
Homologous Recombination Deficiency Testing for BRCA-Like Tumors: The Road to Clinical Validation



Abstract
Simple Summary
Abstract
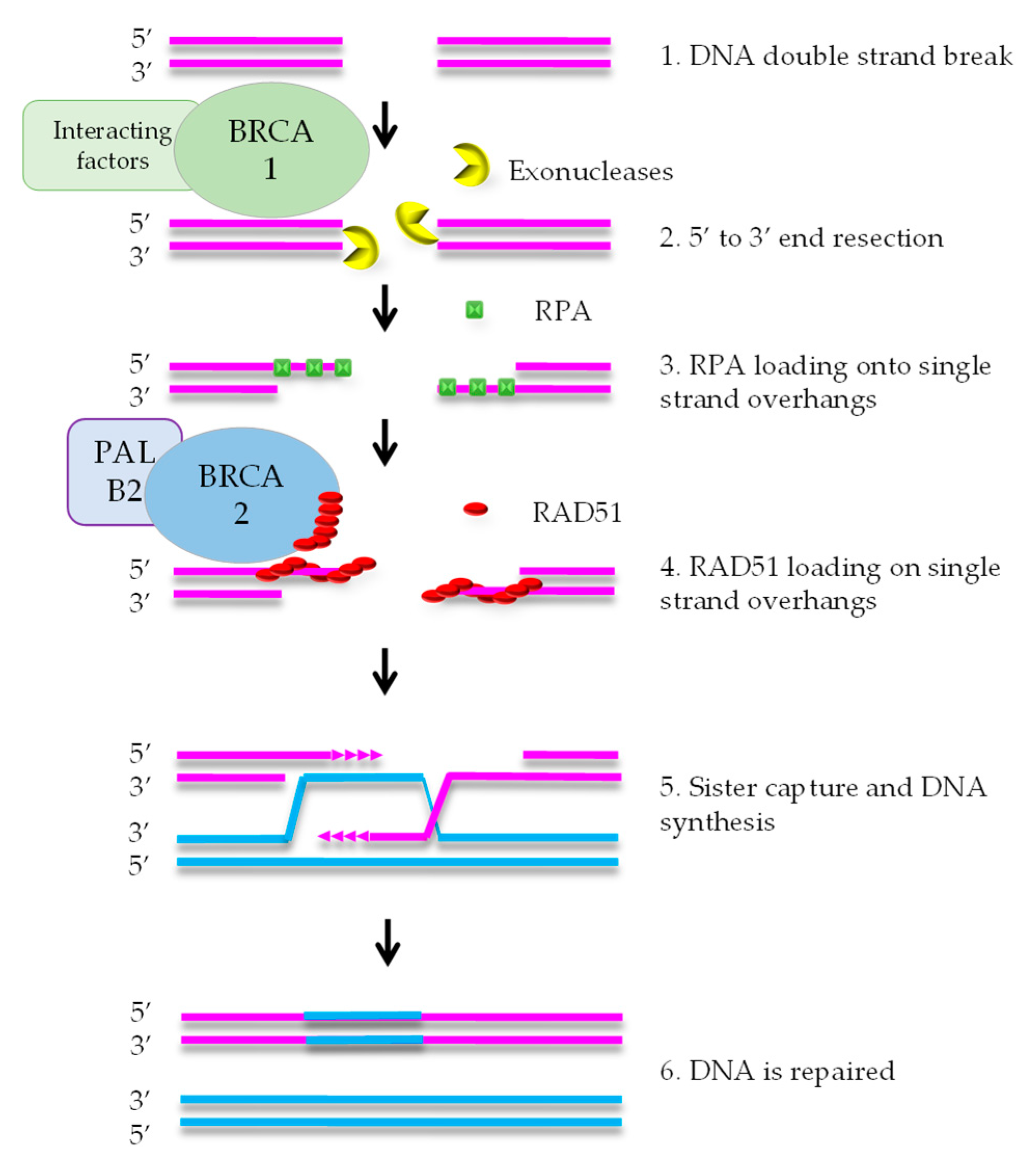
1. Homologous Recombination Deficiency and Cancer
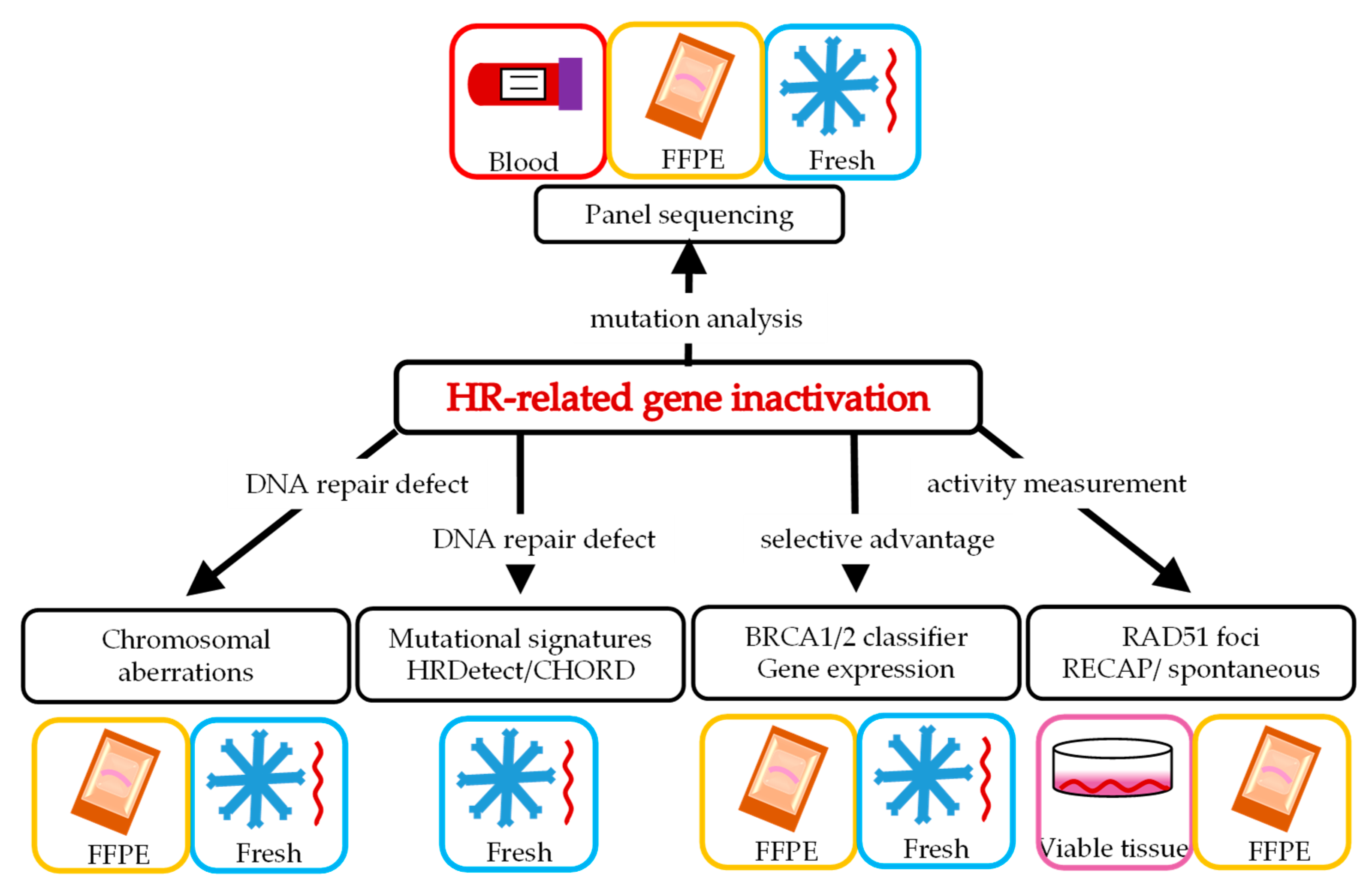
2. Various Ways to Define HRD
3. Panel Sequencing of Potential Causal Genetic Changes in HR Genes
4. Chromosomal Aberrations as a Consequence of HRD
5. Genomic Scars Resulting from HRD
6. The BRCA-Like Classifier as Predictor for HRD
7. Gene Expression Profiles as Predictor for HRD
8. Functional Tests
8.1. HR Pathway Proteins after In Vivo Treatment
8.2. RAD51 Foci after Ex Vivo Treatment
8.3. RAD51 Foci in Untreated Tumors
9. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Curtin, N.J. DNA repair dysregulation from cancer driver to therapeutic target. Nat. Rev. Cancer 2012, 12, 801–817. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. The DNA damage response and cancer therapy. Nature 2012, 481, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Parmigiani, G. Meta-analysis of brca1 and brca2 penetrance. J. Clin. Oncol. 2007, 25, 1329–1333. [Google Scholar] [CrossRef] [PubMed]
- Mylavarapu, S.; Das, A.; Roy, M. Role of brca mutations in the modulation of response to platinum therapy. Front. Oncol. 2018, 8, 16. [Google Scholar] [CrossRef]
- Keung, M.Y.T.; Wu, Y.; Vadgama, J.V. Parp inhibitors as a therapeutic agent for homologous recombination deficiency in breast cancers. J. Clin. Med. 2019, 8, 435. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, J.F.; Kanaar, R. Targeting homologous recombination-mediated DNA repair in cancer. Expert Opin. Ther. Targets 2014, 18, 427–458. [Google Scholar] [CrossRef]
- Wyman, C.; Kanaar, R. DNA double-strand break repair: All’s well that ends well. Annu. Rev. Genet. 2006, 40, 363–383. [Google Scholar] [CrossRef]
- Wright, W.D.; Shah, S.S.; Heyer, W.D. Homologous recombination and the repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10524–10535. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Southey, M.C.; Teo, Z.L.; Winship, I. Palb2 and breast cancer: Ready for clinical translation! Appl Clin Genet. 2013, 6, 43–52. [Google Scholar] [CrossRef][Green Version]
- Riaz, N.; Blecua, P.; Lim, R.S.; Shen, R.; Higginson, D.S.; Weinhold, N.; Norton, L.; Weigelt, B.; Powell, S.N.; Reis-Filho, J.S. Pan-cancer analysis of bi-allelic alterations in homologous recombination DNA repair genes. Nat. Commun. 2017, 8, 857. [Google Scholar] [CrossRef] [PubMed]
- Mateo, J.; Carreira, S.; Sandhu, S.; Miranda, S.; Mossop, H.; Perez-Lopez, R.; Nava Rodrigues, D.; Robinson, D.; Omlin, A.; Tunariu, N.; et al. DNA-repair defects and olaparib in metastatic prostate cancer. N. Engl. J. Med. 2015, 373, 1697–1708. [Google Scholar] [CrossRef] [PubMed]
- Mateo, J.; Porta, N.; Bianchini, D.; McGovern, U.; Elliott, T.; Jones, R.; Syndikus, I.; Ralph, C.; Jain, S.; Varughese, M.; et al. Olaparib in patients with metastatic castration-resistant prostate cancer with DNA repair gene aberrations (toparp-b): A multicentre, open-label, randomised, phase 2 trial. Lancet Oncol. 2020, 21, 162–174. [Google Scholar] [CrossRef]
- de Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for metastatic castration-resistant prostate cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Lin, Y.; Sun, X.J.; Wang, B.Y.; Wang, Z.H.; Luo, J.F.; Wang, L.P.; Zhang, S.; Cao, J.; Tao, Z.H.; et al. Biomarker assessment of the cbcsg006 trial: A randomized phase iii trial of cisplatin plus gemcitabine compared with paclitaxel plus gemcitabine as first-line therapy for patients with metastatic triple-negative breast cancer. Ann. Oncol. 2018, 29, 1741–1747. [Google Scholar] [CrossRef]
- Tung, N.M.; Robson, M.E.; Ventz, S.; Santa-Maria, C.A.; Nanda, R.; Marcom, P.K.; Shah, P.D.; Ballinger, T.J.; Yang, E.S.; Vinayak, S.; et al. Tbcrc 048: Phase ii study of olaparib for metastatic breast cancer and mutations in homologous recombination-related genes. J. Clin. Oncol. 2020, 38, 4274–4282. [Google Scholar] [CrossRef]
- Eikesdal, H.P.; Yndestad, S.; Elzawahry, A.; Llop-Guevara, A.; Gilje, B.; Blix, E.S.; Espelid, H.; Lundgren, S.; Geisler, J.; Vagstad, G.; et al. Olaparib monotherapy as primary treatment in unselected triple negative breast cancer. Ann. Oncol. 2021, 32, 240–249. [Google Scholar] [CrossRef]
- Hodgson, D.R.; Dougherty, B.A.; Lai, Z.; Fielding, A.; Grinsted, L.; Spencer, S.; O’Connor, M.J.; Ho, T.W.; Robertson, J.D.; Lanchbury, J.S.; et al. Candidate biomarkers of parp inhibitor sensitivity in ovarian cancer beyond the brca genes. Br. J. Cancer 2018, 119, 1401–1409. [Google Scholar] [CrossRef]
- Pennington, K.P.; Walsh, T.; Harrell, M.I.; Lee, M.K.; Pennil, C.C.; Rendi, M.H.; Thornton, A.; Norquist, B.M.; Casadei, S.; Nord, A.S.; et al. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin. Cancer Res. 2014, 20, 764–775. [Google Scholar] [CrossRef]
- Swisher, E.M.; Lin, K.K.; Oza, A.M.; Scott, C.L.; Giordano, H.; Sun, J.; Konecny, G.E.; Coleman, R.L.; Tinker, A.V.; O’Malley, D.M.; et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ariel2 part 1): An international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 75–87. [Google Scholar] [CrossRef]
- Coleman, R.L.; Oza, A.M.; Lorusso, D.; Aghajanian, C.; Oaknin, A.; Dean, A.; Colombo, N.; Weberpals, J.I.; Clamp, A.; Scambia, G.; et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ariel3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 1949–1961. [Google Scholar] [CrossRef]
- Telli, M.L.; Jensen, K.C.; Vinayak, S.; Kurian, A.W.; Lipson, J.A.; Flaherty, P.J.; Timms, K.; Abkevich, V.; Schackmann, E.A.; Wapnir, I.L.; et al. Phase ii study of gemcitabine, carboplatin, and iniparib as neoadjuvant therapy for triple-negative and brca1/2 mutation-associated breast cancer with assessment of a tumor-based measure of genomic instability: Precog 0105. J. Clin. Oncol. 2015, 33, 1895–1901. [Google Scholar] [CrossRef]
- Coleman, R.L.; Fleming, G.F.; Brady, M.F.; Swisher, E.M.; Steffensen, K.D.; Friedlander, M.; Okamoto, A.; Moore, K.N.; Efrat Ben-Baruch, N.; Werner, T.L.; et al. Veliparib with first-line chemotherapy and as maintenance therapy in ovarian cancer. N. Engl. J. Med. 2019, 381, 2403–2415. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Martin, A.; Pothuri, B.; Vergote, I.; DePont Christensen, R.; Graybill, W.; Mirza, M.R.; McCormick, C.; Lorusso, D.; Hoskins, P.; Freyer, G.; et al. Niraparib in patients with newly diagnosed advanced ovarian cancer. N. Engl. J. Med. 2019, 381, 2391–2402. [Google Scholar] [CrossRef]
- Stronach, E.A.; Paul, J.; Timms, K.M.; Hughes, E.; Brown, K.; Neff, C.; Perry, M.; Gutin, A.; El-Bahrawy, M.; Steel, J.H.; et al. Biomarker assessment of hr deficiency, tumor brca1/2 mutations, and ccne1 copy number in ovarian cancer: Associations with clinical outcome following platinum monotherapy. Mol. Cancer Res. 2018, 16, 1103–1111. [Google Scholar] [CrossRef] [PubMed]
- Ray-Coquard, I.; Pautier, P.; Pignata, S.; Perol, D.; Gonzalez-Martin, A.; Berger, R.; Fujiwara, K.; Vergote, I.; Colombo, N.; Maenpaa, J.; et al. Olaparib plus bevacizumab as first-line maintenance in ovarian cancer. N. Engl. J. Med. 2019, 381, 2416–2428. [Google Scholar] [CrossRef] [PubMed]
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. N. Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.N.; Secord, A.A.; Geller, M.A.; Miller, D.S.; Cloven, N.; Fleming, G.F.; Wahner Hendrickson, A.E.; Azodi, M.; DiSilvestro, P.; Oza, A.M.; et al. Niraparib monotherapy for late-line treatment of ovarian cancer (quadra): A multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol. 2019, 20, 636–648. [Google Scholar] [CrossRef]
- Mayer, E.L.; Abramson, V.; Jankowitz, R.; Falkson, C.; Marcom, P.K.; Traina, T.; Carey, L.; Rimawi, M.; Specht, J.; Miller, K.; et al. Tbcrc 030: A phase ii study of preoperative cisplatin versus paclitaxel in triple-negative breast cancer: Evaluating the homologous recombination deficiency (hrd) biomarker. Ann. Oncol. 2020, 31, 1518–1525. [Google Scholar] [CrossRef]
- Loibl, S.; Weber, K.E.; Timms, K.M.; Elkin, E.P.; Hahnen, E.; Fasching, P.A.; Lederer, B.; Denkert, C.; Schneeweiss, A.; Braun, S.; et al. Survival analysis of carboplatin added to an anthracycline/taxane-based neoadjuvant chemotherapy and hrd score as predictor of response-final results from geparsixto. Ann. Oncol. 2018, 29, 2341–2347. [Google Scholar] [CrossRef]
- Tutt, A.; Tovey, H.; Cheang, M.C.U.; Kernaghan, S.; Kilburn, L.; Gazinska, P.; Owen, J.; Abraham, J.; Barrett, S.; Barrett-Lee, P.; et al. Carboplatin in brca1/2-mutated and triple-negative breast cancer brcaness subgroups: The tnt trial. Nat. Med. 2018, 24, 628–637. [Google Scholar] [CrossRef] [PubMed]
- Isakoff, S.J.; Mayer, E.L.; He, L.; Traina, T.A.; Carey, L.A.; Krag, K.J.; Rugo, H.S.; Liu, M.C.; Stearns, V.; Come, S.E.; et al. Tbcrc009: A multicenter phase ii clinical trial of platinum monotherapy with biomarker assessment in metastatic triple-negative breast cancer. J. Clin. Oncol. 2015, 33, 1902–1909. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Barlow, W.E.; Godwin, A.K.; Pathak, H.; Isakova, K.; Williams, D.; Timms, K.M.; Hartman, A.R.; Wenstrup, R.J.; Linden, H.M.; et al. Impact of homologous recombination deficiency biomarkers on outcomes in patients with triple-negative breast cancer treated with adjuvant doxorubicin and cyclophosphamide (swog s9313). Ann. Oncol. 2018, 29, 654–660. [Google Scholar] [CrossRef]
- Abkevich, V.; Timms, K.M.; Hennessy, B.T.; Potter, J.; Carey, M.S.; Meyer, L.A.; Smith-McCune, K.; Broaddus, R.; Lu, K.H.; Chen, J.; et al. Patterns of genomic loss of heterozygosity predict homologous recombination repair defects in epithelial ovarian cancer. Br. J. Cancer 2012, 107, 1776–1782. [Google Scholar] [CrossRef]
- Popova, T.; Manie, E.; Rieunier, G.; Caux-Moncoutier, V.; Tirapo, C.; Dubois, T.; Delattre, O.; Sigal-Zafrani, B.; Bollet, M.; Longy, M.; et al. Ploidy and large-scale genomic instability consistently identify basal-like breast carcinomas with brca1/2 inactivation. Cancer Res. 2012, 72, 5454–5462. [Google Scholar] [CrossRef] [PubMed]
- Telli, M.L.; Timms, K.M.; Reid, J.; Hennessy, B.; Mills, G.B.; Jensen, K.C.; Szallasi, Z.; Barry, W.T.; Winer, E.P.; Tung, N.M.; et al. Homologous recombination deficiency (hrd) score predicts response to platinum-containing neoadjuvant chemotherapy in patients with triple-negative breast cancer. Clin. Cancer Res. 2016, 22, 3764–3773. [Google Scholar] [CrossRef]
- Watkins, J.A.; Irshad, S.; Grigoriadis, A.; Tutt, A.N. Genomic scars as biomarkers of homologous recombination deficiency and drug response in breast and ovarian cancers. Breast Cancer Res. 2014, 16, 211. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Hollstein, M.; Alexandrov, L.B.; Wild, C.P.; Ardin, M.; Zavadil, J. Base changes in tumour DNA have the power to reveal the causes and evolution of cancer. Oncogene 2017, 36, 158–167. [Google Scholar] [CrossRef]
- Polak, P.; Kim, J.; Braunstein, L.Z.; Karlic, R.; Haradhavala, N.J.; Tiao, G.; Rosebrock, D.; Livitz, D.; Kubler, K.; Mouw, K.W.; et al. A mutational signature reveals alterations underlying deficient homologous recombination repair in breast cancer. Nat. Genet. 2017, 49, 1476–1486. [Google Scholar] [CrossRef]
- Nik-Zainal, S.; Alexandrov, L.B.; Wedge, D.C.; Van Loo, P.; Greenman, C.D.; Raine, K.; Jones, D.; Hinton, J.; Marshall, J.; Stebbings, L.A.; et al. Mutational processes molding the genomes of 21 breast cancers. Cell 2012, 149, 979–993. [Google Scholar] [CrossRef]
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.B.; Martin, S.; Wedge, D.C.; et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2016, 534, 47–54. [Google Scholar] [CrossRef]
- Davies, H.; Glodzik, D.; Morganella, S.; Yates, L.R.; Staaf, J.; Zou, X.; Ramakrishna, M.; Martin, S.; Boyault, S.; Sieuwerts, A.M.; et al. Hrdetect is a predictor of brca1 and brca2 deficiency based on mutational signatures. Nat. Med. 2017, 23, 517–525. [Google Scholar] [CrossRef]
- Staaf, J.; Glodzik, D.; Bosch, A.; Vallon-Christersson, J.; Reutersward, C.; Hakkinen, J.; Degasperi, A.; Amarante, T.D.; Saal, L.H.; Hegardt, C.; et al. Whole-genome sequencing of triple-negative breast cancers in a population-based clinical study. Nat. Med. 2019, 25, 1526–1533. [Google Scholar] [CrossRef]
- Chopra, N.; Tovey, H.; Pearson, A.; Cutts, R.; Toms, C.; Proszek, P.; Hubank, M.; Dowsett, M.; Dodson, A.; Daley, F.; et al. Homologous recombination DNA repair deficiency and parp inhibition activity in primary triple negative breast cancer. Nat. Commun. 2020, 11, 2662. [Google Scholar] [CrossRef]
- Nguyen, L.; Martens, J.W.M.; Van Hoeck, A.; Cuppen, E. Pan-cancer landscape of homologous recombination deficiency. Nat. Commun. 2020, 11, 5584. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Holstege, H.; van der Gulden, H.; Treur-Mulder, M.; Zevenhoven, J.; Velds, A.; Kerkhoven, R.M.; van Vliet, M.H.; Wessels, L.F.; Peterse, J.L.; et al. Somatic loss of brca1 and p53 in mice induces mammary tumors with features of human brca1-mutated basal-like breast cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 12111–12116. [Google Scholar] [CrossRef]
- Joosse, S.A.; van Beers, E.H.; Tielen, I.H.; Horlings, H.; Peterse, J.L.; Hoogerbrugge, N.; Ligtenberg, M.J.; Wessels, L.F.; Axwijk, P.; Verhoef, S.; et al. Prediction of brca1-association in hereditary non-brca1/2 breast carcinomas with array-cgh. Breast Cancer Res. Treat. 2009, 116, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Vollebergh, M.A.; Lips, E.H.; Nederlof, P.M.; Wessels, L.F.A.; Schmidt, M.K.; van Beers, E.H.; Cornelissen, S.; Holtkamp, M.; Froklage, F.E.; de Vries, E.G.E.; et al. An acgh classifier derived from brca1-mutated breast cancer and benefit of high-dose platinum-based chemotherapy in her2-negative breast cancer patients. Ann. Oncol. 2011, 22, 1561–1570. [Google Scholar] [CrossRef] [PubMed]
- Vollebergh, M.A.; Lips, E.H.; Nederlof, P.M.; Wessels, L.F.; Wesseling, J.; Vd Vijver, M.J.; de Vries, E.G.; van Tinteren, H.; Jonkers, J.; Hauptmann, M.; et al. Genomic patterns resembling brca1- and brca2-mutated breast cancers predict benefit of intensified carboplatin-based chemotherapy. Breast Cancer Res. 2014, 16, R47. [Google Scholar] [CrossRef] [PubMed]
- Schouten, P.C.; Marme, F.; Aulmann, S.; Sinn, H.P.; van Essen, H.F.; Ylstra, B.; Hauptmann, M.; Schneeweiss, A.; Linn, S.C. Breast cancers with a brca1-like DNA copy number profile recur less often than expected after high-dose alkylating chemotherapy. Clin. Cancer Res. 2015, 21, 763–770. [Google Scholar] [CrossRef] [PubMed]
- van Rossum, A.G.J.; Schouten, P.C.; Weber, K.E.; Nekljudova, V.; Denkert, C.; Solbach, C.; Kohne, C.H.; Thomssen, C.; Forstbauer, H.; Hoffmann, G.; et al. Brca1-like profile is not significantly associated with survival benefit of non-myeloablative intensified chemotherapy in the gain randomized controlled trial. Breast Cancer Res. Treat. 2017, 166, 775–785. [Google Scholar] [CrossRef] [PubMed]
- Konstantinopoulos, P.A.; Spentzos, D.; Karlan, B.Y.; Taniguchi, T.; Fountzilas, E.; Francoeur, N.; Levine, D.A.; Cannistra, S.A. Gene expression profile of brcaness that correlates with responsiveness to chemotherapy and with outcome in patients with epithelial ovarian cancer. J. Clin. Oncol. 2010, 28, 3555–3561. [Google Scholar] [CrossRef]
- Severson, T.M.; Wolf, D.M.; Yau, C.; Peeters, J.; Wehkam, D.; Schouten, P.C.; Chin, S.F.; Majewski, I.J.; Michaut, M.; Bosma, A.; et al. The brca1ness signature is associated significantly with response to parp inhibitor treatment versus control in the i-spy 2 randomized neoadjuvant setting. Breast Cancer Res. 2017, 19, 99. [Google Scholar] [CrossRef] [PubMed]
- Meijer, T.G.; Naipal, K.A.; Jager, A.; van Gent, D.C. Ex vivo tumor culture systems for functional drug testing and therapy response prediction. Future Sci. OA 2017, 3, FSO190. [Google Scholar] [CrossRef]
- Scully, R.; Chen, J.; Ochs, R.L.; Keegan, K.; Hoekstra, M.; Feunteun, J.; Livingston, D.M. Dynamic changes of brca1 subnuclear location and phosphorylation state are initiated by DNA damage. Cell 1997, 90, 425–435. [Google Scholar] [CrossRef]
- Graeser, M.; McCarthy, A.; Lord, C.J.; Savage, K.; Hills, M.; Salter, J.; Orr, N.; Parton, M.; Smith, I.E.; Reis-Filho, J.S.; et al. A marker of homologous recombination predicts pathologic complete response to neoadjuvant chemotherapy in primary breast cancer. Clin. Cancer Res. 2010, 16, 6159–6168. [Google Scholar] [CrossRef]
- Asakawa, H.; Koizumi, H.; Koike, A.; Takahashi, M.; Wu, W.; Iwase, H.; Fukuda, M.; Ohta, T. Prediction of breast cancer sensitivity to neoadjuvant chemotherapy based on status of DNA damage repair proteins. Breast Cancer Res. 2010, 12, R17. [Google Scholar] [CrossRef]
- Willers, H.; Taghian, A.G.; Luo, C.M.; Treszezamsky, A.; Sgroi, D.C.; Powell, S.N. Utility of DNA repair protein foci for the detection of putative brca1 pathway defects in breast cancer biopsies. Mol. Cancer Res. 2009, 7, 1304–1309. [Google Scholar] [CrossRef]
- Mutter, R.W.; Riaz, N.; Ng, C.K.; Delsite, R.; Piscuoglio, S.; Edelweiss, M.; Martelotto, L.G.; Sakr, R.A.; King, T.A.; Giri, D.D.; et al. Bi-allelic alterations in DNA repair genes underpin homologous recombination DNA repair defects in breast cancer. J. Pathol. 2017, 242, 165–177. [Google Scholar] [CrossRef]
- McCabe, N.; Turner, N.C.; Lord, C.J.; Kluzek, K.; Bialkowska, A.; Swift, S.; Giavara, S.; O’Connor, M.J.; Tutt, A.N.; Zdzienicka, M.Z.; et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(adp-ribose) polymerase inhibition. Cancer Res. 2006, 66, 8109–8115. [Google Scholar] [CrossRef]
- Naipal, K.A.; Verkaik, N.S.; Ameziane, N.; van Deurzen, C.H.; Ter Brugge, P.; Meijers, M.; Sieuwerts, A.M.; Martens, J.W.; O’Connor, M.J.; Vrieling, H.; et al. Functional ex vivo assay to select homologous recombination-deficient breast tumors for parp inhibitor treatment. Clin. Cancer Res. 2014, 20, 4816–4826. [Google Scholar] [CrossRef] [PubMed]
- Meijer, T.G.; Verkaik, N.S.; Sieuwerts, A.M.; van Riet, J.; Naipal, K.A.T.; van Deurzen, C.H.M.; den Bakker, M.A.; Sleddens, H.; Dubbink, H.J.; den Toom, T.D.; et al. Functional ex vivo assay reveals homologous recombination deficiency in breast cancer beyond brca gene defects. Clin. Cancer Res. 2018, 24, 6277–6287. [Google Scholar] [CrossRef] [PubMed]
- Meijer, T.G.; Verkaik, N.S.; Van Deurzen, C.H.; Dubbink, H.-J.; Den Toom, T.D.; Sleddens, H.F.B.M.; Oomen de Hoop, E.; Dinjens, W.N.M.; Kanaar, R.; Van Gent, D.C.; et al. Direct ex vivo observation of homologous recombination defect reversal after DNA-damaging chemotherapy in patients with metastatic breast cancer. JCO Precis. Oncol. 2019, 1–12. [Google Scholar] [CrossRef]
- van Wijk, L.M.; Vermeulen, S.; Meijers, M.; van Diest, M.F.; Ter Haar, N.T.; de Jonge, M.M.; Solleveld-Westerink, N.; van Wezel, T.; van Gent, D.C.; Kroep, J.R.; et al. The recap test rapidly and reliably identifies homologous recombination-deficient ovarian carcinomas. Cancers 2020, 12, 2805. [Google Scholar] [CrossRef] [PubMed]
- de Jonge, M.M.; Auguste, A.; van Wijk, L.M.; Schouten, P.C.; Meijers, M.; Ter Haar, N.T.; Smit, V.; Nout, R.A.; Glaire, M.A.; Church, D.N.; et al. Frequent homologous recombination deficiency in high-grade endometrial carcinomas. Clin. Cancer Res. 2019, 25, 1087–1097. [Google Scholar] [CrossRef]
- Gentles, L.; Goranov, B.; Matheson, E.; Herriott, A.; Kaufmann, A.; Hall, S.; Mukhopadhyay, A.; Drew, Y.; Curtin, N.J.; O’Donnell, R.L. Exploring the frequency of homologous recombination DNA repair dysfunction in multiple cancer types. Cancers 2019, 11, 354. [Google Scholar] [CrossRef]
- Birkelbach, M.; Ferraiolo, N.; Gheorghiu, L.; Pfaffle, H.N.; Daly, B.; Ebright, M.I.; Spencer, C.; O’Hara, C.; Whetstine, J.R.; Benes, C.H.; et al. Detection of impaired homologous recombination repair in nsclc cells and tissues. J. Thorac. Oncol. 2013, 8, 279–286. [Google Scholar] [CrossRef]
- Castroviejo-Bermejo, M.; Cruz, C.; Llop-Guevara, A.; Gutierrez-Enriquez, S.; Ducy, M.; Ibrahim, Y.H.; Gris-Oliver, A.; Pellegrino, B.; Bruna, A.; Guzman, M.; et al. A rad51 assay feasible in routine tumor samples calls parp inhibitor response beyond brca mutation. EMBO Mol. Med. 2018, 10, e9172. [Google Scholar] [CrossRef]
- Cruz, C.; Castroviejo-Bermejo, M.; Gutierrez-Enriquez, S.; Llop-Guevara, A.; Ibrahim, Y.H.; Gris-Oliver, A.; Bonache, S.; Morancho, B.; Bruna, A.; Rueda, O.M.; et al. Rad51 foci as a functional biomarker of homologous recombination repair and parp inhibitor resistance in germline brca-mutated breast cancer. Ann. Oncol. 2018, 29, 1203–1210. [Google Scholar] [CrossRef]
- Noordermeer, S.M.; van Attikum, H. Parp inhibitor resistance: A tug-of-war in brca-mutated cells. Trends Cell Biol. 2019, 29, 820–834. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Type of Study + Ref. | Patients | Number of Genes in Panel + Definition HRD | Subgroups and Distribution of Mutations | Treatment (Number Treated) | Primary Endpoint and Results |
|---|---|---|---|---|---|
| TOPARP A trial phase 2; single arm [12] | Men with metastatic castration-resistant prostate cancer who had disease progression after one or two regimens of chemotherapy | 113 gene panel on tumor samples HRD: homozygous deletion or deleterious mutation or both in ≥1 HRR genes | 16/49 (33%) HRD: 7× BRCA2, 5× ATM, 3× FANCA, 2× CHEK2, all others genes 1× | Olaparib 400 mg bid (n = 50) | PE: RR defined as CR/PR (RECIST 1.1) or 50% decrease in PSA or CTC count < 5CTCs/7.5 mL Overall RR 16/49 (33%) RR in HRD: 14/16 (88%) RR in HRD excluding BRCA1/2: 7/9 (78%) RR in ATMm only 4/5 (80%) RR in FANC2/3m only (67%) RR non-HRD: 2/33 (6%) |
| TOPARP-B trial Randomized phase 2 [13] | Men with metastatic castration-resistant prostate cancer who have been treated with one or two taxane regimens | 113 gene panel on tumor samples HRD: aberrations in ≥1 HRR genes | 161/592 (27%) HRD: 44× BRCA2; 40× ATM, 33× CDK12 Of all 161 HRD patients, 98 participated in the study of whom: 32× BRCA1/2, 21× ATM; 20× CDK12; 7× PALB2; 18× other | Olaparib 400 mg bid (n = 49) vs. olaparib 300 mg bid (n = 49) | PE: CCR defined as CR/PR (RECIST 1.1) or 50% decrease in PSA or CTC count < 5CTCs/7.5 mL In both (300 + 400 mg) cohorts together: CCR BRCA1/2m: 83.3% (25/30); PFS 8.3 months CCR non-BRCA HRD group: 30.3% (20/66) CCR PALB2m only: 57.1% (4/7); PFS 5.3 months CCR ATMm only: 36.8% (7/19); PFS 5.8 months CCR CDK12m only; 25% (5/20); PFS 2.8 months CCR of other gene mutations: 20% (2/10) |
| PROFOUND study Randomized phase 3 trial [14] | Men with metastatic castration-resistant prostate cancer who had disease progression during enzalutamide or abiraterone treatment | 15 genes panel on tumor samples HRD: suspected deleterious alterations in at least 1 gene | Cohort A: at least one alteration in BRCA1/2, or ATM Cohort B: at least one alteration in any of the other 12 genes Main gene mutations: 160× BRCA1/2; 92× ATM; 94× CDK12; 12× CHEK2; 8× PALB2. | Olaparib vs. nzalutamide or abiraterone (2:1) | PE: iPFS, median FU between 5.4–7.5 months Cohort A (n = 256) iPFS 7.4 vs. 3.6 months; HR 0.34 (0.25–0.47) Cohort B (n = 142) * estimated iPFS 2.8 vs. 3.2 months iPFS ATMm only (n = 86): 5.4 vs. 4.7 months; HR 1.04 (0.61–1.87) iPFS CDK12m only (n = 89): 5.1 vs. 2.2 months; HR 0.74 (0.44–1.31). |
| CBCSG006 trial Randomized phase 3; post-hoc biomarker analyses [15] | Advanced TNBC, first line, no genomic preselection | 28 genes panel in blood samples. HRD defined as at least one gHRR gene mutations | Two groups I: HRD (incl. gBRCA1/2; n = 63) II: no HRD (n = 69) Main gene mutations: 14× BRCA1/2, 10× BARD1, 9× ATM, 6× BRIP1, 6× RAD51C/D, 26× FANC, 5× PALB2, CDH1, MSH2/6, 3× CHEK2 | Paclitaxel/gemcitabine (GP, n = 68) vs. cisplatin/gemcitabine (GT, n = 64) | PE: PFS; median FU 54.7 months Group I HRD (incl. gBRCA1/2m): PFS: GP vs. GT 10.4 vs. 4.3 months (p = 0.011) HRD (excl. gBRCA1/2m): * estimated PFS: GP vs. GT: 10.9 vs. 4.7 months Group II; no HRD: PFS GP vs. GT: 6.0 vs. 7.1 months (p = 0.154) |
| TBCRC 048 phase 2, single arm [16] | Metastatic breast cancer, maximum of 2 lines of chemotherapy for advanced disease, no prior PARPi or progression on platinum, and a mutation ≥1 g/sHRRm | 20 genes panel on tumor samples and in blood HRD: mutation in ≥1 s/g HRR genes | Cohort 1: Germline mutation in gHRRm genes (excl. gBRCA1/2m); n = 27 Cohort 2: Somatic mutations in HRR genes (incl. sBRCA1/2m); n = 27 | Olaparib 300 mg bid (n = 54) | PE: ORR = PR or CR according to RECIST v1.1 Cohort 1 ORR (gHRRm, excl. gBRCA1/2m) 33% (9/27); all 9 had gPALB2m ORR in gPALB2m only: 82% (9/11) Cohort 2 ORR (sHRRm; incl. sBRCA1/2m): 31% (8/26) ORR (sHRRm, excl. sBRCA1/2m): 0% (0/10) |
| Pretermac study Neoadjuvant study, single arm, post-hoc biomarker analyses in TNBC only [17] | Stage II-II primary breast cancer, subset TNBC only | 360 genes panel on tumor samples among which HRR genes. HRD: not specifically defined | Three subgroups I all TNBC excl. gBRCA1/2m and gPALB2mII g/sBRCAm and gPALBm2 only III sHRRm (excl. g/sBRCAm) Main gene mutations 6× g/s BRCA1/2, 1× gPALB2; 5× non-BRCA sHRR gene mutation (16%): all 1× ATR, EMSY, MEN1, SETD2, PTEN | Olaparib 300 mg bid for up to 10 weeks (n = 32) | PE: ORR = PR or CR according to RECIST1.1 ORR all TNBC: 56.3% (18/32) I ORR all TNBC excl. gBRCA1/2m and gPALB2m: 51.9% (14/27) II: ORR g/sBRCAm and gPALBm2 only: 83% (5/6) III: ORR non-BRCA sHRRm: 100% (5/5) |
| Study 19 Randomized phase 2; post-hoc biomarker analyses [18] | Platinum sensitive grade 2/3 serous ovarian carcinoma, platinum sensitive, 2–3 lines of platinum and an objective response on last platinum-based therapy | 287 cancer related genes + select introns from 27 HRR genes + gBRCA1/2 testing HRD: not specifically defined | Four subgroups (n = 209) I: g/s BRCAm (n = 111); II: g/sBRCAwt + sHRRm (n = 21); III: g/sBRCAwt + sHRR mutation unknown (n = 16) IV: g/sBRCAwt en sHRRwt (n = 58) 209/265 (79%) | Olaparib 400 mg bid vs. placebo as maintenance therapy | PE: PFS olaparib versus placebo PFS in subgroups I: HR 0.16 (0.08–0.30) II: HR 0.21 (0.04–0.86) III: not mentioned IV: HR 0.71 (0.37–1.35) |
| Prospective cohort study; post hoc biomarker analyses in subgroup of primary tumors only [19] | Ovarian, fallopian tube, or primary peritoneal carcinoma (primary (n = 304), recurrent (n = 34), paired primary and recurrent (n = 24) | 13 gene panel in blood and tumor samples. HRD: a deleterious germline and/or somatic mutation in at least one HRR gene | No subgroups Main gene mutations 68× (55%) BRCA1; 23× (19%) BRCA2; 32× (26%) in the other HRR genes: ATM, BARD1, BRIP1, CHEK1/2, FAM175A, MRE11A, NBN, PALB2, RAD51C/D. | Adjuvant 6× carboplatin/paclitaxel treatment (n = 304) | PE: platinum sensitivity (PS) defined as complete response (CR) during adjuvant chemotherapy maintenance of CR >6 months post completion chemotherapy. Analysis in 243 patients with sufficient clinical response data PPS in gBRCA1/2m: 81% (38/47) PPS in g/sHRRm (excl. gBRCA1/2m, incl. sBRCA1/2m) 87% (33/38) PPS in g/sHRRm (excl. g/sBRCA1/2m): 78% (14/18) PPS in non-HRD: PPS 60% (95/158) |
| Type of Study + ref. | Patients | Test Used + Definition HRD | Subgroups | Treatment (Total Treated) | Primary Endpoint and Results |
| LOSS OF HETEROZYGOSITY (LOH) | |||||
| ARIEL2 study, phase 2, single arm [20] | Recurrent, platinum sensitive (minimal 1 line platinum-based CTx and PFI >6 months after last platinum dose), high grade serous or endometrioid ovarian, fallopian, or primary peritoneal carcinoma | LOH by Next Generation Sequencing (NGS) HRD = LOH high, i.e., genomic LOH ≥14% on archival or pretreatment biopsies | Three groups I: g/s BRCA1/2m (n = 40); II: g/sBRCA1/2wt + LOH high (n = 82); III: g/sBRCA1/2wt + LOH low (n = 70) | Rucaparib (n = 192) | PE: PFS I vs. II vs. III: PFS 12.8 vs. 5.7 vs. 5.2 months I vs. III HR PFS 0.27 (0.16–0.44) II vs. III HR PFS 0.63 (0.42–0.90) |
| ARIEL3 Randomized phase 3 [21] | Recurrent, platinum sensitive (minimal 2 line platinum-based therapy and PFI >6 months after last platinum dose), high grade serous or endometrioid ovarian, fallopian, or primary peritoneal carcinoma | LOH by NGS HRD = LOH high, i.e., genomic LOH score ≥16% on archival or pretreatment biopsies | Three subgroups I: g/s BRCAm (n = 196); II: BRCAwt + LOH high (n = 158); III: BRCAwt + LOH low (n = 210) | Rucaparib (n = 375) vs. placebo (n = 189) | PE: iaPFS rucaparib vs. placebo I: iaPFS; 16.6 vs. 5.4 months; HR 0.23 (0.16–0.34) II: iaPFS: 9.7 vs. 5.4 months; HR 0.44 (0.29–0.66) III: iaPFS: 6.7 vs. 5.4 months; HR 0.58 (0.40–0.85) |
| PrECOG trial Neoadjuvant, Phase 2, single arm [22] | Primary triple negative breast cancer | LOH DNA copy number was determined using genome-wide SNP data HRD = LOH-high, i.e., LOH score of ≥10% in pre-treatment breast biopsies | Two groups I: HRD: LOH high (n = 50) II: no-HRD: LOH low (n = 15) | 6× carboplatin/gemcitabine/iniparib | PE: RCB = residual cancer burden 0 or 1 I: RCB 0/1: 66% II: RCB0/1: 20% |
| CHROMOSOMAL ABERRATIONS | |||||
| Type of study + ref. | Patients | Test used + definition HRD | Subgroups | Treatment (total treated) | primary endpoint and results |
| VELIA study Randomized Phase 3, first line [23] | High grade serous epithelial ovarian, fallopian tube or primary peritoneal carcinoma; No preselection on platinum sensitivity | MyChoice assay = sum of LOH, TAI and LTS score; HRD = total score ≥42 Second definition HRD = total score ≥33 | Three groups I: g/sBRCA mutated (298) II: BRCAwt + HRD high (329) III: BRCAwt with HRD low (372) Of note: 47% HRD within BRCAwt | CTx + PB + PB maintance (n = 375) vs. CTx + veliparib + PB maintance (n = 383) vs. CTx + veliparib + veliparib maintance (n = 382) | PE: ia PFS, median FU 28 months I: PFS 34.7 vs. 22.0 months; HR 0.44 (0.28–0.68) II: * estimated PFS 21.5 vs. 18.5 months; HR 0.80 (0.64–1.00) III: PFS 15.0 vs. 11.5 months; HR 0.81 (0.60–1.09) |
| PRIMA study Randomized phase 3, maintenance therapy after first line therapy [24] | Platinum sensitive high grade serous or endometrioid ovarian carcinoma, stage III or IV, platinum sensitive (i.e., partial or complete response after first line platinum-based chemotherapy) | MyChoice assay = sum of LOH, TAI, and LTS score; sBRCA1/2m based on NGS HRD = total score ≥42 | Three groups I: g/sBRCAm (n = 223); II: g/sBRCAwt + HRD high (n = 150); III: g/sBRCAwt + HRD low (n = 360) Of note: 29% HRD within BRCAwt | Maintenance therapy for 36 months Niraparib (n = 484) vs. PB (n = 244) | PE: PFS, median FU 13.8 months I: PFS 22.1 vs. 10.9 months; HR 0.40 (0.27–0.62) II: PFS 19.6 vs. 8.2 months; HR 0.50 (0.31–0.83) III: * estimated PFS 6.1 vs. 6.4 months; HR 0.68 (0.49–0.94) |
| SCOTROC4 study (n = 964); randomized phase 3; first line; a prespecified subgroup analyses [25] | Untreated stage III/IV epithelial ovarian, fallopian tube, or primary peritoneal carcinoma; first line | Genome-wide SNP data; sum of LOH, TAI, and LTS score HRD = total score ≥42 Second definition HRD = total score ≥33 | Three subgroups I: HRD (incl. sBRCAm) (n = 79) II: sBRCAm only (n = 47) III: CCNE1 amplified | Carboplatin/ paclitaxel standard vs. dose intensified based on nadir platelet and neutrophil counts | One of the three PE: PFS (n = 225), median FU not mentioned I: PFS 16.5 vs. 9.5 months; HR 0.50 (0.34–0.73) If HRD is defined as ≥33: HR 0.51 (0.36–0.72) II: PFS 18.9 vs. 11.0 months; HR 0.48 (0.29–0.79) III: PFS 9.5 vs. 13.2 months; HR 1.56 (1.04–2.34) |
| PAOLA 1 study, randomized phase 3, second line [26] | High grade serous or endometrioid ovarian, fallopian, or primary peritoneal carcinoma independent of BRCA status or non-mucinous epithelial ovarian cancer with a deleterious gBRCAm; platinum sensitive after first line carboplatin/taxane bevacizumab | MyChoice assay = sum of LOH, TAI, and LTS score. HRD = total score ≥44 | Three subgroups I: g/sBRCAm (n = 237) II: BRCAwt + HRD high (n = 152) III: BRCAwt + HRD low/unknown (n = 419) Of note: 27% HRD within BRCAwt | Olaparib + bevacizumab (n = 537) vs. PB + bevacizumab (n = 269) for a maximum of 24 months | PE: iaPFS, median FU 22.7 months I: iaPFS 37.2 vs. 21.7 months; HR 0.31 (0.20–0.47) II: iaPFS 28.1 vs. 16.6 months; HR 0.43 (0.28–0.66) III: iaPFS 16.9 vs. 16.0 months; HR 0.92 (0.72–1.17) |
| NOVA trial, randomized phase 3 [27] | High grade serous epithelial ovarian, fallopian tube or primary peritoneal carcinoma; platinum-sensitive (PR or CR and ≥6 months PFI of last platinum containing regimen), ≥2 line of CTx | MyChoice assay = sum of LOH, TAI, and LTS score. HR = total score ≥42 | Three subgroups I: gBRCAm (n = 201) II g/sBRCAwt + HRD high (n = 115) III g/sBRCAwt with HRD low (n = 134) Of note: 46% HRD within BRCAwt | Niraparib (n = 372) vs. PB (n = 181) Continue until PD or unacceptable toxicity | PE: PFS, median FU 16.9 months I: PFS 21.0 vs. 5.5 months; HR 0.27 (0.17–0.41) II: *estimated PFS 9.0 vs. 4.0 months; HR 0.38 (0.23–0.63) III: * estimated PFS 5.8 vs. 3.9 months; HR 0.58 (0.36–0.92) |
| Study 19 Randomized phase 2 study [18] | Platinum sensitive grade 2/3 serous ovarian carcinoma, platinum sensitive (2–3 lines of platinum and objective response on last platinum-based therapy) maintenance therapy. | MyChoice assay = sum of LOH, TAI and LTS score. HRD = total score ≥42 Second definition HRD = total score ≥33 | Three subgroups I: g/s BRCAm (n = 106); II: BRCAwt + HRD (n = 139); III: BRCAwt + no HRD (n = 60) Of note: 70% HRD within BRCAwt | Olaparib vs. PB n = 265 in total; continue until PD or unacceptable toxicity | PE: PFS, exact median FU not mentioned I: PFS in months not mentioned; HR 0.18 (0.10–0.31) II: PFS in months not mentioned; HR 0.48 (0.18–1.27) III: PFS in months not mentioned; HR 0.60 (0.31–1.17) Of note: In subgroup with >6 y PFS, no specific molecular aberrations other than high percentage of g/sBRCA2m |
| Quadra study Phase 2, single arm [28] | High grade serous (grade 2 or 3) epithelial ovarian, fallopian tube or primary peritoneal carcinoma; ≥3 lines of CTx, measurable disease, first line platinum-based CTx PFI must be ≥6 months | MyChoice assay = sum of LOH, TAI, and LTS score. HRD = total score ≥42 | Three subgroups I: g/sBRCAm (n = 87) II g/sBRCAwt + HRD (n = 135) III g/sBRCAwt + no HRD (n = 195) Of note: 41% HRD within BRCAwt | Niraparib (n = 463) until PD or unacceptable toxicity | PE: ia-ORR; median FU 12.2 months I: ia-ORR 29% (39 vs. 27%) II: ia-ORR 15% (26 vs. 10%) III: ia-ORR 3% (3 vs. 4%) secondary outcome: median OS: I: median OS 26.0 (18.1-NR) months II: estimated median OS 14.5 months III: median OS 15.5 (11.6–19.0) months |
| TBCRC 030 trial neoadjuvant, randomized phase 2 [29] | Stage II-III, TNBC, patients with known gBRCA1/2m were excluded | MyChoice assay = sum of LOH, TAI, and LTS score. BRCA testing on blood and tumor HRD = total score ≥33 or g/sBRCA1/2m | Two subgroups I: HRD (incl g/sBRCAm) (n = 74) II: no HRD (n = 30) | 4× Cisplatin (n = 56) vs. 12× paclitaxel (n = 48) | Two PE: 1st RCB0/1; 2nd pCR I: RCB0/1: 23% vs. 12%; OR 2.22 II: RCB0/1: 29% vs. 31%%; OR 0.90 Of note, 6 (of the 7) sBRCAm tumors were randomly allocated to CDDP with only 1 RCB 0/1 (17%) I: pCR: 13% vs. 6%; OR 2.32 II: pCR: 14% vs. 23%%; OR 0.55 |
| GEPARSIXTO trial, randomized phase 3, post hoc analyses in TNBC only [30] | TNBC, stage II-III | MyChoice assay = sum of LOH, TAI, and LTS score. HRD = total score ≥42 or gBRCA1/2m | Two subgroups I: HRD or g/sBRCAm (n = 136) II; no HRD and BRCAwt (n = 57) | Paclitaxel/ pegylated doxorubicine/ bevacicumab (n = 157) vs. paclitaxel/ pegylated doxorubicine/ bevacicumab/carboplatin (n = 158) | PE: pCR (ypT0N0) I: pCR 33.9% vs. 63.5%; OR 3.4 (1.7–6.9) II: pCR 20.0% vs. 29.6%; OR 1.7 (0.5–5.7) |
| TNT Trial. Randomized phase 3, first line [31] | TNBC advanced setting, no CTx for advanced setting | MyChoice assay = sum of LOH, TAI, and LTS score. HRD = total score ≥42 | Two subgroups I: gBRCAm (n = 43) II: HRD (independent of gBRCA status) (n = 81) III: HRD (incl gBRCAm) (n = 86) | Carboplatin (n = 188) ns Docetaxel (n = 188) | PE ORR difference between carboplatin vs. docetaxel per group I: ORR difference: 34.6% vs. −6.4% p interaction 0.01 II: ORR difference: −2.2% vs. 2.2% p interaction 0.75 III: ORR difference: 5.1% vs. −1.8% p interaction 0.63 |
| TBCRC009 study, phase 2, single arm, posthoc analysis [32] | TNBC, maximum of one line CTx in advanced setting | LOH by NGS assay; chromosomal breaks by another assay Sum scores of the two assays HRD-LOH and HRD-LST was used as continues variable; no HRD definition. | Posthoc analysis of difference between responder (CR and PR) vs. non-responders (SD or PD) | carboplatin (n = 43) and cisplatin (n = 43) | PE ORR Sum score HRD-LOH and HRD-LST was statistically significantly higher among responder than non-responders gBRCAwt TNBC patients (12.7 vs. 5.1; p = 0.032) No data on ORR for HRD-LOH or HRD-LST scores separately |
| SWOG S9313 study randomized phase 3, post hoc analyses in TNBC [33] | High risk N0, or low risk n+ primary breast cancer, adjuvant chemotherapy. | MyChoice assay = sum of LOH, TAI, and LTS score. HRD = total score ≥42 or sBRCA1/2m | Two subgroups I: HRD (incl. sBRCA1/2m) II: HRD (all sBRCA1/2wt) | Doxorubicine/ Cyclophosphamide concomitant vs. sequential | PE: (5-y) DFS, exact median FU not mentioned I: 5-y DFS: 65.2% vs. 78.7%; HR 0.72 (0.51–1.00) II: 5-y DFS: 65.2% vs. 80.5%; HR 0.64 (0.43–0.94) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ladan, M.M.; van Gent, D.C.; Jager, A. Homologous Recombination Deficiency Testing for BRCA-Like Tumors: The Road to Clinical Validation. Cancers 2021, 13, 1004. https://doi.org/10.3390/cancers13051004
Ladan MM, van Gent DC, Jager A. Homologous Recombination Deficiency Testing for BRCA-Like Tumors: The Road to Clinical Validation. Cancers. 2021; 13(5):1004. https://doi.org/10.3390/cancers13051004
Chicago/Turabian StyleLadan, Marjolijn M., Dik C. van Gent, and Agnes Jager. 2021. "Homologous Recombination Deficiency Testing for BRCA-Like Tumors: The Road to Clinical Validation" Cancers 13, no. 5: 1004. https://doi.org/10.3390/cancers13051004
APA StyleLadan, M. M., van Gent, D. C., & Jager, A. (2021). Homologous Recombination Deficiency Testing for BRCA-Like Tumors: The Road to Clinical Validation. Cancers, 13(5), 1004. https://doi.org/10.3390/cancers13051004