
The Fusion Gene Landscape in Taiwanese Patients with Non-Small Cell Lung Cancer


Abstract
Simple Summary
Abstract
1. Introduction
2. Results
2.1. Identification of Fusion Genes in NSCLC
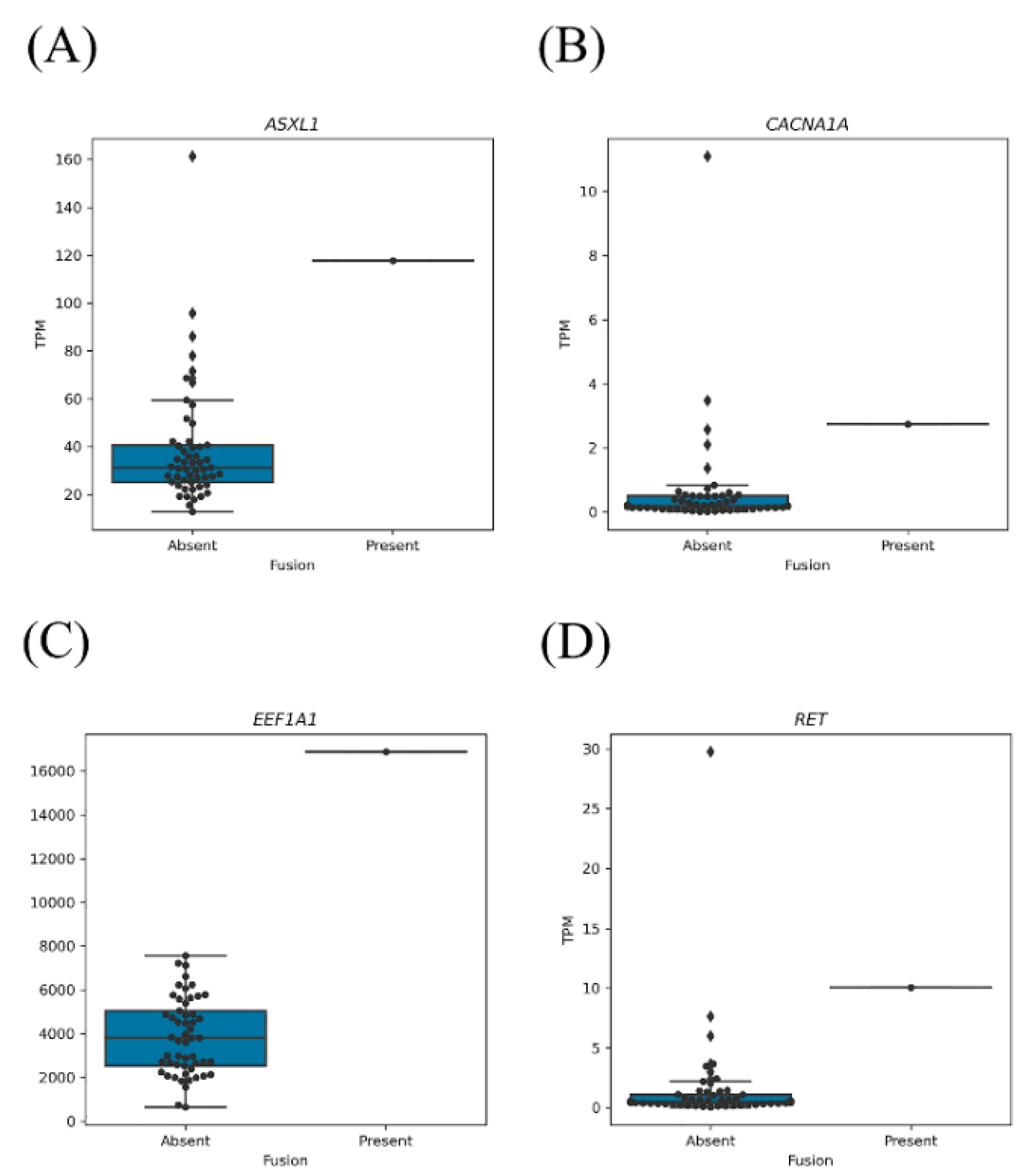
2.2. The Clinical Significance of Fusion Genes Involving Oncogenes or TSGs
2.3. Mutual Exclusivity of Gene Fusions and Gene Mutations
2.4. Correlation between EGFR Mutations and the Number of Fusion Genes
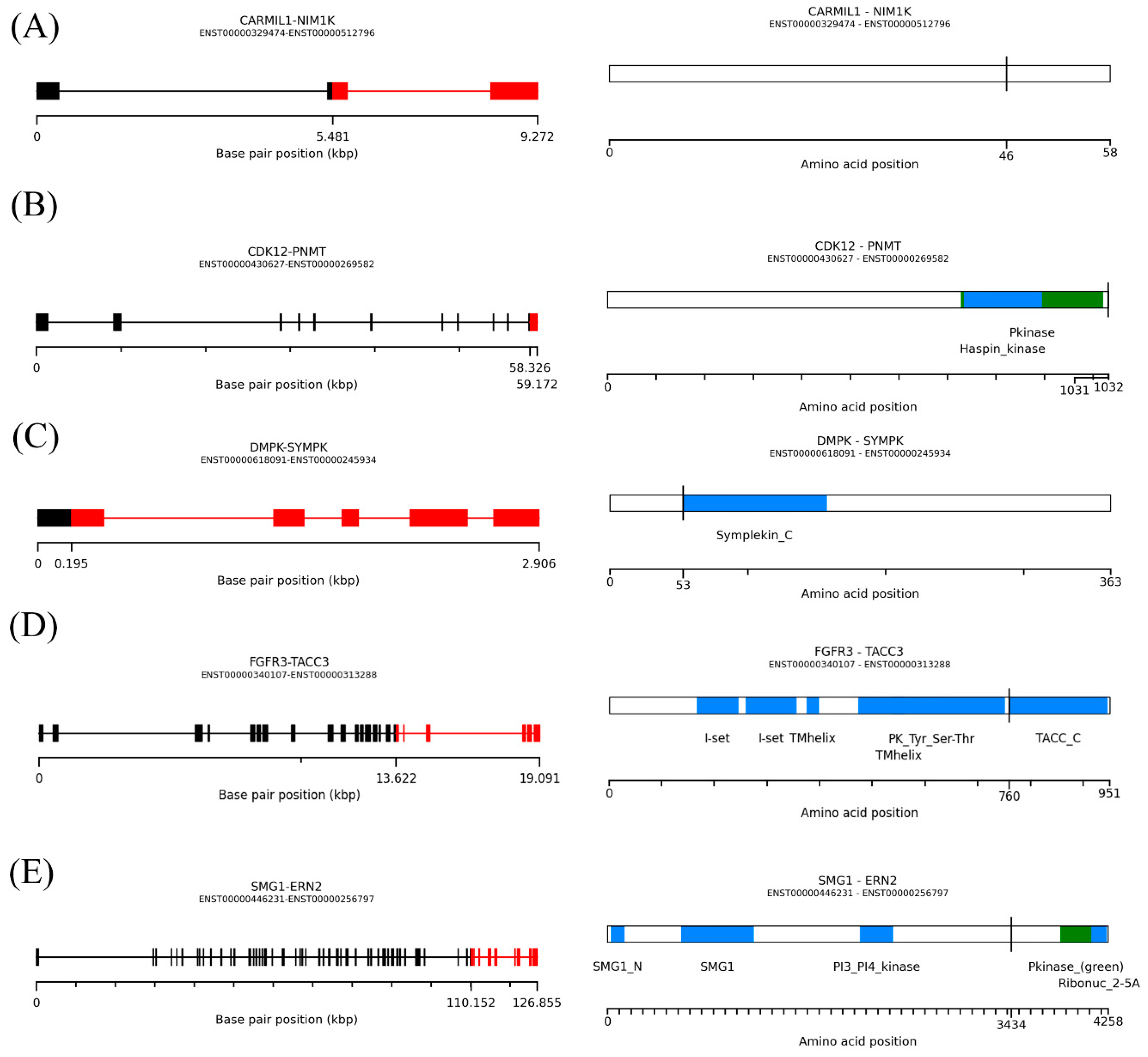
2.5. Structure and Spectrum of Kinase Gene Fusions
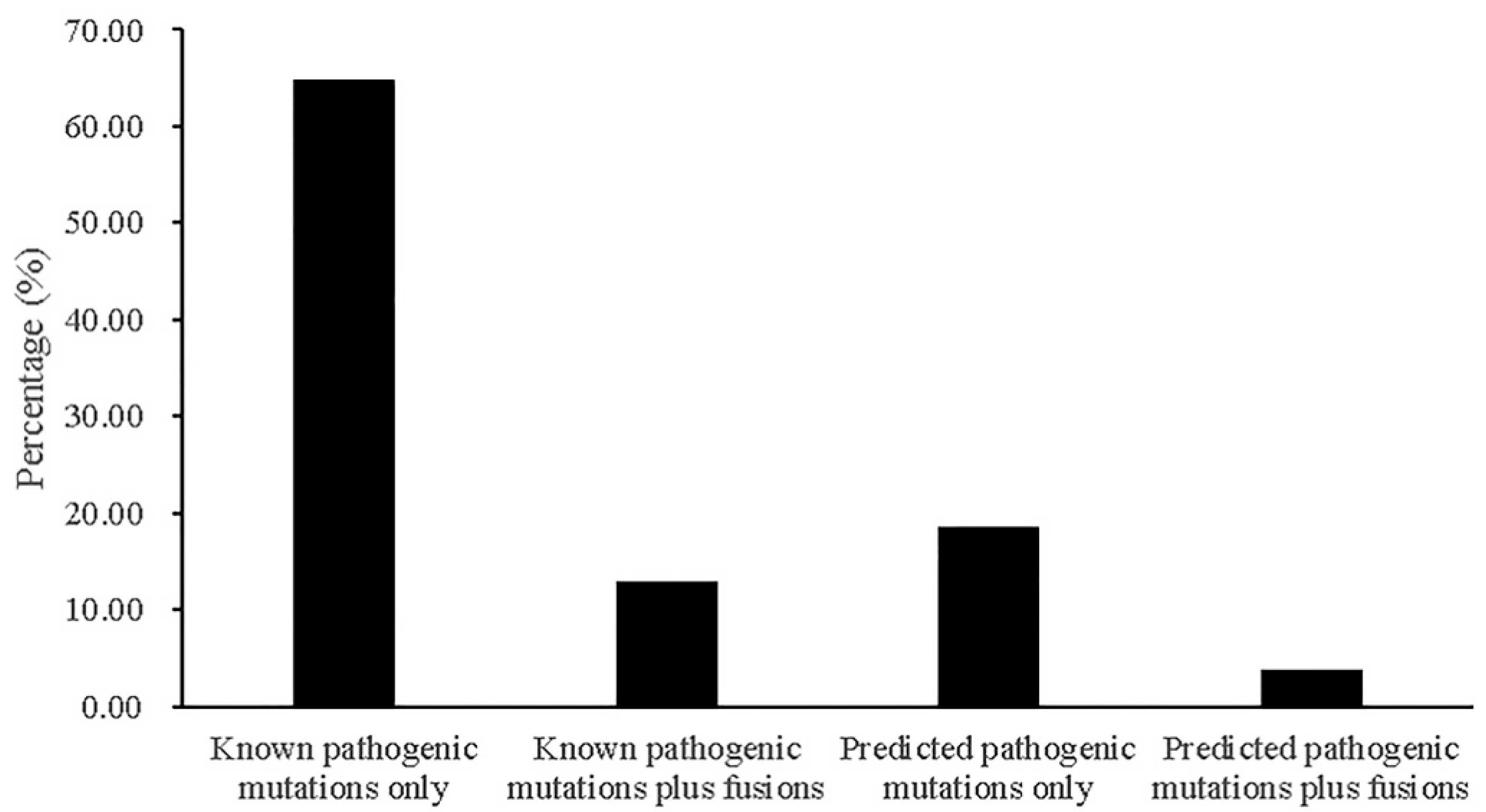
2.6. Contributions of Fusions to Cancer Treatment
3. Discussion
4. Materials and Methods
4.1. Patient Samples
4.2. RNA-Seq
4.3. RNA-Seq Data Analysis
4.4. Reverse Transcription-Polymerase Chain Reaction and Sanger Sequencing
4.5. Whole Exome Sequencing and Data Analysis
4.6. Copy Number Variation Analysis
4.7. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Bach, P.B. Smoking as a factor in causing lung cancer. JAMA 2009, 301, 539–541. [Google Scholar] [CrossRef] [PubMed]
- Gould, M.K.; Silvestri, G.A.; Detterbeck, F. Multidisciplinary management of lung cancer. N. Engl. J. Med. 2004, 350, 2008–2010. [Google Scholar] [PubMed]
- Ettinger, D.S.; Akerley, W.; Bepler, G.; Blum, M.G.; Chang, A.; Cheney, R.T.; Chirieac, L.R.; D’Amico, T.A.; Demmy, T.L.; Ganti, A.K.; et al. Non-small cell lung cancer. J. Natl. Compr. Canc. Netw. 2010, 8, 740–801. [Google Scholar] [CrossRef]
- Zhang, X.C.; Wang, J.; Shao, G.G.; Wang, Q.; Qu, X.; Wang, B.; Moy, C.; Fan, Y.; Albertyn, Z.; Huang, X.; et al. Comprehensive genomic and immunological characterization of Chinese non-small cell lung cancer patients. Nat. Commun. 2019, 10, 1772. [Google Scholar] [CrossRef]
- Ruiz-Cordero, R.; Devine, W.P. Targeted therapy and checkpoint immunotherapy in lung cancer. Surg. Pathol. Clin. 2020, 13, 17–33. [Google Scholar] [CrossRef]
- Gao, Q.; Liang, W.W.; Foltz, S.M.; Mutharasu, G.; Jayasinghe, R.G.; Cao, S.; Liao, W.W.; Reynolds, S.M.; Wyczalkowski, M.A.; Yao, L.; et al. Driver fusions and their implications in the development and treatment of human cancers. Cell Rep. 2018, 23, 227–238 e223. [Google Scholar] [CrossRef] [PubMed]
- Aurias, A.; Rimbaut, C.; Buffe, D.; Dubousset, J.; Mazabraud, A. Translocation of chromosome 22 in Ewing’s sarcoma. C. R. Seances Acad. Sci. III 1983, 296, 1105–1107. [Google Scholar] [PubMed]
- Rowley, J.D. Letter: A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature 1973, 243, 290–293. [Google Scholar] [CrossRef] [PubMed]
- Dinh, T.A.; Vitucci, E.C.; Wauthier, E.; Graham, R.P.; Pitman, W.A.; Oikawa, T.; Chen, M.; Silva, G.O.; Greene, K.G.; Torbenson, M.S.; et al. Comprehensive analysis of The Cancer Genome Atlas reveals a unique gene and non-coding RNA signature of fibrolamellar carcinoma. Sci. Rep. 2017, 7, 44653. [Google Scholar] [CrossRef]
- Nikanjam, M.; Okamura, R.; Barkauskas, D.A.; Kurzrock, R. Targeting fusions for improved outcomes in oncology treatment. Cancer 2020, 126, 1315–1321. [Google Scholar] [CrossRef]
- Lee, M.; Lee, K.; Yu, N.; Jang, I.; Choi, I.; Kim, P.; Jang, Y.E.; Kim, B.; Kim, S.; Lee, B.; et al. ChimerDB 3.0: An enhanced database for fusion genes from cancer transcriptome and literature data mining. Nucleic Acids Res. 2017, 45, D784–D789. [Google Scholar] [CrossRef]
- Hu, X.; Wang, Q.; Tang, M.; Barthel, F.; Amin, S.; Yoshihara, K.; Lang, F.M.; Martinez-Ledesma, E.; Lee, S.H.; Zheng, S.; et al. TumorFusions: An integrative resource for cancer-associated transcript fusions. Nucleic Acids Res. 2018, 46, D1144–D1149. [Google Scholar] [CrossRef]
- Stransky, N.; Cerami, E.; Schalm, S.; Kim, J.L.; Lengauer, C. The landscape of kinase fusions in cancer. Nat. Commun. 2014, 5, 4846. [Google Scholar] [CrossRef] [PubMed]
- Rudin, C.M.; Durinck, S.; Stawiski, E.W.; Poirier, J.T.; Modrusan, Z.; Shames, D.S.; Bergbower, E.A.; Guan, Y.; Shin, J.; Guillory, J.; et al. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nat. Genet. 2012, 44, 1111–1116. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.S.; Ju, Y.S.; Lee, W.C.; Shin, J.Y.; Lee, J.K.; Bleazard, T.; Lee, J.; Jung, Y.J.; Kim, J.O.; Shin, J.Y.; et al. The transcriptional landscape and mutational profile of lung adenocarcinoma. Genome Res. 2012, 22, 2109–2119. [Google Scholar] [CrossRef]
- Kim, S.C.; Jung, Y.; Park, J.; Cho, S.; Seo, C.; Kim, J.; Kim, P.; Park, J.; Seo, J.; Kim, J.; et al. A high-dimensional, deep-sequencing study of lung adenocarcinoma in female never-smokers. PLoS ONE 2013, 8, e55596. [Google Scholar] [CrossRef] [PubMed]
- Miyanaga, A.; Masuda, M.; Motoi, N.; Tsuta, K.; Nakamura, Y.; Nishijima, N.; Watanabe, S.I.; Asamura, H.; Tsuchida, A.; Seike, M.; et al. Whole-exome and RNA sequencing of pulmonary carcinoid reveals chromosomal rearrangements associated with recurrence. Lung Cancer 2020, 145, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive characterization of cancer driver genes and mutations. Cell 2018, 173, 371–385. [Google Scholar] [CrossRef]
- Choi, Y.; Kwon, C.H.; Lee, S.J.; Park, J.; Shin, J.Y.; Park, D.Y. Integrative analysis of oncogenic fusion genes and their functional impact in colorectal cancer. Br. J. Cancer 2018, 119, 230–240. [Google Scholar] [CrossRef] [PubMed]
- Manier, S.; Salem, K.Z.; Park, J.; Landau, D.A.; Getz, G.; Ghobrial, I.M. Genomic complexity of multiple myeloma and its clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 100–113. [Google Scholar] [CrossRef]
- Lu, H.; Villafane, N.; Dogruluk, T.; Grzeskowiak, C.L.; Kong, K.; Tsang, Y.H.; Zagorodna, O.; Pantazi, A.; Yang, L.; Neill, N.J.; et al. Engineering and functional characterization of fusion genes identifies novel oncogenic drivers of cancer. Cancer Res. 2017, 77, 3502–3512. [Google Scholar] [CrossRef]
- Shaw, A.T.; Kim, D.W.; Mehra, R.; Tan, D.S.; Felip, E.; Chow, L.Q.; Camidge, D.R.; Vansteenkiste, J.; Sharma, S.; De Pas, T.; et al. Ceritinib in ALK-rearranged non-small-cell lung cancer. N. Engl. J. Med. 2014, 370, 1189–1197. [Google Scholar] [CrossRef]
- Deininger, M.W.; Druker, B.J. Specific targeted therapy of chronic myelogenous leukemia with imatinib. Pharmacol. Rev. 2003, 55, 401–423. [Google Scholar] [CrossRef] [PubMed]
- Font-Tello, A.; Juanpere, N.; de Muga, S.; Lorenzo, M.; Lorente, J.A.; Fumado, L.; Serrano, L.; Serrano, S.; Lloreta, J.; Hernandez, S. Association of ERG and TMPRSS2-ERG with grade, stage, and prognosis of prostate cancer is dependent on their expression levels. Prostate 2015, 75, 1216–1226. [Google Scholar] [CrossRef] [PubMed]
- Kurozumi, K.; Nakano, Y.; Ishida, J.; Tanaka, T.; Doi, M.; Hirato, J.; Yoshida, A.; Washio, K.; Shimada, A.; Kohno, T.; et al. High-grade glioneuronal tumor with an ARHGEF2-NTRK1 fusion gene. Brain Tumor Pathol. 2019, 36, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Rosenkranz, A.A.; Slastnikova, T.A. Epidermal Growth Factor Receptor: Key to Selective Intracellular Delivery. Biochemistry 2020, 85, 967–1092. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.S.; Tu, S.J.; Chiang, H.S.; Yen, J.C.; Lee, Y.T.; Fang, H.Y.; Chang, J.G. Genome-wide analysis of prognostic alternative splicing signature and splicing factors in lung adenocarcinoma. Genes 2020, 11, 1300. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Haas, B.J.; Dobin, A.; Li, B.; Stransky, N.; Pochet, N.; Regev, A. Accuracy assessment of fusion transcript detection via read-mapping and de novo fusion transcript assembly-based methods. Genome Biol. 2019, 20, 213. [Google Scholar] [CrossRef] [PubMed]
- Yoshihara, K.; Wang, Q.; Torres-Garcia, W.; Zheng, S.; Vegesna, R.; Kim, H.; Verhaak, R.G. The landscape and therapeutic relevance of cancer-associated transcript fusions. Oncogene 2015, 34, 4845–4854. [Google Scholar] [CrossRef]
- Klijn, C.; Durinck, S.; Stawiski, E.W.; Haverty, P.M.; Jiang, Z.; Liu, H.; Degenhardt, J.; Mayba, O.; Gnad, F.; Liu, J.; et al. A comprehensive transcriptional portrait of human cancer cell lines. Nat. Biotechnol. 2015, 33, 306–312. [Google Scholar] [CrossRef]
- Meszaros, B.; Erdos, G.; Dosztanyi, Z. IUPred2A: Context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res. 2018, 46, W329–W337. [Google Scholar] [CrossRef]
- Murphy, C.; Elemento, O. AGFusion: Annotate and visualize gene fusions. bioRxiv 2016. [Google Scholar] [CrossRef]
- Sun, S.Q.; Mashl, R.J.; Sengupta, S.; Scott, A.D.; Wang, W.; Batra, P.; Wang, L.B.; Wyczalkowski, M.A.; Ding, L. Database of evidence for precision oncology portal. Bioinformatics 2018, 34, 4315–4317. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Chang, Y.S.; Huang, H.Y.; Yeh, K.T.; Liu, T.C.; Chang, J.G. Determination of the mutational landscape in Taiwanese patients with papillary thyroid cancer by whole-exome sequencing. Hum. Pathol. 2018, 78, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Geoffroy, V.; Herenger, Y.; Kress, A.; Stoetzel, C.; Piton, A.; Dollfus, H.; Muller, J. AnnotSV: An integrated tool for structural variations annotation. Bioinformatics 2018, 34, 3572–3574. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Features | Fusion Genes Involving Oncogenes | p-Value | Fusion Genes Involving TSGs | p-Value | |||||
|---|---|---|---|---|---|---|---|---|---|
| No | Yes | Total | No | Yes | Total | ||||
| Gender | Male | 26 | 2 | 28 | 0.6633 | 26 | 2 | 28 | 0.6633 |
| Female | 23 | 3 | 26 | 23 | 3 | 26 | |||
| Age | <70 | 25 | 3 | 28 | 1.000 | 28 | 0 | 28 | 0.0208 |
| ≥70 | 24 | 2 | 26 | 21 | 5 | 26 | |||
| Stage | I-II | 34 | 2 | 36 | 0.1626 | 33 | 3 | 36 | 0.6374 |
| III-IV | 13 | 3 | 16 | 14 | 2 | 16 | |||
| Grade | 1-2 | 38 | 1 | 39 | 0.0086 | 38 | 1 | 39 | 0.0363 |
| 3-4 | 8 | 4 | 12 | 9 | 3 | 12 | |||
| Histology type | ADC | 41 | 3 | 44 | 0.2273 | 39 | 5 | 44 | 0.5707 |
| SqCC | 8 | 2 | 10 | 10 | 0 | 10 | |||
| Smoking status | Non-smokers | 32 | 3 | 35 | 1.000 | 31 | 4 | 35 | 0.6461 |
| Smokers | 17 | 2 | 19 | 18 | 1 | 17 | |||
| Sample | Gains of Oncogenes | Losses of TSGs |
|---|---|---|
| Lung-T151 | KIT, CEBPA, PLCB4 | ATM, KMT2A, MEN1, WT1, STK11 |
| Lung-T218 | DICER1, GNAS, DMD | none |
| Lung-T227 | SMARCA4 | none |
| Lung-T236 | KIT, RET, DICER1, GNAS, COL5A1 | none |
| Lung-T291 | KIT, CARD11, WT1, DICER1, MAX, CDH1, EP300 | none |
| Lung-T95 | MSH3, PTPN11, DICER1, CDH1, EP300, MYH9 | BAP1, ATM, CHD8, LZTR1, CDKN2A |
| Lung-T430 | GNAS, MACF1 | none |
| Features | Genotype | Fusion Genes Involving 299 CDGs | p-Value | Number of Fusion Genes | p-Value | ||||
|---|---|---|---|---|---|---|---|---|---|
| No | Yes | Total | <5 | ≥5 | Total | ||||
| EGFR | Mutation | 22 | 5 | 27 | 1.000 | 12 | 15 | 27 | 0.0038 |
| Wildtype | 23 | 4 | 27 | 23 | 4 | 27 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, Y.-S.; Tu, S.-J.; Yen, J.-C.; Lee, Y.-T.; Fang, H.-Y.; Chang, J.-G. The Fusion Gene Landscape in Taiwanese Patients with Non-Small Cell Lung Cancer. Cancers 2021, 13, 1343. https://doi.org/10.3390/cancers13061343
Chang Y-S, Tu S-J, Yen J-C, Lee Y-T, Fang H-Y, Chang J-G. The Fusion Gene Landscape in Taiwanese Patients with Non-Small Cell Lung Cancer. Cancers. 2021; 13(6):1343. https://doi.org/10.3390/cancers13061343
Chicago/Turabian StyleChang, Ya-Sian, Siang-Jyun Tu, Ju-Chen Yen, Ya-Ting Lee, Hsin-Yuan Fang, and Jan-Gowth Chang. 2021. "The Fusion Gene Landscape in Taiwanese Patients with Non-Small Cell Lung Cancer" Cancers 13, no. 6: 1343. https://doi.org/10.3390/cancers13061343
APA StyleChang, Y.-S., Tu, S.-J., Yen, J.-C., Lee, Y.-T., Fang, H.-Y., & Chang, J.-G. (2021). The Fusion Gene Landscape in Taiwanese Patients with Non-Small Cell Lung Cancer. Cancers, 13(6), 1343. https://doi.org/10.3390/cancers13061343