Simple Summary
There is growing evidence that Ph-negative myeloproliferative neoplasms are disorders in which multiple signaling pathways are significantly disturbed. The heterogeneous phenotypes observed among patients have highlighted the importance of having a comprehensive knowledge of the molecular mechanisms behind these diseases. This review aims to show a broad overview of the signaling involved in myeloproliferative neoplasms (MPNs) and other processes that can modify them, which could be helpful to better understand these diseases and develop more effective targeted treatments.
Abstract
Ph-negative myeloproliferative neoplasms (polycythemia vera (PV), essential thrombocythemia (ET) and primary myelofibrosis (PMF)) are infrequent blood cancers characterized by signaling aberrations. Shortly after the discovery of the somatic mutations in JAK2, MPL, and CALR that cause these diseases, researchers extensively studied the aberrant functions of their mutant products. In all three cases, the main pathogenic mechanism appears to be the constitutive activation of JAK2/STAT signaling and JAK2-related pathways (MAPK/ERK, PI3K/AKT). However, some other non-canonical aberrant mechanisms derived from mutant JAK2 and CALR have also been described. Moreover, additional somatic mutations have been identified in other genes that affect epigenetic regulation, tumor suppression, transcription regulation, splicing and other signaling pathways, leading to the modification of some disease features and adding a layer of complexity to their molecular pathogenesis. All of these factors have highlighted the wide variety of cellular processes and pathways involved in the pathogenesis of MPNs. This review presents an overview of the complex signaling behind these diseases which could explain, at least in part, their phenotypic heterogeneity.
1. Introduction
Myeloproliferative neoplasms (MPNs) are rare hematological malignancies characterized by the clonal expansion of mature myeloid cells. MPNs arise from certain somatic mutations in hematopoietic stem cells (HSCs) which provide a selective advantage and lead to the expansion of aberrant clones.
Classic MPNs consist of chronic myeloid leukemia (CML), polycythemia vera (PV), essential thrombocythemia (ET) and primary myelofibrosis (PMF). In the last few years, the advances in molecular biology have provided key insights into the molecular mechanisms behind these diseases. CML is genetically defined by the Philadelphia (Ph) chromosome, the result of t(9;22)(q34;q11). This translocation leads to the production of a chimeric BCR-ABL1 protein with constitutive tyrosine kinase activity. The description of the Ph chromosome as a disease-initiating event in CML revolutionized the diagnosis and treatment of this disease [1]. The targeted therapy imatinib showed a specific inhibitory capacity against the tyrosine kinase activity of BCR-ABL1 [2,3,4] that, despite not being curative [5], increased the 10-year survival of CML patients in chronic phase to more than 83%–84% [6,7].
This review is focused on PV, ET and PMF, all of them Ph-negative MPNs that share similar and mostly mutually exclusive driver mutations affecting JAK2, MPL and CALR. The aberrant functions of the mutant products encoded by these genes have been extensively studied and the main mechanisms that lead to the myeloproliferation described. Currently, it is considered that the major hallmark of Ph-negative MPNs is the constitutive activation of JAK2-related signaling pathways. In fact, at this time, the only targeted therapy approved in MPNs is the JAK1/2 inhibitor ruxolitinib, which can reduce splenomegaly and other common symptoms in patients with PMF, post-PV/ET MF [8,9] and PV resistant or intolerant to hydroxyurea [9,10]. Although a reduction in the mutant allele burden is rare [9], it could be achieved in long-term treatment [11]. However, the improvement in the overall survival of ruxolitinib-treated patients has been questioned [12,13,14]. Actually, malignant cells can still survive in these patients and the clinical response could be mainly due to the downmodulation of proinflammatory cytokines derived from the JAK2 inhibition [15]. These arguments have led researchers to question whether JAK2 is really the best drug target in these diseases or not [16].
In the meantime, some non-canonical mechanisms of mutant JAK2 [17,18,19,20,21,22,23,24] and CALR [25,26,27,28,29,30,31,32,33] have been described. Chronic inflammation [34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62] and the bone marrow microenvironment [63,64,65,66,67,68,69,70,71,72] also seem to contribute to the heterogeneous phenotypes found among MPN patients.
Additionally, mutations in disease-modifying genes that seem to increase the risk of leukemic transformation or progression from ET to myelofibrosis have also been identified [73,74,75]. The products encoded by these genes are involved in epigenetic modification, tumor suppression, transcription regulation, splicing, and some other signaling pathways [76,77]. Other factors, such as genetic predisposition, age or environment have also been shown to influence the heterogeneity of MPN phenotypes [78].
This review presents an overview of the signaling behind Ph-negative MPNs attending not only to the activation of JAK2-related canonical signaling pathways, but also to other non-canonical pathways, disease-modifying signaling, and additional factors that have been found to be involved in the pathogenesis of these diseases.
2. JAK2-Related Canonical Signaling Pathways
JAK2 signaling is activated through a variety of receptors such as those for erythropoietin (EPOR), thrombopoietin (TPOR), and granulocyte/macrophage colony-stimulating factor (GM-CSFR). They regulate the production of the erythroid, megakaryocytic, and granulocytic lineages, respectively. When stimulated by ligands, receptors dimerize and bring JAK2 kinases into proximity. JAK2 is phosphorylated upon receptor binding and induces the phosphorylation of the cytoplasmic portion of the receptor and downstream factors.
In 2005, several research groups simultaneously published the presence of the somatic mutation p.V617F (JAK2V617F) in the exon 14 of JAK2 in patients with PV (96%), PMF (65%) and ET (55%) [79,80,81,82,83,84]. This mutation impairs the physiological inhibitory function of the JH2 pseudokinase domain upon the JH1 kinase domain, which acquires a constitutive activation that promotes JAK2 phosphorylation in the absence of ligand stimulation (Figure 1). In 2007, four additional gain-of-function somatic mutations in the exon 12 of JAK2 were detected in 3% of patients with PV [84,85]: p.N542-E543del (30%), p.K539L (14%), p.E543-D544del (12%), and p.F537-K539delinsL (10%). All of them are located upstream of the JH2 pseudokinase domain and promote an increased phosphorylation of JAK2 compared to p.V617F [86].
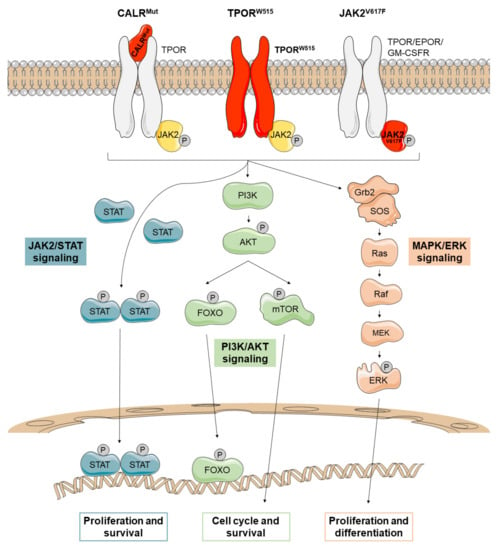
Figure 1.
JAK2-related canonical signaling pathways active in Ph-negative myeloproliferative neoplasms (MPNs). Mutations in CALR (CALRMUT), JAK2 (JAK2V617F), and MPL (TPORW515) lead to the constitutive activation of JAK2/STAT, PI3K/AKT, and MAPK/ERK signaling that promotes the transport to the nucleus of several transcription factors such as STATs and FOXO. There, they regulate transcription of their target genes, causing increased proliferation and survival of mutant cells. Mutant proteins are depicted in red.
In 2006, the gain-of-function mutation p.W515L in the exon 10 of MPL was identified in a minor proportion of MPN patients [87]. p.W515L and p.W515K are the most commonly reported mutations, identified in approximately 5% of PMF patients and 1% of ET patients [88]. MPL encodes the thrombopoietin receptor (TPOR), which depends on JAKs to mediate signal transduction. MPL mutations (TPORW515) promote the dimerization and activation of TPOR, leading to transphosphorylation and activation of the previously bound JAK2 proteins (Figure 1) [89].
The molecular alteration that causes the 60–90% of PMF and ET cases in patients not harboring JAK2/MPL mutations was described in 2013 [90]. During that year, two research groups identified mutations in CALR [91,92], a gene that encodes calreticulin, a ubiquitous protein found in the endoplasmic reticulum (ER) of all nucleated cells with multiple functions inside and outside this organelle. CALR is a Ca2+-binding chaperone mainly involved in the regulation of intracellular Ca2+ homeostasis and a regulator of protein folding in the cellular response to ER stress (unfolding protein response (UPR)) [93]. However, this protein has been also found associated with other cytoplasmic, nuclear and extracellular proteins, so it could be involved in a wide variety of signaling pathways [94]. In fact, CALR has been associated with cellular stress responses, adipocyte differentiation, cardiogenesis, proliferation, wound healing, apoptosis and immunogenic cell death [90,94].
The structure of wild-type CALR consists of a signal peptide and three domains: an amino-terminal N-domain, a proline-rich P-domain and a carboxy-terminal C-domain, which contains an ER retention signal (KDEL). The CALR mutations described to date are insertions or deletions in exon 9 that shift the reading frame by one base pair (+1), mainly a 52-bp deletion (c.1902_1143del) or type 1 mutation (CALRdel52), and a 5-bp insertion (c.1154_1155insTTGTC) or type 2 mutation (CALRins5). As a result, mutant CALRs (CALRMut) show a novel C-terminal end that lacks the ER retention motif (KDEL) [91,92] and some Ca2+-binding sites [95]. In 2016, it was published that CALRMut is transported to the cellular membrane where it activates TPOR in a ligand-independent manner (Figure 1) [96,97,98,99]. The characterization of the TPOR binding capacity has revealed that the C-terminal end of CALRMut blocks the P-domain of the protein, which constitutively exerts an inhibitory effect on the N-domain. Consequently, the N-domain can bind to immature N-glycans on TPOR [96]. This mechanism is consistent with the observation that the N-glycan binding motif located in N-domain of CALRMut is required for TPOR activation [97]. In fact, blocking N-glycosylation on asparagine 117 of TPOR diminishes CALR-dependent TPOR activation [97,100]. Both wild-type and mutant CALR recognize immature forms of N-glycans and fold the protein correctly, but CALRMut fails to dissociate from the targeted protein [101]. Thus, the CALRMut-TPOR complex moves from the ER to the plasma membrane through the Golgi apparatus and is secreted out of cells [102]. However, secreted CALRMut is only capable to activate TPOR on the cell surface of cells expressing CALRMut since only these cells have the immature N-glycans on TPOR [96,102,103]. Stimulation of TPOR leads to the activation of JAK2-dependent signaling in a similar way to the rest of the Ph-negative MPNs.
In conclusion, the mutations described to date in JAK2, MPL and CALR lead to a constitutive activation of JAK2, which ultimately causes the aberrant proliferation and survival of malignant myeloid clones. The three major downstream signaling pathways that are activated by JAK2 are JAK2/STATs, MAPK/ERK, and PI3K/AKT (Figure 1). The evidence suggests that each of these pathways plays an important role in MPNs, although the JAK2/STAT pathway appears to be the main one. In fact, dysregulation of JAK2/STAT signaling has been identified in all MPNs regardless of mutational status [104].
2.1. JAK2/STAT Pathway
In MPNs, JAK2 phosphorylates and activates STATs (mainly STAT1, STAT3 and STAT5). It seems that STATs are differentially activated depending on the type of MPN. For example, MPL mutations increase STAT3 and STAT5 signaling. In PV patients, JAK2V617F binds to EPOR promoting STAT5 activation. In ET patients, both JAK2V617F and CALRMut bind to TPOR; JAK2V617F enhances the phosphorylation of STAT1 and STAT3 but CALRMut promotes STAT3 and STAT5 activation. However, in PMF, phosphorylation of STAT3 is decreased in both JAK2V617F and mutant CALRs. To date, the precise mechanisms that explain differential activation of the STATs remain unclear [78].
Once the STATs are phosphorylated, they form a dimer that enters the nucleus to activate the transcription of target genes (Figure 1). In this way, JAK2/STAT signaling stimulates cell proliferation, differentiation and survival.
2.2. MAPK/ERK Pathway
The activated JAK2 can also lead to the phosphorylation of ERK, a serine threonine kinase that activates multiple proteins in both the cytoplasm and the nucleus. ERK is a key regulator of a wide variety of signaling pathways, so its deregulation could disrupt multiple processes. In the cytoplasm, ERK contributes to ion transport, apoptosis, and regulation of metabolism, among others. In the nucleus, it targets regulators of cell cycle and multiple transcription factors (Figure 1) [105].
2.3. PI3K/AKT Pathway
JAK2 activation also stimulates the PI3K/AKT pathway. AKT is a cell survival kinase which inhibits apoptosis by phosphorylating the proapoptotic protein BAD and the transcription factor FOXO3A. In addition, AKT can activate a wide range of mechanisms such as protein translation through mTOR or the cell cycle machinery (Figure 1) [105].
3. Non-Canonical Signaling Pathways
3.1. JAK2-Related Non-Canonical Signaling
In 2009, activated JAK2 was described to be in the nucleus of hematopoietic cells and to phosphorylate Y41 on histone 3 (H3Y41). This event prevents the binding of heterochromatin protein 1 alpha (HP1α) to H3Y41 [17]. HP1α shows a proliferation-dependent regulation and is involved in gene silencing, genome stability, and chromosome segregation (Figure 2). It has been found overexpressed in some tumors, and it has been proposed as a potential hallmark of cell proliferation that could be relevant in clinical oncology [18].
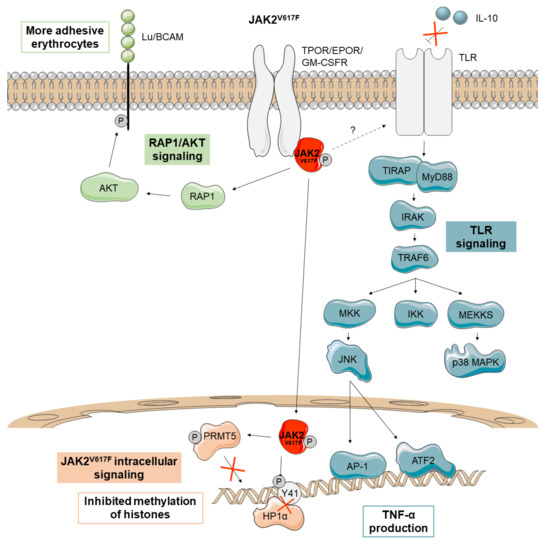
Figure 2.
Main non-canonical signaling pathways activated by JAK2V617F in Ph-negative MPNs. In PV patients, JAK2V617F (depicted in red) activates the adhesion receptor Lu/BCAM through the RAP1-AKT signaling pathway, making their erythrocytes more adhesive. JAK2V617F has also been described to promote aberrant signaling in the nucleus, where it prevents the binding of heterochromatin protein 1 alpha (HP1α) and inhibits the methylation of histones via protein arginine methyltransferase 5 (PRMT5) impairment. MPN patients with JAK2V617F also seem to be insensitive to the anti-inflammatory cytokine IL-10, increasing TNF-α production through Toll-Like Receptor (TLR) signaling.
JAK2V617F also acquires the ability to phosphorylate the protein arginine methyltransferase 5 (PRMT5) leading to an impairment in its ability to methylate histones (Figure 2). When PRMT5 is knocked down in CD34+ cells, an increased colony formation and erythroid differentiation can be observed [19].
A recent study also suggests that erythrocytes from PV patients are more adhesive since JAK2V617F activates the erythrocyte adhesion receptor Lu/BCAM through an EPOR-independent RAP1/AKT signaling pathway (Figure 2) [20].
Finally, monocytes from MPN patients with JAK2V617F have been found to have a defective negative regulation of toll-like receptor (TLR) signaling leading to increased production of the inflammatory cytokine TNF-α. These monocytes are insensitive to the anti-inflammatory cytokine IL-10, which in turn negatively regulates TNF-α production through TLR (Figure 2) [21]. Studies on TNF-α knockout mice have demonstrated that this cytokine is required for the development of an MPN-like disease [22]. Unrestrained production of TNF-α has been observed in an MPN patient but also in his identical twin, suggesting that it may be a genetic feature rather than a consequence of the disease [21]. In any case, the inflammatory environment can favor the maintenance and expansion of the JAK2V617F mutant clone since these cells are resistant to inflammation whereas non-mutant cells are not [22]. Thus, the JAK2V617F mutant clone seems to induce non-mutant cells to produce inflammatory cytokines, reinforcing the self-perpetuating environment for its continuous selection [23]. Finally, CD34+ progenitors of a PV patient with JAK2V617F have been reported to use dual-specificity phosphatase 1 (DUSP1) to protect themselves against inflammatory stress and DNA damage, promoting their proliferation and survival in this microenvironment (Figure 2) [24].
3.2. CALR-Related Non-Canonical Signaling
Several studies have identified novel mechanisms that collaborate with the activation of TPOR in CALR-mediated cellular transformation (Figure 3). CALRMut seems to cause reduced activation of the UPR pro-apoptotic pathway and to have an increased sensitivity to oxidative stress by the down-modulation of oxidation resistance 1 (OXR1) in K562 cells. These mechanisms lead to resistance to UPR-induced apoptosis and genomic instability, respectively [25]. Moreover, CALRdel52 causes increased recruitment of the friend leukemia integration 1 (FLI1) transcription factor to the MPL promoter to enhance transcription [26], which suggests a promotion of tumorigenesis by modulating transcription through interactions with transcription factors in the nucleus.
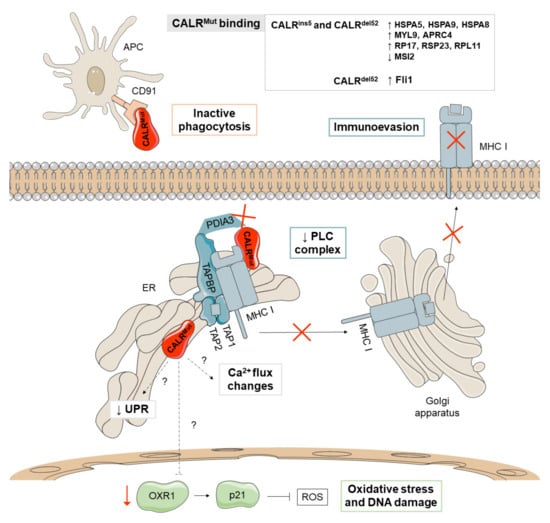
Figure 3.
Major non-canonical mechanisms derived from CALRMut. CALRMut (depicted in red) shows different binding affinities for proteins implicated in the unfolding protein response (UPR) (HSPA5, HSPA9, and HSPA8), proteins of the cytoskeleton (MYL9 and APRC4), and ribosomal proteins (RP17, RSP23, and RPL11), as well as reduced binding to MSI2, a transcriptional regulator that target genes mainly involved in cell cycle regulation. Additionally, CALRMut seems to reduce the activation of the pro-apoptotic pathway of the UPR and increases oxidative stress and DNA damage through the downmodulation of oxidation resistance 1 (OXR1). CALRMut also shows decreased binding affinities for PDIA3 and has a loss-of-function effect on the peptide loading complex (PLC), which mediates the loading of cellular antigens onto major histocompatibility complex class I (MHC-I) molecules, favoring immunoevasion. Mutations in CALR increase the secretion of the protein and bind to CALR receptors in antigen presenting cells (APCs), limiting their ability to phagocytize wild-type CALR-exposing cancer cells. The main differences between the phenotypes observed in patients with type 1 (del52) and type 2 (ins5) mutations have been attributed to thrombopoietin receptor (TPOR)-independent cytosolic calcium fluxes and the binding affinity for the transcription factor FLI1.
Bioinformatic analyses of CALRMut revealed the appearance of potential phosphorylation sites for kinases that may have a role in the regulation of multiple cellular activities [27] and recent studies have shown that CALRMut causes increased binding affinities for proteins involved in the activation of the UPR (HSPA5, HSPA9, and HSPA8) and cytoskeletal (MYL9 and APRC4) and ribosomal proteins (RP17, RSP23, and RPL11), as well as reduced binding to MSI2, a transcriptional regulator that targets genes mainly involved in cell cycle regulation [26].
On the other hand, CALR is an integral part of the peptide loading complex (PLC), which mediates the loading of cellular antigens onto major histocompatibility complex class I (MHC-I) molecules. In addition to CALR, the PLC is composed of PDIA3, TAP-binding protein, TAP1, and TAP2. Specifically, CALR interacts with PDIA3 in a glycan-dependent manner and preserves steady-state levels of TAP-binding protein and MHC-I heavy chains. Besides, it rescues suboptimally assembled MHC-I molecules from post-ER compartments [28]. HEK293T cells lacking CALR expression show a reduction of properly loaded MHC-I on the cell surface, a defect that is not restored by expression of CALRMut [29]. Consistent with this, cells with CALRMut show reduced antigen presentation on MHC-I (Figure 3) [54] and decreased binding affinities for PDIA3 [26]. These results suggest that CALR mutations have a loss-of-function effect on PLC and, therefore, may contribute to the development of MPN by promoting immunoevasion after loss of tumor antigenicity [28]. Additionally, CALR operates as a key damage-associated molecular pattern (DAMP) when it is translocated to the outer cell membrane of dying cancer cells. CALR-exposing cancer cells deliver pro-phagocytic signals to antigen presenting cells (APCs) and activate dendritic cell efferocytosis. Mutations in CALR increase the secretion of the protein both in vitro and in vivo since the ER retention motif (KDEL) is compromised. Soluble CALR binds to CALR receptors in the APCs and limit their ability to phagocytise, leading to immunosuppressive effects (Figure 3) [30].
The wild-type CALR protein also regulates the activation of the stored-operated calcium entry (SOCE) machinery by interacting with PDIA3 and STIM1. Concretely, STIM1 is a protein of the SOCE machinery that leads to calcium mobilization. CALRMut has been shown to trigger TPOR-independent cytosolic calcium fluxes in megakaryocytes through defective interactions between CALRMut, PDIA3 and the SOCE machinery. This results in uncontrolled proliferation of megakaryocytes that can be reversed with a SOCE inhibitor [31].
The type of CALR mutation has been associated with different disease features. Thus, type 1 mutations are more often associated with PMF (70%) or progression from ET to a myelofibrotic state [32], while type 2 mutations are more often associated with ET [91]. The mechanisms underlying this phenomenon have not been fully elucidated, but it has been demonstrated that type 2 mutants retain longer stretches of the negatively charged amino acids of wild-type CALR than type 2 mutants, which may neutralize the positive electronic charge generated at the C-terminal end. Additionally, type 1 mutant C-terminus generates greater changes in megakaryocyte cytosolic calcium flux than type 2 mutants [33].
3.3. Additional Non-Canonical Signaling
Non-canonical mechanisms affecting inflammatory signaling pathways and the bone marrow microenvironment have been widely observed in all MPNs, regardless of subtype and driver mutation.
3.3.1. Inflammatory Signaling Pathways
As previously noted, chronic inflammation is a characteristic feature of MPNs (Figure 4). In fact, MPN patients typically exhibit increased levels of inflammatory cytokines [34,106]. The impaired JAK2/STAT signaling is not the only contributor to inflammation in these diseases, as the inhibition of JAK2 is not sufficient to normalize the levels of inflammatory cytokines [35]. On the contrary, a significant enrichment of the NF-κB signaling pathway has been observed in both malignant and non-malignant cells in MPNs [36].
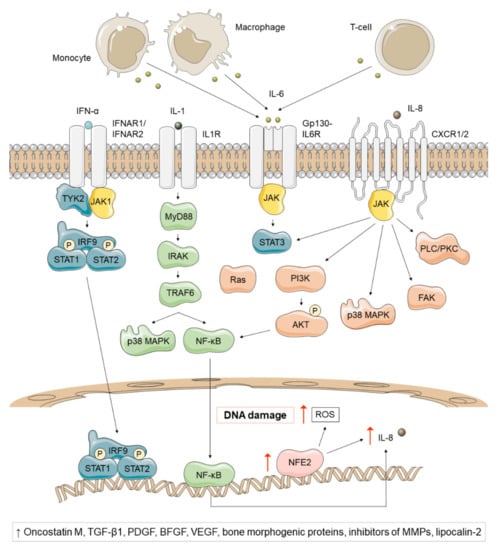
Figure 4.
Non-canonical inflammatory signaling pathways affected in Ph-negative MPNs, regardless of subtype and driver mutation. The preleukemic niche of MPNs secretes high levels of IL-1, a proinflammatory cytokine that activates multiple downstream pathways, such as p38 MAPK and NF-κB. NF-κB, in turn, generates high levels of IL-8, a proinflammatory cytokine that binds to CXCR1 or CXCR2 and activates STAT3, PI3K/AKT, p38 MAPK, FAK and PLC/PKC. NFE2 overexpression has also been reported in most MPN patients and has been associated with high IL-8 levels and increased ROS and DNA damage. On the other hand, IL-6 is a proinflammatory cytokine produced by monocytes, macrophages and T-cells that signals via JAK1/STAT3, whose levels have been found elevated in JAK2V617F PV and PMF patients. Finally, IFN-α is a key regulator of hematopoietic stem cells (HSCs) that depletes previously dormant hematopoietic stem progenitor cells (HSPCs) and enhances the immune response. A pathogenic role of oncostatin-M, TGF-β1, platelet-derived growth factor (PDGF), basic fibroblast growth factor (BFGF), VEGF, bone morphogenic proteins, inhibitors of matrix metalloproteinases (MMPs) and lipocalin-2 has been suggested in Ph-negative MPNs.
NFE2 overexpression has also been reported in most patients and seems to play a role in chronic inflammation [37,38]. NFE2 participates in inflammatory cascades by increasing IL-8 transcription and promotes proliferation by activating the expression of CDK4, CDK6 and cyclin D3 [39,40]. In addition, it produces reactive oxygen species (ROS), a group of highly reactive oxygen-containing molecules which participate in numerous biological processes [41]. This results in lipid and protein oxidation, increased oxidative DNA damage (8-oxo-G), and subsequent double-stranded DNA breaks that induce instability [38]. Excessive ROS production and subsequent oxidative stress confer a proliferative advantage to JAK2V617F clones and activate proinflammatory pathways (NF-κB) that create more ROS. In this way, MPNs have recently been described as “a human inflammation model for cancer development”, as they are characterized by a self-perpetuating circle in which inflammation creates ROS which in turn creates more inflammation [42].
Multiple inflammatory signaling pathways such as IFN-α and IL-1β have been also found to be involved in the pathogenesis of MPN. Interferons are key regulators of HSCs. Data from murine PV JAK2V617F models have shown that hematopoietic stem progenitor cells (HSPCs) become more proliferative and lose quiescence when treated with IFN-α, leading to their depletion [43,44]. The ability to deplete previously dormant malignant stem cells together with the enhancement of the immune response have made IFN-α one of the most efficient treatment options in MPNs [107]. On the other hand, IL-1β is a proinflammatory cytokine released by myeloid cells in response to TLR stimulation, that activates multiple downstream pathways such as NF-kB and p38 MAPK [45]. The preleukemic niche of MPNs secretes high levels of IL-1, which drives granulocyte/macrophage differentiation [46]. IL-6 and IL-8 also seem to participate in MPN pathogenesis. IL-6 is a proinflammatory cytokine produced by monocytes, macrophages and T-cells that signals via JAK1/STAT3 [45]. Several mouse models for MPNs have shown a high expression of IL-6 in both mutant and wild-type HSCs [23]. Additionally, elevated IL-6 levels have been observed in JAK2V617F PV and PMF patients [47]. In fact, some studies point that IL-6 may participate in the progression of MPN to AML [45]. IL-8 is also a proinflammatory chemokine released in response to IL-1 or TNF-α that binds to CXCR1 or CXCR2 and activate JAK/STAT, PI3K/AKT, MAPK, PLC/PKC and FAK [45]. Elevated levels of IL-8 have been found in PV and ET patients [48].
Numerous cytokines have been implicated in mediating fibrosis, osteosclerosis and angiogenesis in PMF patients. Thus, several studies have suggested a pathogenic role for oncostatin-M [49], TGF-β1 [50,51], platelet-derived growth factor [51], basic fibroblast growth factor [50], VEGF [52], bone morphogenetic proteins [53], and inhibitors of matrix metalloproteinases [54,55].
Myeloid cells have been reported also to produce elevated levels of lipocalin-2 in PV, ET, and PMF patients. This protein increases the growth of bone marrow cells in PMF patients, but not in healthy donors. On the contrary, it increases reactive oxygen species, DNA damage, and apoptosis in normal cells, but not in PMF patients. Lipocalin-2 also induces the expression of factors that contribute to fibrosis, such as VEGF, TGF-β1, bone morphogenetic protein-2, RUNX2, osteoprotegerin and collagen type I [56,108].
Heat shock proteins (HSPs) are key players during inflammation. HSP90 stabilizes numerous proteins, such as JAK2. The HSP70 family is composed of some proteins (HSPA5, HSPA8, and HSPA8) that have been found to be enriched in fractions bound to CALRMut [26]. HSP70 also seems to contribute to cell proliferation through regulation of JAK2/STAT signaling. In fact, the inhibition of HSP70 expression in an ex vivo model of PV and ET increased apoptosis of the erythroid lineage and decreased JAK2 signaling [57]. HSP70 also activates TLR2 and TLR4, leading to NF-κB activation, rapid calcium flux, and TNF-α, IL1-β and IL-6 production [58]. Moreover, HSP70 can be secreted as a “danger signal” and bind peptides to form a complex that binds to cell surface receptors, such as CD91 and Lox-1 [59].
Finally, there is also evidence for a link between inflammation and thrombosis. Thrombosis in MPN patients has been associated with an increased platelet-leukocyte interaction. While MPN leukocytes overexpress the surface protein CD11b, its receptor (CD62p) is upregulated on platelets. This results in increased formation of leukocyte-platelet complexes [60,61,62].
3.3.2. Bone Marrow Microenvironment
The bone marrow microenvironment is a complex and dynamic structure composed of multiple cell types. Clonal HSCs in MPNs interact with other cells in this microenvironment and remodel it allowing further malignant expansion (Figure 5). There is a growing evidence that endothelial cells, mesenchymal stem cells, stromal cells, osteoblasts, and osteoclasts may contribute to the pathogenesis of these diseases in the bone marrow [63].
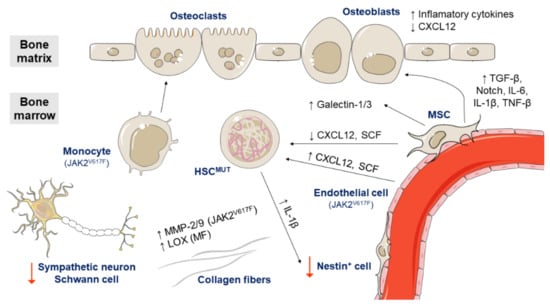
Figure 5.
Role of the bone marrow microenvironment in the pathogenesis of Ph-negative MPNs. Endothelial cells expressing JAK2V617F increase the expression of CXCL12 and stem cell factor (SCF) and cause the expansion of HSCs and progenitor cells. On the other hand, mesenchymal stem cells (MSCs) negative for JAK2V617F show a reduced expression of CXCL12 and SCF. MSCs also overexpress galectin-1 in all MPN subtypes and galectin-3 in PV patients, and promote the expansion of osteoblasts by cell contact and excessive TGF-1β, Notch, IL-6, IL-1β, and TNF-β signaling. Osteoblasts overproduce inflammatory cytokines and reduce CXCL12 expression. By contrast, monocytes with JAK2V617F seem to increase osteoclast forming ability and favor the survival of clonal HSCs. Meanwhile, clonal HSCs produce high levels of IL-1β, which induces nestin-positive MSCs death. Additionally, the sympathetic nerve fibers supporting Schwann cells are reduced in the bone marrow of MPN patients. Regarding the extracellular matrix, MPN patients with JAK2V617F show increased levels of MMP-2 and MMP-9 and patients with primary myelofibrosis (PMF) have increased levels of all LOX family members.
In a mouse model, endothelial cells expressing JAK2V617F have been shown to be capable of causing the expansion of hematopoietic stem and progenitor cells, which could be caused by increased expression of the cytokines CXCL12 (C-X-C motif chemokine ligand 12) and SCF (stem cell factor) by endothelial cells [64,65].
Mesenchymal stem cells (MSCs) also seem to be important in the pathogenesis of MPNs. In contrast to endothelial cells expressing JAK2V617F, MSCs negative for JAK2V617F have been reported to reduce the expression of CXCL12 and SCF [109,110]. They also support HPSC proliferation [66] and overexpress galectin-1 in all MPN subtypes and galectin-3 in PV patients [67]. Galectins mediate cell adhesion and stimulate cell migration, proliferation and apoptosis through interactions with integrins, laminin and fibronectin. In addition, MSCs promote the expansion of osteoblasts by cell contact and excessive TGF-β1, Notch, IL-6, IL-1β, and TNF-β signaling. Abnormal osteoblasts overproduce inflammatory cytokines, promote fibrogenesis and reduce CXCL12 expression [88]. By contrast, monocytes with JAK2V617F seem to increase osteoclast forming ability in MPN patients, favoring the survival of clonal HSCs [68].
A recent study has recently found numerous differences between the bone marrow niche of ET and PV patients. In ET, the HSPCs move faster and more frequently towards the endosteal niche and the number of osteoblasts and osteoclasts increases. However, in PV, only the non-endosteal sinusoids are dilated [69]. Other studies have demonstrated that the sympathetic nervous system has a role in the bone marrow niche of MPN patients. Specifically, sympathetic nerve fibers supporting Schwann cells and nestin-positive MSCs are reduced in the bone marrow of MPN patients. In a murine MPN model harboring JAK2V617F, stem cells secreted IL-1β, which induces nestin-positive MSCs death and enables disease expansion [70].
Regarding the extracellular matrix, several studies have also pointed to a role of matrix metalloproteinases (MMPs) and lysyl oxidase (LOX) in the pathogenesis of MPNs. MPN patients with JAK2V617F show increased levels of MMP-2 and MMP-9 [71] and patients with PMF have increased levels of all LOX family members. LOX is involved in collagen cross-linking and promotes fibrogenesis [72].
4. Disease Modifiers
Several non-driver somatic mutations have been identified in MPN patients. According to recent studies, more than 80% of patients with PMF [73] and over 50% of PV/ET patients have at least one additional somatic mutation of this type [74]. These mutations occur in genes affecting a wide variety of processes like epigenetic regulation, tumor suppression, transcription regulation or splicing, but also additional signaling pathways (Figure 6). They often modify the course of the disease and the presence of more than one such aberration has been associated with a worse survival [75].
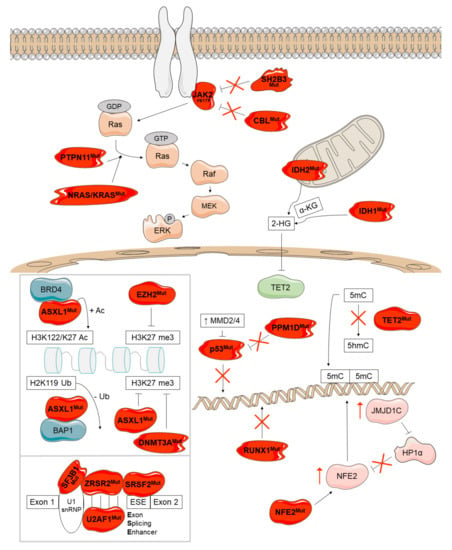
Figure 6.
Overview of the disease-modifying genes mutated in Ph-negative MPNs and their molecular consequences. These mutations occur in genes affecting epigenetic regulation (ASXL1, EZH2, DNMT3A, TET2, IDH1, and IDH2), tumor suppression (TP53 and PPM1D), transcription regulation (RUNX1 and NFE2), splicing (SRSF2, U2AF1, SF3B1, and ZRSR2), and other signaling pathways (SH2B3, CBL, NRAS/KRAS, PTPN11). Mutant proteins are depicted in red.
4.1. Epigenetic Regulation
The most common non-driver somatic mutations affect epigenetic regulation and have been identified in ASXL1 (ASXL transcriptional regulator 1), EZH2 (enhancer of zeste polycomb repressive complex 2 subunit), DNMT3A (DNA methyltransferase 3 alpha), TET2 (TET methylcitosine dioxygenase 2), IDH1 and IDH2 (isocitrate dehydrogenase NADP+, 1 and 2).
The products of ASXL1 and EZH2 are involved in chromatin modification (Figure 6, upper box). Normal ASXL1 interacts with the polycomb repressor complex 2 (PRC2) and enhances its function as methylator of H3K27. H3K27 trimethylation results in the silencing of the HOXA gene family which participates in chromatin remodeling. Additionally, ASXL1 interacts with BRCA1-associated protein 1 (BAP1), creating the polycomb group repressive deubiquitinase complex, which globally removes monoubiquitin from H2AK119 and locally at HOXA and IRF8 in HSCs [76,111,112]. ASXL1 mutations are almost exclusively frameshift and nonsense mutations in exon 12, decrease H3K27 trimethylation [111] and enhance the activity of the ASXL1-BAP deubiquitinase complex [113]. This causes the deregulated expression of genes critical for HSC self-renewal and differentiation, as well as more open chromatin in c-Kit+ cells. Mutant ASXL1 also binds to the bromodomain-containing protein 4 (BDR4), resulting in the phosphorylation of RNA polymerase II and the acetylation of H3K27 and H3K122, which lead to the upregulation of genes governing myeloid differentiation [76,114]. Another mechanism reported for mutant ASXL1 consists of the repression of TGF-β pathway through H3K and H4K deacetylation [115]. Although normal ASXL1 activates the retinoic acid receptor [116] and interacts with the peroxisome proliferator activated receptor gamma (PPARγ) to repress lipogenesis [117], the effects of ASXL1 mutations on these mechanisms are still unknown. In summary, the consequences of ASXL1 mutations are diverse and are not fully elucidated; the mutant protein shows a loss of function in some mechanisms but a gain of function in others.
EZH2 encodes a histone lysine N-methyltransferase that constitutes the catalytic component of PRC2. The majority of EZH2 mutations are missense with loss of function effects resulting in the silencing of HOXA9. This supports myeloid progenitor self-renewal and leukemic transformation [118,119].
DNMT3A, TET2, IDH1 and IDH2 encode DNA methylation modifiers (Figure 6). DNMT3A encodes a de novo DNA methyltransferase responsible for DNA methylation at CpG dinucleotides. The mutation most frequently observed is p.R882H, that impairs the CpG specificity, flanking sequence preference and DNMT3A enzymatic activity [120]. Mechanistic studies in mice indicate that mutant DNMT3A decreases PRC2 recruitment at H3K27 favoring accessibility at enhancer chromatin marks and persistent HSC gene expression. JAK2V617F patients also harboring DNMT3A mutations show aberrant self-renewal and altered inflammatory signaling pathways [121].
TET2 encodes an enzyme that catalyzes the oxidation of 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5-hmC). Mutations in TET2 are nonsense or missense changes that lead to a loss of function [122] and DNA hypermethylation due to decreased production of 5-hmC. Mutant TET2 increases the expression of HSC self-renewal genes and sensitizes hematopoietic cells to acquire other mutations and leads to significant myeloid lineage skewing [123] and increased IL-6 production [124]. The order of mutation acquisition can influence the MPN phenotype; mutations in TET2 arising prior to JAK2V617F favors the ET phenotype, but the acquisition of JAK2V617F in a TET2 non-mutated background is more likely to result in the PV phenotype [125].
IDH1 and IDH2 encode isocitrate dehydrogenases that catalyze decarboxylation of isocitrate into alpha ketoglutarate (α-KG). While IDH1 acts in the cytosol, IDH2 works in the mitochondria. The most common IDH1 (p.R132H and p.R132C) and IDH2 mutations (p.R140Q) increase 2-hydroxyglutarate (2-HG) production. 2-HG prevents histone demethylation and the expression of lineage-specific differentiation genes, leading to a block to cell differentiation [126,127,128]. This compound can also bind ten-eleven translocation (TET) and Jumonji proteins, inhibiting their functions [129]. IDH mutations have also been associated to enhanced aberrant splicing of mutant SRSF2, leading to genomic instability and risk of leukemic transformation [130].
4.2. Tumor Suppression
TP53 (tumor protein P53) and PPM1D (protein phosphatase, Mg2+/Mn2+ dependent 1D or P53-induced protein phosphatase 1) are involved in tumor suppression (Figure 6). TP53 is a transcription factor that responds to DNA damage inducing transcriptional programs that result in cell cycle arrest or apoptosis [131]. TP53 mutations are missense changes with several non-mutually exclusive effects: loss of function, gain of function, and dominant-negative effect on normal TP53 [77]. It has been also demonstrated that mutant TP53 increases HSC self-renewal and resistance to cellular stress [132]. There are several upstream regulators of TP53, which are overexpressed in MPNs, such as MDM2 and MDM4. Both of them inhibit TP53 function by facilitating nuclear export and by inducing its degradation [133].
PPM1D is a serin-threonine phosphatase which negatively regulates TP53 and is transcriptionally upregulated on TP53 induction [134]. Mutations in PPM1D are truncating and frameshift changes in exon 6 that lead to a protein that lacks a carboxyterminal degradation domain. This results in altered cell cycle progression, decreased apoptosis and reduced mitochondrial priming [135].
4.3. Regulation of Transcription
RUNX1 (RUNX family transcription factor 1) and NFE2 (nuclear factor, erythroid 2) encode transcription factors and have been also found mutated in MPNs (Figure 6). RUNX1 contains a runt homology domain (RHD) responsible for DNA binding and heterodimerization with core binding factor β (CBF-β). Through this interaction, RUNX1 controls key hematopoietic transcriptional programs. Specifically, RUNX1 participates in hematopoietic differentiation, cell cycle regulation, ribosome biogenesis, and p53 and TGF-β pathways [136]. RUNX1 mutations are missense, frameshift, and non-sense changes that inactivate the protein leading to a reduced myeloid differentiation and an increase in HSC self-renewal [77].
Mutations described in NFE2 are a 4-amino acid in-frame deletion and frameshift changes that lead to a carboxy-terminally truncated protein [40]. Mutant NFE2 promotes myelopoiesis and causes elevated expression of wild-type NFE2 and histone demethylase JMJD1C maybe by a decreased binding of the repressor HP1α [137].
4.4. Splicing
Pre-mRNA splicing is catalyzed by the spliceosome, a complex of five snRNPs and multiple proteins. Mutually exclusive mutations in RNA splicing factors encoded by SRSF2 (serine and arginine rich splicing factor 2), U2AF1 (U2 small nuclear RNA auxiliary factor 1), SF3B1 (splicing factor 3b subunit 1) and ZRSR2 (zinc finger CCCH-type, RNA binding motif and serine/arginine rich 2) have been reported in MPNs (Figure 6, lower box).
SRSF2 contains a ribonucleoprotein with an RNA binding motif and a carboxyl-terminal serine/arginine rich domain [138], both involved in the recognition and binding to the RNA sequences GGNG and CCNG in exon splicing enhancers (ESEs). The most frequent mutation is p.P95H, that leads to a preferential recognition of CCNG motifs and alters the balance of splicing of multiple pre-mRNAs, which cause downregulation of EZH2 [139], as well as the mis-splicing of CASP8, which activates NF-κB signaling [140]. The expression of mutant SRSF2 has also been demonstrated to cause accumulation of R loops, replication stress and activation of ATR-Chk1 signaling [141,142]. Additionally, mutant SRSF2 seems to predominantly form RUNX1a over RUNX1b and regulate DNA stability [143,144].
U2AF1 recognizes pyrimidine-rich tracts with a conserved terminal AG in 3′ splice sites [145]. The most prevalent somatic mutations affect Q157 and its surroundings; p.Q157 mutants generate mis-splicing of ARID2 and EZH2 [123] and are associated with a worse outcome [146]. Patients can also harbor mutations in serine 34 (p.S34F/Y) that cause different expression and splicing patterns than p.Q157 mutations and have been associated with increased splicing, accumulation of R loops and exon skipping [142,147]. Both types of mutations are located within the CCCH zinc fingers of U2AF1, that are critical for RNA binding [148]. This protein has also been shown to bind to mRNA and repress translation; p.S34F mutation seems to affect the translation of hundreds of mRNAs, but the effect of the other mutations on translation is still unknown [149].
ZRSR2 heterodimerizes with U2AF2 and participates in the recognition of the 3′ splice site. Mutations in this gene are mostly frameshift and nonsense loss-of-function changes that affect splicing and lead to intron retention. Mutant ZRSR2 has been reported to cause increased MAPK and ERBB signaling in myelodysplastic syndromes [150].
SF3B1 forms part of the spliceosome complex. Mutations in SF3B1 are missense changes (p.K700E and p.H662Q) that cause alternative 3′ splice site selection [151]. These mutations block erythroid maturation [152] and modify the expression of genes involved in RNA processing, cell cycle, heme metabolism and nonsense-mediated RNA decay [77].
4.5. Additional Signaling Pathways
Finally, other mutations have been found in SH2B3 (SH2B adaptor protein 3, previously known as LNK), CBL (CBL proto-oncogene), NRAS and KRAS (NRAS and KRAS proto-oncogene, GTPase) and PTPN11 (protein tyrosine phosphatase non-receptor type 11), all of them encoding elements involved in signaling (Figure 6).
SH2B3 is an adaptor protein that interacts with and inhibits signaling through cytokine receptors and kinases such JAK2 [153,154,155] decreasing the proliferation of hematopoietic cells [156,157,158]. In addition, this protein can recruit the E3-ubiquitin ligase CBL for degradation of receptors and other molecules [157]. Mutations in SH2B3 are mainly missense changes that disrupt the negative-feedback loops on growth stimulation [155,157].
CBL recognizes and ubiquitinates activated tyrosine kinase receptors and JAK2 leading to their proteasomal degradation. Mutations in CBL are mostly missense changes that reduce the E3 ligase activity and the degradation of its substrates [159,160,161]. However, they are not merely loss-of-function mutations since CBL knockout cells show increased cytokine sensitivity [162].
Missense substitutions affecting NRAS/KRAS favor the GTP-bound state of RAS, causing a constitutive activation of growth signaling [163].
Finally, PTPN11 encodes a protein tyrosine phosphatase which dephosphorylates RAS [164]. PTPN11 mutations increase its phosphatase activity [165], leading to a high dephosphorylation of RAS which increases the activation of RAS-RAF-MEK-ERK pathway [166].
5. Additional Factors Involved in Disease
There are several factors that have been shown to influence heterogeneity in MPN phenotypes, such as the HSC in which the mutation appears first, genetic background, gender, age, and environmental factors.
HSCs are highly heterogeneous and carry a lineage-bias [167]. It has been demonstrated that the acquisition of a driver mutation in a platelet-biased HSC may drive to an ET phenotype, whereas the PV phenotype is more probable when mutation is acquired in balanced/myeloid-biased HSCs [168].
It is well known that there is an association between the JAK2 haplotype 46/1 or GGCC and MPNs. This haplotype is found in 24% of the population and in the 56% of MPN patients [169] increasing the susceptibility of developing a JAK2 mutation, but also to CALR mutations and weakly to MPL mutations [169,170]. Recent studies have identified several SNPs in different loci which have been associated with an increased risk of developing some MPN subtypes [171,172,173].
Regarding gender, the ET phenotype has been mostly reported in women, while PV/PMF are more prevalent in men [174,175]. Women seem to have a greater symptom burden than men [175], but the male gender has been associated with a higher likelihood of myelofibrotic transformation in ET patients [176].
The incidence of MPNs also increases with age, and this factor is the strongest predictor of death in PV and ET [177,178]. This phenomenon has been related to the influence of aging in hematopoiesis, maybe due to a greater probability of acquiring somatic mutations in HSCs [78] favored by a pro-inflammatory state due to the accumulation of inflammatory cytokines associated with age [45]. This higher probability would also explain the increased risk of disease progression in these patients [78].
Retrospective observational studies have reported that the occupational exposure to benzene and/or petroleum, prior blood donation (specifically for PV) [179], and smoking [180,181] are associated with a higher risk of MPNs.
6. Conclusions
The understanding of the pathogenesis of MPNs has undergone a complete revolution in the last 15 years, especially since the p.V617F mutation in JAK2 was characterized. Since then, MPNs have basically been considered signaling disorders, especially affecting the JAK2/STAT pathway, but also the MAPK/ERK and PI3K/AKT pathways. Further characterization of mutations in MPL, and the mechanism by which CALR mutations activate TPOR, reinforced this view. However, although the pathogenic mechanisms of the JAK2, TPOR, and CALR mutants seem quite straightforward and simple, various studies have shown that these alterations can cause more complex disturbances in the cell through non-canonical mechanisms. This, together with the characterization of new somatic genetic alterations that affect genes involved in other processes and signaling pathways, have revealed the complexity of the pathogenesis of MPN, which could partly explain the phenotypic heterogeneity observed among patients.
Author Contributions
Conceptualization: J.L.V. and A.G.-H.; review of the literature: A.G.-H.; writing—original draft preparation: A.G.-H.; writing—review and editing: J.L.V. All authors have read and agreed to the published version of the manuscript.
Funding
J.L.V. and A.G.-H. research are supported by PIUNA 2020 program of the University of Navarra (Code 15058203).
Acknowledgments
The authors acknowledge Cristina Hurtado by her technical support.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Tefferi, A.; Vardiman, J.W. Classification and diagnosis of myeloproliferative neoplasms: The 2008 World Health Organization criteria and point-of-care diagnostic algorithms. Leukemia 2008, 22, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Druker, B.J.; Sawyers, C.L.; Kantarjian, H.; Resta, D.J.; Reese, S.F.; Ford, J.M.; Capdeville, R.; Talpaz, M. Activity of a specific inhibitor of the bcr-abl tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N. Engl. J. Med. 2001, 344, 1038–1042. [Google Scholar] [CrossRef] [PubMed]
- Druker, B.J.; Talpaz, M.; Resta, D.J.; Peng, B.; Buchdunger, E.; Ford, J.M.; Lydon, N.B.; Kantarjian, H.; Capdeville, R.; Ohno-Jones, S.; et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N. Engl. J. Med. 2001, 344, 1031–1037. [Google Scholar] [CrossRef]
- Druker, B.J. Inhibition of the Bcr-Abl tyrosine kinase as a therapeutic strategy for CML. Oncogene 2002, 21, 8541–8546. [Google Scholar] [CrossRef][Green Version]
- Redner, R.L. Why doesn’t imatinib cure chronic myeloid leukemia? Oncologist 2010, 15, 182–186. [Google Scholar] [CrossRef] [PubMed]
- Kalmanti, L.; Saussele, S.; Lauseker, M.; Müller, M.C.; Dietz, C.; Heinrich, L.; Hanfstein, B.; Proetel, U.; Fabarius, A.; Krause, S.W.; et al. Safety and efficacy of imatinib in CML over a period of 10 years: Data from the randomized CML-study IV. Leukemia 2015, 29, 1123–1132. [Google Scholar] [CrossRef] [PubMed]
- Hochhaus, A.; Larson, R.A.; Guilhot, F.; Radich, J.P.; Branford, S.; Hughes, T.P.; Baccarani, M.; Deininger, M.W.; Cervantes, F.; Fujihara, S.; et al. Long-Term Outcomes of Imatinib Treatment for Chronic Myeloid Leukemia. N. Engl. J. Med. 2017, 376, 917–927. [Google Scholar] [CrossRef]
- Verstovsek, S.; Mesa, R.A.; Gotlib, J.; Levy, R.S.; Gupta, V.; DiPersio, J.F.; Catalano, J.V.; Deininger, M.; Miller, C.; Silver, R.T.; et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N. Engl. J. Med. 2012, 366, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Brkic, S.; Meyer, S.C. Challenges and perspectives for therapeutic targeting of myeloproliferative neoplasms. Hemasphere 2020, 5, e516. [Google Scholar] [CrossRef]
- Vannucchi, A.M.; Kiladjian, J.J.; Griesshammer, M.; Masszi, T.; Durrant, S.; Passamonti, F.; Harrison, C.N.; Pane, F.; Zachee, P.; Mesa, R.; et al. Ruxolitinib versus standard therapy for the treatment of polycythemia vera. N. Engl. J. Med. 2015, 372, 426–435. [Google Scholar] [CrossRef]
- Deininger, M.; Radich, J.; Burn, T.C.; Huber, R.; Paranagama, D.; Verstovsek, S. The effect of long-term ruxolitinib treatment on JAK2p.V617F allele burden in patients with myelofibrosis. Blood 2015, 126, 1551–1554. [Google Scholar] [CrossRef] [PubMed]
- Greenfield, G.; McPherson, S.; Mills, K.; McMullin, M.F. The ruxolitinib effect: Understanding how molecular pathogenesis and epigenetic dysregulation impact therapeutic efficacy in myeloproliferative neoplasms. J. Transl. Med. 2018, 16, 1–16. [Google Scholar] [CrossRef]
- Martí-Carvajal, A.J.; Anand, V.; Solà, I. Janus kinase-1 and Janus kinase-2 inhibitors for treating myelofibrosis. Cochrane Database Syst. Rev. 2015, CD010298. [Google Scholar] [CrossRef]
- Cervantes, F.; Pereira, A. Does ruxolitinib prolong the survival of patients with myelofibrosis? Blood 2017, 129, 832–838. [Google Scholar] [CrossRef]
- Verstovsek, S.; Kantarjian, H.; Mesa, R.A.; Pardanani, A.D.; Cortes-Franco, J.; Thomas, D.A.; Estrov, Z.; Fridman, J.S.; Bradley, E.C.; Erickson-Viitanen, S.; et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N. Engl. J. Med. 2010, 363, 1117–1127. [Google Scholar] [CrossRef]
- Kaplan, J.B.; Stein, B.L.; McMahon, B.; Giles, F.J.; Platanias, L.C. Evolving therapeutic strategies for the classic philadelphia-negative myeloproliferative neoplasms. EBioMedicine 2016, 3, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A.; Bannister, A.J.; Göttgens, B.; Foster, S.D.; Bartke, T.; Green, A.R.; Kouzarides, T. JAK2 phosphorylates histone H3Y41 and excludes HP1α from chromatin. Nature 2009, 461, 819–822. [Google Scholar] [CrossRef]
- De Koning, L.; Sauignoni, A.; Boumendil, C.; Rehman, H.; Asselain, B.; Sastre-Garau, X.; Almouzni, G. Heterochromatin protein lα: A hallmark of cell proliferation relevant to clinical oncology. EMBO Mol. Med. 2009, 1, 178–191. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Zhao, X.; Perna, F.; Wang, L.; Koppikar, P.; Abdel-Wahab, O.; Harr, M.W.; Levine, R.L.; Xu, H.; Tefferi, A.; et al. JAK2V617F-mediated phosphorylation of PRMT5 downregulates its methyltransferase activity and promotes myeloproliferation. Cancer Cell 2011, 19, 283–294. [Google Scholar] [CrossRef] [PubMed]
- De Grandis, M.; Cambot, M.; Wautier, M.P.; Cassinat, B.; Chomienne, C.; Colin, Y.; Wautier, J.-L.; Le Van Kim, C.; El Nemer, W. JAK2V617F activates Lu/BCAM-mediated red cell adhesion in polycythemia vera through an EpoR-independent Rap1/Akt pathway. Blood 2013, 121, 658–665. [Google Scholar] [CrossRef] [PubMed]
- Lai, H.Y.; Brooks, S.A.; Craver, B.M.; Morse, S.J.; Nguyen, T.K.; Haghighi, N.; Garbati, M.R.; Fleischman, A.G. Defective negative regulation of Toll-like receptor signaling leads to excessive TNF-α in myeloproliferative neoplasm. Blood Adv. 2019, 3, 122–131. [Google Scholar] [CrossRef]
- Fleischman, A.G.; Aichberger, K.J.; Luty, S.B.; Bumm, T.G.; Petersen, C.L.; Doratotaj, S.; Vasudevan, K.B.; LaTocha, D.H.; Yang, F.; Press, R.D.; et al. TNFα facilitates clonal expansion of JAK2V617F positive cells in myeloproliferative neoplasms. Blood 2011, 118, 6392–6398. [Google Scholar] [CrossRef] [PubMed]
- Kleppe, M.; Kwak, M.; Koppikar, P.; Riester, M.; Keller, M.; Bastian, L.; Hricik, T.; Bhagwat, N.; McKenney, A.S.; Papalexi, E.; et al. JAK-STAT pathway activation in malignant and non-malignant cells contributes to MPN pathogenesis and therapeutic response. Cancer Discov. 2015, 5, 316–331. [Google Scholar] [CrossRef]
- Stetka, J.; Vyhlidalova, P.; Lanikova, L.; Koralkova, P.; Gursky, J.; Hlusi, A.; Flodr, P.; Hubackova, S.; Bartek, J.; Hodny, Z.; et al. Addiction to DUSP1 protects JAK2V617F-driven polycythemia vera progenitors against inflammatory stress and DNA damage, allowing chronic proliferation. Oncogene 2019, 38, 5627–5642. [Google Scholar] [CrossRef]
- Salati, S.; Genovese, E.; Carretta, C.; Zini, R.; Bartalucci, N.; Prudente, Z.; Pennucci, V.; Ruberti, S.; Rossi, C.; Rontauroli, S.; et al. Calreticulin Ins5 and Del52 mutations impair unfolded protein and oxidative stress responses in K562 cells expressing CALR mutants. Sci. Rep. 2019, 9, 10558. [Google Scholar] [CrossRef]
- Pronier, E.; Cifani, P.; Merlinsky, T.R.; Berman, K.B.; Somasundara, A.V.H.; Rampal, R.K.; LaCava, J.; Wei, K.E.; Pastore, F.; Maag, J.L.; et al. Targeting the CALR interactome in myeloproliferative neoplasms. JCI Insight. 2018, 3, e122703. [Google Scholar] [CrossRef] [PubMed]
- Eder-Azanza, L.; Navarro, D.; Aranaz, P.; Novo, F.J.; Cross, N.C.P.; Vizmanos, J.L. Bioinformatic analyses of CALR mutations in myeloproliferative neoplasms support a role in signaling. Leukemia 2014, 28, 2106–2109. [Google Scholar] [CrossRef]
- Fucikova, J.; Spisek, R.; Kroemer, G.; Galluzzi, L. Calreticulin and cancer. Cell Res. 2021, 31, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Arshad, N.; Cresswell, P. Tumor-associated calreticulin variants functionally compromise the peptide loading complex and impair its recruitment of MHC-I. J. Biol. Chem. 2018, 293, 9555–9569. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Zhao, L.; Loos, F.; Marty, C.; Xie, W.; Martins, I.; Lachkar, S.; Qu, B.; Waeckel-Énée, E.; Plo, I.; et al. Immunosuppression by mutated calreticulin released from malignant cells. Mol. Cell. 2020, 77, 748–760.e9. [Google Scholar] [CrossRef] [PubMed]
- Di Buduo, C.A.; Abbonante, V.; Marty, C.; Moccia, F.; Rumi, E.; Pietra, D.; Soprano, P.M.; Lim, D.; Cattaneo, D.; Iurlo, A.; et al. Defective interaction of mutant calreticulin and SOCE in megakaryocytes from patients with myeloproliferative neoplasms. Blood 2020, 135, 133–144. [Google Scholar] [CrossRef]
- Cabagnols, X.; Defour, J.P.; Ugo, V.; Ianotto, J.C.; Mossuz, P.; Mondet, J.; Girodon, F.; Alexandre, J.H.; Mansier, O.; Viallard, J.F.; et al. Differential association of calreticulin type 1 and type 2 mutations with myelofibrosis and essential thrombocytemia: Relevance for disease evolution. Leukemia 2015, 29, 249–252. [Google Scholar] [CrossRef]
- Pietra, D.; Rumi, E.; Ferretti, V.V.; Di Buduo, C.A.; Milanesi, C.; Cavalloni, C.; Sant’Antonio, E.; Abbonante, V.; Moccia, F.; Casetti, I.C.; et al. Differential clinical effects of different mutation subtypes in CALR-mutant myeloproliferative neoplasms. Leukemia 2016, 30, 431–438. [Google Scholar] [CrossRef]
- Hoermann, G.; Greiner, G.; Valent, P. Cytokine regulation of microenvironmental cells in myeloproliferative neoplasms. Mediators Inflamm. 2015, 2015, 869242. [Google Scholar] [CrossRef] [PubMed]
- Fisher, D.A.C.; Miner, C.A.; Engle, E.K.; Hu, H.; Collins, T.B.; Zhou, A.; Allen, M.J.; Malkova, O.; Oh, S.T. Cytokine production in myelofibrosis exhibits differential responsiveness to JAK-STAT, MAP Kinase, and NFκB signaling. Leukemia 2019, 33, 1978–1995. [Google Scholar] [CrossRef] [PubMed]
- Kleppe, M.; Koche, R.; Zou, L.; van Galen, P.; Hill, C.E.; Dong, L.; De Groote, S.; Papalexi, E.; Hanasoge Somasundara, A.V.; Cordner, K.; et al. Dual targeting of oncogenic activation and inflammatory signaling increases therapeutic efficacy in myeloproliferative neoplasms. Cancer Cell 2018, 33, 785–787. [Google Scholar] [CrossRef]
- Wang, W.; Schwemmers, S.; Hexner, E.O.; Pahl, H.L. AML1 is overexpressed in patients with myeloproliferative neoplasms and mediates JAK2V617F-independent overexpression of NF-E2. Blood 2010, 116, 254–266. [Google Scholar] [CrossRef] [PubMed]
- Hasselbalch, H.C.; Thomassen, M.; Riley, C.H.; Kjær, L.; Larsen, T.S.; Jensen, M.K.; Bjerrum, O.W.; Kruse, T.A.; Skov, V. Whole blood transcriptional profiling reveals deregulation of oxidative and antioxidative defence genes in myelofibrosis and related neoplasms. Potential implications of downregulation of Nrf2 for genomic instability and disease progression. PLoS ONE 2014, 9, e112786. [Google Scholar] [CrossRef] [PubMed]
- Wehrle, J.; Seeger, T.S.; Schwemmers, S.; Pfeifer, D.; Bulashevska, A.; Pahl, H.L. Transcription factor nuclear factor erythroid-2 mediates expression of the cytokine interleukin 8, a known predictor of inferior outcome in patients with myeloproliferative Neoplasms. Haematologica 2013, 98, 1073–1080. [Google Scholar] [CrossRef] [PubMed]
- Jutzi, J.S.; Bogeska, R.; Nikoloski, G.; Schmid, C.A.; Seeger, T.S.; Stegelmann, F.; Schwemmers, S.; Gründer, A.; Peeken, J.C.; Gothwal, M.; et al. MPN patients harbor recurrent truncating mutations in transcription factor NF-E2. J. Exp. Med. 2013, 210, 1003–1019. [Google Scholar] [CrossRef] [PubMed]
- Motohashi, H.; Kimura, M.; Fujita, R.; Inoue, A.; Pan, X.; Takayama, M.; Katsuoka, F.; Aburatani, H.; Bresnick, E.H.; Yamamoto, M. NF-E2 domination over Nrf2 promotes ROS accumulation and megakaryocytic maturation. Blood 2010, 115, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Hasselbalch, H.C. Chronic inflammation as a promotor of mutagenesis in essential thrombocythemia, polycythemia vera and myelofibrosis. A human inflammation model for cancer development? Leuk. Res. 2013, 37, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Hasan, S.; Lacout, C.; Marty, C.; Cuingnet, M.; Solary, E.; Vainchenker, W.; Villeval, J.-L. JAK2V617F expression in mice amplifies early hematopoietic cells and gives them a competitive advantage that is hampered by IFNα. Blood 2013, 22, 1464–1477. [Google Scholar] [CrossRef] [PubMed]
- Mullally, A.; Bruedigam, C.; Poveromo, L.; Heidel, F.H.; Purdon, A.; Vu, T.; Austin, R.; Heckl, D.; Breyfogle, L.J.; Kuhn, C.P.; et al. Depletion of Jak2V617F myeloproliferative neoplasm-propagating stem cells by interferon-α in a murine model of polycythemia vera. Blood 2013, 121, 3692–3702. [Google Scholar] [CrossRef] [PubMed]
- Hemmati, S.; Haque, T.; Gritsman, K. Inflammatory signaling pathways in preleukemic and leukemic stem cells. Front. Oncol. 2017, 7, 265. [Google Scholar] [CrossRef]
- Hérault, A.; Binnewies, M.; Leong, S.; Calero-Nieto, F.J.; Zhang, Y.; Kang, Y.; Wang, X.; Pietras, E.M.; Chu, S.H.; Barry-Holson, K.; et al. Myeloid progenitor cluster formation drives emergency and leukemic myelopoiesis. Nature 2017, 544, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Geyer, H.L.; Dueck, A.C.; Scherber, R.M.; Mesa, R.A. Impact of inflammation on myeloproliferative neoplasm symptom development. Mediators Inflamm. 2015, 2015, 284706. [Google Scholar] [CrossRef]
- Pourcelot, E.; Trocme, C.; Mondet, J.; Bailly, S.; Toussaint, B.; Mossuz, P. Cytokine profiles in polycythemia vera and essential thrombocythemia patients: Clinical implications. Exp. Hematol. 2014, 42, 360–368. [Google Scholar] [CrossRef]
- Hoermann, G.; Cerny-Reiterer, S.; Herrmann, H.; Blatt, K.; Bilban, M.; Gisslinger, H.; Müllauer, L.; Kralovics, R.; Mannhalter, C.; Valent, P.; et al. Identification of oncostatin M as a JAK2 V617F-dependent amplifier of cytokine production and bone marrow remodeling in myeloproliferative neoplasms. FASEB J. 2012, 26, 894–906. [Google Scholar] [CrossRef]
- Le Bousse-Kerdilès, M.C.; Chevillard, S.; Charpentier, A.; Romquin, N.; Clay, D.; Smadja-Joffe, F.; Praloran, V.; Dupriez, B.; Demory, J.L.; Jasmin, C.; et al. Differential expression of transforming growth factor-beta, basic fibroblast growth factor, and their receptors in CD34+ hematopoietic progenitor cells from patients with myelofibrosis and myeloid metaplasia. Blood 1996, 88, 4534–4546. [Google Scholar] [CrossRef] [PubMed]
- Martyré, M.-C.; Magdelenat, H.; Bryckaert, M.-C.; Laine-Bidron, C.; Calvo, F. Increased intraplatelet levels of platelet-derived growth factor and transforming growth factor-β in patients with myelofibrosis with myeloid metaplasia. Br. J. Haematol. 1991, 77, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Di Raimondo, F.; Azzaro, M.P.; Palumbo, G.A.; Bagnato, S.; Stagno, F.; Giustolisi, G.M.; Cacciola, E.; Sortino, G.; Guglielmo, P.; Giustolisi, R. Elevated vascular endothelial growth factor [VEGF] serum levels in idiopathic myelofibrosis. Leukemia 2001, 15, 976–980. [Google Scholar] [CrossRef]
- Bock, O.; Höftmann, J.; Theophile, K.; Hussein, K.; Wiese, B.; Schlué, J.; Kreipe, H. Bone morphogenetic proteins are overexpressed in the bone marrow of primary myelofibrosis and are apparently induced by fibrogenic cytokines. Am. J. Pathol. 2008, 172, 951–960. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.C.; Novetsky, A.; Chen, C.; Novetsky, A.D. Plasma matrix metalloproteinase and tissue inhibitor of metalloproteinase in patients with agnogenic myeloid metaplasia or idiopathic primary myelofibrosis. Br. J. Haematol. 2002, 119, 709–712. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.K.; Holten-Andersen, M.N.; Riisbro, R.; De Nully Brown, P.; Larsen, M.B.; Kjeldsen, L.; Heickendorff, L.; Brünner, N.; Hasselbalch, H.C. Elevated plasma levels of TIMP-1 correlate with plasma suPAR/uPA in patients with chronic myeloproliferative disorders. Eur. J. Haematol. 2003, 71, 377–384. [Google Scholar] [CrossRef]
- Lu, M.; Xia, L.; Liu, Y.C.; Hochman, T.; Bizzari, L.; Aruch, D.; Lew, J.; Weinberg, R.; Goldberg, J.D.; Hoffman, R. Lipocalin produced by myelofibrosis cells affects the fate of both hematopoietic and marrow microenvironmental cells. Blood 2015, 126, 972–982. [Google Scholar] [CrossRef]
- Gallardo, M.; Barrio, S.; Fernandez, M.; Paradela, A.; Arenas, A.; Toldos, O.; Ayala, R.; Albizua, E.; Jimenez, A.; Redondo, S.; et al. Proteomic analysis reveals heat shock protein 70 has a key role in polycythemia vera. Mol. Cancer 2013, 12, 142. [Google Scholar] [CrossRef] [PubMed]
- Asea, A.; Kraeft, S.K.; Kurt-Jones, E.A.; Stevenson, M.A.; Chen, L.B.; Finberg, R.W.; Koo, G.C.; Calderwood, S.K. HSP70 stimulates cytokine production through a CD 14-dependant pathway, demonstrating its dual role as a chaperone and cytokine. Nat. Med. 2000, 6, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Sevin, M.; Girodon, F.; Garrido, C.; De Thonel, A. HSP90 and HSP70: Implication in inflammation processes and therapeutic approaches for myeloproliferative neoplasms. Mediators Inflamm. 2015, 2015, 970242. [Google Scholar] [CrossRef]
- Falanga, A.; Marchetti, M.; Evangelista, V.; Vignoli, A.; Licini, M.; Balicco, M.; Manarini, S.; Finazzi, G.; Cerletti, C.; Barbui, T. Polymorphonuclear leukocyte activation and hemostasis in patients with essential thrombocythemia and polycythemia vera. Blood 2000, 96, 4261–4266. [Google Scholar] [CrossRef]
- Villmow, T.; Kemkes-Matthes, B.; Matzdorff, A.C. Markers of platelet activation and platelet-leukocyte interaction in patients with myeloproliferative syndromes. Thromb. Res. 2002, 108, 139–145. [Google Scholar] [CrossRef]
- Falanga, A.; Marchetti, M.; Vignoli, A.; Balducci, D.; Barbui, T. Leukocyte-platelet interaction in patients with essential thrombocythemia and polycythemia vera. Exp. Hematol. 2005, 33, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Curto-Garcia, N.; Harrison, C.; McLornan, D.P. Bone marrow niche dysregulation in myeloproliferative neoplasms. Haematologica 2020, 105, 1189–1200. [Google Scholar] [CrossRef] [PubMed]
- Zhan, H.; Lin, C.H.S.; Segal, Y.; Kaushansky, K. The JAK2V617F-bearing vascular niche promotes clonal expansion in myeloproliferative neoplasms. Leukemia 2018, 32, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.H.S.; Zhang, Y.; Kaushansky, K.; Zhan, H. JAK2V617F-bearing vascular niche enhances malignant hematopoietic regeneration following radiation injury. Haematologica 2018, 103, 1160–1168. [Google Scholar] [CrossRef]
- Ramos, T.L.; Sánchez-Abarca, L.I.; Rosón-Burgo, B.; Redondo, A.; Rico, A.; Preciado, S.; Ortega, R.; Rodríguez, C.; Muntión, S.; Hernández-Hernández, A.; et al. Mesenchymal stromal cells [MSC] from JAK2+ myeloproliferative neoplasms differ from normal MSC and contribute to the maintenance of neoplastic hematopoiesis. PLoS ONE 2017, 12, 1–21. [Google Scholar] [CrossRef]
- Koopmans, S.M.; Bot, F.J.; Schouten, H.C.; Janssen, J.; van Marion, A.M. The involvement of Galectins in the modulation of the JAK/STAT pathway in myeloproliferative neoplasia. Am. J. Blood Res. 2012, 2, 119–127. [Google Scholar] [PubMed]
- Spanoudakis, E.; Papoutselis, M.; Bazdiara, I.; Lamprianidi, E.; Kordella, X.; Tilkeridis, C.; Tsatalas, C.; Kotsianidis, I. The JAK2V617F point mutation increases the osteoclast forming ability of monocytes in patients with chronic myeloproliferative neoplasms and makes their osteoclasts more susceptible to JAK2 inhibition. Mediterr. J. Hematol. Infect. Dis. 2018, 10, e2018058. [Google Scholar] [CrossRef]
- Korn, C.; Rak, J.; García-García, A.; Fielding, C.; Khorshed, R.; González-Antón, S.; Li, J.; Norfo, R.; Baxter, E.J.; McKerrell, T.; et al. Niche heterogeneity impacts evolution of myeloproliferative neoplasms driven by the same oncogenic pathway. Blood 2018, 132 (Suppl. 1), 98. [Google Scholar] [CrossRef]
- Arranz, L.; Sánchez-Aguilera, A.; Martín-Pérez, D.; Isern, J.; Langa, X.; Tzankov, A.; Lundberg, P.; Muntión, S.; Tzeng, Y.-S.; Lai, D.-M.; et al. Neuropathy of haematopoietic stem cell niche is essential for myeloproliferative neoplasms. Nature 2014, 512, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.M.; Zhang, L.J.; Fu, J.Z.; Liang, W.T.; Cheng, Z.Y.; Bai, P.; Bian, Y.S.; Wan, J.S. Regulation of Ruxolitinib on matrix metalloproteinase in JAK2V617F positive myeloroliferative neoplasms cells. Zhonghua Xue Ye Xue Za Zhi 2017, 38, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Tadmor, T.; Bejar, J.; Attias, D.; Mischenko, E.; Sabo, E.; Neufeld, G.; Vadasz, Z. The expression of lysyl-oxidase gene family members in myeloproliferative neoplasms. Am. J. Hematol. 2013, 88, 355–358. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Lasho, T.L.; Finke, C.M.; Elala, Y.; Hanson, C.A.; Ketterling, R.P.; Gangat, N.; Pardanani, A. Targeted deep sequencing in primary myelofibrosis. Blood Adv. 2016, 1, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Lasho, T.L.; Guglielmelli, P.; Finke, C.M.; Rotunno, G.; Elala, Y.; Pacilli, A.; Hanson, C.A.; Pancrazzi, A.; Ketterling, R.P.; et al. Targeted deep sequencing in polycythemia vera and essential thrombocythemia. Blood Adv. 2016, 1, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Guglielmelli, P.; Lasho, T.L.; Rotunno, G.; Score, J.; Mannarelli, C.; Pancrazzi, A.; Biamonte, F.; Pardanani, A.; Zoi, K.; Reiter, A.; et al. The number of prognostically detrimental mutations and prognosis in primary myelofibrosis: An international study of 797 patients. Leukemia 2014, 28, 1804–1810. [Google Scholar] [CrossRef]
- Grabek, J.; Straube, J.; Bywater, M.; Lane, S.W. MPN: The molecular drivers of disease initiation, progression and transformation and their effect on treatment. Cells 2020, 9, 1901. [Google Scholar] [CrossRef] [PubMed]
- Marneth, A.E.; Mullally, A. The molecular genetics of myeloproliferative neoplasms. Cold Spring Harb. Perspect. Med. 2020, 10, a034876. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, J.; Mead, A.J. Heterogeneity in myeloproliferative neoplasms: Causes and consequences. Adv. Biol. Regul. 2019, 71, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Baxter, E.J.; Scott, L.M.; Campbell, P.J.; East, C.; Fourouclas, N.; Swanton, S.; Vassiliou, G.S.; Bench, A.J.; Boyd, E.M.; Curtin, N.; et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005, 365, 1054–1061. [Google Scholar] [CrossRef]
- James, C.; Ugo, V.; Le Couédic, J.P.; Staerk, J.; Delhommeau, F.; Lacout, C.; Garçon, L.; Raslova, H.; Berger, R.; Bennaceur-Griscelli, A.; et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005, 434, 1144–1148. [Google Scholar] [CrossRef]
- Kralovics, R.; Teo, S.-S.; Li, S.; Theocharides, A.; Buser, A.S.; Tichelli, A.; Skoda, R.C. Acquisition of the V617F mutation of JAK2 is a late genetic event in a subset of patients with myeloproliferative disorders. Blood 2006, 108, 1377–1380. [Google Scholar] [CrossRef] [PubMed]
- Levine, R.L.; Wadleigh, M.; Cools, J.; Ebert, B.L.; Wernig, G.; Huntly, B.J.P.; Boggon, T.J.; Wlodarska, I.; Clark, J.J.; Moore, S.; et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005, 7, 387–397. [Google Scholar] [CrossRef]
- Zhao, R.; Xing, S.; Li, Z.; Fu, X.; Li, Q.; Krantz, S.B.; Zhao, Z.J. Identification of an acquired JAK2 mutation in polycythemia vera. J. Biol. Chem. 2005, 280, 22788–22792. [Google Scholar] [CrossRef]
- Tefferi, A.; Barbui, T. Polycythemia vera and essential thrombocythemia: 2019 update on diagnosis, risk-stratification and management. Am. J. Hematol. 2019, 94, 133–143. [Google Scholar] [CrossRef]
- Scott, L.M.; Tong, W.; Levine, R.L.; Scott, M.A.; Beer, P.A.; Stratton, M.R.; Futreal, P.A.; Erber, W.N.; McMullin, M.F.; Harrison, C.N.; et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N. Engl. J. Med. 2007, 356, 459–468. [Google Scholar] [CrossRef]
- Passamonti, F.; Elena, C.; Schnittger, S.; Skoda, R.C.; Green, A.R.; Girodon, F.; Kiladjian, J.-J.; McMullin, M.F.; Ruggeri, M.; Besses, C.; et al. Molecular and clinical features of the myeloproliferative neoplasm associated with JAK2 exon 12 mutations. Blood 2011, 117, 2813–2816. [Google Scholar] [CrossRef] [PubMed]
- Pikman, Y.; Lee, B.H.; Mercher, T.; McDowell, E.; Ebert, B.L.; Gozo, M.; Cuker, A.; Wernig, G.; Moore, S.; Galinsky, I.; et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006, 3, e270. [Google Scholar] [CrossRef]
- Pardanani, A.D.; Levine, R.L.; Lasho, T.; Pikman, Y.; Mesa, R.A.; Wadleigh, M.; Steensma, D.P.; Elliott, M.A.; Wolanskyj, A.P.; Hogan, W.J.; et al. MPL515 mutations in myeloproliferative and other myeloid disorders: A study of 1182 patients. Blood 2006, 108, 3472–3476. [Google Scholar] [CrossRef]
- Defour, J.P.; Itaya, M.; Gryshkova, V.; Brett, I.C.; Pecquet, C.; Sato, T.; Smith, S.O.; Constantinescu, S.N. Tryptophan at the transmembrane-cytosolic junction modulates thrombopoietin receptor dimerization and activation. Proc. Natl. Acad. Sci. USA 2013, 110, 2540–2545. [Google Scholar] [CrossRef] [PubMed]
- Nangalia, J.; Grinfeld, J.; Green, A.R. Pathogenesis of myeloproliferative disorders. Annu. Rev. Pathol. 2016, 11, 101–126. [Google Scholar] [CrossRef] [PubMed]
- Nangalia, J.; Massie, C.E.; Baxter, E.J.; Nice, F.L.; Gundem, G.; Wedge, D.C.; Avezov, E.; Li, J.; Kollmann, K.; Kent, D.G.; et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N. Engl. J. Med. 2013, 369, 2391–2405. [Google Scholar] [CrossRef]
- Klampfl, T.; Gisslinger, H.; Harutyunyan, A.S.; Nivarthi, H.; Rumi, E.; Milosevic, J.D.; Them, N.C.C.; Berg, T.; Gisslinger, B.; Pietra, D.; et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N. Engl. J. Med. 2013, 369, 2379–2390. [Google Scholar] [CrossRef] [PubMed]
- Michalak, M.; Corbett, E.F.; Mesaeli, N.; Nakamura, K.; Opas, M. Calreticulin: One protein, one gene, many functions. Biochem. J. 1999, 344, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.A.; Groenendyk, J.; Michalak, M. Calreticulin signaling in health and disease. Int. J. Biochem. Cell Biol. 2012, 44, 842–846. [Google Scholar] [CrossRef] [PubMed]
- Shivarov, V.; Ivanova, M.; Tiu, R.V. Mutated calreticulin retains structurally disordered C terminus that cannot bind Ca[2+]: Some mechanistic and therapeutic implications. Blood Cancer J. 2014, 4, e185. [Google Scholar] [CrossRef]
- Araki, M.; Yang, Y.; Masubuchi, N.; Hironaka, Y.; Takei, H.; Morishita, S.; Mizukami, Y.; Kan, S.; Shirane, S.; Edahiro, Y.; et al. Activation of the thrombopoietin receptor by mutant calreticulin in CALR-mutant myeloproliferative neoplasms. Blood 2016, 127, 1307–1316. [Google Scholar] [CrossRef]
- Chachoua, I.; Pecquet, C.; El-Khoury, M.; Nivarthi, H.; Albu, R.I.; Marty, C.; Gryshkova, V.; Defour, J.-P.; Vertenoeil, G.; Ngo, A.; et al. Thrombopoietin receptor activation by myeloproliferative neoplasm associated calreticulin mutants. Blood 2016, 127, 1325–1335. [Google Scholar] [CrossRef] [PubMed]
- Elf, S.; Abdelfattah, N.S.; Chen, E.; Perales-Patón, J.; Rosen, E.A.; Ko, A.; Peisker, F.; Florescu, N.; Giannini, S.; Wolach, O.; et al. Mutant calreticulin requires both its mutant C-terminus and the thrombopoietin receptor for oncogenic transformation. Cancer Discov. 2016, 6, 368–381. [Google Scholar] [CrossRef] [PubMed]
- Vainchenker, W.; Kralovics, R. Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood 2017, 129, 667–679. [Google Scholar] [CrossRef]
- Elf, S.; Abdelfattah, N.S.; Baral, A.J.; Beeson, D.; Rivera, J.F.; Ko, A.; Florescu, N.; Birrane, G.; Chen, E.; Mullally, A. Defining the requirements for the pathogenic interaction between mutant calreticulin and MPL in MPN. Blood 2018, 131, 782–786. [Google Scholar] [CrossRef] [PubMed]
- Araki, M.; Komatsu, N. The role of calreticulin mutations in myeloproliferative neoplasms. Int. J. Hematol. 2020, 111, 200–205. [Google Scholar] [CrossRef]
- Han, L.; Schubert, C.; Köhler, J.; Schemionek, M.; Isfort, S.; Brümmendorf, T.H.; Koschmieder, S.; Chatain, N. Calreticulin-mutant proteins induce megakaryocytic signaling to transform hematopoietic cells and undergo accelerated degradation and Golgi-mediated secretion. J. Hematol. Oncol. 2016, 9, 45. [Google Scholar] [CrossRef]
- Pecquet, C.; Balligand, T.; Chachoua, I.; Roy, A.; Vertenoeil, G.; Colau, D.; Fertig, E.; Marty, C.; Nivarthi, H.; Defour, J.-P.; et al. Secreted mutant calreticulins as rogue cytokines trigger thrombopoietin receptor activation specifically in CALR mutated cells: Perspectives for MPN therapy. Blood 2018, 132 (Suppl. 1), 4. [Google Scholar] [CrossRef]
- Rampal, R.; Al-Shahrour, F.; Abdel-Wahab, O.; Patel, J.; Brunel, J.-P.; Mermel, C.H.; Bass, A.J.; Pretz, J.; Ahn, J.; Hricik, T.; et al. Integrated genomic analysis illustrates the central role of JAK-STAT pathway activation in myeloproliferative neoplasm pathogenesis. Blood 2014, 123, e123–e133. [Google Scholar] [CrossRef] [PubMed]
- Reuther, G.W. Myeloproliferative neoplasms: Molecular drivers and therapeutics. Prog. Mol. Biol. Transl. Sci. 2016, 144, 437–484. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Vaidya, R.; Caramazza, D.; Finke, C.; Lasho, T.; Pardanani, A. Circulating interleukin (IL)-8, IL-2R, IL-12, and IL-15 levels are independently prognostic in primary myelofibrosis: A comprehensive cytokine profiling study. J. Clin. Oncol. 2011, 29, 1356–1363. [Google Scholar] [CrossRef] [PubMed]
- Kiladjian, J.J.; Giraudier, S.; Cassinat, B. Interferon-alpha for the therapy of myeloproliferative neoplasms: Targeting the malignant clone. Leukemia 2016, 30, 776–781. [Google Scholar] [CrossRef] [PubMed]
- Kagoya, Y.; Yoshimi, A.; Tsuruta-Kishino, T.; Arai, S.; Satoh, T.; Akira, S.; Kurokawa, M. JAK2V617F+ myeloproliferative neoplasm clones evoke paracrine DNA damage to adjacent normal cells through secretion of lipocalin-2. Blood 2014, 124, 2996–3006. [Google Scholar] [CrossRef] [PubMed]
- Decker, M.; Martinez-Morentin, L.; Wang, G.; Lee, Y.; Liu, Q.; Leslie, J.; Ding, L. Leptin-receptor-expressing bone marrow stromal cells are myofibroblasts in primary myelofibrosis. Nat. Cell Biol. 2017, 19, 677–688. [Google Scholar] [CrossRef] [PubMed]
- Schneider, R.K.; Mullally, A.; Dugourd, A.; Peisker, F.; Hoogenboezem, R.; Van Strien, P.M.H.; Bindels, E.M.; Heckl, D.; Büsche, G.; Fleck, D.; et al. Gli1+ mesenchymal stromal cells are a key driver of bone marrow fibrosis and an important cellular therapeutic target. Cell. Stem Cell. 2017, 20, 785–800. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Wahab, O.; Adli, M.; LaFave, L.M.; Gao, J.; Hricik, T.; Shih, A.H.; Pandey, S.; Patel, J.P.; Chung, Y.R.; Koche, R.; et al. ASXL1 mutations promote myeloid transformation through loss of PRC2-mediated gene repression. Cancer Cell 2012, 22, 180–193. [Google Scholar] [CrossRef]
- Asada, S.; Goyama, S.; Inoue, D.; Shikata, S.; Takeda, R.; Fukushima, T.; Yonezawa, T.; Fujino, T.; Hayashi, Y.; Kawabata, K.C.; et al. Mutant ASXL1 cooperates with BAP1 to promote myeloid leukaemogenesis. Nat. Commun. 2018, 9, 2733. [Google Scholar] [CrossRef] [PubMed]
- Balasubramani, A.; Larjo, A.; Bassein, J.A.; Chang, X.; Hastie, R.B.; Togher, S.M.; Lähdesmäki, H.; Rao, A. Cancer-associated ASXL1 mutations may act as gain-of-function mutations of the ASXL1-BAP1 complex. Nat. Commun. 2015, 6, 7307. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Kurtenbach, S.; Guo, Y.; Lohse, I.; Durante, M.A.; Li, J.; Li, Z.; Al-Ali, H.; Li, L.; Chen, Z.; et al. Gain of function of ASXL1 truncating protein in the pathogenesis of myeloid malignancies. Blood 2018, 131, 328–341. [Google Scholar] [CrossRef] [PubMed]
- Saika, M.; Inoue, D.; Nagase, R.; Sato, N.; Tsuchiya, A.; Yabushita, T.; Kitamura, T.; Goyama, S. ASXL1 and SETBP1 mutations promote leukaemogenesis by repressing TGFβ pathway genes through histone deacetylation. Sci. Rep. 2018, 8, 15873. [Google Scholar] [CrossRef]
- Cho, Y.-S.; Kim, E.-J.; Park, U.-H.; Sin, H.-S.; Um, S.-J. Additional sex comb-like 1 [ASXL1], in cooperation with SRC-1, acts as a ligand-dependent coactivator for retinoic acid receptor. J. Biol. Chem. 2006, 281, 17588–17598. [Google Scholar] [CrossRef] [PubMed]
- Park, U.-H.; Yoon, S.-K.; Park, T.; Kim, E.-J.; Um, S.-J. Additional sex comb-like [ASXL] proteins 1 and 2 play opposite roles in adipogenesis via reciprocal regulation of peroxisome proliferator-activated receptor γ. J. Biol. Chem. 2011, 286, 1354–1363. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.N.; Jankowska, A.M.; Mahfouz, R.; Dunbar, A.J.; Sugimoto, Y.; Hosono, N.; Hu, Z.; Cheriyath, V.; Vatolin, S.; Przychodzen, B.; et al. Multiple mechanisms deregulate EZH2 and histone H3 lysine 27 epigenetic changes in myeloid malignancies. Leukemia 2013, 27, 1301–1309. [Google Scholar] [CrossRef] [PubMed]
- Rinke, J.; Chase, A.; Cross, N.C.P.; Hochhaus, A.; Ernst, T. EZH2 in myeloid malignancies. Cells 2020, 9, 1639. [Google Scholar] [CrossRef]
- Anteneh, H.; Fang, J.; Song, J. Structural basis for impairment of DNA methylation by the DNMT3A R882H mutation. Nat. Commun. 2020, 11, 2294. [Google Scholar] [CrossRef] [PubMed]
- Jacquelin, S.; Straube, J.; Cooper, L.; Vu, T.; Song, A.; Bywater, M.; Baxter, E.; Heidecker, M.; Wackrow, B.; Porter, A.; et al. Jak2V617F and Dnmt3a loss cooperate to induce myelofibrosis through activated enhancer-driven inflammation. Blood 2018, 132, 2707–2721. [Google Scholar] [CrossRef]
- Abdel-Wahab, O.; Mullally, A.; Hedvat, C.; Garcia-Manero, G.; Patel, J.; Wadleigh, M.; Malinge, S.; Yao, J.; Kilpivaara, O.; Bhat, R.; et al. Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood 2009, 114, 144–147. [Google Scholar] [CrossRef] [PubMed]
- Ostrander, E.L.; Kramer, A.C.; Mallaney, C.; Celik, H.; Koh, W.K.; Fairchild, J.; Haussler, E.; Zhang, C.R.C.; Challen, G.A. Divergent effects of Dnmt3a and Tet2 mutations on hematopoietic progenitor cell fitness. Stem Cell Reports 2020, 14, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhao, K.; Shen, Q.; Han, Y.; Gu, Y.; Li, X.; Zhao, D.; Liu, Y.; Wang, C.; Zhang, X.; et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature 2015, 525, 389–393. [Google Scholar] [CrossRef]
- Ortmann, C.A.; Kent, D.G.; Nangalia, J.; Silber, Y.; Wedge, D.C.; Grinfeld, J.; Baxter, E.J.; Massie, C.E.; Papaemmanuil, E.; Menon, S.; et al. Effect of mutation order on myeloproliferative neoplasms. N. Engl. J. Med. 2015, 372, 601–612. [Google Scholar] [CrossRef]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Lasho, T.L.; Abdel-Wahab, O.; Guglielmelli, P.; Patel, J.; Caramazza, D.; Pieri, L.; Finke, C.M.; Kilpivaara, O.; Wadleigh, M.; et al. IDH1 and IDH2 mutation studies in 1473 patients with chronic-, fibrotic- or blast-phase essential thrombocythemia, polycythemia vera or myelofibrosis. Leukemia 2010, 24, 1302–1309. [Google Scholar] [CrossRef]
- Pardanani, A.; Lasho, T.L.; Finke, C.M.; Mai, M.; McClure, R.F.; Tefferi, A. IDH1 and IDH2 mutation analysis in chronic-and blast-phase myeloproliferative neoplasms. Leukemia 2010, 24, 1146–1151. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.T.; et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef]
- Yoshimi, A.; Lin, K.T.; Wiseman, D.H.; Rahman, M.A.; Pastore, A.; Wang, B.; Lee, S.C.; Micol, J.B.; Zhang, X.J.; de Botton, S.; et al. Coordinated alterations in RNA splicing and epigenetic regulation drive leukaemogenesis. Nature 2019, 574, 273–277. [Google Scholar] [CrossRef]
- Hafner, A.; Bulyk, M.L.; Jambhekar, A.; Lahav, G. The multiple mechanisms that regulate p53 activity and cell fate. Nat. Rev. Mol. Cell Biol. 2019, 20, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Elf, S.E.; Miyata, Y.; Sashida, G.; Liu, Y.; Huang, G.; Di Giandomenico, S.; Lee, J.M.; Deblasio, A.; Menendez, S.; et al. p53 regulates hematopoietic stem cell quiescence. Cell Stem Cell. 2009, 4, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Marcellino, B.K.; Hoffman, R.; Tripodi, J.; Lu, M.; Kosiorek, H.; Mascarenhas, J.; Rampal, R.K.; Dueck, A.; Najfeld, V. Advanced forms of MPNs are accompanied by chromosomal abnormalities that lead to dysregulation of TP53. Blood Adv. 2018, 2, 3581–3589. [Google Scholar] [CrossRef]
- Fiscella, M.; Zhang, H.; Fan, S.; Sakaguchi, K.; Shen, S.; Mercer, W.E.; Vande Woude, G.F.; O’Connor, P.M.; Appella, E. Wip1, a novel human protein phosphatase that is induced in response to ionizing radiation in a p53-dependent manner. Proc. Natl. Acad. Sci. USA 1997, 94, 6048–6053. [Google Scholar] [CrossRef] [PubMed]
- Kahn, J.D.; Miller, P.G.; Silver, A.J.; Sellar, R.S.; Bhatt, S.; Gibson, C.; McConkey, M.; Adams, D.; Mar, B.; Mertins, P.; et al. PPM1D-truncating mutations confer resistance to chemotherapy and sensitivity to PPM1D inhibition in hematopoietic cells. Blood 2018, 132, 1095–1105. [Google Scholar] [CrossRef]
- Sood, R.; Kamikubo, Y.; Liu, P. Role of RUNX1 in hematological malignancies. Blood 2017, 129, 2070–2082. [Google Scholar] [CrossRef] [PubMed]
- Peeken, J.C.; Jutzi, J.S.; Wehrle, J.; Koellerer, C.; Staehle, H.F.; Becker, H.; Schoenwandt, E.; Seeger, T.S.; Schanne, D.H.; Gothwal, M.; et al. Epigenetic regulation of NFE2 overexpression in myeloproliferative neoplasms. Blood 2018, 131, 2065–2073. [Google Scholar] [CrossRef]
- Will, C.L.; Lührmann, R. Spliceosome structure and function. Cold Spring Harb. Perspect. Biol. 2011, 3, a003707. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Ilagan, J.O.; Liang, Y.; Daubner, G.M.; Lee, S.C.; Ramakrishnan, A.; Li, Y.; Chung, Y.R.; Micol, J.B.; Murphy, M.E.; et al. SRSF2 mutations contribute to myelodysplasia by mutant-specific effects on exon recognition. Cancer Cell 2015, 27, 617–630. [Google Scholar] [CrossRef]
- Lee, S.C.; North, K.; Kim, E.; Jang, E.; Obeng, E.; Lu, S.X.; Liu, B.; Inoue, D.; Yoshimi, A.; Ki, M.; et al. Synthetic lethal and convergent biological effects of cancer-associated spliceosomal gene mutations. Cancer Cell 2018, 34, 225–241.e8. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Chen, J.Y.; Huang, Y.J.; Gu, Y.; Qiu, J.; Qian, H.; Shao, C.; Zhang, X.; Hu, J.; Li, H.; et al. The augmented R-loop is a unifying mechanism for myelodysplastic syndromes induced by high-risk splicing factor mutations. Mol. Cell 2018, 69, 412–425.e6. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.D.; Leong, W.Y.; Li, W.; Reddy, P.N.G.; Sullivan, J.D.; Walter, M.J.; Zou, L.; Graubert, T.A. Spliceosome mutations induce R loop-associated sensitivity to ATR inhibition in myelodysplastic syndromes. Cancer Res. 2018, 78, 5363–5374. [Google Scholar] [CrossRef]
- Sakurai, H.; Harada, Y.; Ogata, Y.; Kagiyama, Y.; Shingai, N.; Doki, N.; Ohashi, K.; Kitamura, T.; Komatsu, N.; Harada, H. Overexpression of RUNX1 short isoform has an important role in the development of myelodysplastic/myeloproliferative neoplasms. Blood Adv. 2017, 1, 1382–1386. [Google Scholar] [CrossRef][Green Version]
- Xiao, R.; Sun, Y.; Ding, J.H.; Lin, S.; Rose, D.W.; Rosenfeld, M.G.; Fu, X.D.; Li, X. Splicing regulator SC35 is essential for genomic stability and cell proliferation during mammalian organogenesis. Mol. Cell Biol. 2007, 27, 5393–5402. [Google Scholar] [CrossRef]
- Soares, L.M.; Zanier, K.; Mackereth, C.; Sattler, M.; Valcárcel, J. Intron removal requires proofreading of U2AF/3′ splice site recognition by DEK. Science 2006, 312, 1961–1965. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Finke, C.M.; Lasho, T.L.; Hanson, C.A.; Ketterling, R.P.; Gangat, N.; Pardanani, A. U2AF1 mutation types in primary myelofibrosis: Phenotypic and prognostic distinctions. Leukemia 2018, 32, 2274–2278. [Google Scholar] [CrossRef] [PubMed]
- Graubert, T.A.; Shen, D.; Ding, L.; Okeyo-Owuor, T.; Lunn, C.L.; Shao, J.; Krysiak, K.; Harris, C.C.; Koboldt, D.C.; Larson, D.E.; et al. Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nat. Genet. 2011, 44, 53–57. [Google Scholar] [CrossRef] [PubMed]
- Webb, C.J.; Wise, J.A. The splicing factor U2AF small subunit is functionally conserved between fission yeast and humans. Mol. Cell Biol. 2004, 24, 4229–4240. [Google Scholar] [CrossRef] [PubMed]
- Palangat, M.; Anastasakis, D.G.; Fei, D.L.; Lindblad, K.E.; Bradley, R.; Hourigan, C.S.; Hafner, M.; Larson, D.R. The splicing factor U2AF1 contributes to cancer progression through a noncanonical role in translation regulation. Genes Dev. 2019, 33, 482–497. [Google Scholar] [CrossRef] [PubMed]
- Madan, V.; Kanojia, D.; Li, J.; Okamoto, R.; Sato-Otsubo, A.; Kohlmann, A.; Sanada, M.; Grossmann, V.; Sundaresan, J.; Shiraishi, Y.; et al. Aberrant splicing of U12-type introns is the hallmark of ZRSR2 mutant myelodysplastic syndrome. Nat. Commun. 2015, 6, 6042. [Google Scholar] [CrossRef]
- Cazzola, M.; Rossi, M.; Malcovati, L. Biologic and clinical significance of somatic mutations of SF3B1 in myeloid and lymphoid neoplasms. Blood 2013, 121, 260–269. [Google Scholar] [CrossRef]
- Obeng, E.A.; Chappell, R.J.; Seiler, M.; Chen, M.C.; Campagna, D.R.; Schmidt, P.J.; Schneider, R.K.; Lord, A.M.; Wang, L.; Gambe, R.G.; et al. Physiologic expression of Sf3b1[K700E] causes impaired erythropoiesis, aberrant splicing, and sensitivity to therapeutic spliceosome modulation. Cancer Cell 2016, 30, 404–417. [Google Scholar] [CrossRef]
- Tong, W.; Zhang, J.; Lodish, H.F. Lnk inhibits erythropoiesis and Epo-dependent JAK2 activation and downstream signaling pathways. Blood 2005, 105, 4604–4612. [Google Scholar] [CrossRef] [PubMed]
- Bersenev, A.; Wu, C.; Balcerek, J.; Tong, W. Lnk controls mouse hematopoietic stem cell self-renewal and quiescence through direct interactions with JAK2. J. Clin. Investig. 2008, 118, 2832–2844. [Google Scholar] [CrossRef]
- Simon, C.; Dondi, E.; Chaix, A.; de Sepulveda, P.; Kubiseski, T.J.; Varin-Blank, N.; Velazquez, L. Lnk adaptor protein down-regulates specific Kit-induced signaling pathways in primary mast cells. Blood 2008, 112, 4039–4047. [Google Scholar] [CrossRef] [PubMed]
- Takaki, S.; Sauer, K.; Iritani, B.M.; Chien, S.; Ebihara, Y.; Tsuji, K.; Takatsu, K.; Perlmutter, R.M. Control of B cell production by the adaptor protein Lnk: Definition of a conserved family of signal-modulating proteins. Immunity 2000, 13, 599–609. [Google Scholar] [CrossRef]
- Takaki, S.; Morita, H.; Tezuka, Y.; Takatsu, K. Enhanced hematopoiesis by hematopoietic progenitor cells lacking intracellular adaptor protein, Lnk. J. Exp. Med. 2002, 195, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Velazquez, L.; Cheng, A.M.; Fleming, H.E.; Furlonger, C.; Vesely, S.; Bernstein, A.; Paige, C.J.; Pawson, T. Cytokine signaling and hematopoietic homeostasis are disrupted in Lnk-deficient mice. J. Exp. Med. 2002, 195, 1599–1611. [Google Scholar] [CrossRef] [PubMed]
- Thien, C.B.; Langdon, W.Y. Negative regulation of PTK signalling by Cbl proteins. Growth Factors. 2005, 23, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Mohapatra, B.; Ahmad, G.; Nadeau, S.; Zutshi, N.; An, W.; Scheffe, S.; Dong, L.; Feng, D.; Goetz, B.; Arya, P.; et al. Protein tyrosine kinase regulation by ubiquitination: Critical roles of Cbl-family ubiquitin ligases. Biochim. Biophys. Acta. 2013, 1833, 122–139. [Google Scholar] [CrossRef] [PubMed]
- Lv, K.; Jiang, J.; Donaghy, R.; Riling, C.R.; Cheng, Y.; Chandra, V.; Rozenova, K.; An, W.; Mohapatra, B.C.; Goetz, B.T.; et al. CBL family E3 ubiquitin ligases control JAK2 ubiquitination and stability in hematopoietic stem cells and myeloid malignancies. Genes Dev. 2017, 31, 1007–1023. [Google Scholar] [CrossRef] [PubMed]
- Sanada, M.; Suzuki, T.; Shih, L.Y.; Otsu, M.; Kato, M.; Yamazaki, S.; Tamura, A.; Honda, H.; Sakata-Yanagimoto, M.; Kumano, K.; et al. Gain-of-function of mutated C-CBL tumour suppressor in myeloid neoplasms. Nature 2009, 460, 904–908. [Google Scholar] [CrossRef] [PubMed]
- Schubbert, S.; Shannon, K.; Bollag, G. Hyperactive Ras in developmental disorders and cancer. Nat. Rev. Cancer. 2007, 7, 295–308. [Google Scholar] [CrossRef] [PubMed]
- Bunda, S.; Burrell, K.; Heir, P.; Zeng, L.; Alamsahebpour, A.; Kano, Y.; Raught, B.; Zhang, Z.Y.; Zadeh, G.; Ohh, M. Inhibition of SHP2-mediated dephosphorylation of Ras suppresses oncogenesis. Nat. Commun. 2015, 6, 8859. [Google Scholar] [CrossRef]
- Niihori, T.; Aoki, Y.; Ohashi, H.; Kurosawa, K.; Kondoh, T.; Ishikiriyama, S.; Kawame, H.; Kamasaki, H.; Yamanaka, T.; Takada, F.; et al. Functional analysis of PTPN11/SHP-2 mutants identified in Noonan syndrome and childhood leukemia. J. Hum. Genet. 2005, 50, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.Q.; Yu, D.H.; Park, M.; Marshall, M.; Feng, G.S. Molecular mechanism for the Shp-2 tyrosine phosphatase function in promoting growth factor stimulation of Erk activity. Mol. Cell Biol. 2000, 20, 1526–1536. [Google Scholar] [CrossRef]
- Eaves, C.J. Hematopoietic stem cells: Concepts, definitions, and the new reality. Blood 2015, 125, 2605–2613. [Google Scholar] [CrossRef]
- Mead, A.J.; Mullally, A. Myeloproliferative neoplasm stem cells. Blood 2017, 129, 1607–1616. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.V.; Chase, A.; Silver, R.T.; Oscier, D.; Zoi, K.; Wang, Y.L.; Cario, H.; Pahl, H.L.; Collins, A.; Reiter, A.; et al. JAK2 haplotype is a major risk factor for the development of myeloproliferative neoplasms. Nat. Genet. 2009, 41, 446–449. [Google Scholar] [CrossRef]
- Jones, A.V.; Campbell, P.J.; Beer, P.A.; Schnittger, S.; Vannucchi, A.M.; Zoi, K.; Percy, M.J.; McMullin, M.F.; Scott, L.M.; Tapper, W.; et al. The JAK2 46/1 haplotype predisposes to MPL-mutated myeloproliferative neoplasms. Blood 2010, 115, 4517–4523. [Google Scholar] [CrossRef]
- Tapper, W.; Jones, A.V.; Kralovics, R.; Harutyunyan, A.S.; Zoi, K.; Leung, W.; Godfrey, A.L.; Guglielmelli, P.; Callaway, A.; Ward, D.; et al. Genetic variation at MECOM, TERT, JAK2 and HBS1L-MYB predisposes to myeloproliferative neoplasms. Nat. Commun. 2015, 6, 6691. [Google Scholar] [CrossRef] [PubMed]
- Trifa, A.P.; Bănescu, C.; Bojan, A.S.; Voina, C.M.; Popa, Ș.; Vișan, S.; Ciubean, A.D.; Tripon, F.; Dima, D.; Popov, V.M.; et al. MECOM, HBS1L-MYB, THRB-RARB, JAK2, and TERT polymorphisms defining the genetic predisposition to myeloproliferative neoplasms: A study on 939 patients. Am. J. Hematol. 2018, 93, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Hinds, D.A.; Barnholt, K.E.; Mesa, R.A.; Kiefer, A.K.; Do, C.B.; Eriksson, N.; Mountain, J.L.; Francke, U.; Tung, J.Y.; Nguyen, H.M.; et al. Germ line variants predispose to both JAK2 V617F clonal hematopoiesis and myeloproliferative neoplasms. Blood 2016, 128, 1121–1128. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, A.L.; Chen, E.; Pagano, F.; Silber, Y.; Campbell, P.J.; Green, A.R. Clonal analyses reveal associations of JAK2V617F homozygosity with hematologic features, age and gender in polycythemia vera and essential thrombocythemia. Haematologica 2013, 98, 718–721. [Google Scholar] [CrossRef] [PubMed]
- Geyer, H.L.; Kosiorek, H.; Dueck, A.C.; Scherber, R.; Slot, S.; Zweegman, S.; Te Boekhorst, P.A.; Senyak, Z.; Schouten, H.C.; Sackmann, F.; et al. Associations between gender, disease features and symptom burden in patients with myeloproliferative neoplasms: An analysis by the MPN QOL International Working Group. Haematologica 2017, 102, 85–93. [Google Scholar] [CrossRef]
- Alvarez-Larrán, A.; Cervantes, F.; Bellosillo, B.; Giralt, M.; Juliá, A.; Hernández-Boluda, J.C.; Bosch, A.; Hernández-Nieto, L.; Clapés, V.; Burgaleta, C.; et al. Essential thrombocythemia in young individuals: Frequency and risk factors for vascular events and evolution to myelofibrosis in 126 patients. Leukemia 2007, 21, 1218–2123. [Google Scholar] [CrossRef] [PubMed]
- Srour, S.A.; Devesa, S.S.; Morton, L.M.; Check, D.P.; Curtis, R.E.; Linet, M.S.; Dores, G.M. Incidence and patient survival of myeloproliferative neoplasms and myelodysplastic/myeloproliferative neoplasms in the United States, 2001–2012. Br. J. Haematol. 2016, 174, 382–396. [Google Scholar] [CrossRef]
- Grinfeld, J.; Nangalia, J.; Baxter, E.J.; Wedge, D.C.; Angelopoulos, N.; Cantrill, R.; Godfrey, A.L.; Papaemmanuil, E.; Gundem, G.; MacLean, C.; et al. Classification and personalized prognosis in myeloproliferative neoplasms. N. Engl. J. Med. 2018, 379, 1416–1430. [Google Scholar] [CrossRef] [PubMed]
- Anderson, L.A.; Duncombe, A.S.; Hughes, M.; Mills, M.E.; Wilson, J.C.; McMullin, M.F. Environmental, lifestyle, and familial/ethnic factors associated with myeloproliferative neoplasms. Am. J. Hematol. 2012, 87, 175–182. [Google Scholar] [CrossRef]
- Jayasuriya, N.A.; Ellervik, C.; Hasselbalch, H.C.; Sørensen, A. Cigarette smoking, complete blood count, and myeloproliferative neoplasms—A meta-analysis. Blood 2017, 130, 4199. [Google Scholar] [CrossRef]
- Jayasuriya, N.A.; Kjaergaard, A.D.; Pedersen, K.M.; Sørensen, A.L.; Bak, M.; Larsen, M.K.; Nordestgaard, B.G.; Bojesen, S.E.; Çolak, Y.; Skov, V.; et al. Smoking, blood cells and myeloproliferative neoplasms: Meta-analysis and Mendelian randomization of 2·3 million people. Br. J. Haematol. 2020, 189, 323–334. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).