
The Role of Cancer Stem Cells in Colorectal Cancer: From the Basics to Novel Clinical Trials



Abstract
Simple Summary
Abstract
1. Introduction
2. Colorectal Cancer Stem Cells
2.1. Colorectal Cancer Stem Cell Origin
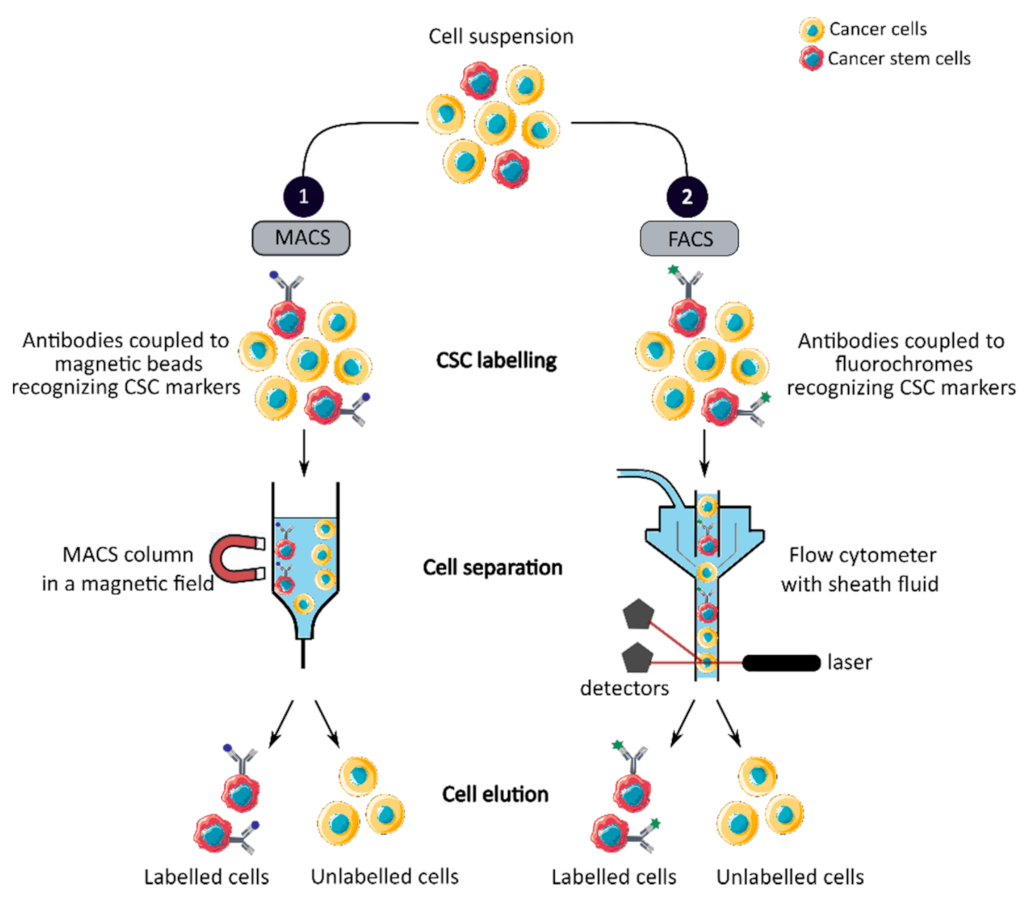
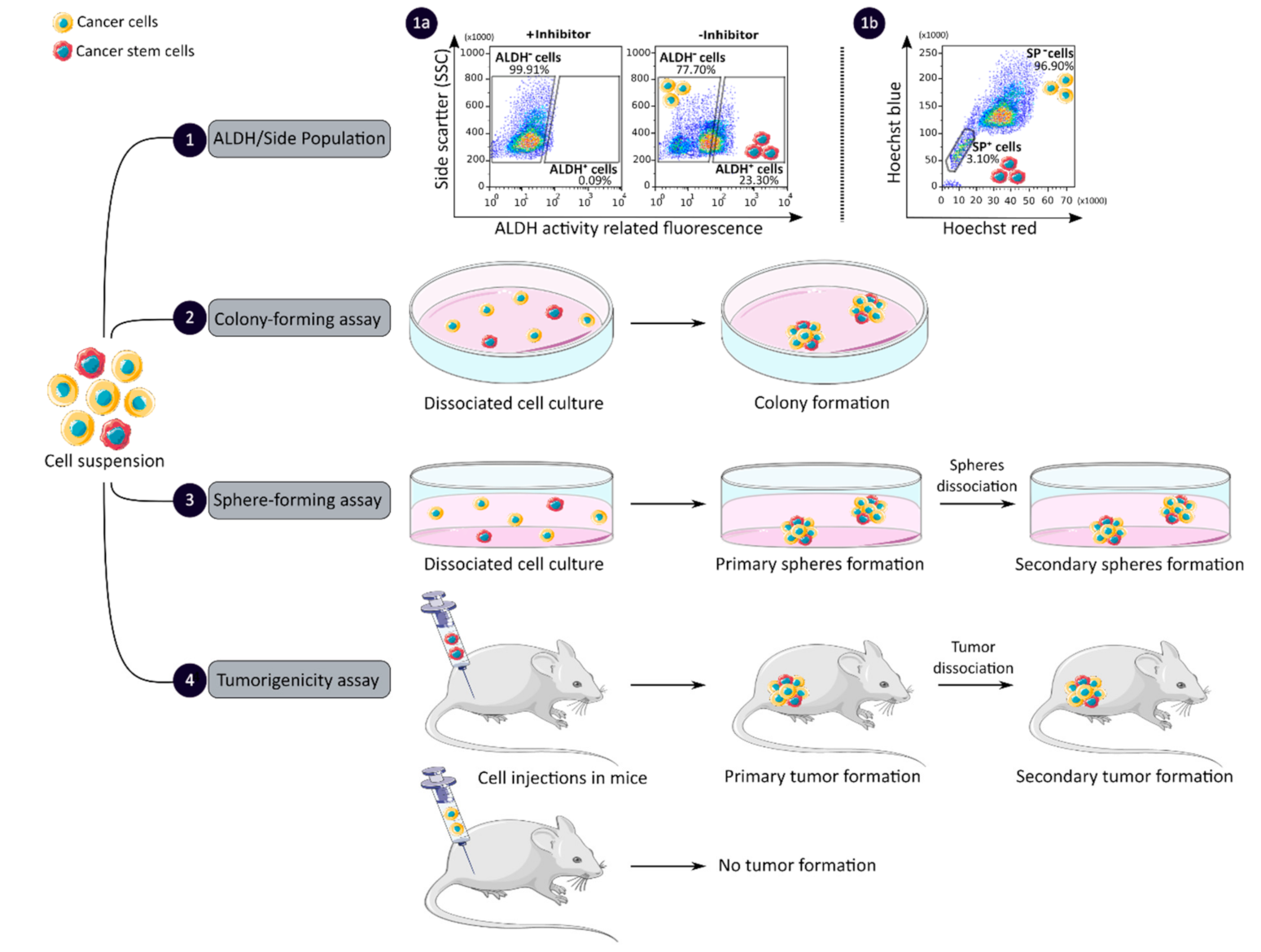
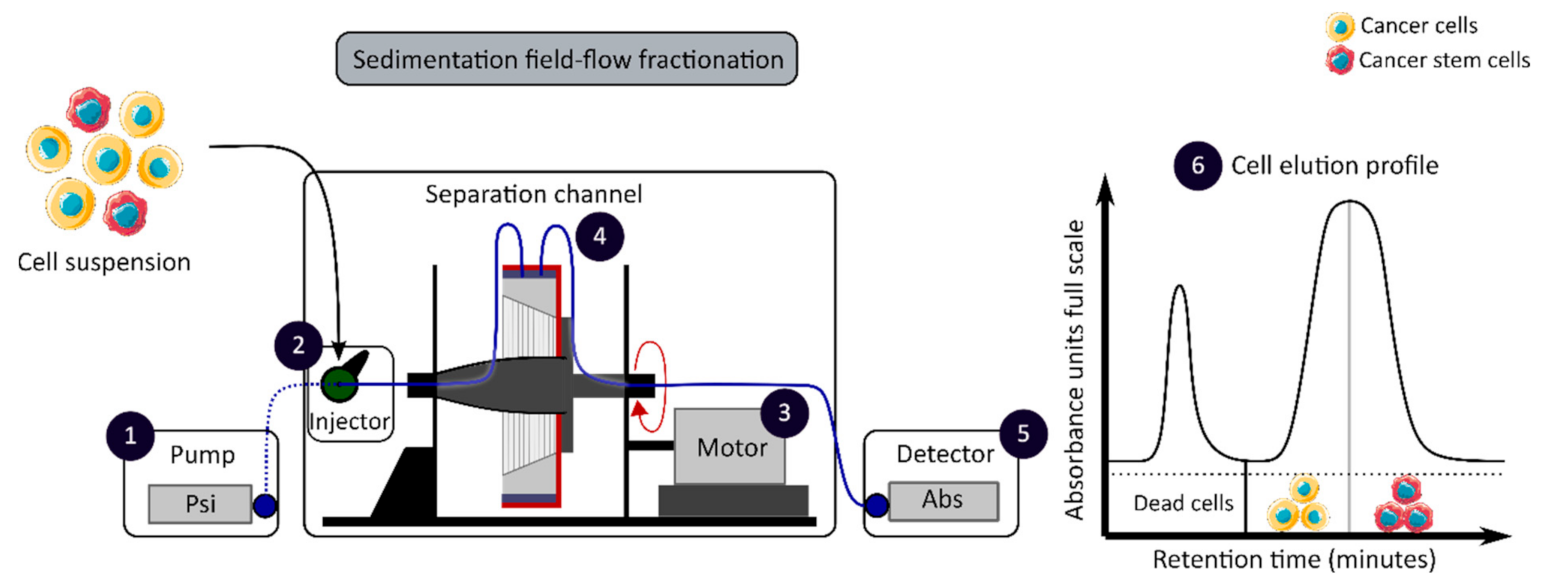
2.2. Colorectal Cancer Stem Cell Isolation Methods
2.2.1. CCSC Isolation Based on Phenotypic Features
2.2.2. CCSC Isolation Based on Functional Features
2.2.3. CCSC Isolation Based on Biophysical Features
2.2.4. CCSC Isolation Methods: Discussion
3. Clinical Relevance of Colorectal Cancer Stem Cells
3.1. Clinical Management of Colorectal Cancer
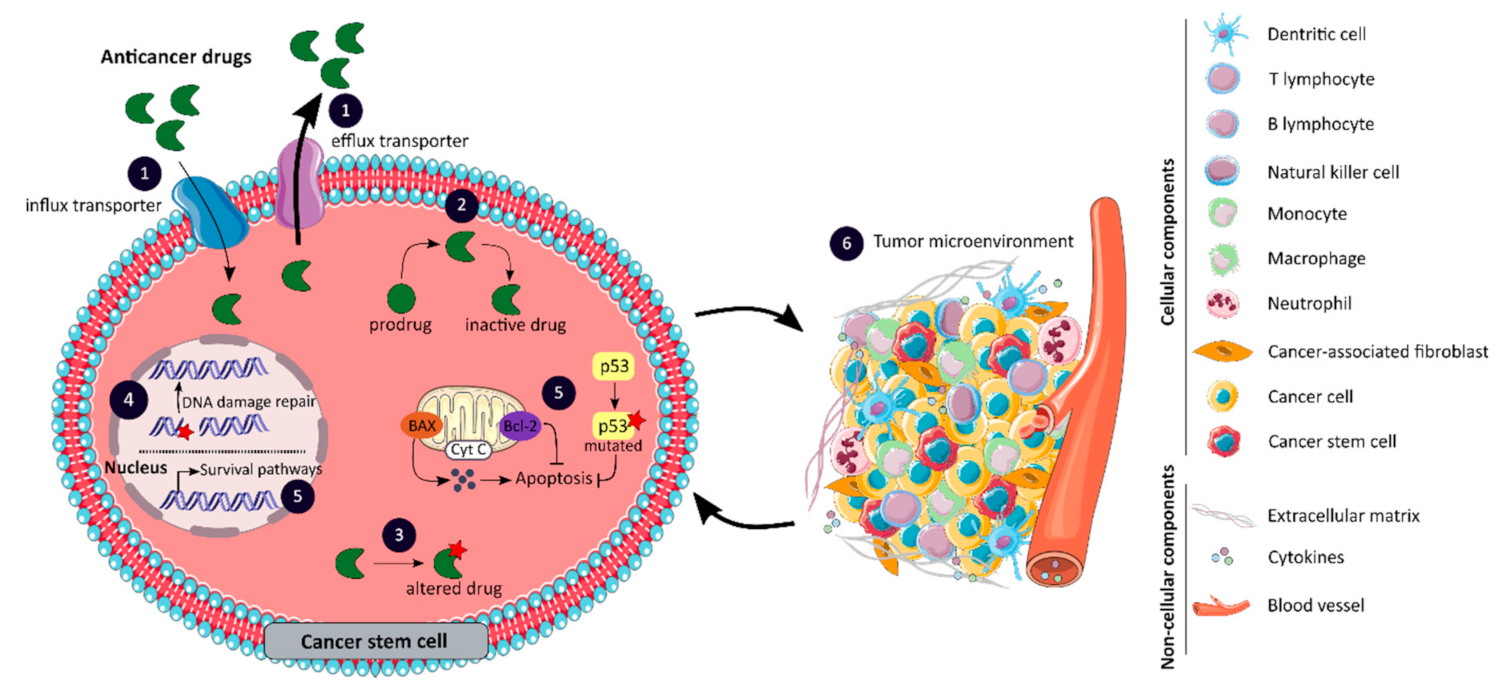
3.2. Mechanisms of Drug Resistance Associated with Colorectal Cancer Stem Cells
3.2.1. Changes in Drug Transport
3.2.2. Impaired Drug Metabolism
3.2.3. Alterations in Drug Targets
3.2.4. Enhanced DNA Damage Repair
3.2.5. Impaired Balance between Apoptosis and Survival Pathways
3.2.6. Role of the Tumor Microenvironment
3.2.7. Mechanisms of Drug Resistance Associated with CCSCs: Discussion
4. Clinical Trials on Colorectal Cancer Stem Cells
5. Future Perspectives
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Dekker, E.; Tanis, P.J.; Vleugels, J.L.A.; Kasi, P.M.; Wallace, M.B. Colorectal Cancer. Lancet 2019, 394, 1467–1480. [Google Scholar] [CrossRef]
- World Health Organization: Regional Office for Europe. World Cancer Report: Cancer Research for Cancer Development; IARC: Lyon, France, 2020; ISBN 978-92-832-0447-3. [Google Scholar]
- Brierley, J.D.; Gospodarowicz, M.K.; Wittekind, C. TNM Classification of Malignant Tumours; John Wiley & Sons: Hoboken, NJ, USA, 2017; ISBN 978-1-119-26357-9. [Google Scholar]
- Amin, M.B.; Edge, S.; Greene, F.; Byrd, D.R.; Brookland, R.K.; Washington, M.K.; Gershenwald, J.E.; Compton, C.C.; Hess, K.R.; Sullivan, D.C.; et al. AJCC Cancer Staging Manual, 8th ed.; Springer International Publishing: Cham, Switzerland, 2017. [Google Scholar]
- Benson, A.B.; Venook, A.P.; Al-Hawary, M.M.; Cederquist, L.; Chen, Y.-J.; Ciombor, K.K.; Cohen, S.; Cooper, H.S.; Deming, D.; Engstrom, P.F.; et al. NCCN Guidelines Insights: Colon Cancer, Version 2.2018. J. Natl. Compr. Canc. Netw. 2018, 16, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Cervantes, A.; Adam, R.; Sobrero, A.; Van Krieken, J.H.; Aderka, D.; Aranda Aguilar, E.; Bardelli, A.; Benson, A.; Bodoky, G.; et al. ESMO Consensus Guidelines for the Management of Patients with Metastatic Colorectal Cancer. Ann. Oncol. 2016, 27, 1386–1422. [Google Scholar] [CrossRef] [PubMed]
- Van der Heijden, M.; Vermeulen, L. A Cancer Stem Cell Perspective on Minimal Residual Disease in Solid Malignancies. In Cancer Stem Cell Resistance to Targeted Therapy; Maccalli, C., Todaro, M., Ferrone, S., Eds.; Resistance to Targeted Anti-Cancer Therapeutics; Springer International Publishing: Cham, Switzerland, 2019; Volume 19, pp. 31–49. ISBN 978-3-030-16623-6. [Google Scholar]
- Batlle, E.; Clevers, H. Cancer Stem Cells Revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef]
- Desbats, M.A.; Giacomini, I.; Prayer-Galetti, T.; Montopoli, M. Metabolic Plasticity in Chemotherapy Resistance. Front. Oncol. 2020, 10, 281. [Google Scholar] [CrossRef] [PubMed]
- Serpa, J. Tumor Microenvironment: The Main Driver of Metabolic Adaptation; Serpa, J., Ed.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2020; Volume 1219, ISBN 978-3-030-34024-7. [Google Scholar]
- Jordan, C.T. Cancer Stem Cells. N. Engl. J. Med. 2006, 355, 1253–1261. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, D.; Dick, J.E. Human Acute Myeloid Leukemia Is Organized as a Hierarchy That Originates from a Primitive Hematopoietic Cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef]
- Schwitalla, S.; Fingerle, A.A.; Cammareri, P.; Nebelsiek, T.; Göktuna, S.I.; Ziegler, P.K.; Canli, O.; Heijmans, J.; Huels, D.J.; Moreaux, G.; et al. Intestinal Tumorigenesis Initiated by Dedifferentiation and Acquisition of Stem-Cell-like Properties. Cell 2013, 152, 25–38. [Google Scholar] [CrossRef]
- Jahanafrooz, Z.; Mosafer, J.; Akbari, M.; Hashemzaei, M.; Mokhtarzadeh, A.; Baradaran, B. Colon Cancer Therapy by Focusing on Colon Cancer Stem Cells and Their Tumor Microenvironment. J. Cell. Physiol. 2020, 235, 4153–4166. [Google Scholar] [CrossRef]
- Najafi, M.; Farhood, B.; Mortezaee, K. Cancer Stem Cells (CSCs) in Cancer Progression and Therapy. J. Cell. Physiol. 2019, 234, 8381–8395. [Google Scholar] [CrossRef] [PubMed]
- Mélin, C.; Perraud, A.; Akil, H.; Jauberteau, M.-O.; Cardot, P.; Mathonnet, M.; Battu, S. Cancer Stem Cell Sorting from Colorectal Cancer Cell Lines by Sedimentation Field Flow Fractionation. Anal. Chem. 2012, 84, 1549–1556. [Google Scholar] [CrossRef] [PubMed]
- Hirata, A.; Hatano, Y.; Niwa, M.; Hara, A.; Tomita, H. Heterogeneity of Colon Cancer Stem Cells. In Stem Cells Heterogeneity in Cancer; Birbrair, A., Ed.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2019; Volume 1139, pp. 115–126. ISBN 978-3-030-14365-7. [Google Scholar]
- Akbarzadeh, M.; Maroufi, N.F.; Tazehkand, A.P.; Akbarzadeh, M.; Bastani, S.; Safdari, R.; Farzane, A.; Fattahi, A.; Nejabati, H.R.; Nouri, M.; et al. Current Approaches in Identification and Isolation of Cancer Stem Cells. J. Cell. Physiol. 2019, 234, 14759–14772. [Google Scholar] [CrossRef] [PubMed]
- Ricci-Vitiani, L.; Lombardi, D.G.; Pilozzi, E.; Biffoni, M.; Todaro, M.; Peschle, C.; De Maria, R. Identification and Expansion of Human Colon-Cancer-Initiating Cells. Nature 2007, 445, 111–115. [Google Scholar] [CrossRef]
- O’Brien, C.A.; Pollett, A.; Gallinger, S.; Dick, J.E. A Human Colon Cancer Cell Capable of Initiating Tumour Growth in Immunodeficient Mice. Nature 2007, 445, 106–110. [Google Scholar] [CrossRef]
- Kemper, K.; Sprick, M.R.; de Bree, M.; Scopelliti, A.; Vermeulen, L.; Hoek, M.; Zeilstra, J.; Pals, S.T.; Mehmet, H.; Stassi, G.; et al. The AC133 Epitope, but Not the CD133 Protein, Is Lost upon Cancer Stem Cell Differentiation. Cancer Res. 2010, 70, 719–729. [Google Scholar] [CrossRef]
- Dalerba, P.; Dylla, S.J.; Park, I.-K.; Liu, R.; Wang, X.; Cho, R.W.; Hoey, T.; Gurney, A.; Huang, E.H.; Simeone, D.M.; et al. Phenotypic Characterization of Human Colorectal Cancer Stem Cells. Proc. Natl. Acad. Sci. USA 2007, 104, 10158–10163. [Google Scholar] [CrossRef] [PubMed]
- Barker, N.; Ridgway, R.A.; van Es, J.H.; van de Wetering, M.; Begthel, H.; van den Born, M.; Danenberg, E.; Clarke, A.R.; Sansom, O.J.; Clevers, H. Crypt Stem Cells as the Cells-of-Origin of Intestinal Cancer. Nature 2009, 457, 608–611. [Google Scholar] [CrossRef]
- Sangiorgi, E.; Capecchi, M.R. Bmi1 Is Expressed in Vivo in Intestinal Stem Cells. Nat. Genet. 2008, 40, 915–920. [Google Scholar] [CrossRef]
- Vermeulen, L.; Todaro, M.; de Sousa Mello, F.; Sprick, M.R.; Kemper, K.; Perez Alea, M.; Richel, D.J.; Stassi, G.; Medema, J.P. Single-Cell Cloning of Colon Cancer Stem Cells Reveals a Multi-Lineage Differentiation Capacity. Proc. Natl. Acad. Sci. USA 2008, 105, 13427–13432. [Google Scholar] [CrossRef]
- Pang, R.; Law, W.L.; Chu, A.C.Y.; Poon, J.T.; Lam, C.S.C.; Chow, A.K.M.; Ng, L.; Cheung, L.W.H.; Lan, X.R.; Lan, H.Y.; et al. A Subpopulation of CD26+ Cancer Stem Cells with Metastatic Capacity in Human Colorectal Cancer. Cell Stem Cell 2010, 6, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Todaro, M.; Gaggianesi, M.; Catalano, V.; Benfante, A.; Iovino, F.; Biffoni, M.; Apuzzo, T.; Sperduti, I.; Volpe, S.; Cocorullo, G.; et al. CD44v6 Is a Marker of Constitutive and Reprogrammed Cancer Stem Cells Driving Colon Cancer Metastasis. Cell Stem Cell 2014, 14, 342–356. [Google Scholar] [CrossRef] [PubMed]
- Barker, N.; van Es, J.H.; Kuipers, J.; Kujala, P.; van den Born, M.; Cozijnsen, M.; Haegebarth, A.; Korving, J.; Begthel, H.; Peters, P.J.; et al. Identification of Stem Cells in Small Intestine and Colon by Marker Gene Lgr5. Nature 2007, 449, 1003–1007. [Google Scholar] [CrossRef]
- Saito, S.; Okabe, H.; Watanabe, M.; Ishimoto, T.; Iwatsuki, M.; Baba, Y.; Tanaka, Y.; Kurashige, J.; Miyamoto, Y.; Baba, H. CD44v6 Expression Is Related to Mesenchymal Phenotype and Poor Prognosis in Patients with Colorectal Cancer. Oncol. Rep. 2013, 29, 1570–1578. [Google Scholar] [CrossRef]
- Ma, L.; Dong, L.; Chang, P. CD44v6 Engages in Colorectal Cancer Progression. Cell Death Dis. 2019, 10, 30. [Google Scholar] [CrossRef]
- Yanai, H.; Atsumi, N.; Tanaka, T.; Nakamura, N.; Komai, Y.; Omachi, T.; Tanaka, K.; Ishigaki, K.; Saiga, K.; Ohsugi, H.; et al. Intestinal Cancer Stem Cells Marked by Bmi1 or Lgr5 Expression Contribute to Tumor Propagation via Clonal Expansion. Sci. Rep. 2017, 7, 41838. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Li, Y.; Li, J.; Zheng, J. Polycomb Group Protein Bmi1 Expression in Colon Cancers Predicts the Survival. Med. Oncol. 2010, 27, 1273–1276. [Google Scholar] [CrossRef]
- Weichert, W. Cytoplasmic CD24 Expression in Colorectal Cancer Independently Correlates with Shortened Patient Survival. Clin. Cancer Res. 2005, 11, 6574–6581. [Google Scholar] [CrossRef] [PubMed]
- Lam, C.S.-C.; Cheung, A.H.-K.; Wong, S.K.-M.; Wan, T.M.-H.; Ng, L.; Chow, A.K.-M.; Cheng, N.S.-M.; Pak, R.C.-H.; Li, H.-S.; Man, J.H.-W.; et al. Prognostic Significance of CD26 in Patients with Colorectal Cancer. PLoS ONE 2014, 9, e98582. [Google Scholar] [CrossRef]
- Liu, Q.-Z.; Gao, X.-H.; Chang, W.-J.; Gong, H.-F.; Fu, C.-G.; Zhang, W. Expression of ITGB1 Predicts Prognosis in Colorectal Cancer: A Large Prospective Study Based on Tissue Microarray. Int. J. Clin. Exp. Pathol. 2015, 8, 12802. [Google Scholar]
- Du, L.; Wang, H.; He, L.; Zhang, J.; Ni, B.; Wang, X.; Jin, H.; Cahuzac, N.; Mehrpour, M.; Lu, Y.; et al. CD44 Is of Functional Importance for Colorectal Cancer Stem Cells. Clin. Cancer Res. 2008, 14, 6751–6760. [Google Scholar] [CrossRef]
- Leng, Z.; Xia, Q.; Chen, J.; Li, Y.; Xu, J.; Zhao, E.; Zheng, H.; Ai, W.; Dong, J. Lgr5+CD44+EpCAM+ Strictly Defines Cancer Stem Cells in Human Colorectal Cancer. Cell. Physiol. Biochem. 2018, 46, 860–872. [Google Scholar] [CrossRef]
- Wang, Z.; Tang, Y.; Xie, L.; Huang, A.; Xue, C.; Gu, Z.; Wang, K.; Zong, S. The Prognostic and Clinical Value of CD44 in Colorectal Cancer: A Meta-Analysis. Front. Oncol. 2019, 9, 309. [Google Scholar] [CrossRef]
- Zhu, L.; Gibson, P.; Currle, D.S.; Tong, Y.; Richardson, R.J.; Bayazitov, I.T.; Poppleton, H.; Zakharenko, S.; Ellison, D.W.; Gilbertson, R.J. Prominin 1 Marks Intestinal Stem Cells That Are Susceptible to Neoplastic Transformation. Nature 2009, 457, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Glumac, P.M.; LeBeau, A.M. The Role of CD133 in Cancer: A Concise Review. Clin. Transl. Med. 2018, 7, 18. [Google Scholar] [CrossRef] [PubMed]
- Weichert, W. ALCAM/CD166 Is Overexpressed in Colorectal Carcinoma and Correlates with Shortened Patient Survival. J. Clin. Pathol. 2004, 57, 1160–1164. [Google Scholar] [CrossRef]
- Han, S.; Zong, S.; Shi, Q.; Li, H.; Liu, S.; Yang, W.; Li, W.; Hou, F. Is Ep-CAM Expression a Diagnostic and Prognostic Biomarker for Colorectal Cancer? A Systematic Meta-Analysis. EBioMedicine 2017, 20, 61–69. [Google Scholar] [CrossRef]
- Han, Y.; Xue, X.; Jiang, M.; Guo, X.; Li, P.; Liu, F.; Yuan, B.; Shen, Y.; Guo, X.; Zhi, Q.; et al. LGR5, a Relevant Marker of Cancer Stem Cells, Indicates a Poor Prognosis in Colorectal Cancer Patients: A Meta-Analysis. Clin. Res. Hepatol. Gastroenterol. 2015, 39, 267–273. [Google Scholar] [CrossRef]
- Cammareri, P.; Lombardo, Y.; Francipane, M.G.; Bonventre, S.; Todaro, M.; Stassi, G. Isolation and Culture of Colon Cancer Stem Cells. In Methods in Cell Biology; Elsevier: Amsterdam, The Netherlands, 2008; Volume 86, pp. 311–324. ISBN 978-0-12-373876-9. [Google Scholar]
- Korkusuz, P.; Köse, S.; Yersal, N.; Önen, S. Magnetic-Based Cell Isolation Technique for the Selection of Stem Cells. In Skin Stem Cells; Turksen, K., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2018; Volume 1879, pp. 153–163. ISBN 978-1-4939-8869-3. [Google Scholar]
- Mathonnet, M. Hallmarks in Colorectal Cancer: Angiogenesis and Cancer Stem-like Cells. World J. Gastroenterol. 2014, 20, 4189. [Google Scholar] [CrossRef] [PubMed]
- Mele, L.; Liccardo, D.; Tirino, V. Evaluation and Isolation of Cancer Stem Cells Using ALDH Activity Assay. In Cancer Stem Cells; Papaccio, G., Desiderio, V., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2018; Volume 1692, pp. 43–48. ISBN 978-1-4939-7400-9. [Google Scholar]
- Huang, E.H.; Hynes, M.J.; Zhang, T.; Ginestier, C.; Dontu, G.; Appelman, H.; Fields, J.Z.; Wicha, M.S.; Boman, B.M. Aldehyde Dehydrogenase 1 Is a Marker for Normal and Malignant Human Colonic Stem Cells (SC) and Tracks SC Overpopulation during Colon Tumorigenesis. Cancer Res. 2009, 69, 3382–3389. [Google Scholar] [CrossRef]
- Golebiewska, A.; Brons, N.H.C.; Bjerkvig, R.; Niclou, S.P. Critical Appraisal of the Side Population Assay in Stem Cell and Cancer Stem Cell Research. Cell Stem Cell 2011, 8, 136–147. [Google Scholar] [CrossRef]
- Franken, N.A.P.; Rodermond, H.M.; Stap, J.; Haveman, J.; van Bree, C. Clonogenic Assay of Cells in Vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef] [PubMed]
- Shaheen, S.; Ahmed, M.; Lorenzi, F.; Nateri, A.S. Spheroid-Formation (Colonosphere) Assay for in Vitro Assessment and Expansion of Stem Cells in Colon Cancer. Stem Cell Rev. Rep. 2016, 12, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Borowicz, S.; Van Scoyk, M.; Avasarala, S.; Karuppusamy Rathinam, M.K.; Tauler, J.; Bikkavilli, R.K.; Winn, R.A. The Soft Agar Colony Formation Assay. J. Vis. Exp. 2014, 51998. [Google Scholar] [CrossRef] [PubMed]
- Costa, E.C.; Moreira, A.F.; de Melo-Diogo, D.; Gaspar, V.M.; Carvalho, M.P.; Correia, I.J. 3D Tumor Spheroids: An Overview on the Tools and Techniques Used for Their Analysis. Biotechnol. Adv. 2016, 34, 1427–1441. [Google Scholar] [CrossRef]
- Relier, S.; Yazdani, L.; Ayad, O.; Choquet, A.; Bourgaux, J.-F.; Prudhomme, M.; Pannequin, J.; Macari, F.; David, A. Antibiotics Inhibit Sphere-Forming Ability in Suspension Culture. Cancer Cell Int. 2016, 16, 6. [Google Scholar] [CrossRef]
- Visvader, J.E.; Lindeman, G.J. Cancer Stem Cells in Solid Tumours: Accumulating Evidence and Unresolved Questions. Nat. Rev. Cancer 2008, 8, 755–768. [Google Scholar] [CrossRef]
- Nguyen, L.V.; Vanner, R.; Dirks, P.; Eaves, C.J. Cancer Stem Cells: An Evolving Concept. Nat. Rev. Cancer 2012, 12, 133–143. [Google Scholar] [CrossRef]
- Aiken, C.; Werbowetski-Ogilvie, T. Animal Models of Cancer Stem Cells: What Are They Really Telling Us? Curr. Pathobiol. Rep. 2013, 1, 91–99. [Google Scholar] [CrossRef][Green Version]
- O’Brien, C.A.; Kreso, A.; Jamieson, C.H.M. Cancer Stem Cells and Self-Renewal. Clin. Cancer Res. 2010, 16, 3113–3120. [Google Scholar] [CrossRef]
- Mélin, C.; Perraud, A.; Bounaix Morand du Puch, C.; Loum, E.; Giraud, S.; Cardot, P.; Jauberteau, M.-O.; Lautrette, C.; Battu, S.; Mathonnet, M. Sedimentation Field Flow Fractionation Monitoring of in Vitro Enrichment in Cancer Stem Cells by Specific Serum-Free Culture Medium. J. Chromatogr. B 2014, 963, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Lacroix, A.; Deluche, E.; Zhang, L.Y.; Dalmay, C.; Mélin, C.; Leroy, J.; Babay, M.; Morand Du Puch, C.; Giraud, S.; Bessette, B.; et al. A New Label-Free Approach to Glioblastoma Cancer Stem Cell Sorting and Detection. Anal. Chem. 2019, 91, 8948–8957. [Google Scholar] [CrossRef]
- Vedrenne, N.; Sarrazy, V.; Battu, S.; Bordeau, N.; Richard, L.; Billet, F.; Coronas, V.; Desmoulière, A. Neural Stem Cell Properties of an Astrocyte Subpopulation Sorted by Sedimentation Field-Flow Fractionation. Rejuvenation Res. 2016, 19, 362–372. [Google Scholar] [CrossRef]
- Faye, P.-A.; Vedrenne, N.; De la Cruz-Morcillo, M.A.; Barrot, C.-C.; Richard, L.; Bourthoumieu, S.; Sturtz, F.; Funalot, B.; Lia, A.-S.; Battu, S. New Method for Sorting Endothelial and Neural Progenitors from Human Induced Pluripotent Stem Cells by Sedimentation Field Flow Fractionation. Anal. Chem. 2016, 88, 6696–6702. [Google Scholar] [CrossRef] [PubMed]
- Mélin, C.; Perraud, A.; Christou, N.; Bibes, R.; Cardot, P.; Jauberteau, M.-O.; Battu, S.; Mathonnet, M. New Ex-Ovo Colorectal-Cancer Models from Different SdFFF-Sorted Tumor-Initiating Cells. Anal. Bioanal. Chem. 2015, 407, 8433–8443. [Google Scholar] [CrossRef] [PubMed]
- Shmelkov, S.V.; Butler, J.M.; Hooper, A.T.; Hormigo, A.; Kushner, J.; Milde, T.; St. Clair, R.; Baljevic, M.; White, I.; Jin, D.K.; et al. CD133 Expression Is Not Restricted to Stem Cells, and Both CD133+ and CD133− Metastatic Colon Cancer Cells Initiate Tumors. J. Clin. Investig. 2008, JCI34401. [Google Scholar] [CrossRef]
- Ricci-Vitiani, L.; Fabrizi, E.; Palio, E.; De Maria, R. Colon Cancer Stem Cells. J. Mol. Med. 2009, 87, 1097–1104. [Google Scholar] [CrossRef] [PubMed]
- Labianca, R.; Nordlinger, B.; Beretta, G.D.; Mosconi, S.; Mandalà, M.; Cervantes, A.; Arnold, D. Early Colon Cancer: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-up. Ann. Oncol. 2013, 24, vi64–vi72. [Google Scholar] [CrossRef] [PubMed]
- Costas-Chavarri, A.; Nandakumar, G.; Temin, S.; Lopes, G.; Cervantes, A.; Cruz Correa, M.; Engineer, R.; Hamashima, C.; Ho, G.F.; Huitzil, F.D.; et al. Treatment of Patients with Early-Stage Colorectal Cancer: ASCO Resource-Stratified Guideline. J. Glob. Oncol. 2019, 1–19. [Google Scholar] [CrossRef]
- FOxTROT Collaborative Group Feasibility of Preoperative Chemotherapy for Locally Advanced, Operable Colon Cancer: The Pilot Phase of a Randomised Controlled Trial. Lancet Oncol. 2012, 13, 1152–1160. [CrossRef]
- André, T.; de Gramont, A.; Vernerey, D.; Chibaudel, B.; Bonnetain, F.; Tijeras-Raballand, A.; Scriva, A.; Hickish, T.; Tabernero, J.; Van Laethem, J.L.; et al. Adjuvant Fluorouracil, Leucovorin, and Oxaliplatin in Stage II to III Colon Cancer: Updated 10-Year Survival and Outcomes According to BRAF Mutation and Mismatch Repair Status of the MOSAIC Study. J. Clin. Oncol. 2015, 33, 4176–4187. [Google Scholar] [CrossRef] [PubMed]
- André, T.; Boni, C.; Navarro, M.; Tabernero, J.; Hickish, T.; Topham, C.; Bonetti, A.; Clingan, P.; Bridgewater, J.; Rivera, F.; et al. Improved Overall Survival with Oxaliplatin, Fluorouracil, and Leucovorin as Adjuvant Treatment in Stage II or III Colon Cancer in the MOSAIC Trial. J. Clin. Oncol. 2009, 27, 3109–3116. [Google Scholar] [CrossRef] [PubMed]
- Schmoll, H.-J.; Cartwright, T.; Tabernero, J.; Nowacki, M.P.; Figer, A.; Maroun, J.; Price, T.; Lim, R.; Van Cutsem, E.; Park, Y.-S.; et al. Phase III Trial of Capecitabine Plus Oxaliplatin as Adjuvant Therapy for Stage III Colon Cancer: A Planned Safety Analysis in 1,864 Patients. J. Clin. Oncol. 2007, 26, 102–109. [Google Scholar] [CrossRef]
- Twelves, C.; Wong, A.; Nowacki, M.; Abt, M.; Burris, H., 3rd; Carrato, A.; Cassidy, J.; Cervantes, A.; Fagerberg, J.; Georgoulias, V.; et al. Capecitabine as Adjuvant Treatment for Stage III Colon Cancer. N. Engl. J. Med. 2005, 352, 2696–2704. [Google Scholar] [CrossRef]
- Haller, D.G.; Tabernero, J.; Maroun, J.; de Braud, F.; Price, T.; Van Cutsem, E.; Hill, M.; Gilberg, F.; Rittweger, K.; Schmoll, H.-J. Capecitabine Plus Oxaliplatin Compared with Fluorouracil and Folinic Acid as Adjuvant Therapy for Stage III Colon Cancer. J. Clin. Oncol. 2011, 29, 1465–1471. [Google Scholar] [CrossRef] [PubMed]
- Marin, J.J.G.; Sanchez de Medina, F.; Castaño, B.; Bujanda, L.; Romero, M.R.; Martinez-Augustin, O.; Moral-Avila, R.D.; Briz, O. Chemoprevention, Chemotherapy, and Chemoresistance in Colorectal Cancer. Drug Metab. Rev. 2012, 44, 148–172. [Google Scholar] [CrossRef] [PubMed]
- Chiorean, E.G.; Nandakumar, G.; Fadelu, T.; Temin, S.; Alarcon-Rozas, A.E.; Bejarano, S.; Croitoru, A.-E.; Grover, S.; Lohar, P.V.; Odhiambo, A.; et al. Treatment of Patients with Late-Stage Colorectal Cancer: ASCO Resource-Stratified Guideline. JCO Glob. Oncol. 2020, 414–438. [Google Scholar] [CrossRef] [PubMed]
- Atreya, C.E.; Yaeger, R.; Chu, E. Systemic Therapy for Metastatic Colorectal Cancer: From Current Standards to Future Molecular Targeted Approaches. Am. Soc. Clin. Oncol. Educ. Book 2017, 37, 246–256. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Oliveira, J. Advanced Colorectal Cancer: ESMO Clinical Recommendations for Diagnosis, Treatment and Follow-up. Ann. Oncol. 2009, 20, iv61–iv63. [Google Scholar] [CrossRef]
- Johnston, F.M.; Mavros, M.N.; Herman, J.M.; Pawlik, T.M. Local Therapies for Hepatic Metastases. J. Natl. Compr. Canc. Netw. 2013, 11, 153–160. [Google Scholar] [CrossRef]
- Nosher, J.L.; Ahmed, I.; Patel, A.N.; Gendel, V.; Murillo, P.G.; Moss, R.; Jabbour, S.K. Non-Operative Therapies for Colorectal Liver Metastases. Surg. Treat. Colorectal Liver Metastases 2015, 6, 17. [Google Scholar]
- Sveen, A.; Kopetz, S.; Lothe, R.A. Biomarker-Guided Therapy for Colorectal Cancer: Strength in Complexity. Nat. Rev. Clin. Oncol. 2020, 17, 11–32. [Google Scholar] [CrossRef] [PubMed]
- Advani, S.; Kopetz, S. Ongoing and Future Directions in the Management of Metastatic Colorectal Cancer: Update on Clinical Trials. J. Surg. Oncol. 2019, 119, 642–652. [Google Scholar] [CrossRef]
- IMPACT Investigators. Efficacy of Adjuvant Fluorouracil and Folinic Acid in Colon Cancer. The Lancet 1995, 345, 939–944. [Google Scholar] [CrossRef]
- Saltz, L.B.; Moore, M.J.; Pirotta, N. Irinotecan plus Fluorouracil and Leucovorin for Metastatic Colorectal Cancer. N. Engl. J. Med. 2000, 343, 905–914. [Google Scholar] [CrossRef]
- André, T.; Boni, C.; Mounedji-Boudiaf, L.; Navarro, M.; Tabernero, J.; Hickish, T.; Topham, C.; Zaninelli, M.; Clingan, P.; Bridgewater, J.; et al. Oxaliplatin, Fluorouracil, and Leucovorin as Adjuvant Treatment for Colon Cancer. N. Engl. J. Med. 2004, 350, 2343–2351. [Google Scholar] [CrossRef] [PubMed]
- Mayer, R.J.; Van Cutsem, E.; Falcone, A.; Yoshino, T.; Garcia-Carbonero, R.; Mizunuma, N.; Yamazaki, K.; Shimada, Y.; Tabernero, J.; Komatsu, Y.; et al. Randomized Trial of TAS-102 for Refractory Metastatic Colorectal Cancer. N. Engl. J. Med. 2015, 372, 1909–1919. [Google Scholar] [CrossRef]
- Hurwitz, H.; Fehrenbacher, L.; Novotny, W.; Cartwright, T.; Hainsworth, J.; Heim, W.; Berlin, J.; Baron, A.; Griffing, S.; Holmgren, E.; et al. Bevacizumab plus Irinotecan, Fluorouracil, and Leucovorin for Metastatic Colorectal Cancer. N. Engl. J. Med. 2004, 350, 2335–2342. [Google Scholar] [CrossRef] [PubMed]
- Grothey, A.; Cutsem, E.V.; Sobrero, A.; Siena, S.; Falcone, A.; Ychou, M.; Humblet, Y.; Bouché, O.; Mineur, L.; Barone, C.; et al. Regorafenib Monotherapy for Previously Treated Metastatic Colorectal Cancer (CORRECT): An International, Multicentre, Randomised, Placebo-Controlled, Phase 3 Trial. Lancet 2013, 381, 303–312. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Tabernero, J.; Lakomy, R.; Prenen, H.; Prausová, J.; Macarulla, T.; Ruff, P.; van Hazel, G.A.; Moiseyenko, V.; Ferry, D.; et al. Addition of Aflibercept to Fluorouracil, Leucovorin, and Irinotecan Improves Survival in a Phase III Randomized Trial in Patients with Metastatic Colorectal Cancer Previously Treated with an Oxaliplatin-Based Regimen. J. Clin. Oncol. 2012, 30, 3499–3506. [Google Scholar] [CrossRef] [PubMed]
- Tabernero, J.; Yoshino, T.; Cohn, A.L.; Obermannova, R.; Bodoky, G.; Garcia-Carbonero, R.; Ciuleanu, T.-E.; Portnoy, D.C.; Van Cutsem, E.; Grothey, A.; et al. Ramucirumab versus Placebo in Combination with Second-Line FOLFIRI in Patients with Metastatic Colorectal Carcinoma That Progressed during or after First-Line Therapy with Bevacizumab, Oxaliplatin, and a Fluoropyrimidine (RAISE): A Randomised, Double-Blind, Multicentre, Phase 3 Study. Lancet Oncol. 2015, 16, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Messersmith, W.A.; Ahnen, D.J. Targeting EGFR in Colorectal Cancer. N. Engl. J. Med. 2008, 359, 1834–1836. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef]
- Overman, M.J.; Lonardi, S.; Wong, K.Y.M.; Lenz, H.-J.; Gelsomino, F.; Aglietta, M.; Morse, M.A.; Van Cutsem, E.; McDermott, R.; Hill, A.; et al. Durable Clinical Benefit with Nivolumab Plus Ipilimumab in DNA Mismatch Repair–Deficient/Microsatellite Instability–High Metastatic Colorectal Cancer. J. Clin. Oncol. 2018, 36, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, R.B.; André, T.; Atreya, C.E.; Schellens, J.H.M.; Yoshino, T.; Bendell, J.C.; Hollebecque, A.; McRee, A.J.; Siena, S.; Middleton, G.; et al. Combined BRAF, EGFR, and MEK Inhibition in Patients with BRAFV600E-Mutant Colorectal Cancer. Cancer Discov. 2018, 8, 428–443. [Google Scholar] [CrossRef] [PubMed]
- Kopetz, S.; Grothey, A.; Yaeger, R.; Van Cutsem, E.; Desai, J.; Yoshino, T.; Wasan, H.; Ciardiello, F.; Loupakis, F.; Hong, Y.S.; et al. Encorafenib, Binimetinib, and Cetuximab in BRAF V600E–Mutated Colorectal Cancer. N. Engl. J. Med. 2019, 381, 1632–1643. [Google Scholar] [CrossRef]
- Meric-Bernstam, F.; Hurwitz, H.; Raghav, K.P.S.; McWilliams, R.R.; Fakih, M.; VanderWalde, A.; Swanton, C.; Kurzrock, R.; Burris, H.; Sweeney, C.; et al. Pertuzumab plus Trastuzumab for HER2-Amplified Metastatic Colorectal Cancer (MyPathway): An Updated Report from a Multicentre, Open-Label, Phase 2a, Multiple Basket Study. Lancet Oncol. 2019, 20, 518–530. [Google Scholar] [CrossRef]
- Sartore-Bianchi, A.; Trusolino, L.; Martino, C.; Bencardino, K.; Lonardi, S.; Bergamo, F.; Zagonel, V.; Leone, F.; Depetris, I.; Martinelli, E.; et al. Dual-Targeted Therapy with Trastuzumab and Lapatinib in Treatment-Refractory, KRAS Codon 12/13 Wild-Type, HER2-Positive Metastatic Colorectal Cancer (HERACLES): A Proof-of-Concept, Multicentre, Open-Label, Phase 2 Trial. Lancet Oncol. 2016, 17, 738–746. [Google Scholar] [CrossRef]
- Drilon, A.; Laetsch, T.W.; Kummar, S.; DuBois, S.G.; Lassen, U.N.; Demetri, G.D.; Nathenson, M.; Doebele, R.C.; Farago, A.F.; Pappo, A.S.; et al. Efficacy of Larotrectinib in TRK Fusion–Positive Cancers in Adults and Children. N. Engl. J. Med. 2018, 378, 731–739. [Google Scholar] [CrossRef]
- Demetri, G.D.; Paz-Ares, L.; Farago, A.F.; Liu, S.V.; Chawla, S.P.; Tosi, D.; Kim, E.S.; Blakely, C.M.; Krauss, J.C.; Sigal, D.; et al. Efficacy and Safety of Entrectinib in Patients with NTRK Fusion-Positive Tumours: Pooled Analysis of STARTRK-2, STARTRK-1, and ALKA-372-001. Ann. Oncol. 2018, 29, ix175. [Google Scholar] [CrossRef]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer Drug Resistance: An Evolving Paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef]
- Li, Q.; Shu, Y. Role of Solute Carriers in Response to Anticancer Drugs. Mol. Cell. Ther. 2014, 2, 15. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Li, Z.; Gao, C.-Y.; Cho, C.H. Mechanisms of Drug Resistance in Colon Cancer and Its Therapeutic Strategies. World J. Gastroenterol. 2016, 22, 6876. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Lovejoy, K.S.; Shima, J.E.; Lagpacan, L.L.; Shu, Y.; Lapuk, A.; Chen, Y.; Komori, T.; Gray, J.W.; Chen, X.; et al. Organic Cation Transporters Are Determinants of Oxaliplatin Cytotoxicity. Cancer Res. 2006, 66, 8847–8857. [Google Scholar] [CrossRef]
- Buck, E.; Sprick, M.; Gaida, M.; Grüllich, C.; Weber, T.; Herpel, E.; Bruckner, T.; Koschny, R. Tumor Response to Irinotecan Is Associated with CYP3A5 Expression in Colorectal Cancer. Oncol. Lett. 2019. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.L.; Coyle, K.M.; Sultan, M.; Marcato, P. Cancer Stem Cells and Chemoresistance: Strategies to Overcome Therapeutic Resistance. In Cancer Stem Cells: Emerging Concepts and Future Perspectives in Translational Oncology; Babashah, S., Ed.; Springer International Publishing: Cham, Switzerland, 2015; pp. 477–518. ISBN 978-3-319-21029-2. [Google Scholar]
- Hoskins, J.M.; Goldberg, R.M.; Qu, P.; Ibrahim, J.G.; McLeod, H.L. UGT1A1*28 Genotype and Irinotecan-Induced Neutropenia: Dose Matters. J. Natl. Cancer Inst. 2007, 99, 1290–1295. [Google Scholar] [CrossRef] [PubMed]
- Stoehlmacher, J. Association between Glutathione S-Transferase P1, T1, and M1 Genetic Polymorphism and Survival of Patients with Metastatic Colorectal Cancer. J. Natl. Cancer Inst. 2002, 94, 936–942. [Google Scholar] [CrossRef] [PubMed]
- Ramos, P.; Bentires-Alj, M. Mechanism-Based Cancer Therapy: Resistance to Therapy, Therapy for Resistance. Oncogene 2015, 34, 3617–3626. [Google Scholar] [CrossRef]
- Misale, S.; Yaeger, R.; Hobor, S.; Scala, E.; Janakiraman, M.; Liska, D.; Valtorta, E.; Schiavo, R.; Buscarino, M.; Siravegna, G.; et al. Emergence of KRAS Mutations and Acquired Resistance to Anti-EGFR Therapy in Colorectal Cancer. Nature 2012, 486, 532–536. [Google Scholar] [CrossRef]
- Shirota, Y.; Stoehlmacher, J.; Brabender, J.; Xiong, Y.-P.; Uetake, H.; Danenberg, K.D.; Groshen, S.; Tsao-Wei, D.D.; Danenberg, P.V.; Lenz, H.-J. ERCC1 and Thymidylate Synthase MRNA Levels Predict Survival for Colorectal Cancer Patients Receiving Combination Oxaliplatin and Fluorouracil Chemotherapy. J. Clin. Oncol. 2001, 19, 4298–4304. [Google Scholar] [CrossRef] [PubMed]
- Valeri, N.; Gasparini, P.; Braconi, C.; Paone, A.; Lovat, F.; Fabbri, M.; Sumani, K.M.; Alder, H.; Amadori, D.; Patel, T.; et al. MicroRNA-21 Induces Resistance to 5-Fluorouracil by down-Regulating Human DNA MutS Homolog 2 (HMSH2). Proc. Natl. Acad. Sci. USA 2010, 107, 21098–21103. [Google Scholar] [CrossRef]
- Hosoya, N.; Miyagawa, K. Targeting DNA Damage Response in Cancer Therapy. Cancer Sci. 2014, 105, 370–388. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J. Cancer Stem Cells and Chemoresistance: The Smartest Survives the Raid. Pharmacol. Ther. 2016, 160, 145–158. [Google Scholar] [CrossRef]
- Nehls, O.; Okech, T.; Hsieh, C.-J.; Enzinger, T.; Sarbia, M.; Borchard, F.; Gruenagel, H.-H.; Gaco, V.; Hass, H.G.; Arkenau, H.T.; et al. Studies on P53, BAX and Bcl-2 Protein Expression and Microsatellite Instability in Stage III (UICC) Colon Cancer Treated by Adjuvant Chemotherapy: Major Prognostic Impact of Proapoptotic BAX. Br. J. Cancer 2007, 96, 1409–1418. [Google Scholar] [CrossRef]
- Zhao, Y.; Dong, Q.; Li, J.; Zhang, K.; Qin, J.; Zhao, J.; Sun, Q.; Wang, Z.; Wartmann, T.; Jauch, K.W.; et al. Targeting Cancer Stem Cells and Their Niche: Perspectives for Future Therapeutic Targets and Strategies. Semin. Cancer Biol. 2018, 53, 139–155. [Google Scholar] [CrossRef]
- Vermeulen, L.; De Sousa E Melo, F.; van der Heijden, M.; Cameron, K.; de Jong, J.H.; Borovski, T.; Tuynman, J.B.; Todaro, M.; Merz, C.; Rodermond, H.; et al. Wnt Activity Defines Colon Cancer Stem Cells and Is Regulated by the Microenvironment. Nat. Cell Biol. 2010, 12, 468–476. [Google Scholar] [CrossRef]
- Borovski, T.; De Sousa E Melo, F.; Vermeulen, L.; Medema, J.P. Cancer Stem Cell Niche: The Place to Be. Cancer Res. 2011, 71, 634–639. [Google Scholar] [CrossRef]
- De Sousa e Melo, F.; Kurtova, A.V.; Harnoss, J.M.; Kljavin, N.; Hoeck, J.D.; Hung, J.; Anderson, J.E.; Storm, E.E.; Modrusan, Z.; Koeppen, H.; et al. A Distinct Role for Lgr5+ Stem Cells in Primary and Metastatic Colon Cancer. Nature 2017, 543, 676–680. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, A.; Oost, K.C.; Kester, L.; Morgner, J.; Bornes, L.; Bruens, L.; Spaargaren, L.; Azkanaz, M.; Schelfhorst, T.; Beerling, E.; et al. Plasticity of Lgr5-Negative Cancer Cells Drives Metastasis in Colorectal Cancer. Cell Stem Cell 2020, 26, 569–578.e7. [Google Scholar] [CrossRef]
- Medema, J.P. Targeting the Colorectal Cancer Stem Cell. N. Engl. J. Med. 2017, 377, 888–890. [Google Scholar] [CrossRef]
- Shimokawa, M.; Ohta, Y.; Nishikori, S.; Matano, M.; Takano, A.; Fujii, M.; Date, S.; Sugimoto, S.; Kanai, T.; Sato, T. Visualization and Targeting of LGR5+ Human Colon Cancer Stem Cells. Nature 2017, 545, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Lenos, K.J.; Miedema, D.M.; Lodestijn, S.C.; Nijman, L.E.; van den Bosch, T.; Romero Ros, X.; Lourenço, F.C.; Lecca, M.C.; van der Heijden, M.; van Neerven, S.M.; et al. Stem Cell Functionality Is Microenvironmentally Defined during Tumour Expansion and Therapy Response in Colon Cancer. Nat. Cell Biol. 2018, 20, 1193–1202. [Google Scholar] [CrossRef]
- De Sousa e Melo, F.; de Sauvage, F.J. Cellular Plasticity in Intestinal Homeostasis and Disease. Cell Stem Cell 2019, 24, 54–64. [Google Scholar] [CrossRef]
- Li, Y.; Rogoff, H.A.; Keates, S.; Gao, Y.; Murikipudi, S.; Mikule, K.; Leggett, D.; Li, W.; Pardee, A.B.; Li, C.J. Suppression of Cancer Relapse and Metastasis by Inhibiting Cancer Stemness. Proc. Natl. Acad. Sci. USA 2015, 112, 1839–1844. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ (accessed on 7 October 2020).
- Hubbard, J.M.; Grothey, A. Napabucasin: An Update on the First-in-Class Cancer Stemness Inhibitor. Drugs 2017, 77, 1091–1103. [Google Scholar] [CrossRef] [PubMed]
- Langleben, A.; Supko, J.G.; Hotte, S.J.; Batist, G.; Hirte, H.W.; Rogoff, H.; Li, Y.; Li, W.; Kerstein, D.; Leggett, D.; et al. A Dose-Escalation Phase I Study of a First-in-Class Cancer Stemness Inhibitor in Patients with Advanced Malignancies. J. Clin. Oncol. 2013, 31, 2542. [Google Scholar] [CrossRef]
- Jonker, D.J.; Stephenson, J.; Edenfield, W.J.; Supko, J.G.; Li, Y.; Li, W.; Hitron, M.; Leggett, D.; Kerstein, D.; Li, C. A Phase I Extension Study of BBI608, a First-in-Class Cancer Stem Cell (CSC) Inhibitor, in Patients with Advanced Solid Tumors. J. Clin. Oncol. 2014, 32, 2546. [Google Scholar] [CrossRef]
- Larson, T.; Ortuzar, W.F.; Bekaii-Saab, T.S.; Becerra, C.; Ciombor, K.K.; Hubbard, J.M.; Edenfield, W.J.; Shao, S.H.; Grothey, A.; Borodyansky, L.; et al. BBI608-224: A Phase Ib/II Study of Cancer Stemness Inhibitor Napabucasin (BBI-608) Administered with Panitumumab in KRAS Wild-Type Patients with Metastatic Colorectal Cancer. J. Clin. Oncol. 2017, 35, 677. [Google Scholar] [CrossRef]
- O’Neil, B.H.; Hubbard, J.M.; Starodub, A.; Jonker, D.J.; Edenfield, W.J.; El-Rayes, B.F.; Halfdanarson, T.R.; Ramanathan, R.K.; Pitot, H.C.; Britten, C.D.; et al. Phase 1b Extension Study of Cancer Stemness Inhibitor BB608 (Napabucasin) Administered in Combination with FOLFIRI +/− Bevacizumab (Bev) in Patients (Pts) with Advanced Colorectal Cancer (CRC). J. Clin. Oncol. 2016, 34, 3564. [Google Scholar] [CrossRef]
- Bendell, J.C.; Hubbard, J.M.; O’Neil, B.H.; Jonker, D.J.; Starodub, A.; Peyton, J.D.; Pitot, H.C.; Halfdanarson, T.R.; Nadeau, B.R.; Zubkus, J.D.; et al. Phase 1b/II Study of Cancer Stemness Inhibitor Napabucasin (BBI-608) in Combination with FOLFIRI +/− Bevacizumab (Bev) in Metastatic Colorectal Cancer (MCRC) Patients (Pts). J. Clin. Oncol. 2017, 35, 3529. [Google Scholar] [CrossRef]
- Jonker, D.J.; Nott, L.; Yoshino, T.; Gill, S.; Shapiro, J.; Ohtsu, A.; Zalcberg, J.; Vickers, M.M.; Wei, A.; Gao, Y.; et al. A Randomized Phase III Study of Napabucasin [BBI608] (NAPA) vs Placebo (PBO) in Patients (Pts) with Pretreated Advanced Colorectal Cancer (ACRC): The CCTG/AGITG CO.23 Trial. Ann. Oncol. 2016, 27, vi150. [Google Scholar] [CrossRef]
- Jonker, D.J.; Nott, L.; Yoshino, T.; Gill, S.; Shapiro, J.; Ohtsu, A.; Zalcberg, J.; Vickers, M.M.; Wei, A.C.; Gao, Y.; et al. Napabucasin versus Placebo in Refractory Advanced Colorectal Cancer: A Randomised Phase 3 Trial. Lancet Gastroenterol. Hepatol. 2018, 3, 263–270. [Google Scholar] [CrossRef]
- Grothey, A.; Shah, M.A.; Yoshino, T.; Van Cutsem, E.; Taieb, J.; Xu, R.; Tebbutt, N.C.; Falcone, A.; Cervantes, A.; Borodyansky, L.; et al. CanStem303C Trial: A Phase III Study of Napabucasin (BBI-608) in Combination with 5-Fluorouracil (5-FU), Leucovorin, Irinotecan (FOLFIRI) in Adult Patients with Previously Treated Metastatic Colorectal Cancer (MCRC). J. Clin. Oncol. 2017, 35, TPS3619. [Google Scholar] [CrossRef]
- Sonbol, M.; Ahn, D.; Bekaii-Saab, T. Therapeutic Targeting Strategies of Cancer Stem Cells in Gastrointestinal Malignancies. Biomedicines 2019, 7, 17. [Google Scholar] [CrossRef]
- Fischer, M.; Yen, W.-C.; Kapoun, A.M.; Wang, M.; O’Young, G.; Lewicki, J.; Gurney, A.; Hoey, T. Anti-DLL4 Inhibits Growth and Reduces Tumor-Initiating Cell Frequency in Colorectal Tumors with Oncogenic KRAS Mutations. Cancer Res. 2011, 71, 1520–1525. [Google Scholar] [CrossRef] [PubMed]
- Hoey, T.; Yen, W.-C.; Axelrod, F.; Basi, J.; Donigian, L.; Dylla, S.; Fitch-Bruhns, M.; Lazetic, S.; Park, I.-K.; Sato, A.; et al. DLL4 Blockade Inhibits Tumor Growth and Reduces Tumor-Initiating Cell Frequency. Cell Stem Cell 2009, 5, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Ridgway, J.; Zhang, G.; Wu, Y.; Stawicki, S.; Liang, W.-C.; Chanthery, Y.; Kowalski, J.; Watts, R.J.; Callahan, C.; Kasman, I.; et al. Inhibition of Dll4 Signalling Inhibits Tumour Growth by Deregulating Angiogenesis. Nature 2006, 444, 1083–1087. [Google Scholar] [CrossRef]
- Smith, D.C.; Eisenberg, P.D.; Manikhas, G.; Chugh, R.; Gubens, M.A.; Stagg, R.J.; Kapoun, A.M.; Xu, L.; Dupont, J.; Sikic, B. A Phase I Dose Escalation and Expansion Study of the Anticancer Stem Cell Agent Demcizumab (Anti-DLL4) in Patients with Previously Treated Solid Tumors. Clin. Cancer Res. 2014, 20, 6295–6303. [Google Scholar] [CrossRef]
- Johnson, M.; Rasco, D.; Schneider, B.; Shu, C.; Jotte, R.; Parmer, H.; Stagg, R.; Lopez, J. Abstract A081: A Phase 1b, Open-Label, Dose Escalation and Expansion Study of Demcizumab plus Pembrolizumab in Patients with Locally Advanced or Metastatic Solid Tumors. Mol. Cancer Ther. 2018, 17, A081. [Google Scholar] [CrossRef]
- Quarni, W.; Dutta, R.; Green, R.; Katiri, S.; Patel, B.; Mohapatra, S.S.; Mohapatra, S. Mithramycin A Inhibits Colorectal Cancer Growth by Targeting Cancer Stem Cells. Sci. Rep. 2019, 9, 15202. [Google Scholar] [CrossRef]
- Rao, M.; Atay, S.M.; Shukla, V.; Hong, Y.; Upham, T.; Ripley, R.T.; Hong, J.A.; Zhang, M.; Reardon, E.; Fetsch, P.; et al. Mithramycin Depletes Specificity Protein 1 and Activates P53 to Mediate Senescence and Apoptosis of Malignant Pleural Mesothelioma Cells. Clin. Cancer Res. 2016, 22, 1197–1210. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, W.; Guo, Z.; Ma, F.; Wu, Y.; Bai, Y.; Gong, W.; Chen, Y.; Cheng, T.; Zhi, F.; et al. Inhibition of the Transcription Factor Sp1 Suppresses Colon Cancer Stem Cell Growth and Induces Apoptosis in Vitro and in Nude Mouse Xenografts. Oncol. Rep. 2013, 30, 1782–1792. [Google Scholar] [CrossRef]
- Grohar, P.J.; Glod, J.; Peer, C.J.; Sissung, T.M.; Arnaldez, F.I.; Long, L.; Figg, W.D.; Whitcomb, P.; Helman, L.J.; Widemann, B.C. A Phase I/II Trial and Pharmacokinetic Study of Mithramycin in Children and Adults with Refractory Ewing Sarcoma and EWS–FLI1 Fusion Transcript. Cancer Chemother. Pharmacol. 2017, 80, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Sissung, T.M.; Huang, P.A.; Hauke, R.J.; McCrea, E.M.; Peer, C.J.; Barbier, R.H.; Strope, J.D.; Ley, A.M.; Zhang, M.; Hong, J.A.; et al. Severe Hepatotoxicity of Mithramycin Therapy Caused by Altered Expression of Hepatocellular Bile Transporters. Mol. Pharmacol. 2019, 96, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Takebe, N.; Miele, L.; Harris, P.J.; Jeong, W.; Bando, H.; Kahn, M.; Yang, S.X.; Ivy, S.P. Targeting Notch, Hedgehog, and Wnt Pathways in Cancer Stem Cells: Clinical Update. Nat. Rev. Clin. Oncol. 2015, 12, 445–464. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| References | Experimental Models | Identified CCSC Subpopulations | CCSC Isolation Methods | CCSC CharacterizationAssays |
|---|---|---|---|---|
| O’Brien et al. [20] | CRC patient tissues CRC cells from patient tumors Animal model (mice) | CD133+ | MACS and FACS | Flow cytometry Immunohistochemistry Tumorigenicity assay |
| Ricci-Vitiani et al. [19] | CRC patient tissues CRC cells from patient tumors Primary tumor cell cultures Animal model (mice) | CD133+ | MACS and FACS | Sphere formation assay Flow cytometry Immunohistochemistry Tumorigenicity assay |
| Dalerba et al. [22] | CRC patient tissues CRC xenograft lines Single-cell suspensions | EpCAMhigh/CD44+ EpCAMhigh/CD44+/CD166+ | FACS | ALDH assay Flow cytometry Tumorigenicity assay |
| Barker et al. [23] | Animal model (Ah-cre/Apcflox/flox and Lgr5-EGFP-IRES-creERT2/APCflox/flox mice) | Lgr5+ | / | LacZ analysis Immunohistochemistry |
| Sangiorgi and Capecchi [24] | Animal model (Bmi1-IRES-Cre-ER mice) | Bmi1+ | / | LacZ analysis Immunohistochemistry |
| Vermeulen et al. [25] | CRC patient tissues CRC cells and single-cell-derived cultures from patient tumors Animal model (mice) | CD133+/CD24+ CD44+/CD166+ CD24+/CD29+ | MACS and FACS | Sphere formation assay In vitro differentiation assay Immunohistochemistry Flow cytometry Tumorigenicity assay |
| Pang et al. [26] | CRC patient tissues CRC cells from patient tumors Animal model (mice) | CD133+/CD26+ CD133+/CD26+/CD44+ | MACS and FACS | Sphere formation assay In vitro invasion assays Chemotherapeutic treatments Tumorigenicity assay |
| Todaro et al. [27] | CRC patient tissues Sphere-derived adherent cultures CRC cells from patient tumors or spheres Animal model (mice) | CD44v6+ | MACS and FACS | Immunofluorescence Immunohistochemistry Invasion assay Sphere formation assay Tumorigenicity assay |
| CCSC Markers | Functions | Roles in Prognosis of CRC | References |
|---|---|---|---|
| Bmi-1 | Polycomb-repressor protein Involved in self-renewal | High expression of Bmi-1 is associated with poor survival | [23,24,31,32] |
| CD24 (Heat stable antigen 24) | Cell adhesion molecule Alternative ligand of P-selectin | Strong cytoplasmic expression of CD24 is correlated with shortened patient survival | [25,33] |
| CD26 | Cell adhesion glycoprotein Promote invasion and metastases | Elevated-CD26 expression is associated with advanced tumor staging and worse overall survival | [26,34] |
| CD29 (Integrin-β1) | Transmembrane proteinInvolved in cell adhesion | Overexpression of CD29 is correlated with poor prognosis and aggressive clinicopathological features | [25,35] |
| CD44 | Transmembrane glycoprotein Regulate cell interactions, adhesion and migration | CD44 overexpression is associated with lymph node metastasis, distant metastases and poor prognosis | [36,37,38] |
| CD44v6 | Bind hepatocyte growth factor Promote migration and metastases | High level of CD44v6 has an unfavorable impact on overall survival | [27,29,38] |
| CD133 (Prominin-1) | Cell surface glycoprotein Regulate self-renewal and tumor angiogenesis | CD133 expression is correlated with low survival in CRC patients | [21,39,40] |
| CD166 (Activated leukocyte adhesion molecule) | Cell adhesion molecule Mediate homophilic interactions | Overexpression of CD166 is correlated with shortened patient survival | [22,25,41] |
| EpCAM (Epithelial cell adhesion molecule) | Transmembrane glycoprotein Regulate cell adhesion, proliferation and migration | Loss of EpCAM expression is associated with tumor stage, lymph node and distant metastases and poor prognosis | [22,37,42] |
| Lgr5 (Leucine-rich repeat- containing G-protein coupled receptor 5) | Seven-transmembrane protein Target of Wnt pathway involved in self-renewal | Lgr5 expression is associated with lymph node and distant metastases, and overexpression with reduced overall survival | [23,28,37,43] |
| Features | Isolation Methods | Advantages | Disadvantages | References |
|---|---|---|---|---|
| Phenotypic | MACS | High specificity Fast and easy method | No universal CCSC marker Monoparameter separation | [18,31,32] |
| FACS | High specificity Multiparameter separation | No universal CCSC marker Require large number of cells | [18,31] | |
| Functional | ALDH activity assay | High stability | Low specificity | [47,48] |
| Side population assay | No cell labelling required | Low purity and specificity | [49] | |
| Colony and sphere formation assay | No need for complicated laboratory equipment | Absence of standardized protocol Require proper cell dilution | [50,52,53] | |
| Tumorigenicity assay | Gold standard method | Complicated laboratory equipment Ethical consideration | [56,58] | |
| Biophysical | SdFFF | No cell labelling required Cell size and density separation | Time consuming | [16,46,59] |
| Systemic Therapies | Drug Names | Functions | Recommendations | References |
|---|---|---|---|---|
| Chemotherapy | 5-Fluorouracil | Antimetabolite | Localized and advanced tumors | [82] |
| Capecitabine | Antimetabolite | [72] | ||
| Irinotecan | Topoisomerase inhibitor | [83] | ||
| Oxaliplatin | Alkylating agent | [84] | ||
| Trifluridine/ Tipiracil | Nucleoside analog/ TP inhibitor | [85] | ||
| Targeted therapy | Bevacizumab | mAb anti-VEGF-A | KRAS/NRAS/BRAF Mutated tumors | [86] |
| Regorafenib | Multikinase inhibitor targeting e.g., VEGFR and BRAF | [87] | ||
| Aflibercept | Recombinant fusion protein blocking VEGF-A/B | [88] | ||
| Ramucirumab | mAb anti-VEGFR-2 | [89] | ||
| Cetuximab | mAb anti-EGFR | KRAS/NRAS/BRAF Wild-type tumors | [90] | |
| Panitumumab | [90] | |||
| Immunotherapy | Pembrolizumab | mAb anti-PD-1 | MSI-high tumors | [91] |
| Nivolumab | [92] | |||
| Ipilimumab | mAb anti-CTLA4 | [92] | ||
| Newly developed therapy | Vemurafenib | BRAF inhibitors | BRAF V600E mutated tumors | [93] |
| Dabrafenib | [93] | |||
| Encorafenib | [94] | |||
| Trametinib | MEK inhibitors | [93] | ||
| Binimetinib | [94] | |||
| Trastuzumab | mAb anti-HER2 | HER2 amplified tumors | [95] | |
| Pertuzumab | [95] | |||
| Lapatinib | Dual HER2/EGFR inhibitor | [96] | ||
| Larotrectinib | TRK inhibitors | NTRK gene fusion-positive tumors | [97] | |
| Entrectinib | [98] |
| Trial Registration and Status | Study Titles | Interventions | Phases | Investigators |
|---|---|---|---|---|
| NCT02753127 Active, not recruiting | A Study of Napabucasin (BBI-608) in Combination with FOLFIRI in Adult Patients with Previously Treated Metastatic Colorectal Cancer (CanStem303C) | Drug: Napabucasin | Phase III | Sumitomo Dainippon Pharma Oncology, Inc |
| NCT01189942 Completed | A Study of FOLFIRI Plus OMP-21M18 as 1st or 2nd-line Treatment in Subjects with Metastatic Colorectal Cancer | Drug: OMP-21M18 | Phase I | Mereo BioPharma (OncoMed Pharmaceuticals, Inc.) |
| NCT02859415 Recruiting | Continuous 24 h Intravenous Infusion of Mithramycin, an Inhibitor of Cancer Stem Cell Signaling, in People with Primary Thoracic Malignancies or Carcinomas, Sarcomas or Germ Cell Neoplasms with Pleuropulmonary Metastases | Drug: Mithramycin | Phase I and II | National Cancer Institute |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hervieu, C.; Christou, N.; Battu, S.; Mathonnet, M. The Role of Cancer Stem Cells in Colorectal Cancer: From the Basics to Novel Clinical Trials. Cancers 2021, 13, 1092. https://doi.org/10.3390/cancers13051092
Hervieu C, Christou N, Battu S, Mathonnet M. The Role of Cancer Stem Cells in Colorectal Cancer: From the Basics to Novel Clinical Trials. Cancers. 2021; 13(5):1092. https://doi.org/10.3390/cancers13051092
Chicago/Turabian StyleHervieu, Céline, Niki Christou, Serge Battu, and Muriel Mathonnet. 2021. "The Role of Cancer Stem Cells in Colorectal Cancer: From the Basics to Novel Clinical Trials" Cancers 13, no. 5: 1092. https://doi.org/10.3390/cancers13051092
APA StyleHervieu, C., Christou, N., Battu, S., & Mathonnet, M. (2021). The Role of Cancer Stem Cells in Colorectal Cancer: From the Basics to Novel Clinical Trials. Cancers, 13(5), 1092. https://doi.org/10.3390/cancers13051092