
Cancer-Associated Fibroblast-Induced Resistance to Chemotherapy and Radiotherapy in Gastrointestinal Cancers


Abstract
Simple Summary
Abstract
1. Introduction
2. Clinical Evidence for the Role of CAFs in Chemotherapy and Radiotherapy Resistance in GI Cancer
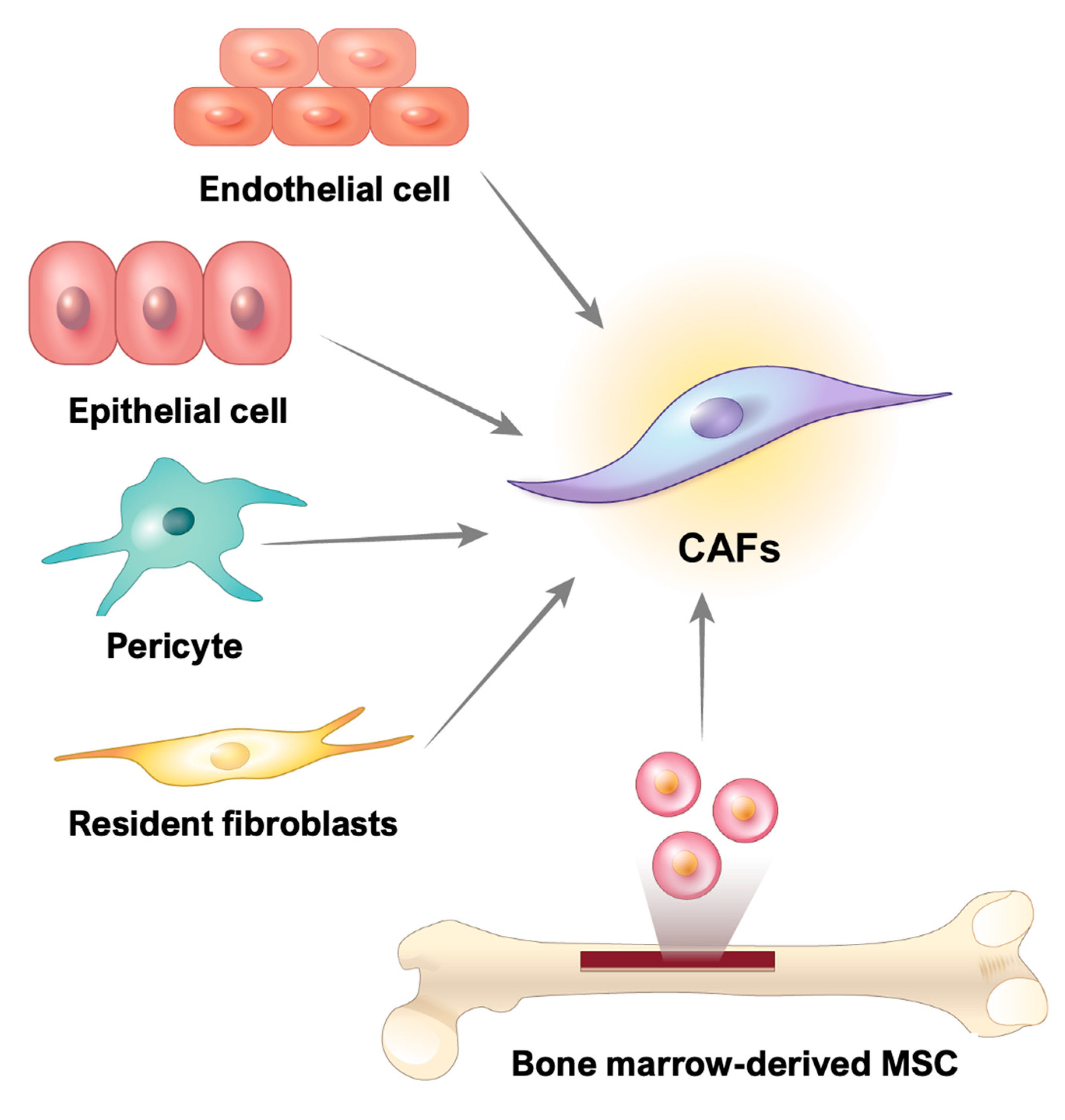
3. Origin of CAFs in GI Cancer
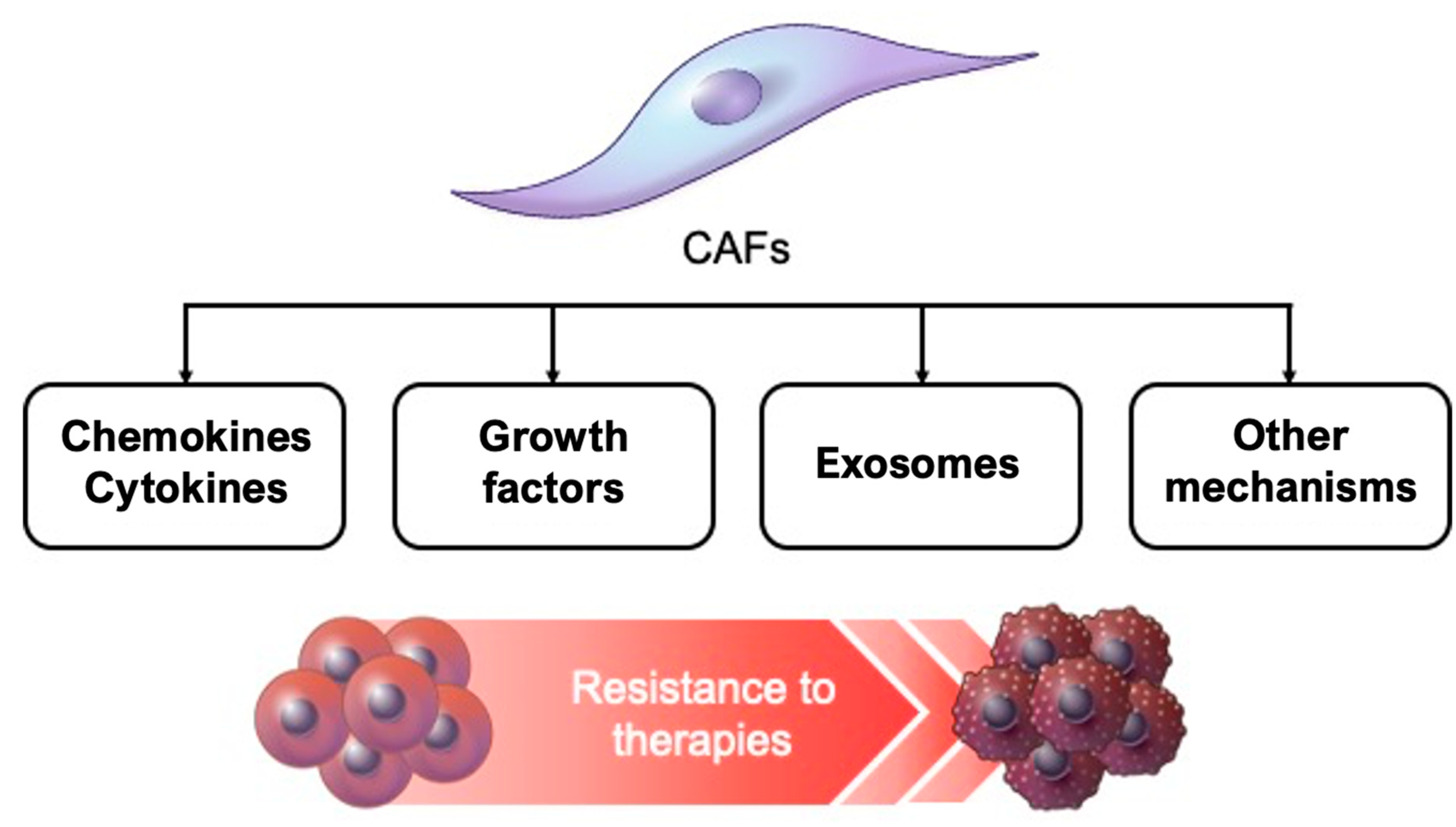
4. Factors Related to CAF-induced Resistance to Cancer Treatment
4.1. Cytokines and Chemokines
4.2. Growth Factors
4.3. Exosomes
4.4. Other Mechanisms
5. Heterogeneity of CAFs in GI Cancers
6. Conclusions and Future Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Global Burden of Disease Cancer, C.; Fitzmaurice, C.; Akinyemiju, T.F.; Al Lami, F.H.; Alam, T.; Alizadeh-Navaei, R.; Allen, C.; Alsharif, U.; Alvis-Guzman, N.; Amini, E.; et al. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived With Disability, and Disability-Adjusted Life-Years for 29 Cancer Groups, 1990 to 2016: A Systematic Analysis for the Global Burden of Disease Study. JAMA Oncol. 2018, 4, 1553–1568. [Google Scholar] [CrossRef]
- Myint, A.S. The role of radiotherapy in the palliative treatment of gastrointestinal cancer. Eur. J. Gastroenterol Hepatol. 2000, 12, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Luqmani, Y.A. Mechanisms of drug resistance in cancer chemotherapy. Med. Princ. Pract. 2005, 14 (Suppl. S1), 35–48. [Google Scholar] [CrossRef]
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug resistance in cancer: An overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef] [PubMed]
- Senthebane, D.A.; Rowe, A.; Thomford, N.E.; Shipanga, H.; Munro, D.; Mazeedi, M.; Almazyadi, H.A.M.; Kallmeyer, K.; Dandara, C.; Pepper, M.S.; et al. The Role of Tumor Microenvironment in Chemoresistance: To Survive, Keep Your Enemies Closer. Int. J. Mol. Sci. 2017, 18, 1586. [Google Scholar] [CrossRef]
- Ansems, M.; Span, P.N. The tumor microenvironment and radiotherapy response; a central role for cancer-associated fibroblasts. Clin. Transl. Radiat Oncol. 2020, 22, 90–97. [Google Scholar] [CrossRef]
- Ribeiro Franco, P.I.; Rodrigues, A.P.; de Menezes, L.B.; Pacheco Miguel, M. Tumor microenvironment components: Allies of cancer progression. Pathol. Res. Pract. 2020, 216, 152729. [Google Scholar] [CrossRef] [PubMed]
- Hinshaw, D.C.; Shevde, L.A. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef]
- Sounni, N.E.; Noel, A. Targeting the tumor microenvironment for cancer therapy. Clin. Chem. 2013, 59, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef]
- Melissari, M.T.; Chalkidi, N.; Sarris, M.E.; Koliaraki, V. Fibroblast Reprogramming in Gastrointestinal Cancer. Front. Cell Dev. Biol. 2020, 8, 630. [Google Scholar] [CrossRef]
- Orimo, A.; Weinberg, R.A. Stromal fibroblasts in cancer: A novel tumor-promoting cell type. Cell Cycle 2006, 5, 1597–1601. [Google Scholar] [CrossRef]
- Mueller, M.M.; Fusenig, N.E. Friends or foes - bipolar effects of the tumour stroma in cancer. Nat. Rev. Cancer 2004, 4, 839–849. [Google Scholar] [CrossRef]
- Ham, I.H.; Oh, H.J.; Jin, H.; Bae, C.A.; Jeon, S.M.; Choi, K.S.; Son, S.Y.; Han, S.U.; Brekken, R.A.; Lee, D.; et al. Targeting interleukin-6 as a strategy to overcome stroma-induced resistance to chemotherapy in gastric cancer. Mol. Cancer 2019, 18, 68. [Google Scholar] [CrossRef] [PubMed]
- Duluc, C.; Moatassim-Billah, S.; Chalabi-Dchar, M.; Perraud, A.; Samain, R.; Breibach, F.; Gayral, M.; Cordelier, P.; Delisle, M.B.; Bousquet-Dubouch, M.P.; et al. Pharmacological targeting of the protein synthesis mTOR/4E-BP1 pathway in cancer-associated fibroblasts abrogates pancreatic tumour chemoresistance. EMBO Mol. Med. 2015, 7, 735–753. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef]
- Liu, H.; Ma, Q.; Xu, Q.; Lei, J.; Li, X.; Wang, Z.; Wu, E. Therapeutic potential of perineural invasion, hypoxia and desmoplasia in pancreatic cancer. Curr. Pharm. Des. 2012, 18, 2395–2403. [Google Scholar] [CrossRef]
- Vennin, C.; Murphy, K.J.; Morton, J.P.; Cox, T.R.; Pajic, M.; Timpson, P. Reshaping the Tumor Stroma for Treatment of Pancreatic Cancer. Gastroenterology 2018, 154, 820–838. [Google Scholar] [CrossRef] [PubMed]
- Ueno, H.; Jones, A.M.; Wilkinson, K.H.; Jass, J.R.; Talbot, I.C. Histological categorisation of fibrotic cancer stroma in advanced rectal cancer. Gut 2004, 53, 581–586. [Google Scholar] [CrossRef] [PubMed]
- Sis, B.; Sarioglu, S.; Sokmen, S.; Sakar, M.; Kupelioglu, A.; Fuzun, M. Desmoplasia measured by computer assisted image analysis: An independent prognostic marker in colorectal carcinoma. J. Clin. Pathol. 2005, 58, 32–38. [Google Scholar] [CrossRef]
- Hofheinz, R.D.; Al-Batran, S.E.; Hartmann, F.; Hartung, G.; Jager, D.; Renner, C.; Tanswell, P.; Kunz, U.; Amelsberg, A.; Kuthan, H.; et al. Stromal antigen targeting by a humanised monoclonal antibody: An early phase II trial of sibrotuzumab in patients with metastatic colorectal cancer. Onkologie 2003, 26, 44–48. [Google Scholar] [CrossRef]
- Murphy, J.E.; Wo, J.Y.; Ryan, D.P.; Clark, J.W.; Jiang, W.; Yeap, B.Y.; Drapek, L.C.; Ly, L.; Baglini, C.V.; Blaszkowsky, L.S.; et al. Total Neoadjuvant Therapy With FOLFIRINOX in Combination With Losartan Followed by Chemoradiotherapy for Locally Advanced Pancreatic Cancer: A Phase 2 Clinical Trial. JAMA Oncol. 2019, 5, 1020–1027. [Google Scholar] [CrossRef]
- Hingorani, S.R.; Zheng, L.; Bullock, A.J.; Seery, T.E.; Harris, W.P.; Sigal, D.S.; Braiteh, F.; Ritch, P.S.; Zalupski, M.M.; Bahary, N.; et al. HALO 202: Randomized Phase II Study of PEGPH20 Plus Nab-Paclitaxel/Gemcitabine Versus Nab-Paclitaxel/Gemcitabine in Patients With Untreated, Metastatic Pancreatic Ductal Adenocarcinoma. J. Clin. Oncol. 2018, 36, 359–366. [Google Scholar] [CrossRef]
- Ma, J.; Song, X.; Xu, X.; Mou, Y. Cancer-Associated Fibroblasts Promote the Chemo-resistance in Gastric Cancer through Secreting IL-11 Targeting JAK/STAT3/Bcl2 Pathway. Cancer Res. Treat. 2019, 51, 194–210. [Google Scholar] [CrossRef]
- Gu, J.; Li, Z.; Zhou, J.; Sun, Z.; Bai, C. Response prediction to oxaliplatin plus 5-fluorouracil chemotherapy in patients with colorectal cancer using a four-protein immunohistochemical model. Oncol. Lett. 2019, 18, 2091–2101. [Google Scholar] [CrossRef] [PubMed]
- Saigusa, S.; Toiyama, Y.; Tanaka, K.; Yokoe, T.; Okugawa, Y.; Kawamoto, A.; Yasuda, H.; Inoue, Y.; Miki, C.; Kusunoki, M. Stromal CXCR4 and CXCL12 expression is associated with distant recurrence and poor prognosis in rectal cancer after chemoradiotherapy. Ann. Surg. Oncol. 2010, 17, 2051–2058. [Google Scholar] [CrossRef] [PubMed]
- Saigusa, S.; Toiyama, Y.; Tanaka, K.; Yokoe, T.; Okugawa, Y.; Fujikawa, H.; Matsusita, K.; Kawamura, M.; Inoue, Y.; Miki, C.; et al. Cancer-associated fibroblasts correlate with poor prognosis in rectal cancer after chemoradiotherapy. Int. J. Oncol. 2011, 38, 655–663. [Google Scholar] [CrossRef]
- Zhang, H.; Yue, J.; Jiang, Z.; Zhou, R.; Xie, R.; Xu, Y.; Wu, S. CAF-secreted CXCL1 conferred radioresistance by regulating DNA damage response in a ROS-dependent manner in esophageal squamous cell carcinoma. Cell Death Dis. 2017, 8, e2790. [Google Scholar] [CrossRef]
- Zhang, H.; Xie, C.; Yue, J.; Jiang, Z.; Zhou, R.; Xie, R.; Wang, Y.; Wu, S. Cancer-associated fibroblasts mediated chemoresistance by a FOXO1/TGFbeta1 signaling loop in esophageal squamous cell carcinoma. Mol. Carcinog 2017, 56, 1150–1163. [Google Scholar] [CrossRef] [PubMed]
- Che, Y.; Wang, J.; Li, Y.; Lu, Z.; Huang, J.; Sun, S.; Mao, S.; Lei, Y.; Zang, R.; Sun, N.; et al. Cisplatin-activated PAI-1 secretion in the cancer-associated fibroblasts with paracrine effects promoting esophageal squamous cell carcinoma progression and causing chemoresistance. Cell Death Dis. 2018, 9, 759. [Google Scholar] [CrossRef]
- Luo, Y.; Mao, Q.; Wang, X.; Yu, J.; Li, M. Radiotherapy for esophageal carcinoma: Dose, response and survival. Cancer Manag. Res. 2018, 10, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Malone, E.R.; Oliva, M.; Sabatini, P.J.B.; Stockley, T.L.; Siu, L.L. Molecular profiling for precision cancer therapies. Genome Med. 2020, 12, 8. [Google Scholar] [CrossRef] [PubMed]
- Ally, A.; Balasundaram, M.; Carlsen, R.; Chuah, E.; Clarke, A.; Dhalla, N.; Holt, R.A.; Jones, S.J.M.; Lee, D.; Ma, Y.; et al. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017, 169, 1327–1341. [Google Scholar] [CrossRef]
- Raphael, B.J.; Hruban, R.H.; Aguirre, A.J.; Moffitt, R.A.; Yeh, J.J.; Stewart, C.; Robertson, A.G.; Cherniack, A.D.; Gupta, M.; Getz, G. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Integrated genomic characterization of oesophageal carcinoma. Nature 2017, 541, 169–175. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef]
- Yoshihara, K.; Shahmoradgoli, M.; Martinez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Trevino, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Steen, C.B.; Liu, C.L.; Gentles, A.J.; Chaudhuri, A.A.; Scherer, F.; Khodadoust, M.S.; Esfahani, M.S.; Luca, B.A.; Steiner, D.; et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 2019, 37, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Racle, J.; de Jonge, K.; Baumgaertner, P.; Speiser, D.E.; Gfeller, D. Simultaneous enumeration of cancer and immune cell types from bulk tumor gene expression data. Elife 2017, 6. [Google Scholar] [CrossRef]
- Becht, E.; Giraldo, N.A.; Lacroix, L.; Buttard, B.; Elarouci, N.; Petitprez, F.; Selves, J.; Laurent-Puig, P.; Sautes-Fridman, C.; Fridman, W.H.; et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016, 17, 218. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Grabsch, H.; Ivanova, T.; Tan, I.B.; Murray, J.; Ooi, C.H.; Wright, A.I.; West, N.P.; Hutchins, G.G.; Wu, J.; et al. Comprehensive genomic meta-analysis identifies intra-tumoural stroma as a predictor of survival in patients with gastric cancer. Gut 2013, 62, 1100–1111. [Google Scholar] [CrossRef] [PubMed]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reynies, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef]
- Calon, A.; Lonardo, E.; Berenguer-Llergo, A.; Espinet, E.; Hernando-Momblona, X.; Iglesias, M.; Sevillano, M.; Palomo-Ponce, S.; Tauriello, D.V.; Byrom, D.; et al. Stromal gene expression defines poor-prognosis subtypes in colorectal cancer. Nat. Genet. 2015, 47, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef]
- Ji, J.; Eggert, T.; Budhu, A.; Forgues, M.; Takai, A.; Dang, H.; Ye, Q.; Lee, J.S.; Kim, J.H.; Greten, T.F.; et al. Hepatic stellate cell and monocyte interaction contributes to poor prognosis in hepatocellular carcinoma. Hepatology 2015, 62, 481–495. [Google Scholar] [CrossRef]
- Li, Z.; Gao, X.; Peng, X.; May Chen, M.J.; Li, Z.; Wei, B.; Wen, X.; Wei, B.; Dong, Y.; Bu, Z.; et al. Multi-omics characterization of molecular features of gastric cancer correlated with response to neoadjuvant chemotherapy. Sci. Adv. 2020, 6, eaay4211. [Google Scholar] [CrossRef]
- Liu, T.; Han, C.; Wang, S.; Fang, P.; Ma, Z.; Xu, L.; Yin, R. Cancer-associated fibroblasts: An emerging target of anti-cancer immunotherapy. J. Hematol. Oncol. 2019, 12, 86. [Google Scholar] [CrossRef]
- Kadel, D.; Zhang, Y.; Sun, H.R.; Zhao, Y.; Dong, Q.Z.; Qin, L.X. Current perspectives of cancer-associated fibroblast in therapeutic resistance: Potential mechanism and future strategy. Cell Biol. Toxicol. 2019, 35, 407–421. [Google Scholar] [CrossRef]
- Yoshida, G.J.; Azuma, A.; Miura, Y.; Orimo, A. Activated Fibroblast Program Orchestrates Tumor Initiation and Progression; Molecular Mechanisms and the Associated Therapeutic Strategies. Int. J. Mol. Sci. 2019, 20, 2256. [Google Scholar] [CrossRef]
- De Wever, O.; Nguyen, Q.D.; Van Hoorde, L.; Bracke, M.; Bruyneel, E.; Gespach, C.; Mareel, M. Tenascin-C and SF/HGF produced by myofibroblasts in vitro provide convergent pro-invasive signals to human colon cancer cells through RhoA and Rac. FASEB J. 2004, 18, 1016–1018. [Google Scholar] [CrossRef]
- Fuyuhiro, Y.; Yashiro, M.; Noda, S.; Kashiwagi, S.; Matsuoka, J.; Doi, Y.; Kato, Y.; Hasegawa, T.; Sawada, T.; Hirakawa, K. Upregulation of cancer-associated myofibroblasts by TGF-beta from scirrhous gastric carcinoma cells. Br. J. Cancer 2011, 105, 996–1001. [Google Scholar] [CrossRef] [PubMed]
- Spaeth, E.L.; Dembinski, J.L.; Sasser, A.K.; Watson, K.; Klopp, A.; Hall, B.; Andreeff, M.; Marini, F. Mesenchymal stem cell transition to tumor-associated fibroblasts contributes to fibrovascular network expansion and tumor progression. PLoS ONE 2009, 4, e4992. [Google Scholar] [CrossRef]
- Apte, M.V.; Haber, P.S.; Darby, S.J.; Rodgers, S.C.; McCaughan, G.W.; Korsten, M.A.; Pirola, R.C.; Wilson, J.S. Pancreatic stellate cells are activated by proinflammatory cytokines: Implications for pancreatic fibrogenesis. Gut 1999, 44, 534–541. [Google Scholar] [CrossRef]
- Bailey, J.M.; Swanson, B.J.; Hamada, T.; Eggers, J.P.; Singh, P.K.; Caffery, T.; Ouellette, M.M.; Hollingsworth, M.A. Sonic hedgehog promotes desmoplasia in pancreatic cancer. Clin. Cancer Res. 2008, 14, 5995–6004. [Google Scholar] [CrossRef] [PubMed]
- Catenacci, D.V.; Junttila, M.R.; Karrison, T.; Bahary, N.; Horiba, M.N.; Nattam, S.R.; Marsh, R.; Wallace, J.; Kozloff, M.; Rajdev, L.; et al. Randomized Phase Ib/II Study of Gemcitabine Plus Placebo or Vismodegib, a Hedgehog Pathway Inhibitor, in Patients With Metastatic Pancreatic Cancer. J. Clin. Oncol. 2015, 33, 4284–4292. [Google Scholar] [CrossRef]
- Quante, M.; Tu, S.P.; Tomita, H.; Gonda, T.; Wang, S.S.; Takashi, S.; Baik, G.H.; Shibata, W.; Diprete, B.; Betz, K.S.; et al. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell 2011, 19, 257–272. [Google Scholar] [CrossRef]
- Direkze, N.C.; Hodivala-Dilke, K.; Jeffery, R.; Hunt, T.; Poulsom, R.; Oukrif, D.; Alison, M.R.; Wright, N.A. Bone marrow contribution to tumor-associated myofibroblasts and fibroblasts. Cancer Res. 2004, 64, 8492–8495. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, E.M.; Tarnavski, O.; Zeisberg, M.; Dorfman, A.L.; McMullen, J.R.; Gustafsson, E.; Chandraker, A.; Yuan, X.; Pu, W.T.; Roberts, A.B.; et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007, 13, 952–961. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, E.M.; Potenta, S.; Xie, L.; Zeisberg, M.; Kalluri, R. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res. 2007, 67, 10123–10128. [Google Scholar] [CrossRef]
- Birbrair, A.; Zhang, T.; Wang, Z.M.; Messi, M.L.; Mintz, A.; Delbono, O. Pericytes at the intersection between tissue regeneration and pathology. Clin. Sci. 2015, 128, 81–93. [Google Scholar] [CrossRef] [PubMed]
- von Tell, D.; Armulik, A.; Betsholtz, C. Pericytes and vascular stability. Exp. Cell Res. 2006, 312, 623–629. [Google Scholar] [CrossRef]
- Hosaka, K.; Yang, Y.; Seki, T.; Fischer, C.; Dubey, O.; Fredlund, E.; Hartman, J.; Religa, P.; Morikawa, H.; Ishii, Y.; et al. Pericyte-fibroblast transition promotes tumor growth and metastasis. Proc. Natl. Acad. Sci. USA 2016, 113, E5618–E5627. [Google Scholar] [CrossRef]
- Rhim, A.D.; Mirek, E.T.; Aiello, N.M.; Maitra, A.; Bailey, J.M.; McAllister, F.; Reichert, M.; Beatty, G.L.; Rustgi, A.K.; Vonderheide, R.H.; et al. EMT and dissemination precede pancreatic tumor formation. Cell 2012, 148, 349–361. [Google Scholar] [CrossRef]
- Landskron, G.; De la Fuente, M.; Thuwajit, P.; Thuwajit, C.; Hermoso, M.A. Chronic inflammation and cytokines in the tumor microenvironment. J. Immunol. Res. 2014, 2014, 149185. [Google Scholar] [CrossRef] [PubMed]
- Reyes, M.E.; de La Fuente, M.; Hermoso, M.; Ili, C.G.; Brebi, P. Role of CC Chemokines Subfamily in the Platinum Drugs Resistance Promotion in Cancer. Front. Immunol. 2020, 11, 901. [Google Scholar] [CrossRef]
- Jones, V.S.; Huang, R.Y.; Chen, L.P.; Chen, Z.S.; Fu, L.; Huang, R.P. Cytokines in cancer drug resistance: Cues to new therapeutic strategies. Biochim. Biophys. Acta 2016, 1865, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Kumari, N.; Dwarakanath, B.S.; Das, A.; Bhatt, A.N. Role of interleukin-6 in cancer progression and therapeutic resistance. Tumour Biol. 2016, 37, 11553–11572. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Y.; Zhang, C.; Li, A.; Wang, D.; Luo, Z.; Ping, Y.; Zhou, B.; Liu, S.; Li, H.; Yue, D.; et al. IL6 derived from cancer-associated fibroblasts promotes chemoresistance via CXCR7 in esophageal squamous cell carcinoma. Oncogene 2018, 37, 873–883. [Google Scholar] [CrossRef]
- Huynh, P.T.; Beswick, E.J.; Coronado, Y.A.; Johnson, P.; O’Connell, M.R.; Watts, T.; Singh, P.; Qiu, S.; Morris, K.; Powell, D.W.; et al. CD90(+) stromal cells are the major source of IL-6, which supports cancer stem-like cells and inflammation in colorectal cancer. Int. J. Cancer 2016, 138, 1971–1981. [Google Scholar] [CrossRef]
- Ebbing, E.A.; van der Zalm, A.P.; Steins, A.; Creemers, A.; Hermsen, S.; Rentenaar, R.; Klein, M.; Waasdorp, C.; Hooijer, G.K.J.; Meijer, S.L.; et al. Stromal-derived interleukin 6 drives epithelial-to-mesenchymal transition and therapy resistance in esophageal adenocarcinoma. Proc. Natl. Acad. Sci. USA 2019, 116, 2237–2242. [Google Scholar] [CrossRef]
- Balkwill, F.R. The chemokine system and cancer. J. Pathol. 2012, 226, 148–157. [Google Scholar] [CrossRef]
- Meng, W.; Xue, S.; Chen, Y. The role of CXCL12 in tumor microenvironment. Gene 2018, 641, 105–110. [Google Scholar] [CrossRef]
- Zhang, H.; Wu, H.; Guan, J.; Wang, L.; Ren, X.; Shi, X.; Liang, Z.; Liu, T. Paracrine SDF-1alpha signaling mediates the effects of PSCs on GEM chemoresistance through an IL-6 autocrine loop in pancreatic cancer cells. Oncotarget 2015, 6, 3085–3097. [Google Scholar] [CrossRef] [PubMed]
- Sleightholm, R.L.; Neilsen, B.K.; Li, J.; Steele, M.M.; Singh, R.K.; Hollingsworth, M.A.; Oupicky, D. Emerging roles of the CXCL12/CXCR4 axis in pancreatic cancer progression and therapy. Pharmacol. Ther. 2017, 179, 158–170. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Srivastava, S.K.; Bhardwaj, A.; Owen, L.B.; Singh, A.P. CXCL12-CXCR4 signalling axis confers gemcitabine resistance to pancreatic cancer cells: A novel target for therapy. Br. J. Cancer 2010, 103, 1671–1679. [Google Scholar] [CrossRef]
- Ghobrial, I.M.; Liu, C.J.; Zavidij, O.; Azab, A.K.; Baz, R.; Laubach, J.P.; Mishima, Y.; Armand, P.; Munshi, N.C.; Basile, F.; et al. Phase I/II trial of the CXCR4 inhibitor plerixafor in combination with bortezomib as a chemosensitization strategy in relapsed/refractory multiple myeloma. Am. J. Hematol. 2019, 94, 1244–1253. [Google Scholar] [CrossRef] [PubMed]
- Venkatesulu, B.P.; Hsieh, C.E.; Sanders, K.L.; Krishnan, S. Recent advances in radiation therapy of pancreatic cancer. F1000Research 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Qu, C.; Ning, Z.; Wang, H.; Zang, K.; Zhuang, L.; Chen, L.; Wang, P.; Meng, Z. Radiation promotes epithelial-to-mesenchymal transition and invasion of pancreatic cancer cell by activating carcinoma-associated fibroblasts. Am. J. Cancer Res. 2016, 6, 2192–2206. [Google Scholar]
- Lotti, F.; Jarrar, A.M.; Pai, R.K.; Hitomi, M.; Lathia, J.; Mace, A.; Gantt, G.A., Jr.; Sukhdeo, K.; DeVecchio, J.; Vasanji, A.; et al. Chemotherapy activates cancer-associated fibroblasts to maintain colorectal cancer-initiating cells by IL-17A. J. Exp. Med. 2013, 210, 2851–2872. [Google Scholar] [CrossRef]
- Moritz, A.; Li, Y.; Guo, A.; Villen, J.; Wang, Y.; MacNeill, J.; Kornhauser, J.; Sprott, K.; Zhou, J.; Possemato, A.; et al. Akt-RSK-S6 kinase signaling networks activated by oncogenic receptor tyrosine kinases. Sci. Signal. 2010, 3, ra64. [Google Scholar] [CrossRef]
- Engelman, J.A.; Settleman, J. Acquired resistance to tyrosine kinase inhibitors during cancer therapy. Curr. Opin. Genet. Dev. 2008, 18, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Regad, T. Targeting RTK Signaling Pathways in Cancer. Cancers 2015, 7, 1758–1784. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Miyata, H.; Sugimura, K.; Fukuda, S.; Kanemura, T.; Yamashita, K.; Miyazaki, Y.; Takahashi, T.; Kurokawa, Y.; Yamasaki, M.; et al. miR-27 is associated with chemoresistance in esophageal cancer through transformation of normal fibroblasts to cancer-associated fibroblasts. Carcinogenesis 2015, 36, 894–903. [Google Scholar] [CrossRef] [PubMed]
- LeRoith, D.; Baserga, R.; Helman, L.; Roberts, C.T., Jr. Insulin-like growth factors and cancer. Ann. Intern. Med. 1995, 122, 54–59. [Google Scholar] [CrossRef]
- Ireland, L.; Santos, A.; Ahmed, M.S.; Rainer, C.; Nielsen, S.R.; Quaranta, V.; Weyer-Czernilofsky, U.; Engle, D.D.; Perez-Mancera, P.A.; Coupland, S.E.; et al. Chemoresistance in Pancreatic Cancer Is Driven by Stroma-Derived Insulin-Like Growth Factors. Cancer Res. 2016, 76, 6851–6863. [Google Scholar] [CrossRef]
- Xiao, Q.; Zhou, D.; Rucki, A.A.; Williams, J.; Zhou, J.; Mo, G.; Murphy, A.; Fujiwara, K.; Kleponis, J.; Salman, B.; et al. Cancer-Associated Fibroblasts in Pancreatic Cancer Are Reprogrammed by Tumor-Induced Alterations in Genomic DNA Methylation. Cancer Res. 2016, 76, 5395–5404. [Google Scholar] [CrossRef]
- Tommelein, J.; De Vlieghere, E.; Verset, L.; Melsens, E.; Leenders, J.; Descamps, B.; Debucquoy, A.; Vanhove, C.; Pauwels, P.; Gespach, C.P.; et al. Radiotherapy-Activated Cancer-Associated Fibroblasts Promote Tumor Progression through Paracrine IGF1R Activation. Cancer Res. 2018, 78, 659–670. [Google Scholar] [CrossRef]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- Lau, E.Y.; Lo, J.; Cheng, B.Y.; Ma, M.K.; Lee, J.M.; Ng, J.K.; Chai, S.; Lin, C.H.; Tsang, S.Y.; Ma, S.; et al. Cancer-Associated Fibroblasts Regulate Tumor-Initiating Cell Plasticity in Hepatocellular Carcinoma through c-Met/FRA1/HEY1 Signaling. Cell Rep. 2016, 15, 1175–1189. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.L.H.; Segelov, E.; Wong, R.S.; Smith, A.; Herbertson, R.A.; Li, B.T.; Tebbutt, N.; Price, T.; Pavlakis, N. Epidermal growth factor receptor (EGFR) inhibitors for metastatic colorectal cancer. Cochrane Database Syst. Rev. 2017, 6, CD007047. [Google Scholar] [CrossRef] [PubMed]
- Luraghi, P.; Reato, G.; Cipriano, E.; Sassi, F.; Orzan, F.; Bigatto, V.; De Bacco, F.; Menietti, E.; Han, M.; Rideout, W.M., 3rd; et al. MET signaling in colon cancer stem-like cells blunts the therapeutic response to EGFR inhibitors. Cancer Res. 2014, 74, 1857–1869. [Google Scholar] [CrossRef] [PubMed]
- Patel, Z.S.; Grugan, K.D.; Rustgi, A.K.; Cucinotta, F.A.; Huff, J.L. Ionizing radiation enhances esophageal epithelial cell migration and invasion through a paracrine mechanism involving stromal-derived hepatocyte growth factor. Radiat Res. 2012, 177, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Kahlert, C.; Kalluri, R. Exosomes in tumor microenvironment influence cancer progression and metastasis. J. Mol. Med. 2013, 91, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Yan, C.; Mu, L.; Huang, K.; Li, X.; Tao, D.; Wu, Y.; Qin, J. Fibroblast-Derived Exosomes Contribute to Chemoresistance through Priming Cancer Stem Cells in Colorectal Cancer. PLoS ONE 2015, 10, e0125625. [Google Scholar] [CrossRef]
- Xie, Y.; Dang, W.; Zhang, S.; Yue, W.; Yang, L.; Zhai, X.; Yan, Q.; Lu, J. The role of exosomal noncoding RNAs in cancer. Mol. Cancer 2019, 18, 37. [Google Scholar] [CrossRef]
- Lin, S.; Gregory, R.I. MicroRNA biogenesis pathways in cancer. Nat. Rev. Cancer 2015, 15, 321–333. [Google Scholar] [CrossRef]
- Bach, D.H.; Hong, J.Y.; Park, H.J.; Lee, S.K. The role of exosomes and miRNAs in drug-resistance of cancer cells. Int. J. Cancer 2017, 141, 220–230. [Google Scholar] [CrossRef]
- Hu, J.L.; Wang, W.; Lan, X.L.; Zeng, Z.C.; Liang, Y.S.; Yan, Y.R.; Song, F.Y.; Wang, F.F.; Zhu, X.H.; Liao, W.J.; et al. CAFs secreted exosomes promote metastasis and chemotherapy resistance by enhancing cell stemness and epithelial-mesenchymal transition in colorectal cancer. Mol. Cancer 2019, 18, 91. [Google Scholar] [CrossRef]
- Zhang, H.; Deng, T.; Liu, R.; Ning, T.; Yang, H.; Liu, D.; Zhang, Q.; Lin, D.; Ge, S.; Bai, M.; et al. CAF secreted miR-522 suppresses ferroptosis and promotes acquired chemo-resistance in gastric cancer. Mol. Cancer 2020, 19, 43. [Google Scholar] [CrossRef]
- Fang, Y.; Zhou, W.; Rong, Y.; Kuang, T.; Xu, X.; Wu, W.; Wang, D.; Lou, W. Exosomal miRNA-106b from cancer-associated fibroblast promotes gemcitabine resistance in pancreatic cancer. Exp. Cell Res. 2019, 383, 111543. [Google Scholar] [CrossRef] [PubMed]
- St Laurent, G.; Wahlestedt, C.; Kapranov, P. The Landscape of long noncoding RNA classification. Trends Genet. 2015, 31, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Ruan, H.; Zhang, X.; Xu, X.; Zhu, Y.; Peng, H.; Zhang, X.; Kong, F.; Guan, M. Long noncoding RNA CCAL transferred from fibroblasts by exosomes promotes chemoresistance of colorectal cancer cells. Int. J. Cancer 2020, 146, 1700–1716. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Ding, L.; Zhang, D.; Shi, G.; Xu, Q.; Shen, S.; Wang, Y.; Wang, T.; Hou, Y. Carcinoma-associated fibroblasts promote the stemness and chemoresistance of colorectal cancer by transferring exosomal lncRNA H. Theranostics 2018, 8, 3932–3948. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, Z.; Zhou, L.; Hu, L.; Yin, C.; Qing, D.; Huang, S.; Cai, X.; Chen, Y. Cancer associated fibroblasts-derived exosomes contribute to radioresistance through promoting colorectal cancer stem cells phenotype. Exp. Cell Res. 2020, 391, 111956. [Google Scholar] [CrossRef] [PubMed]
- Hirahata, M.; Osaki, M.; Kanda, Y.; Sugimoto, Y.; Yoshioka, Y.; Kosaka, N.; Takeshita, F.; Fujiwara, T.; Kawai, A.; Ito, H.; et al. PAI-1, a target gene of miR-143, regulates invasion and metastasis by upregulating MMP-13 expression of human osteosarcoma. Cancer Med. 2016, 5, 892–902. [Google Scholar] [CrossRef] [PubMed]
- Geis, T.; Doring, C.; Popp, R.; Grossmann, N.; Fleming, I.; Hansmann, M.L.; Dehne, N.; Brune, B. HIF-2alpha-dependent PAI-1 induction contributes to angiogenesis in hepatocellular carcinoma. Exp. Cell Res. 2015, 331, 46–57. [Google Scholar] [CrossRef]
- Vennin, C.; Melenec, P.; Rouet, R.; Nobis, M.; Cazet, A.S.; Murphy, K.J.; Herrmann, D.; Reed, D.A.; Lucas, M.C.; Warren, S.C.; et al. CAF hierarchy driven by pancreatic cancer cell p53-status creates a pro-metastatic and chemoresistant environment via perlecan. Nat. Commun. 2019, 10, 3637. [Google Scholar] [CrossRef]
- Lee, J.; Yakubov, B.; Ivan, C.; Jones, D.R.; Caperell-Grant, A.; Fishel, M.; Cardenas, H.; Matei, D. Tissue Transglutaminase Activates Cancer-Associated Fibroblasts and Contributes to Gemcitabine Resistance in Pancreatic Cancer. Neoplasia 2016, 18, 689–698. [Google Scholar] [CrossRef]
- Izumi, D.; Toden, S.; Ureta, E.; Ishimoto, T.; Baba, H.; Goel, A. TIAM1 promotes chemoresistance and tumor invasiveness in colorectal cancer. Cell Death Dis. 2019, 10, 267. [Google Scholar] [CrossRef]
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 418–429. [Google Scholar] [CrossRef]
- Yoshida, G.J. Regulation of heterogeneous cancer-associated fibroblasts: The molecular pathology of activated signaling pathways. J. Exp. Clin. Cancer Res. 2020, 39, 112. [Google Scholar] [CrossRef] [PubMed]
- Pereira, B.A.; Vennin, C.; Papanicolaou, M.; Chambers, C.R.; Herrmann, D.; Morton, J.P.; Cox, T.R.; Timpson, P. CAF Subpopulations: A New Reservoir of Stromal Targets in Pancreatic Cancer. Trends Cancer 2019, 5, 724–741. [Google Scholar] [CrossRef]
- Helms, E.; Onate, M.K.; Sherman, M.H. Fibroblast Heterogeneity in the Pancreatic Tumor Microenvironment. Cancer Discov. 2020, 10, 648–656. [Google Scholar] [CrossRef] [PubMed]
- Ohlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Ozdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef] [PubMed]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H., 2nd; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef]
- Puram, S.V.; Tirosh, I.; Parikh, A.S.; Patel, A.P.; Yizhak, K.; Gillespie, S.; Rodman, C.; Luo, C.L.; Mroz, E.A.; Emerick, K.S.; et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 2017, 171, 1611–1624. [Google Scholar] [CrossRef]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef] [PubMed]
- Yashiro, M.; Matsuoka, T.; Ohira, M. The significance of scirrhous gastric cancer cell lines: The molecular characterization using cell lines and mouse models. Hum. Cell 2018, 31, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Kasashima, H.; Yashiro, M.; Nakamae, H.; Kitayama, K.; Masuda, G.; Kinoshita, H.; Fukuoka, T.; Hasegawa, T.; Nakane, T.; Hino, M.; et al. CXCL1-Chemokine (C-X-C Motif) Receptor 2 Signaling Stimulates the Recruitment of Bone Marrow-Derived Mesenchymal Cells into Diffuse-Type Gastric Cancer Stroma. Am. J. Pathol. 2016, 186, 3028–3039. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Patients | Methods | Marker | Resistance to | Results | References |
|---|---|---|---|---|---|
| 30 pts with gastric cancer | IHC | α-SMA | Chemotherapy | High expression in the non-response group | [25] |
| 71 pts with colorectal cancer | IHC | α-SMA | Palliative 5-FU and oxaliplatin | 5.5 (high) vs. 15.0 (low) months (p = 0.005) | [26] |
| 53 pts with rectal cancer | RT-PCR | High expression of CXCL12 mRNA from microdissection for the stromal region | Neoadjuvant 5-FU and 20-45 cGy radiation | Positive CXCL12: poor overall survival (p < 0.01) | [27] |
| 52 pts with rectal cancer | RT-PCR | High expression of CXCL12 and FAP mRNA from microdissection for the stromal region | Neoadjuvant 5-FU and 20-45 cGy radiation | High two genes: poor overall survival (p < 0.05) | [28] |
| 141 pts with esophageal cancer | IHC | CXCL1 in CAF | Chemoradiation | High expression: HR 3.347 (p = 0.001) | [29] |
| 130 pts with esophageal cancer | IHC | TGF-β in CAF | Chemoradiation | High expression: poor overall survival (p = 0.002) | [30] |
| 68 pts with esophageal cancer | IHC | PAI-1 in CAF | Cisplatin | High expression: poor progression-free survival (p = 0.0267) | [31] |
| 10 pts with gastric cancer | NanoString | ECM-related gene set in pretreated biopsy tissues | 5-FU based palliative chemotherapy | Significantly high in non-response group | [15] |
| CAF-Derived Factors | Mechanism | Resistant to | Cancer Type | References |
|---|---|---|---|---|
| Cytokines and chemokines | ||||
| IL-6 | CXCR7 via STAT3/NF-κB pathway | Cisplatin | ESOC | [70] |
| Upregulation of CSC markers | Paclitaxel Carboplatin Radiotherapy | ESOC | [72] | |
| JAK1/STAT3 signaling pathway | 5-Fluorouracil | GC | [15] | |
| CXCL1 | MEK/ERK pathway | Radiotherapy | ESOC | [29] |
| CXCL12/SDF-1 | FAK/ERK1/2/AKT signaling pathway Activation of β-catenin and NF-κB | Gemcitabine | PDAC | [77] |
| FAK/ERK1/2/AKT signaling pathway Upregulation of IL-6 | Gemcitabine | PDAC | [75] | |
| CXCR4 activation | Radiotherapy | PDAC | [80] | |
| IL-17A | NF-κB pathway | FOLFOX (5-Fluorouracil, oxaliplatin, leucovorin) | CRC | [81] |
| Growth factors | ||||
| TGF-β | FOXO1/TGF-β signaling loop | Cisplatin Taxol Radiotherapy | ESOC | [30] |
| - | Cisplatin | ESOC | [85] | |
| IGF | IGF-insulin/IGF1R paracrine signaling axis | Gemcitabine | PDAC | [87] |
| IGF1R activation | Radiotherapy | CRC | [89] | |
| HGF | c-MET/FRA1/HEY1 signaling | Cisplatin | HCC | [91] |
| MAPK/AKT pathway | EGFR inhibitor (Cetuximab) | CRC | [93] | |
| - | Radiotherapy | ESOC | [94] | |
| Exosomes | ||||
| - | Wnt signaling pathway | 5-Fluorouracil Oxaliplatin | CRC | [96] |
| miR-92a-3p | Inhibition of FBXW7 and MOAP1 | 5-FU/L-OHP (5-Fluorouracil, oxaliplatin) | CRC | [100] |
| miR-522 | Inhibition of ALOX15 and blocking lipid-ROS production | Cisplatin Paclitaxel | GC | [101] |
| miR-106b | Targeting TP53INP1 | Gemcitabine | PDAC | [102] |
| lncRNA CCAL | Wnt/β-catenin pathway | Oxaliplatin | CRC | [104] |
| lncRNA H19 | Activation of β-catenin pathway through blocking miR-141 | Oxaliplatin | CRC | [105] |
| - | TGF-β signaling pathway | Radiotherapy | CRC | [105] |
| Other secreted proteins | ||||
| PAI-1 | AKT/ERK1/2 signaling pathway | Cisplatin | ESOC | [31] |
| Perlecan/HSPG2 | - | Gemcitabine/Abraxane | PDAC | [109] |
| Laminin A1 | - | Gemcitabine | PDAC | [110] |
| TIAM1 | Stemness through Wnt signaling | 5-Fluorouracil Oxaliplatin Irinotecan | CRC | [111] |
| Hyaluronic acid | Increase interstitial fluid pressure | Gemcitabine | PDAC | [112] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ham, I.-H.; Lee, D.; Hur, H. Cancer-Associated Fibroblast-Induced Resistance to Chemotherapy and Radiotherapy in Gastrointestinal Cancers. Cancers 2021, 13, 1172. https://doi.org/10.3390/cancers13051172
Ham I-H, Lee D, Hur H. Cancer-Associated Fibroblast-Induced Resistance to Chemotherapy and Radiotherapy in Gastrointestinal Cancers. Cancers. 2021; 13(5):1172. https://doi.org/10.3390/cancers13051172
Chicago/Turabian StyleHam, In-Hye, Dagyeong Lee, and Hoon Hur. 2021. "Cancer-Associated Fibroblast-Induced Resistance to Chemotherapy and Radiotherapy in Gastrointestinal Cancers" Cancers 13, no. 5: 1172. https://doi.org/10.3390/cancers13051172
APA StyleHam, I.-H., Lee, D., & Hur, H. (2021). Cancer-Associated Fibroblast-Induced Resistance to Chemotherapy and Radiotherapy in Gastrointestinal Cancers. Cancers, 13(5), 1172. https://doi.org/10.3390/cancers13051172