
Genomic Instability Profiles at the Single Cell Level in Mouse Colorectal Cancers of Defined Genotypes

 , and
, and
Abstract
Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
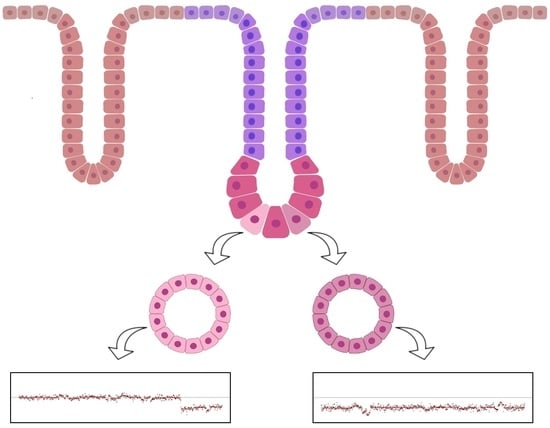
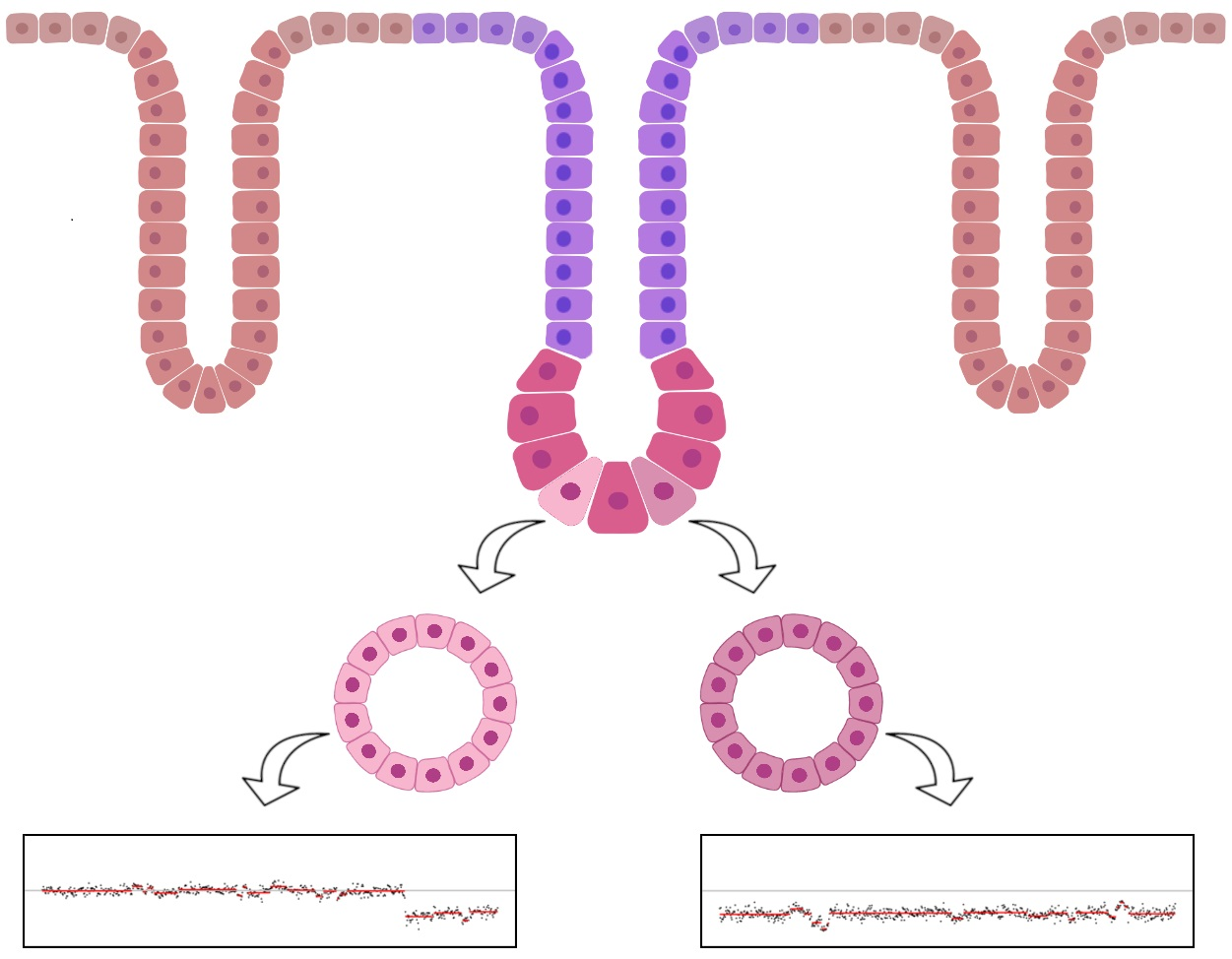
2.1. Clonal Organoid Cultures Derived from Single Tumoral Cells
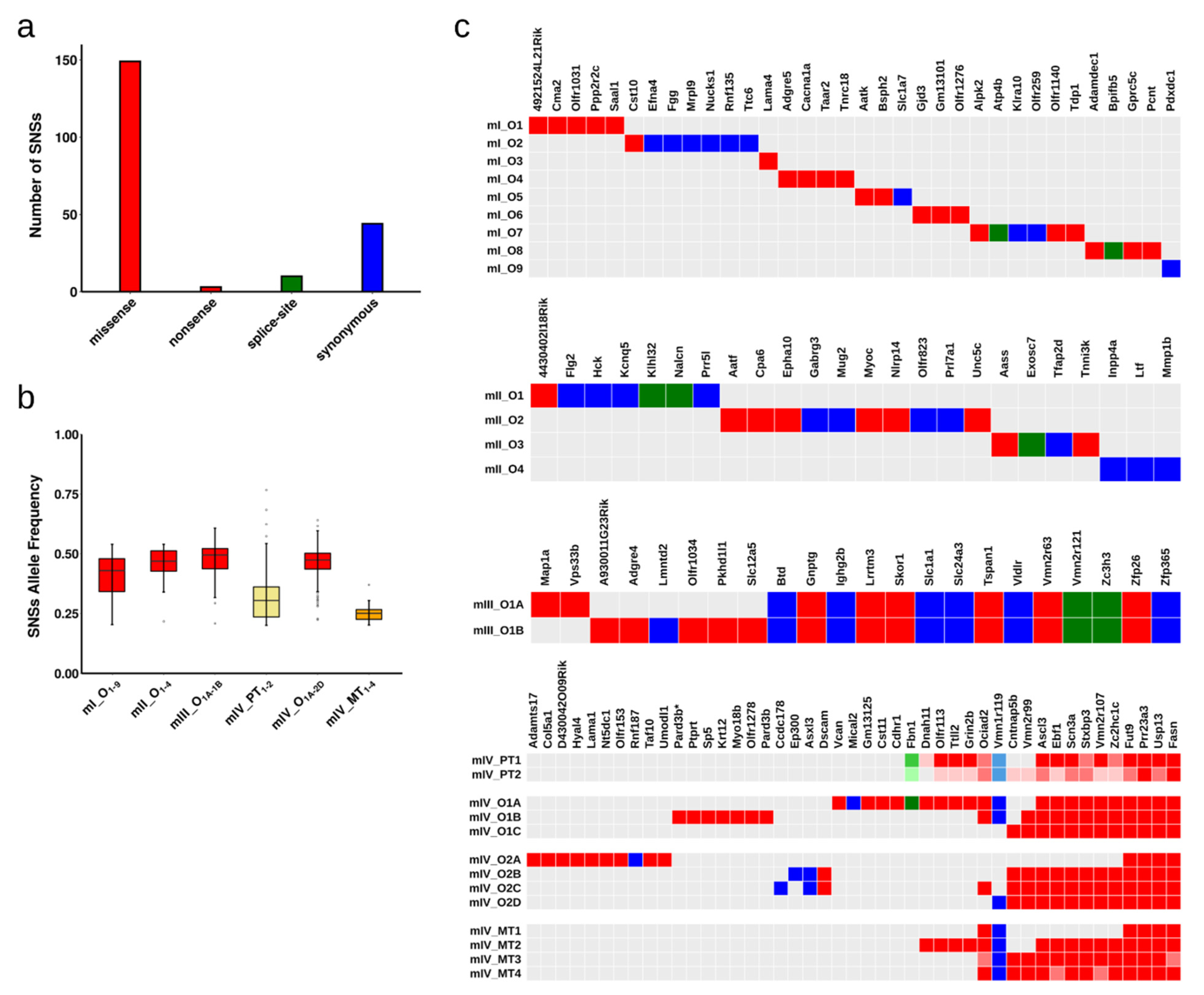
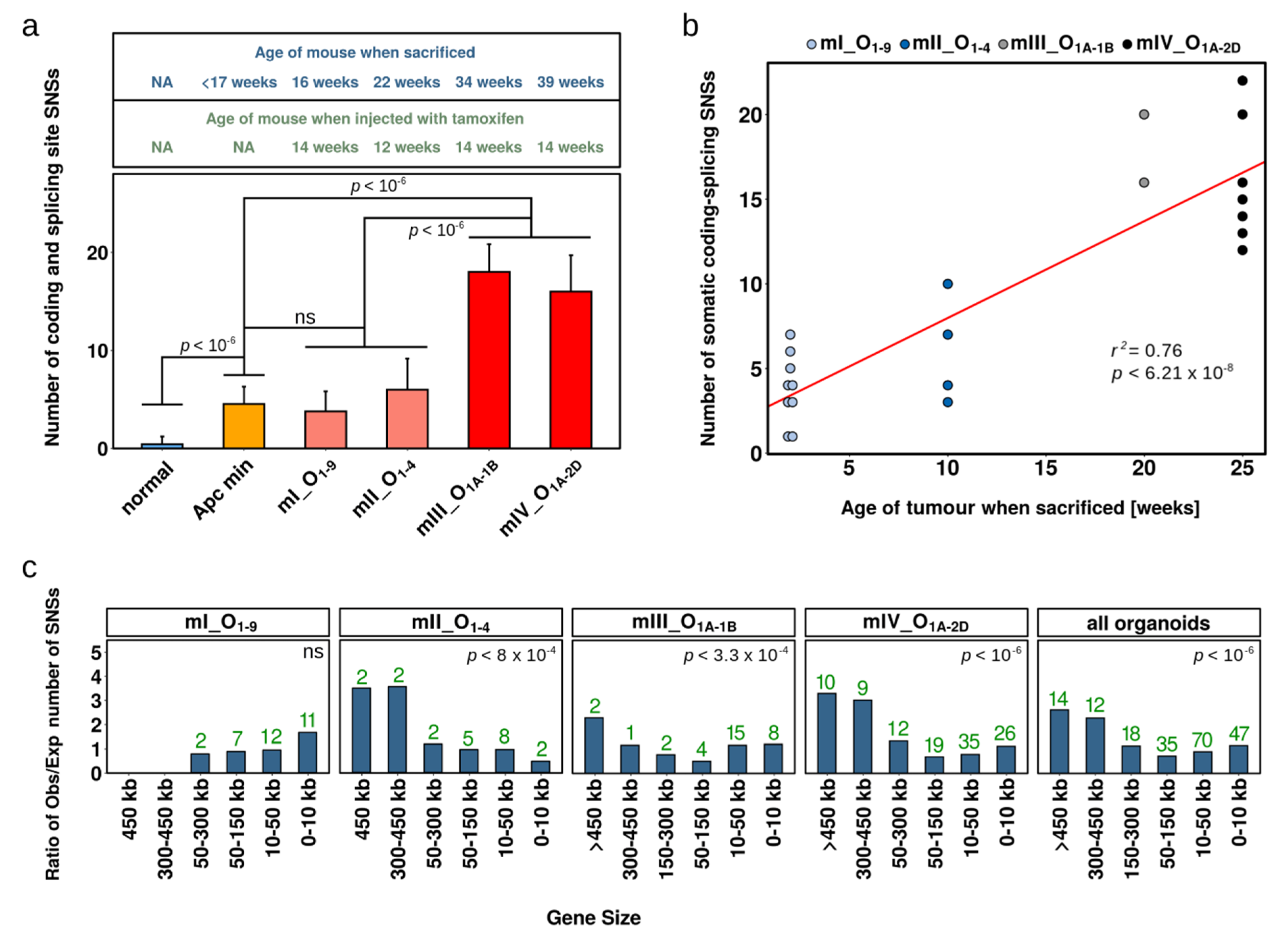
2.2. Single Nucleotide Substitutions—Prevalence
2.3. Single Nucleotide Substitutions—Link to Genotype and Distribution
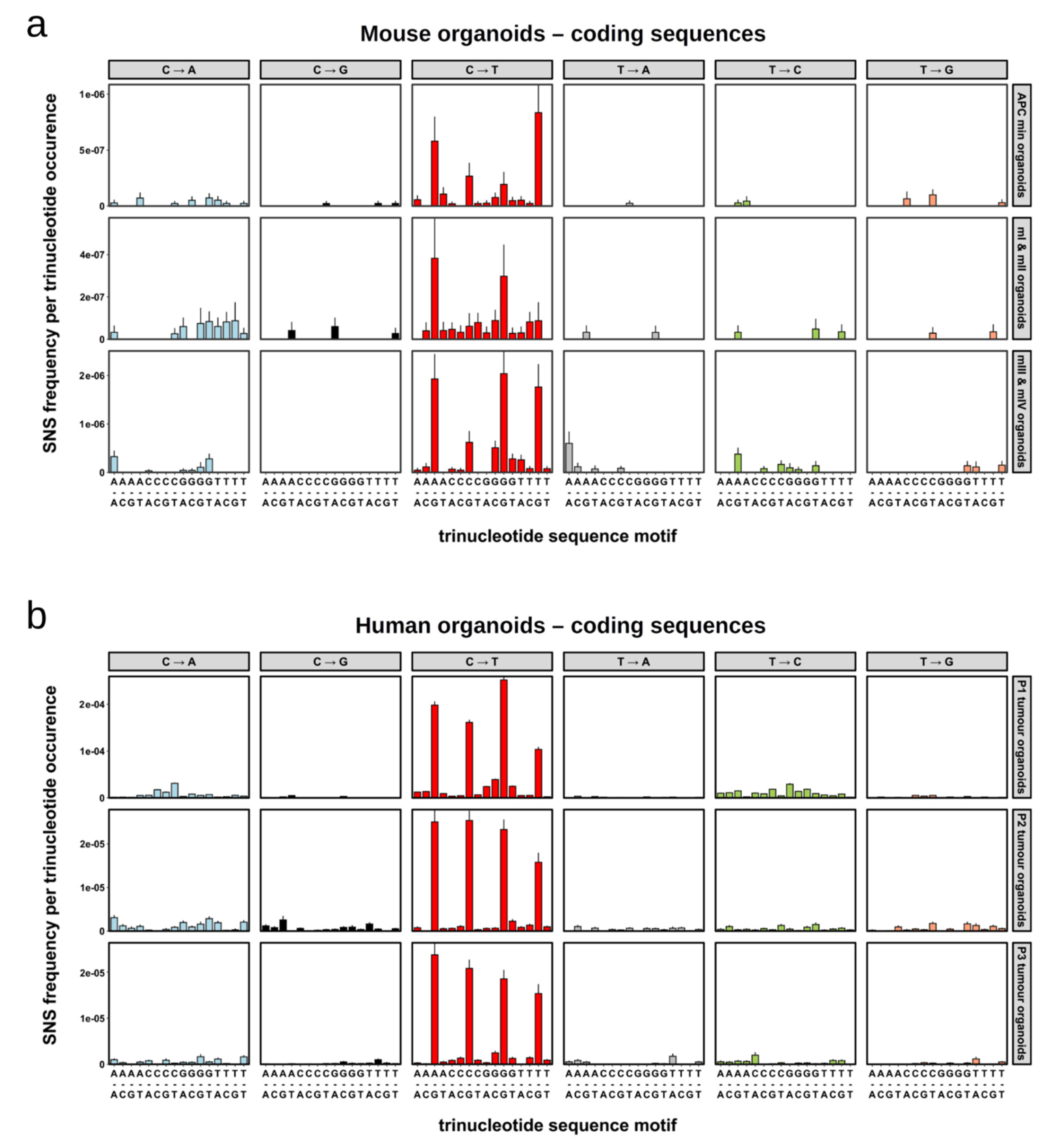
2.4. Single Nucleotide Substitutions—Mutational Signature
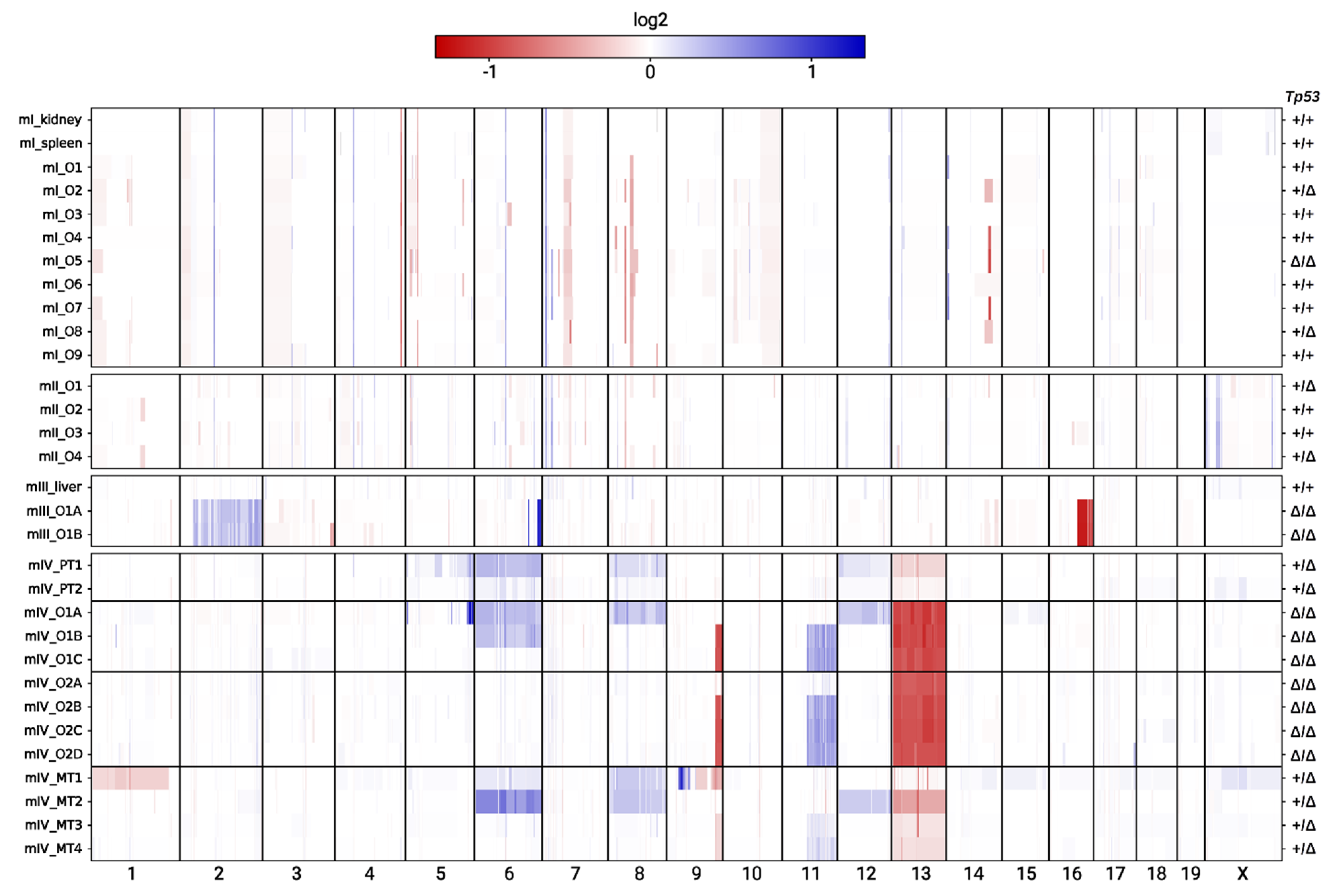
2.5. Copy Number Alterations
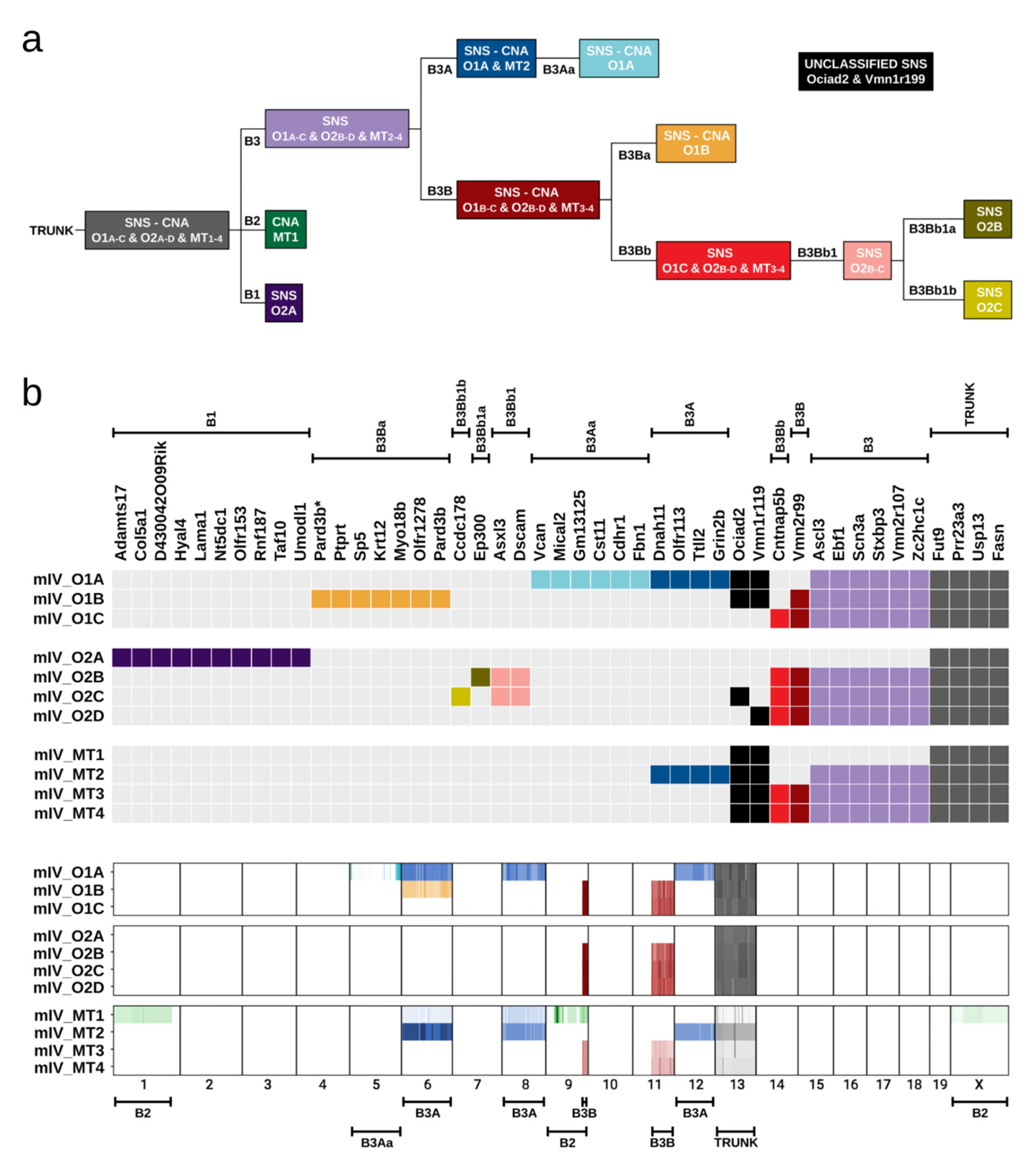
2.6. Evolution of Tumour Clones in Mouse IV
3. Discussion
4. Materials and Methods
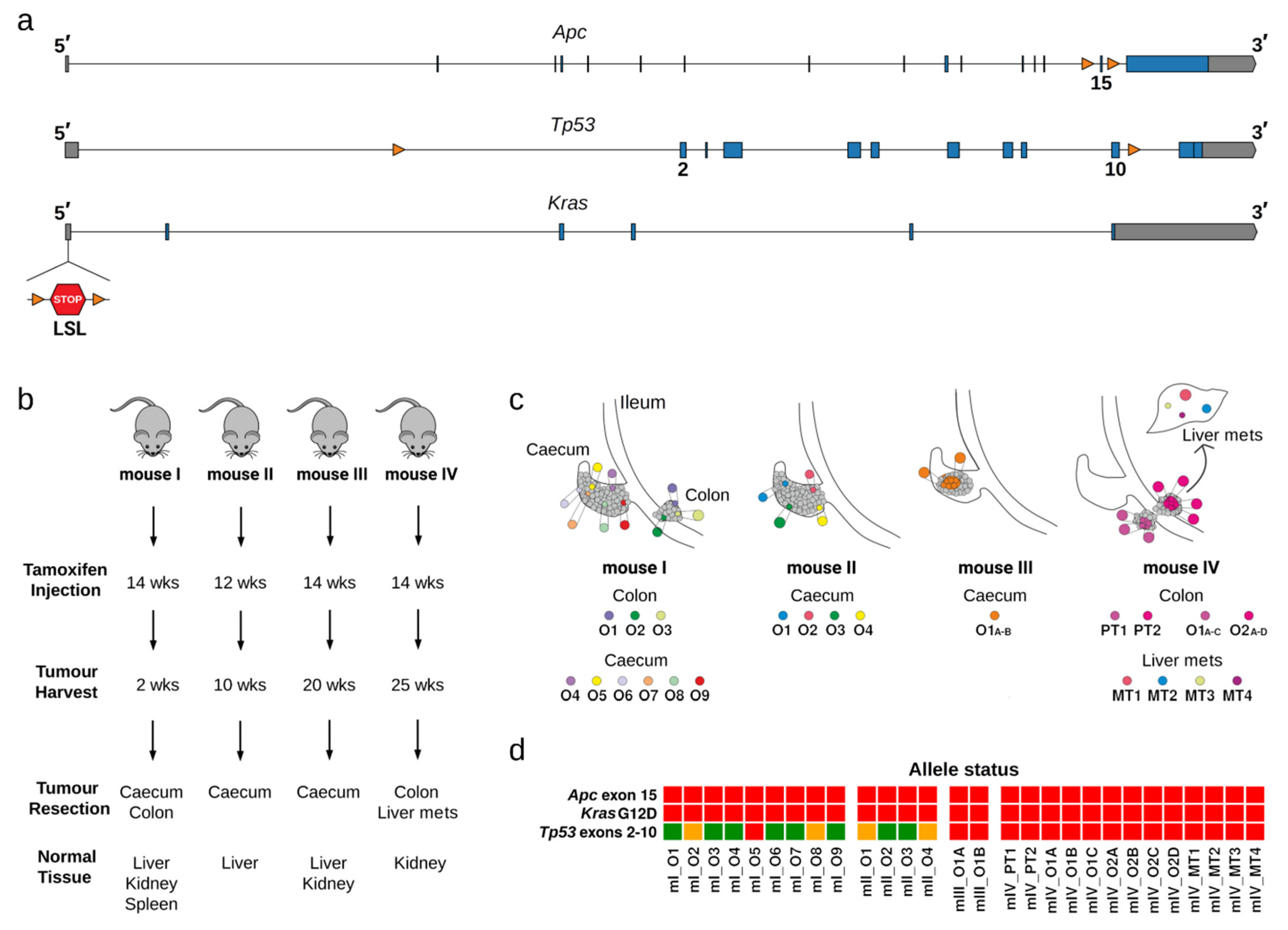
4.1. Mice
4.2. Histopathology of Tumour and Normal Caecum Tissues
4.3. Genotyping
4.4. Induction of Tumour Formation
4.5. Tissue Isolation, Organoid Culture, and Expansion
4.6. Measurement of Cell Doubling Times in Organoids
4.7. DNA Extraction and Exome Sequencing
4.8. Sequence Analysis
4.9. SNS Signature Normalisation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kinzler, K.W.; Vogelstein, B. Lessons from hereditary colorectal cancer. Cell 1996, 87, 159–170. [Google Scholar] [CrossRef]
- Hamilton, S.R.; Powell, S.M.; Papadopoulos, N.; Smith, K.J.; Vogelstein, B.; Kinzler, K.W. Molecular Determinants of Dysplasia in Colorectal Lesions. Cancer Res. 1994, 54, 5523–5526. [Google Scholar]
- Vogelstein, B.; Kinzler, K.W. The multistep nature of cancer. Trends Genet. 1993, 9, 138–141. [Google Scholar] [CrossRef]
- Powell, S.M.; Zilz, N.; Beazer-Barclay, Y.; Bryan, T.M.; Hamilton, S.R.; Thibodeau, S.N.; Vogelstein, B.; Kinzler, K.W. APC mutations occur early during colorectal tumorigenesis. Nature 1992, 359, 235–237. [Google Scholar] [CrossRef] [PubMed]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Vogelstein, B.; Fearon, E.R.; Hamilton, S.R.; Kern, S.E.; Preisinger, A.C.; Leppert, M.; Smits, A.M.M.; Bos, J.L. Genetic Alterations during Colorectal-Tumor Development. N. Engl. J. Med. 1988, 319, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Roberts, D.M.; Pronobis, M.I.; Poulton, J.S.; Kane, E.G.; Peifer, M. Regulation of Wnt signaling by the tumor suppressor adenomatous polyposis coli does not require the ability to enter the nucleus or a particular cytoplasmic localization. Mol. Biol. Cell 2012, 23, 2041–2056. [Google Scholar] [CrossRef]
- Ghazvini, M.; Sonneveld, P.; Kremer, A.; Franken, P.; Sacchetti, A.; Atlasi, Y.; Roth, S.; Joosten, R.; Smits, R.; Fodde, R. Cancer Stemness in Apc- vs. Apc/KRAS-Driven Intestinal Tumorigenesis. PLoS ONE 2013, 8, e73872. [Google Scholar] [CrossRef] [PubMed]
- Schneikert, J.; Behrens, J. The canonical Wnt signalling pathway and its APC partner in colon cancer development. Gut 2007, 56, 417–425. [Google Scholar] [CrossRef]
- Stastna, M.; Janeckova, L.; Hrckulak, D.; Kriz, V.; Korinek, V. Human colorectal cancer from the perspective of mouse models. Genes 2019, 10, 788. [Google Scholar] [CrossRef]
- Tauriello, D.V.F.; Calon, A.; Lonardo, E.; Batlle, E. Determinants of metastatic competency in colorectal cancer. Mol. Oncol. 2017, 11, 97–119. [Google Scholar] [CrossRef]
- Jackstadt, R.; Sansom, O.J. Mouse models of intestinal cancer. J. Pathol. 2016, 238, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Sottoriva, A.; Kang, H.; Ma, Z.; Graham, T.A.; Salomon, M.P.; Zhao, J.; Marjoram, P.; Siegmund, K.; Press, M.F.; Shibata, D.; et al. A big bang model of human colorectal tumor growth. Nat. Genet. 2015, 47, 209–216. [Google Scholar] [CrossRef]
- Rodrigues, N.R.; Rowan, A.; Smith, M.E.F.; Kerr, I.B.; Bodmer, W.F.; Gannon, J.V.; Lane, D.P. p53 mutations in colorectal cancer. Proc. Natl. Acad. Sci. USA 1990, 87, 7555–7559. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, R.A. Oncogenes, Antioncogenes, and the Molecular Bases of Multistep Carcinogenesis. Cancer Res. 1989, 49, 3713–3721. [Google Scholar] [PubMed]
- Janssen, K.P.; Alberici, P.; Fsihi, H.; Gaspar, C.; Breukel, C.; Franken, P.; Rosty, C.; Abal, M.; El Marjou, F.; Smits, R.; et al. APC and Oncogenic KRAS Are Synergistic in Enhancing Wnt Signaling in Intestinal Tumor Formation and Progression. Gastroenterology 2006, 131, 1096–1109. [Google Scholar] [CrossRef]
- Halazonetis, T.D.; Gorgoulis, V.G.; Bartek, J. An oncogene-induced DNA damage model for cancer development. Science 2008, 319, 1352–1355. [Google Scholar] [CrossRef]
- Moser, A.R.; Pitot, H.C.; Dove, W.F. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science 1990, 247, 322–324. [Google Scholar] [CrossRef]
- Shibata, H.; Toyama, K.; Shioya, H.; Ito, M.; Hirota, M.; Hasegawa, S.; Matsumoto, H.; Takano, H.; Akiyama, T.; Toyoshima, K.; et al. Rapid colorectal adenoma formation initiated by conditional targeting of the APC gene. Science 1997, 278, 120–123. [Google Scholar] [CrossRef] [PubMed]
- Sansom, O.J.; Meniel, V.; Wilkins, J.A.; Cole, A.M.; Oien, K.A.; Marsh, V.; Jamieson, T.J.; Guerra, C.; Ashton, G.H.; Barbacid, M.; et al. Loss of Apc allows phenotypic manifestation of the transforming properties of an endogenous K-ras oncogene in vivo. Proc. Natl. Acad. Sci. USA 2006, 103, 14122–14127. [Google Scholar] [CrossRef]
- Hung, K.E.; Maricevich, M.A.; Richard, L.G.; Chen, W.Y.; Richardson, M.P.; Kunin, A.; Bronson, R.T.; Mahmood, U.; Kucherlapati, R. Development of a mouse model for sporadic and metastatic colon tumors and its use in assessing drug treatment. Proc. Natl. Acad. Sci. USA 2010, 107, 1565–1570. [Google Scholar] [CrossRef]
- Martin, E.S.; Belmont, P.J.; Sinnamon, M.J.; Richard, L.G.; Yuan, J.; Coffee, E.M.; Roper, J.; Lee, L.; Heidari, P.; Lunt, S.Y.; et al. Development of a colon cancer GEMM-derived orthotopic transplant model for drug discovery and validation. Clin. Cancer Res. 2013, 19, 2929–2940. [Google Scholar] [CrossRef]
- Tetteh, P.W.; Kretzschmar, K.; Begthel, H.; Van Den Born, M.; Korving, J.; Morsink, F.; Farin, H.; Van Es, J.H.; Offerhaus, G.J.A.; Clevers, H. Generation of an inducible colon-specific Cre enzyme mouse line for colon cancer research. Proc. Natl. Acad. Sci. USA 2016, 113, 11859–11864. [Google Scholar] [CrossRef]
- Roper, J.; Tammela, T.; Cetinbas, N.M.; Akkad, A.; Roghanian, A.; Rickelt, S.; Almeqdadi, M.; Wu, K.; Oberli, M.A.; Sánchez-Rivera, F.; et al. In vivo genome editing and organoid transplantation models of colorectal cancer and metastasis. Nat. Biotechnol. 2017, 35, 569–576. [Google Scholar] [CrossRef]
- Hadjihannas, M.V.; Behrens, J. CIN by Wnt: Growth pathways, mitotic control and chromosomal instability in cancer. Cell Cycle 2006, 5, 2077–2081. [Google Scholar] [CrossRef] [PubMed]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability an evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef]
- Swanton, C. Intratumor heterogeneity: Evolution through space and time. Cancer Res. 2012, 72, 4875–4882. [Google Scholar] [CrossRef]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; Tarpey, P.; et al. Intratumor Heterogeneity and Branched Evolution Revealed by Multiregion Sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef]
- Nikolaev, S.I.; Sotiriou, S.K.; Pateras, I.S.; Santoni, F.; Sougioultzis, S.; Edgren, H.; Almusa, H.; Robyr, D.; Guipponi, M.; Saarela, J.; et al. A single-nucleotide substitution mutator phenotype revealed by exome sequencing of human colon adenomas. Cancer Res. 2012, 72, 6279–6289. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Van Hoeck, A.; Tjoonk, N.H.; Van Boxtel, R.; Cuppen, E. Portrait of a cancer: Mutational signature analyses for cancer diagnostics. BMC Cancer 2019, 19, 457. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef]
- Nik-Zainal, S.; Alexandrov, L.B.; Wedge, D.C.; Van Loo, P.; Greenman, C.D.; Raine, K.; Jones, D.; Hinton, J.; Marshall, J.; Stebbings, L.A.; et al. Mutational processes molding the genomes of 21 breast cancers. Cell 2012, 149, 979–993. [Google Scholar] [CrossRef] [PubMed]
- Kane, A.M.; Fennell, L.J.; Liu, C.; Borowsky, J.; McKeone, D.M.; Bond, C.E.; Kazakoff, S.; Patch, A.M.; Koufariotis, L.T.; Pearson, J.; et al. Alterations in signaling pathways that accompany spontaneous transition to malignancy in a mouse model of BRAF mutant microsatellite stable colorectal cancer. Neoplasia 2020, 22, 120–128. [Google Scholar] [CrossRef]
- Niknafs, N.; Zhong, Y.; Moral, J.A.; Zhang, L.; Shao, M.X.; Lo, A.; Makohon-Moore, A.; Iacobuzio-Donahue, C.A.; Karchin, R. Characterization of genetic subclonal evolution in pancreatic cancer mouse models. Nat. Commun. 2019, 10, 5435. [Google Scholar] [CrossRef]
- Connor, F.; Rayner, T.F.; Aitken, S.J.; Feig, C.; Lukk, M.; Santoyo-Lopez, J.; Odom, D.T. Mutational landscape of a chemically-induced mouse model of liver cancer. J. Hepatol. 2018, 69, 840–850. [Google Scholar] [CrossRef]
- Fantini, D.; Glaser, A.P.; Rimar, K.J.; Wang, Y.; Schipma, M.; Varghese, N.; Rademaker, A.; Behdad, A.; Yellapa, A.; Yu, Y.; et al. A Carcinogen-induced mouse model recapitulates the molecular alterations of human muscle invasive bladder cancer. Oncogene 2018, 37, 1911–1925. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Murphy, C.J.; Karreth, F.A.; Emdal, K.B.; Yang, K.; White, F.M.; Elemento, O.; Toker, A.; Wulf, G.M.; Cantley, L.C. Identifying and targeting sporadic oncogenic genetic aberrations in mouse models of triple-negative breast cancer. Cancer Discov. 2018, 8, 354–369. [Google Scholar] [CrossRef]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Cañellas, A.; Hernando-Momblona, X.; et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef] [PubMed]
- Nassar, D.; Latil, M.; Boeckx, B.; Lambrechts, D.; Blanpain, C. Genomic landscape of carcinogen-induced and genetically induced mouse skin squamous cell carcinoma. Nat. Med. 2015, 21, 946–954. [Google Scholar] [CrossRef]
- Sjöblom, T.; Jones, S.; Wood, L.D.; Parsons, D.W.; Lin, J.; Barber, T.D.; Mandelker, D.; Leary, R.J.; Ptak, J.; Silliman, N.; et al. The consensus coding sequences of human breast and colorectal cancers. Science 2006, 314, 268–274. [Google Scholar] [CrossRef]
- Lindahl, T. Instability and decay of the primary structure of DNA. Nature 1993, 362, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, C.; Vogelstein, B. Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 2015, 347, 78–81. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, C.; Vogelstein, B.; Parmigiani, G. Half or more of the somatic mutations in cancers of self-renewing tissues originate prior to tumor initiation. Proc. Natl. Acad. Sci. USA 2013, 110, 1999–2004. [Google Scholar] [CrossRef]
- McGranahan, N.; Swanton, C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell 2017, 168, 613–628. [Google Scholar] [CrossRef]
- Lugli, N.; Dionellis, V.S.; Ordóñez-Morán, P.; Kamileri, I.; Sotiriou, S.K.; Huelsken, J.; Halazonetis, T.D. Enhanced Rate of Acquisition of Point Mutations in Mouse Intestinal Adenomas Compared to Normal Tissue. Cell Rep. 2017, 19, 2185–2192. [Google Scholar] [CrossRef][Green Version]
- Roerink, S.F.; Sasaki, N.; Lee-Six, H.; Young, M.D.; Alexandrov, L.B.; Behjati, S.; Mitchell, T.J.; Grossmann, S.; Lightfoot, H.; Egan, D.A.; et al. Intra-tumour diversification in colorectal cancer at the single-cell level. Nature 2018, 556, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Drost, J.; Van Jaarsveld, R.H.; Ponsioen, B.; Zimberlin, C.; Van Boxtel, R.; Buijs, A.; Sachs, N.; Overmeer, R.M.; Offerhaus, G.J.; Begthel, H.; et al. Sequential cancer mutations in cultured human intestinal stem cells. Nature 2015, 521, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Matano, M.; Date, S.; Shimokawa, M.; Takano, A.; Fujii, M.; Ohta, Y.; Watanabe, T.; Kanai, T.; Sato, T. Modeling colorectal cancer using CRISPR-Cas9-mediated engineering of human intestinal organoids. Nat. Med. 2015, 21, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Vries, R.G.; Snippert, H.J.; Van De Wetering, M.; Barker, N.; Stange, D.E.; Van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Barker, N.; Van Es, J.H.; Kuipers, J.; Kujala, P.; Van Den Born, M.; Cozijnsen, M.; Haegebarth, A.; Korving, J.; Begthel, H.; Peters, P.J.; et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 2007, 449, 1003–1007. [Google Scholar] [CrossRef]
- Elango, R.; Osia, B.; Harcy, V.; Malc, E.; Mieczkowski, P.A.; Roberts, S.A.; Malkova, A. Repair of base damage within break-induced replication intermediates promotes kataegis associated with chromosome rearrangements. Nucleic Acids Res. 2019, 47, 9666–9684. [Google Scholar] [CrossRef] [PubMed]
- Costantino, L.; Sotiriou, S.K.; Rantala, J.K.; Magin, S.; Mladenov, E.; Helleday, T.; Haber, J.E.; Iliakis, G.; Kallioniemi, O.P.; Halazonetis, T.D. Break-induced replication repair of damaged forks induces genomic duplications in human cells. Science 2014, 343, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Deem, A.; Keszthelyi, A.; Blackgrove, T.; Vayl, A.; Coffey, B.; Mathur, R.; Chabes, A.; Malkova, A. Break-induced replication is highly inaccurate. PLoS Biol. 2011, 9, e1000594. [Google Scholar] [CrossRef]
- McAvoy, S.; Ganapathiraju, S.C.; Ducharme-Smith, A.L.; Pritchett, J.R.; Kosari, F.; Perez, D.S.; Zhu, Y.; James, C.D.; Smith, D.I. Non-random inactivation of large common fragile site genes in different cancers. Cytogenet. Genome Res. 2007, 118, 260–269. [Google Scholar] [CrossRef]
- Helmrich, A.; Stout-Weider, K.; Hermann, K.; Schrock, E.; Heiden, T. Common fragile sites are conserved features of human and mouse chromosomes and relate to large active genes. Genome Res. 2006, 16, 1222–1230. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.I.; Zhu, Y.; McAvoy, S.; Kuhn, R. Common fragile sites, extremely large genes, neural development and cancer. Cancer Lett. 2006, 232, 48–57. [Google Scholar] [CrossRef]
- Gorgoulis, V.G.; Vassiliou, L.V.F.; Karakaidos, P.; Zacharatos, P.; Kotsinas, A.; Liloglou, T.; Venere, M.; DiTullio, R.A.; Kastrinakis, N.G.; Levy, B.; et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 2005, 434, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Secrier, M.; Li, X.; De Silva, N.; Eldridge, M.D.; Contino, G.; Bornschein, J.; Macrae, S.; Grehan, N.; O’Donovan, M.; Miremadi, A.; et al. Mutational signatures in esophageal adenocarcinoma define etiologically distinct subgroups with therapeutic relevance. Nat. Genet. 2016, 48, 1131–1141. [Google Scholar] [CrossRef]
- Dulak, A.M.; Stojanov, P.; Peng, S.; Lawrence, M.S.; Fox, C.; Stewart, C.; Bandla, S.; Imamura, Y.; Schumacher, S.E.; Shefler, E.; et al. Exome and whole-genome sequencing of esophageal adenocarcinoma identifies recurrent driver events and mutational complexity. Nat. Genet. 2013, 45, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Christensen, S.; Van der Roest, B.; Besselink, N.; Janssen, R.; Boymans, S.; Martens, J.W.M.; Yaspo, M.L.; Priestley, P.; Kuijk, E.; Cuppen, E.; et al. 5-Fluorouracil treatment induces characteristic T>G mutations in human cancer. Nat. Commun. 2019, 10, 4571. [Google Scholar] [CrossRef] [PubMed]
- Donehower, L.A.; Soussi, T.; Korkut, A.; Liu, Y.; Schultz, A.; Cardenas, M.; Li, X.; Babur, O.; Hsu, T.K.; Lichtarge, O.; et al. Integrated Analysis of TP53 Gene and Pathway Alterations in The Cancer Genome Atlas. Cell Rep. 2019, 28, 1370–1384.e5. [Google Scholar] [CrossRef] [PubMed]
- Eischen, C.M. Genome stability requires p53. Cold Spring Harb. Perspect. Med. 2016, 6, a026096. [Google Scholar] [CrossRef]
- Helleday, T.; Eshtad, S.; Nik-Zainal, S. Mechanisms underlying mutational signatures in human cancers. Nat. Rev. Genet. 2014, 15, 585–598. [Google Scholar] [CrossRef]
- Sansom, O.J.; Reed, K.R.; Hayes, A.J.; Ireland, H.; Brinkmann, H.; Newton, I.P.; Batlle, E.; Simon-Assmann, P.; Clevers, H.; Nathke, I.S.; et al. Loss of Apc in vivo immediately perturbs Wnt signaling, differentiation, and migration. Genes Dev. 2004, 18, 1385–1390. [Google Scholar] [CrossRef]
- Jackson, E.L.; Willis, N.; Mercer, K.; Bronson, R.T.; Crowley, D.; Montoya, R.; Jacks, T.; Tuveson, D.A. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001, 15, 3243–3248. [Google Scholar] [CrossRef]
- Marino, S.; Vooijs, M.; Van Der Gulden, H.; Jonkers, J.; Berns, A. Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev. 2000, 14, 994–1004. [Google Scholar]
- Feng, Y.; Sentani, K.; Wiese, A.; Sands, E.; Green, M.; Bommer, G.T.; Cho, K.R.; Fearon, E.R. Sox9 induction, ectopic paneth cells, and mitotic spindle axis defects in mouse colon adenomatous epithelium arising from conditional biallelic Apc inactivation. Am. J. Pathol. 2013, 183, 493–503. [Google Scholar] [CrossRef]
- Ordóñez-Morán, P.; Dafflon, C.; Imajo, M.; Nishida, E.; Huelsken, J. HOXA5 Counteracts Stem Cell Traits by Inhibiting Wnt Signaling in Colorectal Cancer. Cancer Cell 2015, 28, 815–829. [Google Scholar] [CrossRef]
- Macheret, M.; Halazonetis, T.D. Intragenic origins due to short G1 phases underlie oncogene-induced DNA replication stress. Nature 2018, 555, 112–116. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dionellis, V.S.; Norkin, M.; Karamichali, A.; Rossetti, G.G.; Huelsken, J.; Ordonez-Moran, P.; Halazonetis, T.D. Genomic Instability Profiles at the Single Cell Level in Mouse Colorectal Cancers of Defined Genotypes. Cancers 2021, 13, 1267. https://doi.org/10.3390/cancers13061267
Dionellis VS, Norkin M, Karamichali A, Rossetti GG, Huelsken J, Ordonez-Moran P, Halazonetis TD. Genomic Instability Profiles at the Single Cell Level in Mouse Colorectal Cancers of Defined Genotypes. Cancers. 2021; 13(6):1267. https://doi.org/10.3390/cancers13061267
Chicago/Turabian StyleDionellis, Vasilis S., Maxim Norkin, Angeliki Karamichali, Giacomo G. Rossetti, Joerg Huelsken, Paloma Ordonez-Moran, and Thanos D. Halazonetis. 2021. "Genomic Instability Profiles at the Single Cell Level in Mouse Colorectal Cancers of Defined Genotypes" Cancers 13, no. 6: 1267. https://doi.org/10.3390/cancers13061267
APA StyleDionellis, V. S., Norkin, M., Karamichali, A., Rossetti, G. G., Huelsken, J., Ordonez-Moran, P., & Halazonetis, T. D. (2021). Genomic Instability Profiles at the Single Cell Level in Mouse Colorectal Cancers of Defined Genotypes. Cancers, 13(6), 1267. https://doi.org/10.3390/cancers13061267