell cycle arrest but not apoptosis or induction of romidepsin effectors. J. Cell Mol. Med. 2019, 23, 670–679. [Google Scholar] [CrossRef]
- Nettersheim, D.; Jostes, S.; Fabry, M.; Honecker, F.; Schumacher, V.; Kirfel, J.; Kristiansen, G.; Schorle, H. A signaling cascade including ARID1A, GADD45B and DUSP1 induces apoptosis and affects the cell cycle of germ cell cancers after romidepsin treatment. Oncotarget 2016, 7, 74931–74946. [Google Scholar] [CrossRef]
- Nettersheim, D.; Gillis, A.; Biermann, K.; Looijenga, L.H.; Schorle, H. The seminoma cell line TCam-2 is sensitive to HDAC inhibitor depsipeptide but tolerates various other chemotherapeutic drugs and loss of NANOG expression. Genes Chromosomes Cancer 2011, 50, 1033–1042. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lu, F.; Ren, Q.; Sun, H.; Xu, Z.; Lan, R.; Liu, Y.; Ward, D.; Quan, J.; Ye, T.; et al. Novel histone demethylase LSD1 inhibitors selectively target cancer cells with pluripotent stem cell properties. Cancer Res. 2011, 71, 7238–7249. [Google Scholar] [CrossRef]
- Jostes, S.; Nettersheim, D.; Fellermeyer, M.; Schneider, S.; Hafezi, F.; Honecker, F.; Schumacher, V.; Geyer, M.; Kristiansen, G.; Schorle, H. The bromodomain inhibitor JQ1 triggers growth arrest and apoptosis in testicular germ cell tumours in vitro and in vivo. J. Cell Mol. Med. 2017, 21, 1300–1314. [Google Scholar] [CrossRef]
- Singh, R.; Fazal, Z.; Corbet, A.K.; Bikorimana, E.; Rodriguez, J.C.; Khan, E.M.; Shahid, K.; Freemantle, S.J.; Spinella, M.J. Epigenetic remodeling through downregulation of polycomb repressive complex 2 mediates chemotherapy resistance in testicular germ cell tumors. Cancers 2019, 11, 796. [Google Scholar] [CrossRef]
- Regouc, M.; Belge, G.; Lorch, A.; Dieckmann, K.P.; Pichler, M. Non-Coding microRNAs as Novel Potential Tumor Markers in Testicular Cancer. Cancers 2020, 12, 749. [Google Scholar] [CrossRef]
- Nappi, L.; Nichols, C. MicroRNAs as biomarkers for germ cell tumors. Urol. Clin. N. Am. 2019, 46, 449–457. [Google Scholar] [CrossRef]
- Radtke, A.; Cremers, J.F.; Kliesch, S.; Riek, S.; Junker, K.; Mohamed, S.A.; Anheuser, P.; Belge, G.; Dieckmann, K.P. Can germ cell neoplasia in situ be diagnosed by measuring serum levels of microRNA371a-3p? J. Cancer Res. Clin. Oncol. 2017, 143, 2383–2392. [Google Scholar] [CrossRef]
- Nappi, L.; Thi, M.; Adra, N.; Hamilton, R.J.; Leao, R.; Lavoie, J.M.; Soleimani, M.; Eigl, B.J.; Chi, K.; Gleave, M.; et al. Integrated expression of circulating mir375 and mir371 to identify teratoma and active germ cell malignancy components in malignant germ cell tumors. Eur. Urol. 2021, 79, 16–19. [Google Scholar] [CrossRef]
- Voorhoeve, P.M.; le Sage, C.; Schrier, M.; Gillis, A.J.; Stoop, H.; Nagel, R.; Liu, Y.P.; van Duijse, J.; Drost, J.; Griekspoor, A.; et al. A genetic screen implicates miRNA-372 and miRNA-373 as oncogenes in testicular germ cell tumors. Cell 2006, 124, 1169–1181. [Google Scholar] [CrossRef] [PubMed]
- Murray, M.J.; Saini, H.K.; Siegler, C.A.; Hanning, J.E.; Barker, E.M.; van Dongen, S.; Ward, D.M.; Raby, K.L.; Groves, I.J.; Scarpini, C.G.; et al. LIN28 Expression in malignant germ cell tumors downregulates let-7 and increases oncogene levels. Cancer Res. 2013, 73, 4872–4884. [Google Scholar] [CrossRef]
- Chen, B.F.; Suen, Y.K.; Gu, S.; Li, L.; Chan, W.Y. A miR-199a/miR-214 self-regulatory network via PSMD10, TP53 and DNMT1 in testicular germ cell tumor. Sci. Rep. 2014, 4, 6413. [Google Scholar] [CrossRef]
- Das, M.K.; Evensen, H.S.F.; Furu, K.; Haugen, T.B. miRNA-302s may act as oncogenes in human testicular germ cell tumours. Sci. Rep. 2019, 9, 9189. [Google Scholar] [CrossRef] [PubMed]
- Karakaidos, P.; Karagiannis, D.; Rampias, T. Resolving DNA Damage: Epigenetic regulation of DNA repair. Molecules 2020, 25, 2496. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H. Chromatin Remodeling and epigenetic regulation in plant DNA damage repair. Int. J. Mol. Sci. 2019, 20, 4093. [Google Scholar] [CrossRef]
- Gong, F.; Miller, K.M. Histone methylation and the DNA damage response. Mutat. Res. 2019, 780, 37–47. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, R.; Fazal, Z.; Freemantle, S.J.; Spinella, M.J. Between a Rock and a Hard Place: An Epigenetic-Centric View of Testicular Germ Cell Tumors. Cancers 2021, 13, 1506. https://doi.org/10.3390/cancers13071506
Singh R, Fazal Z, Freemantle SJ, Spinella MJ. Between a Rock and a Hard Place: An Epigenetic-Centric View of Testicular Germ Cell Tumors. Cancers. 2021; 13(7):1506. https://doi.org/10.3390/cancers13071506
Chicago/Turabian StyleSingh, Ratnakar, Zeeshan Fazal, Sarah J. Freemantle, and Michael J. Spinella. 2021. "Between a Rock and a Hard Place: An Epigenetic-Centric View of Testicular Germ Cell Tumors" Cancers 13, no. 7: 1506. https://doi.org/10.3390/cancers13071506
APA StyleSingh, R., Fazal, Z., Freemantle, S. J., & Spinella, M. J. (2021). Between a Rock and a Hard Place: An Epigenetic-Centric View of Testicular Germ Cell Tumors. Cancers, 13(7), 1506. https://doi.org/10.3390/cancers13071506