
Pancreatic Cancers with High Grade Tumor Budding Exhibit Hallmarks of Diminished Anti-Tumor Immunity

 ,
,  ,
,
Abstract
Simple Summary
Abstract
1. Introduction
2. Results
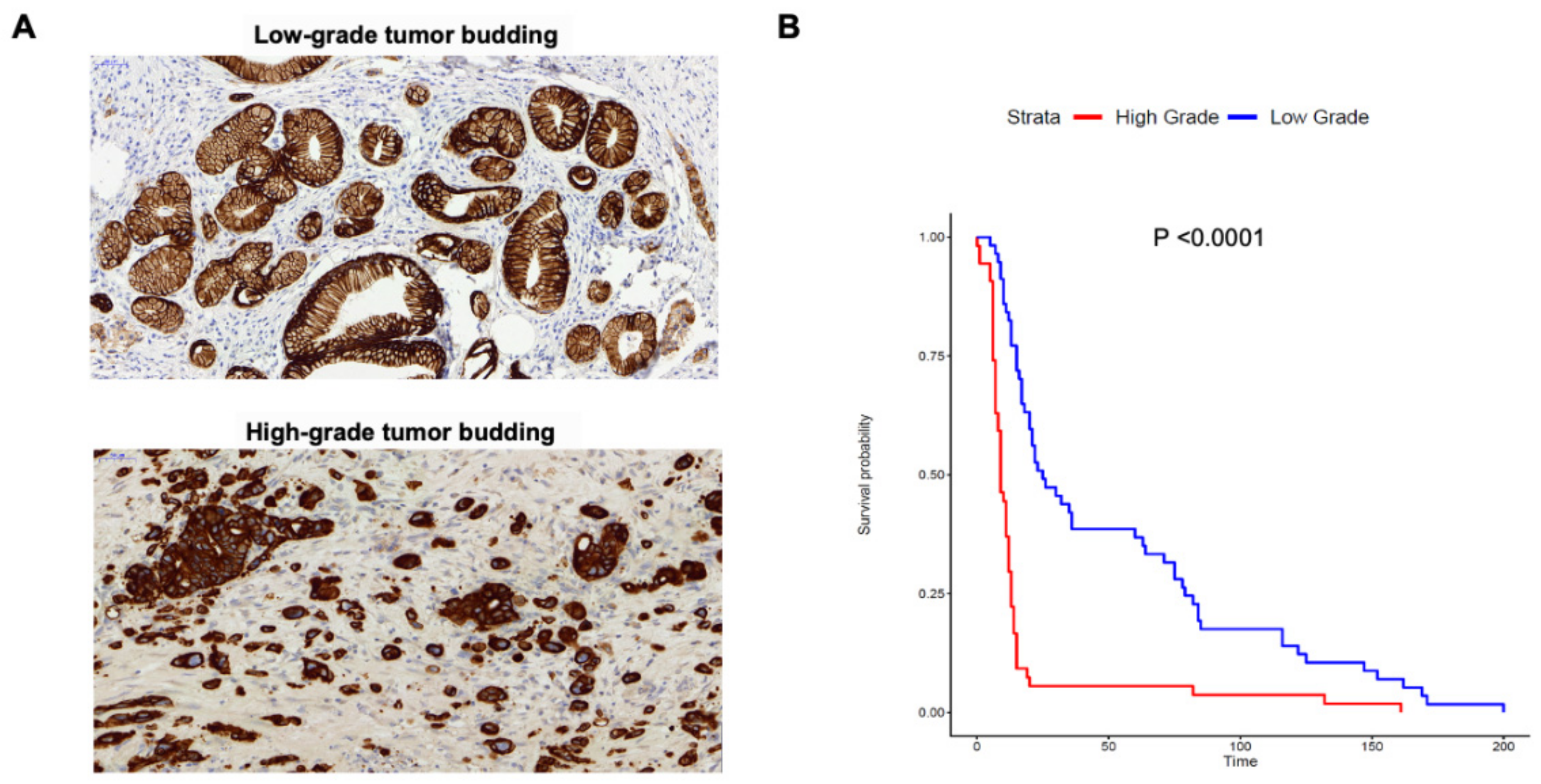
2.1. Tumor Budding Grade Is a Negative Prognostic Factor in PDAC
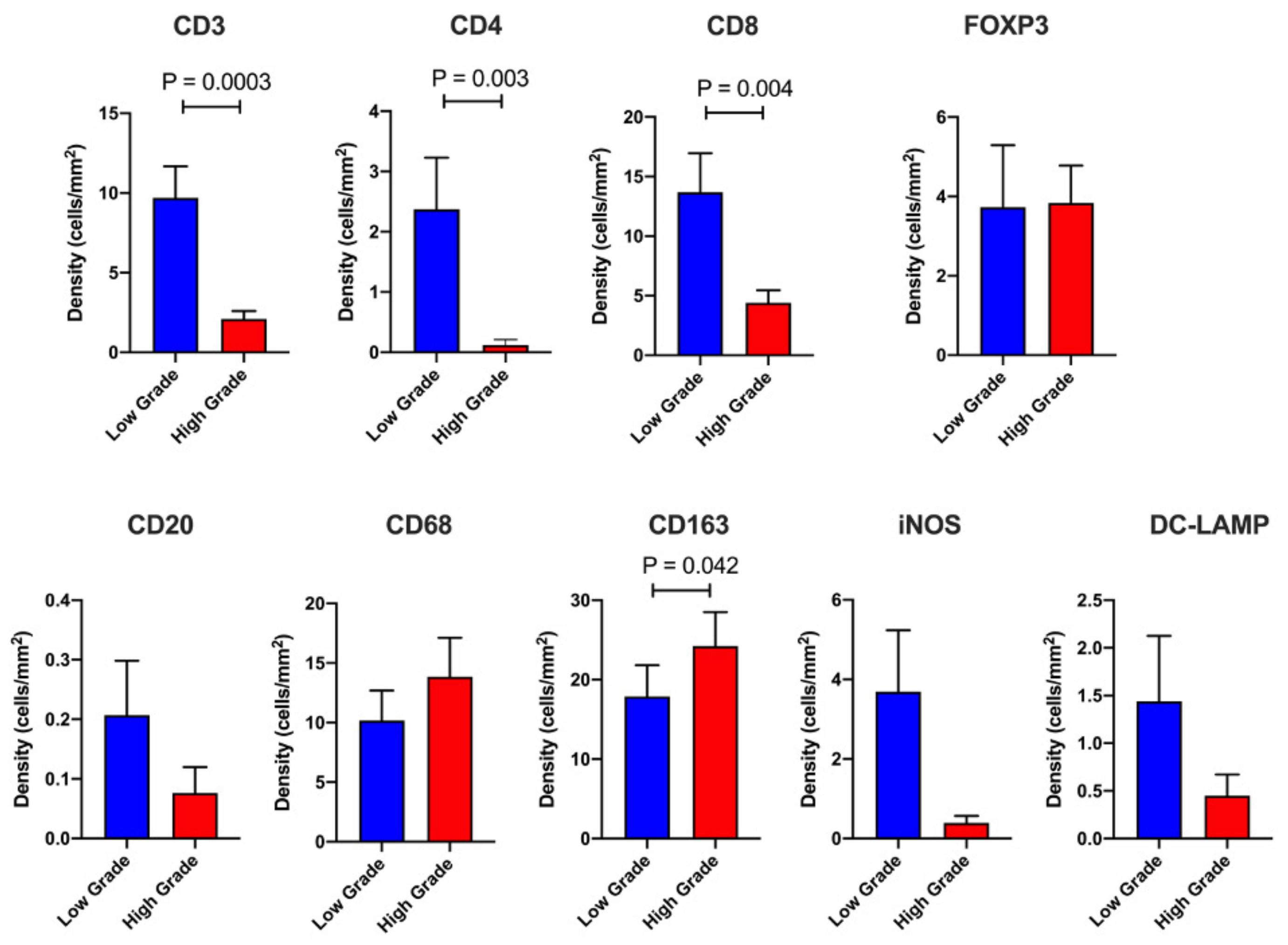
2.2. Dissecting the Immune Landscape of High Grade versus Low Grade Budding Cases
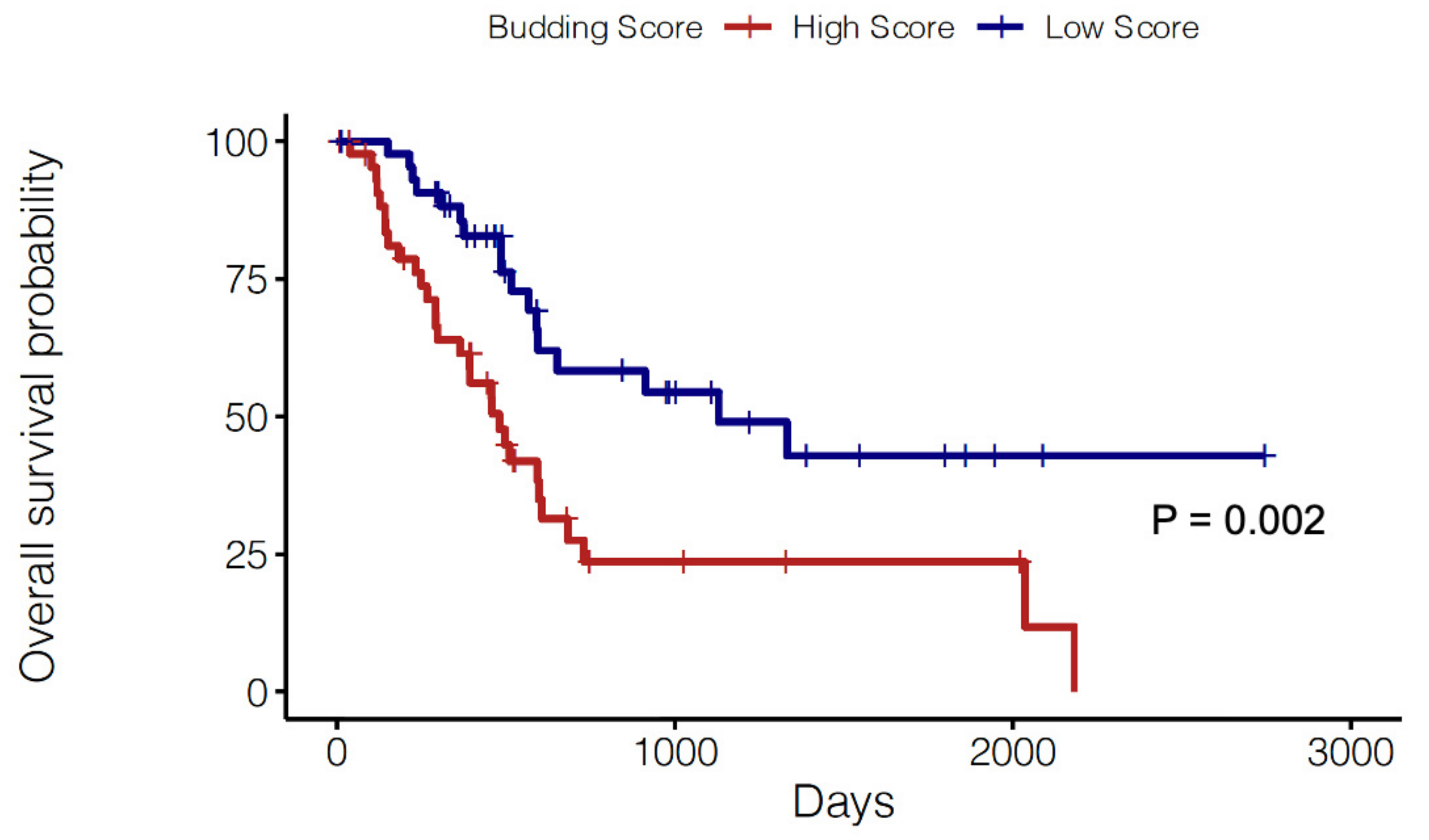
2.3. Prognostic Significance of a Tumor Budding Gene Signature
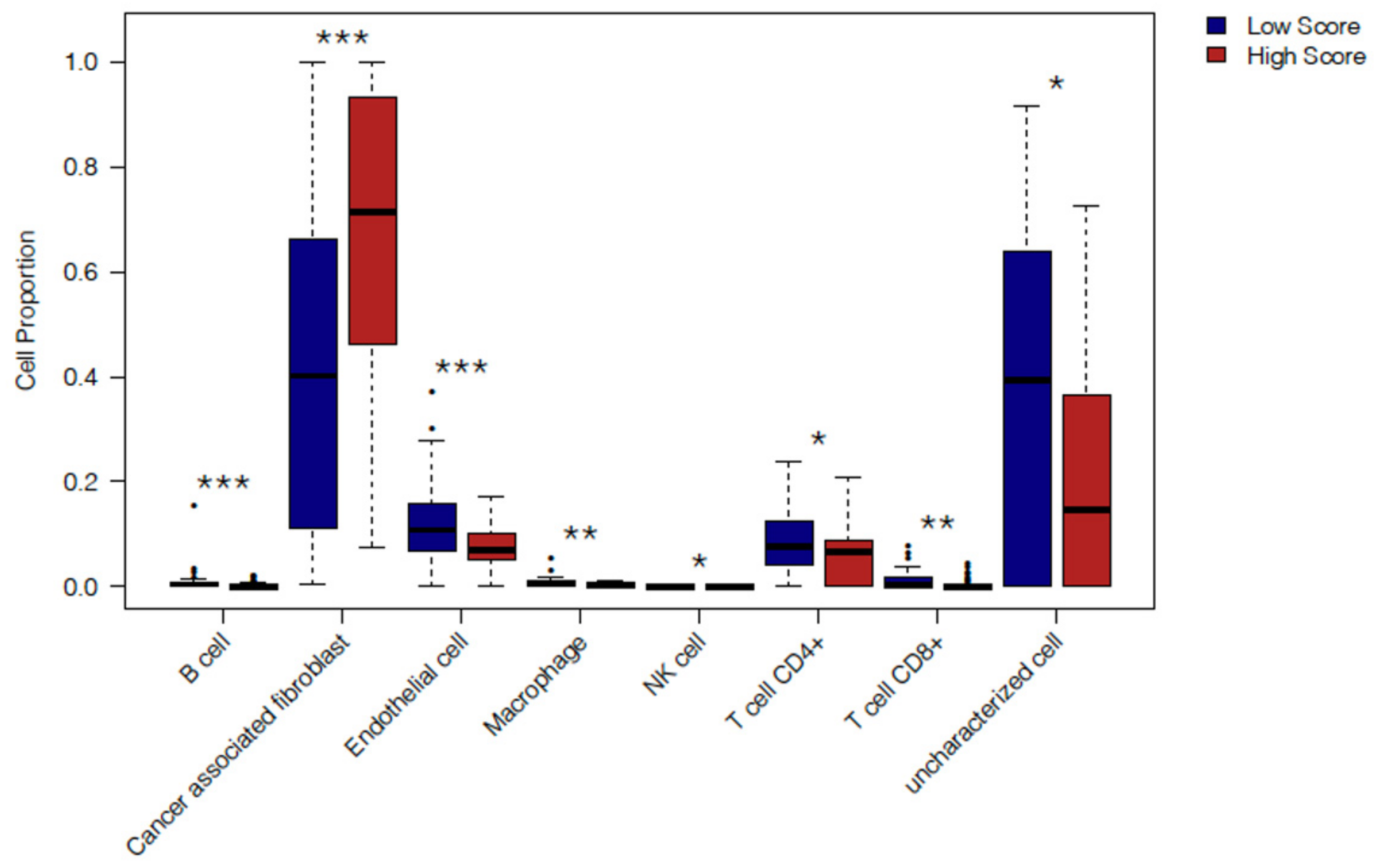
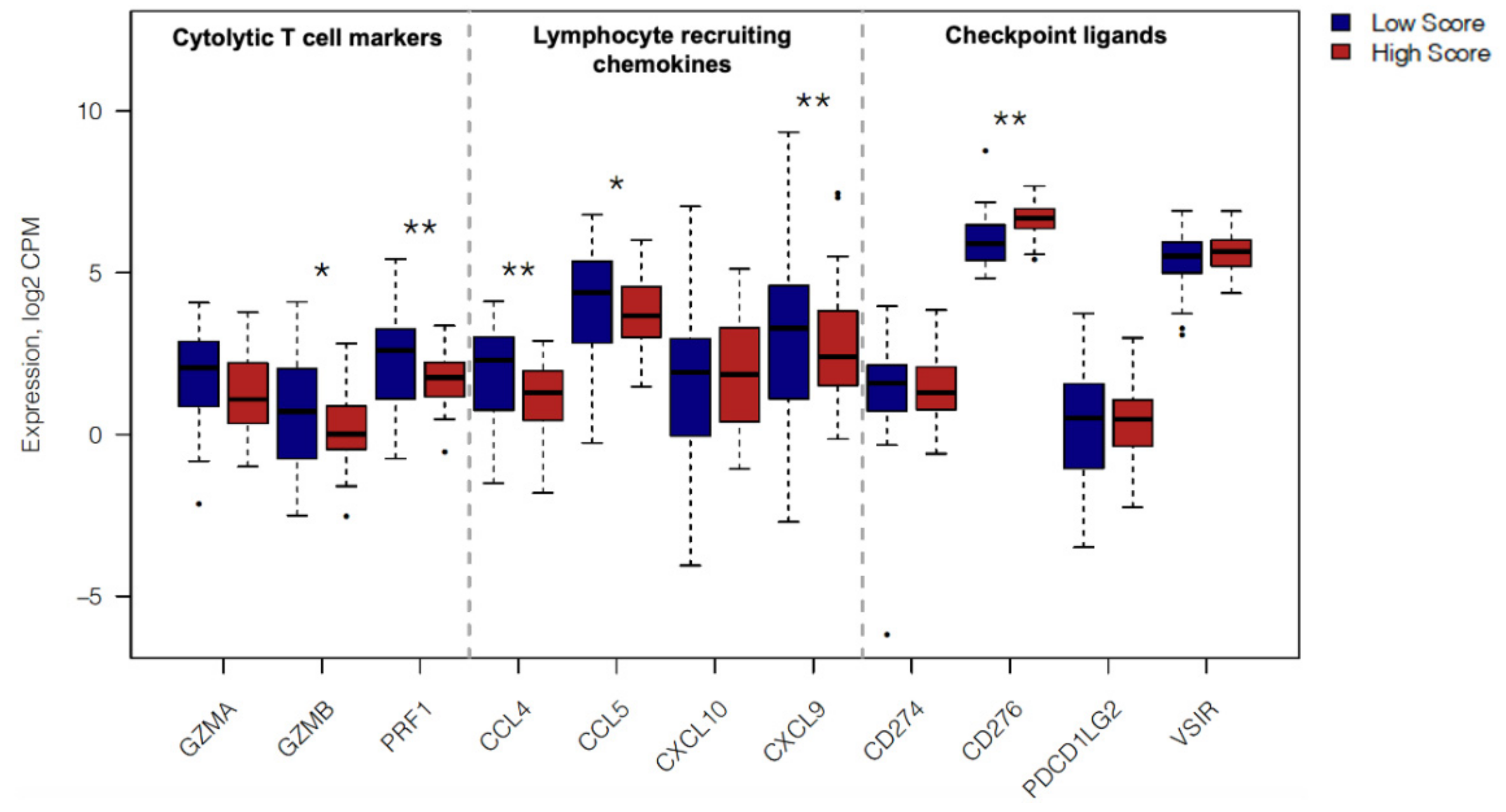
2.4. In Silico Immune Profiling of High Score versus Low Score Budding Cases
3. Discussion
4. Materials and Methods
4.1. Patient Cohort
4.2. Next-Generation Tissue Microarray (ngTMA) Construction
4.3. Assessment of Tumor Budding and TLT Status
4.4. Immunohistochemistry
4.5. Immune Cell Scoring and Normalization
4.6. TCGA Data Acquisition and Analysis
4.7. Cell-Type Deconvolution
4.8. Survival Analysis
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kamisawa, T.; Wood, L.D.; Itoi, T.; Takaori, K. Pancreatic cancer. Lancet 2016, 388, 73–85. [Google Scholar] [CrossRef]
- Strobel, O.; Neoptolemos, J.; Jäger, D.; Büchler, M.W. Optimizing the outcomes of pancreatic cancer surgery. Nat. Rev. Clin. Oncol. 2019, 16, 11–26. [Google Scholar] [CrossRef]
- Collisson, E.A.; Sadanandam, A.; Olson, P.; Gibb, W.J.; Truitt, M.; Gu, S.; Cooc, J.; Weinkle, J.; Kim, G.E.; Jakkula, L.; et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 2011, 17. [Google Scholar] [CrossRef]
- Raut, C.P.; Tseng, J.F.; Sun, C.C.; Wang, H.; Wolff, R.A.; Crane, C.H.; Hwang, R.; Vauthey, J.-N.; Abdalla, E.K.; Lee, J.E.; et al. Impact of Resection Status on Pattern of Failure and Survival After Pancreaticoduodenectomy for Pancreatic Adenocarcinoma. Ann. Surg. 2007, 246, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Sierzega, M.; Popiela, T.; Kulig, J.; Nowak, K. The Ratio of Metastatic/Resected Lymph Nodes is an Independent Prognostic Factor in Patients with Node-positive Pancreatic Head Cancer. Pancreas 2006, 33, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Rau, B.M.; Moritz, K.; Schuschan, S.; Alsfasser, G.; Prall, F.; Klar, E. R1 resection in pancreatic cancer has significant impact on long-term outcome in standardized pathology modified for routine use. Surgery 2012, 152, S103–S111. [Google Scholar] [CrossRef]
- Karamitopoulou, E.; Zlobec, I.; Born, D.; Kondi-Pafiti, A.; Lykoudis, P.; Mellou, A.; Gennatas, K.; Gloor, B.; Lugli, A. Tumour budding is a strong and independent prognostic factor in pancreatic cancer. Eur. J. Cancer 2013, 49, 1032–1039. [Google Scholar] [CrossRef]
- O’Connor, K.; Li-Chang, H.H.; Kalloger, S.E.; Peixoto, R.D.; Webber, D.L.; Owen, D.A.; Driman, D.K.; Kirsch, R.; Serra, S.; Scudamore, C.H.; et al. Tumor Budding Is an Independent Adverse Prognostic Factor in Pancreatic Ductal Adenocarcinoma. Am. J. Surg. Pathol. 2015, 39, 472–478. [Google Scholar] [CrossRef] [PubMed]
- Lohneis, P.; Sinn, M.; Klein, F.; Bischoff, S.; Striefler, J.K.; Wislocka, L.; Sinn, B.V.; Pelzer, U.; Oettle, H.; Riess, H.; et al. Tumour buds determine prognosis in resected pancreatic ductal adenocarcinoma. Br. J. Cancer 2018, 118, 1485–1491. [Google Scholar] [CrossRef]
- Lugli, A.; Zlobec, I.; Berger, M.D.; Kirsch, R.; Nagtegaal, I.D. Tumour budding in solid cancers. Nat. Rev. Clin. Oncol. 2021, 18, 101–115. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Garner, H.; de Visser, K.E. Immune crosstalk in cancer progression and metastatic spread: A complex conversation. Nat. Rev. Immunol. 2020, 20, 483–497. [Google Scholar] [CrossRef] [PubMed]
- Wartenberg, M.; Cibin, S.; Zlobec, I.; Vassella, E.; Eppenberger-Castori, S.; Terracciano, L.; Eichmann, M.D.; Worni, M.; Gloor, B.; Perren, A.; et al. Integrated Genomic and Immunophenotypic Classification of Pancreatic Cancer Reveals Three Distinct Subtypes with Prognostic/Predictive Significance. Clin. Cancer Res. 2018, 24, 4444–4454. [Google Scholar] [CrossRef]
- Karamitopoulou, E.; Wartenberg, M.; Zlobec, I.; Cibin, S.; Worni, M.; Gloor, B.; Lugli, A. Tumour budding in pancreatic cancer revisited: Validation of the ITBCC scoring system. Histopathology 2018, 73, 137–146. [Google Scholar] [CrossRef]
- Lawlor, R.; Veronese, N.; Nottegar, A.; Malleo, G.; Smith, L.; Demurtas, J.; Cheng, L.; Wood, L.; Silvestris, N.; Salvia, R.; et al. Prognostic Role of High-Grade Tumor Budding in Pancreatic Ductal Adenocarcinoma: A Systematic Review and Meta-Analysis with a Focus on Epithelial to Mesenchymal Transition. Cancers 2019, 11, 113. [Google Scholar] [CrossRef] [PubMed]
- Teillaud, J.-L.; Dieu-Nosjean, M.-C. Tertiary Lymphoid Structures: An Anti-tumor School for Adaptive Immune Cells and an Antibody Factory to Fight Cancer? Front. Immunol. 2017, 8. [Google Scholar] [CrossRef]
- Hiraoka, N.; Ino, Y.; Yamazaki-Itoh, R.; Kanai, Y.; Kosuge, T.; Shimada, K. Intratumoral tertiary lymphoid organ is a favourable prognosticator in patients with pancreatic cancer. Br. J. Cancer 2015, 112, 1782–1790. [Google Scholar] [CrossRef]
- Shinto, E.; Yoshida, Y.; Kajiwara, Y.; Okamoto, K.; Mochizuki, S.; Yamadera, M.; Shiraishi, T.; Nagata, K.; Tsuda, H.; Hase, K.; et al. Clinical Significance of a Gene Signature Generated from Tumor Budding Grade in Colon Cancer. Ann. Surg. Oncol. 2020, 27, 4044–4054. [Google Scholar] [CrossRef]
- Foroutan, M.; Bhuva, D.D.; Lyu, R.; Horan, K.; Cursons, J.; Davis, M.J. Single sample scoring of molecular phenotypes. BMC Bioinform. 2018, 19, 404. [Google Scholar] [CrossRef] [PubMed]
- Racle, J.; de Jonge, K.; Baumgaertner, P.; Speiser, D.E.; Gfeller, D. Simultaneous enumeration of cancer and immune cell types from bulk tumor gene expression data. Elife 2017, 6. [Google Scholar] [CrossRef]
- Finotello, F.; Trajanoski, Z. Quantifying tumor-infiltrating immune cells from transcriptomics data. Cancer Immunol. Immunother. 2018, 67, 1031–1040. [Google Scholar] [CrossRef]
- Van der Woude, L.L.; Gorris, M.A.J.; Halilovic, A.; Figdor, C.G.; de Vries, I.J.M. Migrating into the Tumor: A Roadmap for T Cells. Trends Cancer 2017, 3, 797–808. [Google Scholar] [CrossRef] [PubMed]
- Trujillo, J.A.; Sweis, R.F.; Bao, R.; Luke, J.J. T cell–inflamed versus Non-T cell–inflamed tumors: A conceptual framework for cancer immunotherapy drug development and combination therapy selection. Cancer Immunol. Res. 2018, 6, 990–1000. [Google Scholar] [CrossRef]
- Marin-Acevedo, J.A.; Soyano, A.E.; Dholaria, B.; Knutson, K.L.; Lou, Y. Cancer immunotherapy beyond immune checkpoint inhibitors. J. Hematol. Oncol. 2018, 11, 8. [Google Scholar] [CrossRef]
- Neoptolemos, J.P.; Kleeff, J.; Michl, P.; Costello, E.; Greenhalf, W.; Palmer, D.H. Therapeutic developments in pancreatic cancer: Current and future perspectives. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 333–348. [Google Scholar] [CrossRef]
- Balachandran, V.P.; Beatty, G.L.; Dougan, S.K. Broadening the Impact of Immunotherapy to Pancreatic Cancer: Challenges and Opportunities. Gastroenterology 2019, 156, 2056–2072. [Google Scholar] [CrossRef] [PubMed]
- Grigore, A.; Jolly, M.; Jia, D.; Farach-Carson, M.; Levine, H. Tumor Budding: The Name is EMT. Partial EMT. J. Clin. Med. 2016, 5, 51. [Google Scholar] [CrossRef]
- Galván, J.A.; Zlobec, I.; Wartenberg, M.; Lugli, A.; Gloor, B.; Perren, A.; Karamitopoulou, E. Expression of E-cadherin repressors SNAIL, ZEB1 and ZEB2 by tumour and stromal cells influences tumour-budding phenotype and suggests heterogeneity of stromal cells in pancreatic cancer. Br. J. Cancer 2015, 112, 1944–1950. [Google Scholar] [CrossRef]
- Pastushenko, I.; Blanpain, C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 2019, 29, 212–226. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.-C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.-G.; Zhu, L.-C.; Wang, Y.-J.; Li, Y.-Y.; Wang, D. Current Advance of Therapeutic Agents in Clinical Trials Potentially Targeting Tumor Plasticity. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef]
- Ramesh, V.; Brabletz, T.; Ceppi, P. Targeting EMT in Cancer with Repurposed Metabolic Inhibitors. Trends Cancer 2020, 6, 942–950. [Google Scholar] [CrossRef] [PubMed]
- Datar, I.; Schalper, K.A. Epithelial–Mesenchymal Transition and Immune Evasion during Lung Cancer Progression: The Chicken or the Egg? Clin. Cancer Res. 2016, 22, 3422–3424. [Google Scholar] [CrossRef] [PubMed]
- Terry, S.; Savagner, P.; Ortiz-Cuaran, S.; Mahjoubi, L.; Saintigny, P.; Thiery, J.-P.; Chouaib, S. New insights into the role of EMT in tumor immune escape. Mol. Oncol. 2017, 11, 824–846. [Google Scholar] [CrossRef]
- Teng, M.W.L.; Galon, J.; Fridman, W.-H.; Smyth, M.J. From mice to humans: Developments in cancer immunoediting. J. Clin. Invest. 2015, 125, 3338–3346. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Erazo, L.; Rhodes, J.L.; Marion, V.C.; Kemp, R.A. Tertiary lymphoid structures in cancer—Considerations for patient prognosis. Cell. Mol. Immunol. 2020, 17, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Fujiyoshi, K.; Väyrynen, J.P.; Borowsky, J.; Papke, D.J.; Arima, K.; Haruki, K.; Kishikawa, J.; Akimoto, N.; Ugai, T.; Lau, M.C.; et al. Tumour budding, poorly differentiated clusters, and T-cell response in colorectal cancer. EBioMedicine 2020, 57, 102860. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Delin, Z.; Kefei, Y.; Hong, W.; Jiwei, H.; Yange, Z. A classification based on tumor budding and immune score for patients with hepatocellular carcinoma. Oncoimmunology 2020, 9, 1672495. [Google Scholar] [CrossRef]
- O’Donnell, J.S.; Teng, M.W.L.; Smyth, M.J. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 151–167. [Google Scholar] [CrossRef]
- Akalay, I.; Janji, B.; Hasmim, M.; Noman, M.Z.; André, F.; De Cremoux, P.; Bertheau, P.; Badoual, C.; Vielh, P.; Larsen, A.K.; et al. Epithelial-to-Mesenchymal Transition and Autophagy Induction in Breast Carcinoma Promote Escape from T-cell–Mediated Lysis. Cancer Res. 2013, 73, 2418–2427. [Google Scholar] [CrossRef]
- Terry, S.; Buart, S.; Tan, T.Z.; Gros, G.; Noman, M.Z.; Lorens, J.B.; Mami-Chouaib, F.; Thiery, J.P.; Chouaib, S. Acquisition of tumor cell phenotypic diversity along the EMT spectrum under hypoxic pressure: Consequences on susceptibility to cell-mediated cytotoxicity. Oncoimmunology 2017, 6, e1271858. [Google Scholar] [CrossRef] [PubMed]
- Mak, M.P.; Tong, P.; Diao, L.; Cardnell, R.J.; Gibbons, D.L.; William, W.N.; Skoulidis, F.; Parra, E.R.; Rodriguez-Canales, J.; Wistuba, I.I.; et al. A Patient-Derived, Pan-Cancer EMT Signature Identifies Global Molecular Alterations and Immune Target Enrichment Following Epithelial-to-Mesenchymal Transition. Clin. Cancer Res. 2016, 22, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Harlin, H.; Meng, Y.; Peterson, A.C.; Zha, Y.; Tretiakova, M.; Slingluff, C.; McKee, M.; Gajewski, T.F. Chemokine Expression in Melanoma Metastases Associated with CD8 + T-Cell Recruitment. Cancer Res. 2009, 69, 3077–3085. [Google Scholar] [CrossRef]
- Kontos, F.; Michelakos, T.; Kurokawa, T.; Sadagopan, A.; Schwab, J.H.; Ferrone, C.R.; Ferrone, S. B7-H3: An attractive target for antibody-based immunotherapy. Clin. Cancer Res. 2020, 27, clincanres.2584.2020. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Xu, L.; Yi, M.; Yu, S.; Wu, K.; Luo, S. Novel immune checkpoint targets: Moving beyond PD-1 and CTLA-4. Mol. Cancer 2019, 18, 155. [Google Scholar] [CrossRef] [PubMed]
- Wylie, B.; Macri, C.; Mintern, J.; Waithman, J. Dendritic Cells and Cancer: From Biology to Therapeutic Intervention. Cancers 2019, 11, 521. [Google Scholar] [CrossRef]
- DeNardo, D.G.; Ruffell, B. Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 2019, 19, 369–382. [Google Scholar] [CrossRef]
- Wu, K.; Lin, K.; Li, X.; Yuan, X.; Xu, P.; Ni, P.; Xu, D. Redefining Tumor-Associated Macrophage Subpopulations and Functions in the Tumor Microenvironment. Front. Immunol. 2020, 11, 1731. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Killingsworth, M.C.; Myasoedova, V.A.; Orekhov, A.N.; Bobryshev, Y.V. CD68/macrosialin: Not just a histochemical marker. Lab. Investig. 2017, 97, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-Y.; Xu, J.-Y.; Shi, X.-Y.; Huang, W.; Ruan, T.-Y.; Xie, P.; Ding, J.-L. M2-polarized tumor-associated macrophages promoted epithelial–mesenchymal transition in pancreatic cancer cells, partially through TLR4/IL-10 signaling pathway. Lab. Investig. 2013, 93, 844–854. [Google Scholar] [CrossRef]
- Zhu, L.; Fu, X.; Chen, X.; Han, X.; Dong, P. M2 macrophages induce EMT through the TGF-β/Smad2 signaling pathway. Cell Biol. Int. 2017, 41, 960–968. [Google Scholar] [CrossRef]
- Helm, O.; Held-Feindt, J.; Grage-Griebenow, E.; Reiling, N.; Ungefroren, H.; Vogel, I.; Krüger, U.; Becker, T.; Ebsen, M.; Röcken, C.; et al. Tumor-associated macrophages exhibit pro- and anti-inflammatory properties by which they impact on pancreatic tumorigenesis. Int. J. Cancer 2014, 135, 843–861. [Google Scholar] [CrossRef]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Garrido-Martin, E.M.; Mellows, T.W.P.; Clarke, J.; Ganesan, A.-P.; Wood, O.; Cazaly, A.; Seumois, G.; Chee, S.J.; Alzetani, A.; King, E.V.; et al. M1 hot tumor-associated macrophages boost tissue-resident memory T cells infiltration and survival in human lung cancer. J. Immunother. Cancer 2020, 8, e000778. [Google Scholar] [CrossRef]
- Guedj, K.; Khallou-Laschet, J.; Clement, M.; Morvan, M.; Gaston, A.-T.; Fornasa, G.; Dai, J.; Gervais-Taurel, M.; Eberl, G.; Michel, J.-B.; et al. M1 macrophages act as LTβR-independent lymphoid tissue inducer cells during atherosclerosis-related lymphoid neogenesis. Cardiovasc. Res. 2014, 101, 434–443. [Google Scholar] [CrossRef]
- Panni, R.Z.; Herndon, J.M.; Zuo, C.; Hegde, S.; Hogg, G.D.; Knolhoff, B.L.; Breden, M.A.; Li, X.; Krisnawan, V.E.; Khan, S.Q.; et al. Agonism of CD11b reprograms innate immunity to sensitize pancreatic cancer to immunotherapies. Sci. Transl. Med. 2019, 11, eaau9240. [Google Scholar] [CrossRef] [PubMed]
- Roberts, E.W.; Broz, M.L.; Binnewies, M.; Headley, M.B.; Nelson, A.E.; Wolf, D.M.; Kaisho, T.; Bogunovic, D.; Bhardwaj, N.; Krummel, M.F. Critical Role for CD103+ /CD141+ Dendritic Cells Bearing CCR7 for Tumor Antigen Trafficking and Priming of T Cell Immunity in Melanoma. Cancer Cell 2016, 30, 324–336. [Google Scholar] [CrossRef]
- Spranger, S.; Dai, D.; Horton, B.; Gajewski, T.F. Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell 2017, 31, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Argentiero, A.; De Summa, S.; Di Fonte, R.; Iacobazzi, R.M.; Porcelli, L.; Da Vià, M.; Brunetti, O.; Azzariti, A.; Silvestris, N.; Solimando, A.G. Gene Expression Comparison between the Lymph Node-Positive and -Negative Reveals a Peculiar Immune Microenvironment Signature and a Theranostic Role for WNT Targeting in Pancreatic Ductal Adenocarcinoma: A Pilot Study. Cancers 2019, 11, 942. [Google Scholar] [CrossRef] [PubMed]
- Mhaidly, R.; Mechta-Grigoriou, F. Fibroblast heterogeneity in tumor micro-environment: Role in immunosuppression and new therapies. Semin. Immunol. 2020, 48, 101417. [Google Scholar] [CrossRef] [PubMed]
- De Smedt, L.; Palmans, S.; Andel, D.; Govaere, O.; Boeckx, B.; Smeets, D.; Galle, E.; Wouters, J.; Barras, D.; Suffiotti, M.; et al. Expression profiling of budding cells in colorectal cancer reveals an EMT-like phenotype and molecular subtype switching. Br. J. Cancer 2017, 116, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Levitin, H.M.; Yuan, J.; Sims, P.A. Single-Cell Transcriptomic Analysis of Tumor Heterogeneity. Trends Cancer 2018, 4, 264–268. [Google Scholar] [CrossRef]
- Leipold, M.D.; Newell, E.W.; Maecker, H.T. Multiparameter Phenotyping of Human PBMCs Using Mass Cytometry. In Methods in Molecular Biology; Humana Press: New York, NY, USA, 2015; pp. 81–95. [Google Scholar]
- Zlobec, I.; Suter, G.; Perren, A.; Lugli, A. A Next-generation Tissue Microarray (ngTMA) Protocol for Biomarker Studies. J. Vis. Exp. 2014, e51893. [Google Scholar] [CrossRef]
- Lugli, A.; Kirsch, R.; Ajioka, Y.; Bosman, F.; Cathomas, G.; Dawson, H.; El Zimaity, H.; Fléjou, J.-F.; Hansen, T.P.; Hartmann, A.; et al. Recommendations for reporting tumor budding in colorectal cancer based on the International Tumor Budding Consensus Conference (ITBCC) 2016. Mod. Pathol. 2017, 30, 1299–1311. [Google Scholar] [CrossRef] [PubMed]
- Salgado, R.; Denkert, C.; Demaria, S.; Sirtaine, N.; Klauschen, F.; Pruneri, G.; Wienert, S.; Van den Eynden, G.; Baehner, F.L.; Penault-Llorca, F.; et al. The evaluation of tumor-infiltrating lymphocytes (TILs) in breast cancer: Recommendations by an International TILs Working Group 2014. Ann. Oncol. 2015, 26, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Sturm, G.; Finotello, F.; List, M. Immunedeconv: An R Package for Unified Access to Computational Methods for Estimating Immune Cell Fractions from Bulk RNA-Sequencing Data. In Methods in Molecular Biology; Humana Press: New York, NY, USA, 2020; pp. 223–232. [Google Scholar]
- Kassambara, A.; Kosinski, M.; Biecek, P. Survminer: Drawing Survival Curves Using’ggplot2’. R Packag Version 0.3. 2017. Available online: https://cran.r-project.org/web/packages/survminer/index.html.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | High Grade, n = 54 | Low Grade, n = 57 | p-Value |
|---|---|---|---|
| OS (months) | 9 (6–13) | 25 (15–79) | <0.001 |
| PFS (months) | 5 (4–7) | 18 (8–78) | <0.001 |
| Sex | 0.5 | ||
| F | 25 (46%) | 31 (54%) | |
| M | 29 (54%) | 26 (46%) | |
| Age | 64 (56–71) | 67 (61–75) | 0.053 |
| Size (mm) | 30 (25–40) | 30 (25–35) | 0.6 |
| CA19-9 (U/mL) | 687 (220–2123) | 159 (95.5–597) | 0.0005 |
| Anatomic Site | 0.33 | ||
| Head | 42 (78%) | 49 (86%) | |
| Others (Tail/Body) | 12 (22%) | 8 (14%) | |
| Tertiary lymphoid tissue (TLT) | <0.0001 | ||
| Present | 2 (4%) | 24 (42%) | |
| Absent | 52 (96%) | 33 (58%) | |
| Grade | 0.013 | ||
| 1 | 6 (11%) | 19 (33%) | |
| 2 | 23 (43%) | 22 (39%) | |
| 3 | 25 (46%) | 16 (28%) | |
| UICC Stage 8th Edition | 0.065 | ||
| IA | 2 (3.7%) | 3 (5.3%) | |
| IB | 4 (7.4%) | 13 (23%) | |
| IIA | 2 (3.7%) | 3 (5.3%) | |
| IIB | 27 (50%) | 30 (53%) | |
| III | 16 (30%) | 7 (12%) | |
| IV | 3 (5.6%) | 1 (1.8%) |
| Parameters | Hazard Ratio | Lower 0.95 | Upper 0.95 | p-Value |
|---|---|---|---|---|
| CD3 S | 1.000725 | 0.999777 | 1.001674 | 0.133869 |
| CD4 S | 0.998226 | 0.996616 | 0.999839 | 0.031084 |
| CD8 S | 0.998329 | 0.997033 | 0.999626 | 0.011575 |
| CD20 S | 1.00113 | 0.999907 | 1.002355 | 0.070211 |
| CD68 S | 1.000301 | 0.999701 | 1.0009 | 0.325558 |
| CD163 S | 1.000102 | 0.999669 | 1.000535 | 0.644591 |
| iNOS S | 1.000123 | 0.992674 | 1.007629 | 0.974216 |
| DC-LAMP S | 0.998948 | 0.994303 | 1.003614 | 0.657925 |
| FOXP3 S | 1.004488 | 1.000889 | 1.008101 | 0.01449 |
| CD3 IT | 1.01572 | 0.988186 | 1.04402 | 0.265969 |
| CD4 IT | 0.922045 | 0.846725 | 1.004065 | 0.061949 |
| CD8 IT | 1.023522 | 1.001187 | 1.046354 | 0.038888 |
| CD20 IT | 1.050171 | 0.64113 | 1.720179 | 0.84584 |
| CD68 IT | 0.985977 | 0.957906 | 1.01487 | 0.337894 |
| CD163 IT | 1.01417 | 1.002567 | 1.025907 | 0.016547 |
| iNOS IT | 0.997833 | 0.972125 | 1.02422 | 0.870592 |
| DC-LAMP IT | 0.997575 | 0.936183 | 1.062992 | 0.940266 |
| FOXP3 IT | 0.991568 | 0.953417 | 1.031245 | 0.672285 |
| Sex | 0.727182 | 0.435132 | 1.215248 | 0.224019 |
| Age | 0.982572 | 0.959449 | 1.006252 | 0.147893 |
| Size | 1.009228 | 0.98936 | 1.029495 | 0.365219 |
| UICCIB | 1.506231 | 0.330167 | 6.871471 | 0.596841 |
| UICCIIA | 2.930027 | 0.419885 | 20.4462 | 0.278136 |
| UICCIIB | 2.71031 | 0.656752 | 11.18501 | 0.168012 |
| UICCIII | 3.307966 | 0.700725 | 15.61617 | 0.130831 |
| UICCIV | 82.59885 | 10.07591 | 677.117 | 3.92 × 10−5 |
| TLT | 0.541932 | 0.286001 | 1.026886 | 0.060298 |
| Anatomical-HEAD | 0.749378 | 0.142995 | 3.927188 | 0.732818 |
| Anatomical-TAIL | 1.001193 | 0.178253 | 5.623408 | 0.99892 |
| Budding grade | 0.757975 | 0.513434 | 1.118988 | 0.163231 |
| Budding number | 1.057654 | 1.033902 | 1.081952 | 1.32 × 10−6 |
| Model Performance | p value | |||
| logtest | 7.47 × 10−12 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sadozai, H.; Acharjee, A.; Gruber, T.; Gloor, B.; Karamitopoulou, E. Pancreatic Cancers with High Grade Tumor Budding Exhibit Hallmarks of Diminished Anti-Tumor Immunity. Cancers 2021, 13, 1090. https://doi.org/10.3390/cancers13051090
Sadozai H, Acharjee A, Gruber T, Gloor B, Karamitopoulou E. Pancreatic Cancers with High Grade Tumor Budding Exhibit Hallmarks of Diminished Anti-Tumor Immunity. Cancers. 2021; 13(5):1090. https://doi.org/10.3390/cancers13051090
Chicago/Turabian StyleSadozai, Hassan, Animesh Acharjee, Thomas Gruber, Beat Gloor, and Eva Karamitopoulou. 2021. "Pancreatic Cancers with High Grade Tumor Budding Exhibit Hallmarks of Diminished Anti-Tumor Immunity" Cancers 13, no. 5: 1090. https://doi.org/10.3390/cancers13051090
APA StyleSadozai, H., Acharjee, A., Gruber, T., Gloor, B., & Karamitopoulou, E. (2021). Pancreatic Cancers with High Grade Tumor Budding Exhibit Hallmarks of Diminished Anti-Tumor Immunity. Cancers, 13(5), 1090. https://doi.org/10.3390/cancers13051090