
Worm-Based Microfluidic Biosensor for Real-Time Assessment of the Metastatic Status




Abstract
Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
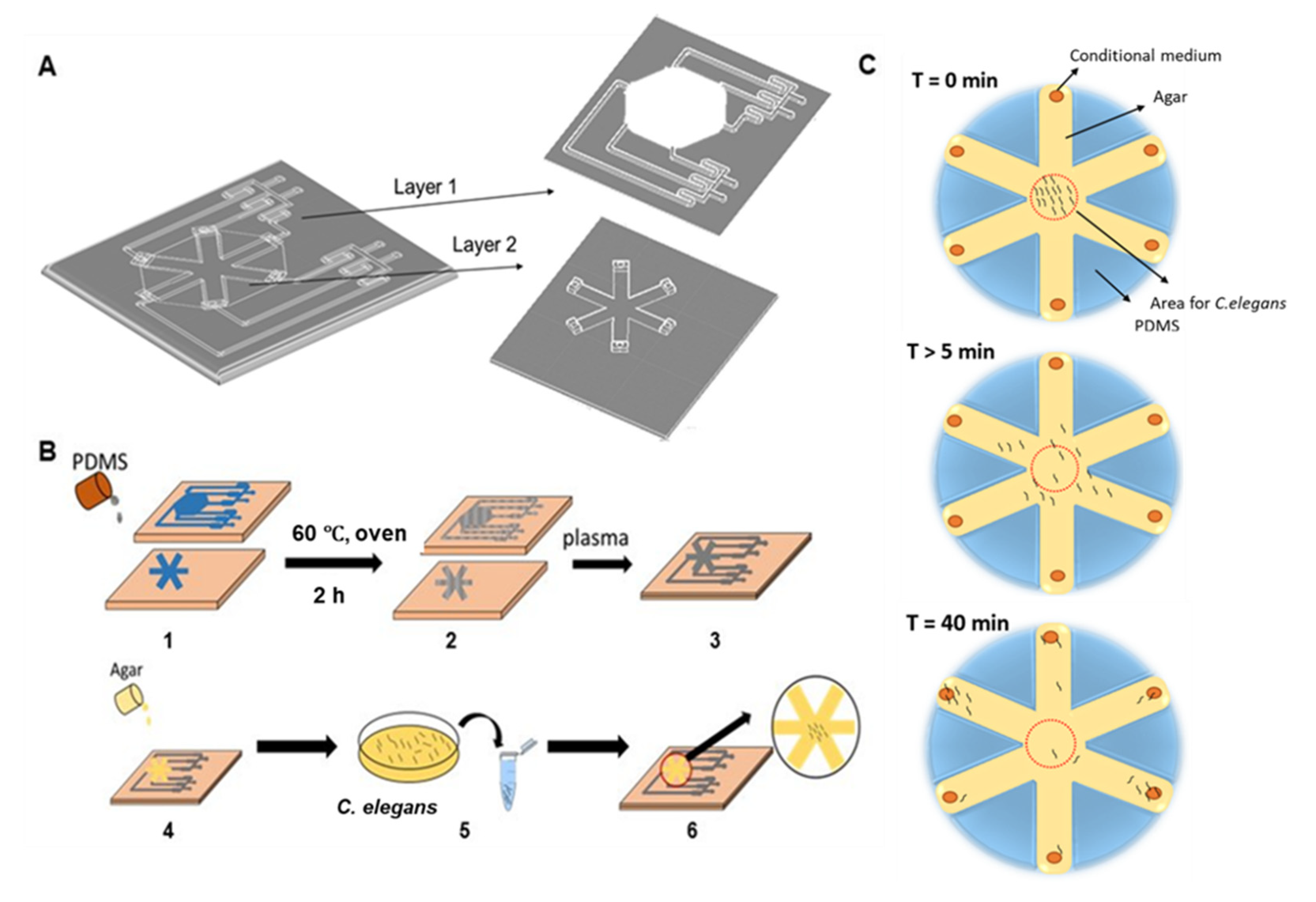
2.1. WB Biosensor Operations
2.1.1. Device Fabrication
2.1.2. C. elegans Loading
2.1.3. Sample Collection from Cancer Cell Cluster Cultures
2.2. C. elegans Culture
2.3. Cell Culture
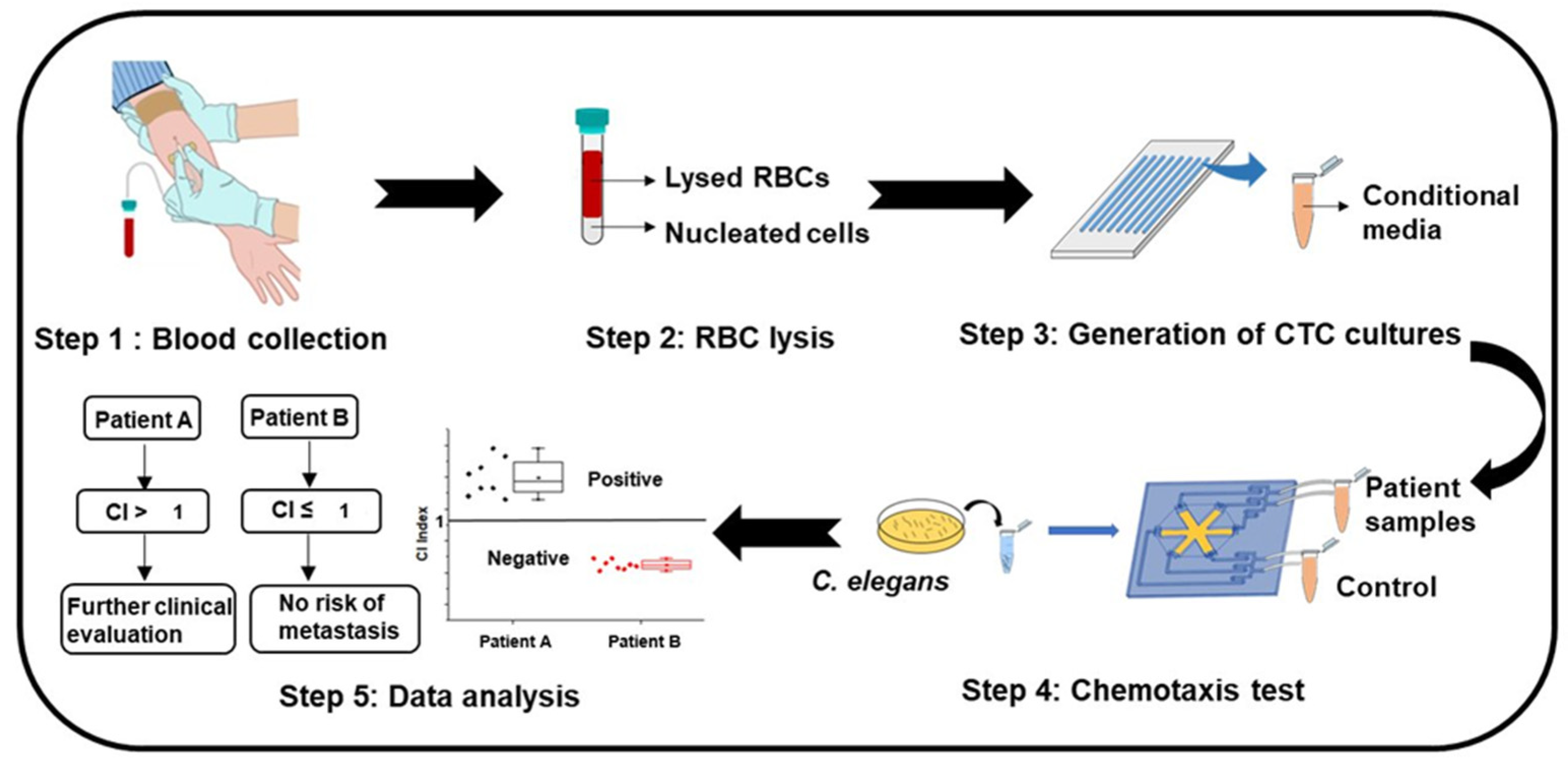
2.4. Screening of Samples with the WB Biosensor
2.5. Glutamate Detection and Degradation Assays
2.6. Statistical Analysis
3. Results
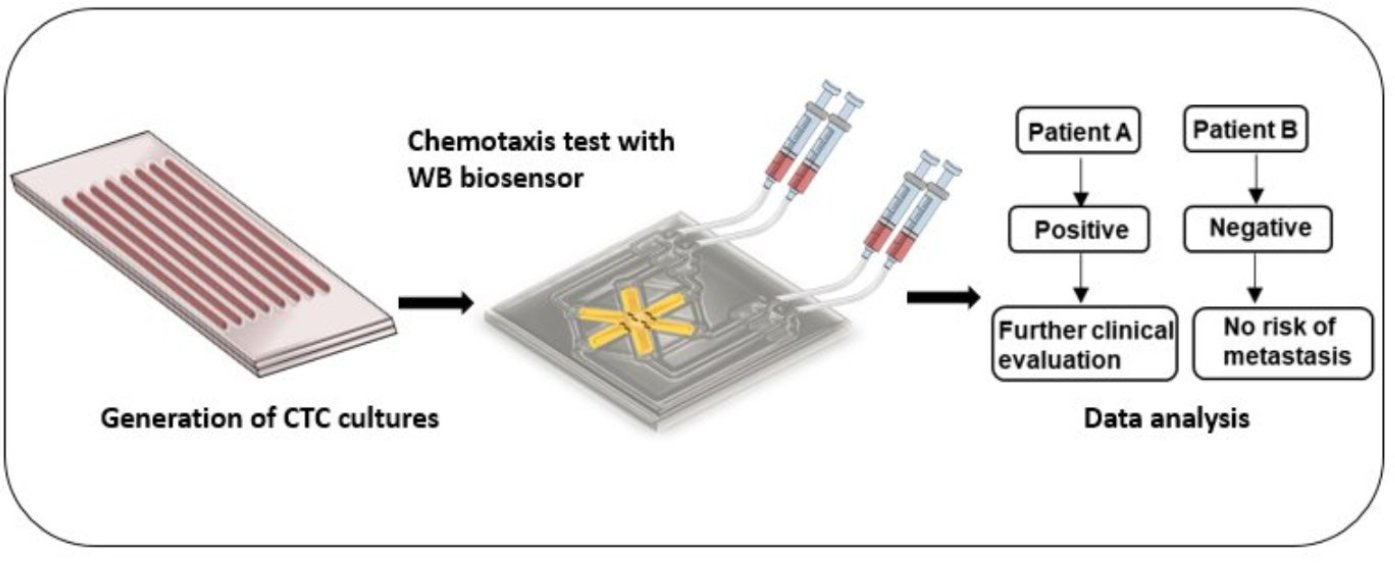
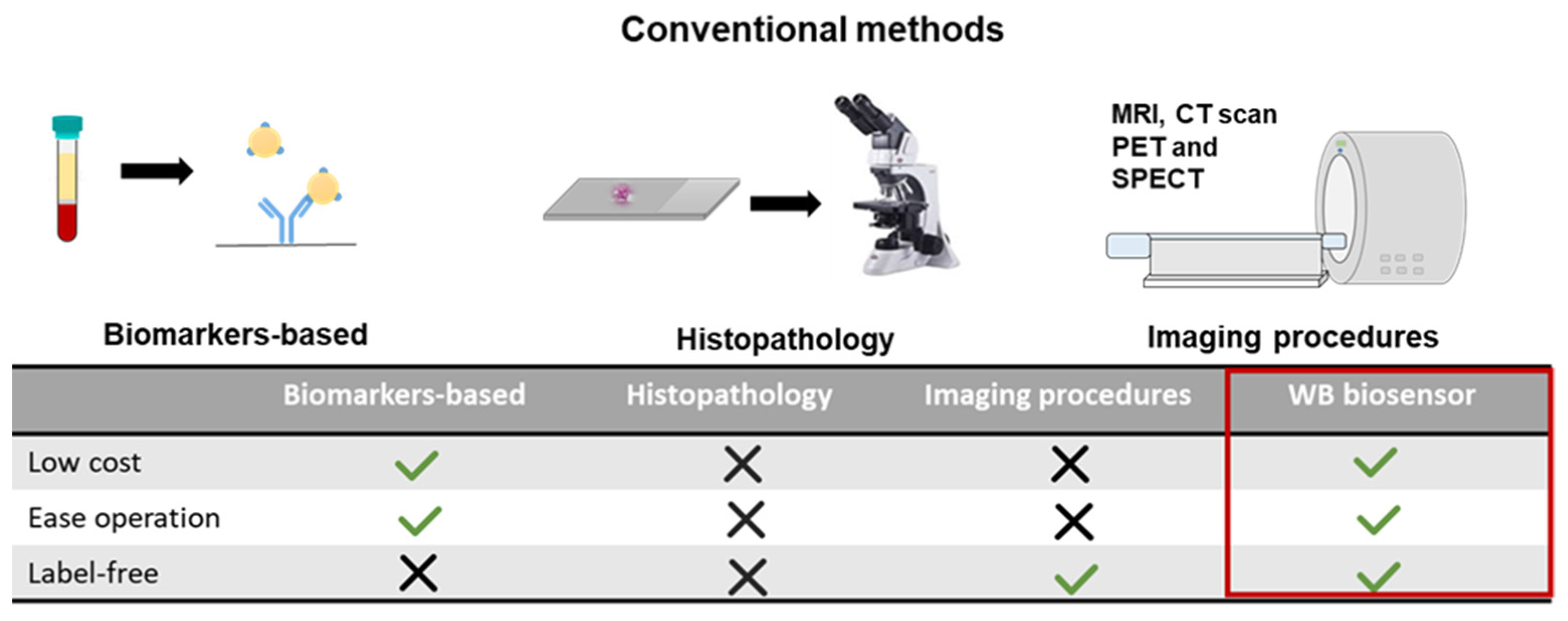
3.1. Worm-Based (WB) Microfluidic Biosensor for the Detection of Metastasis
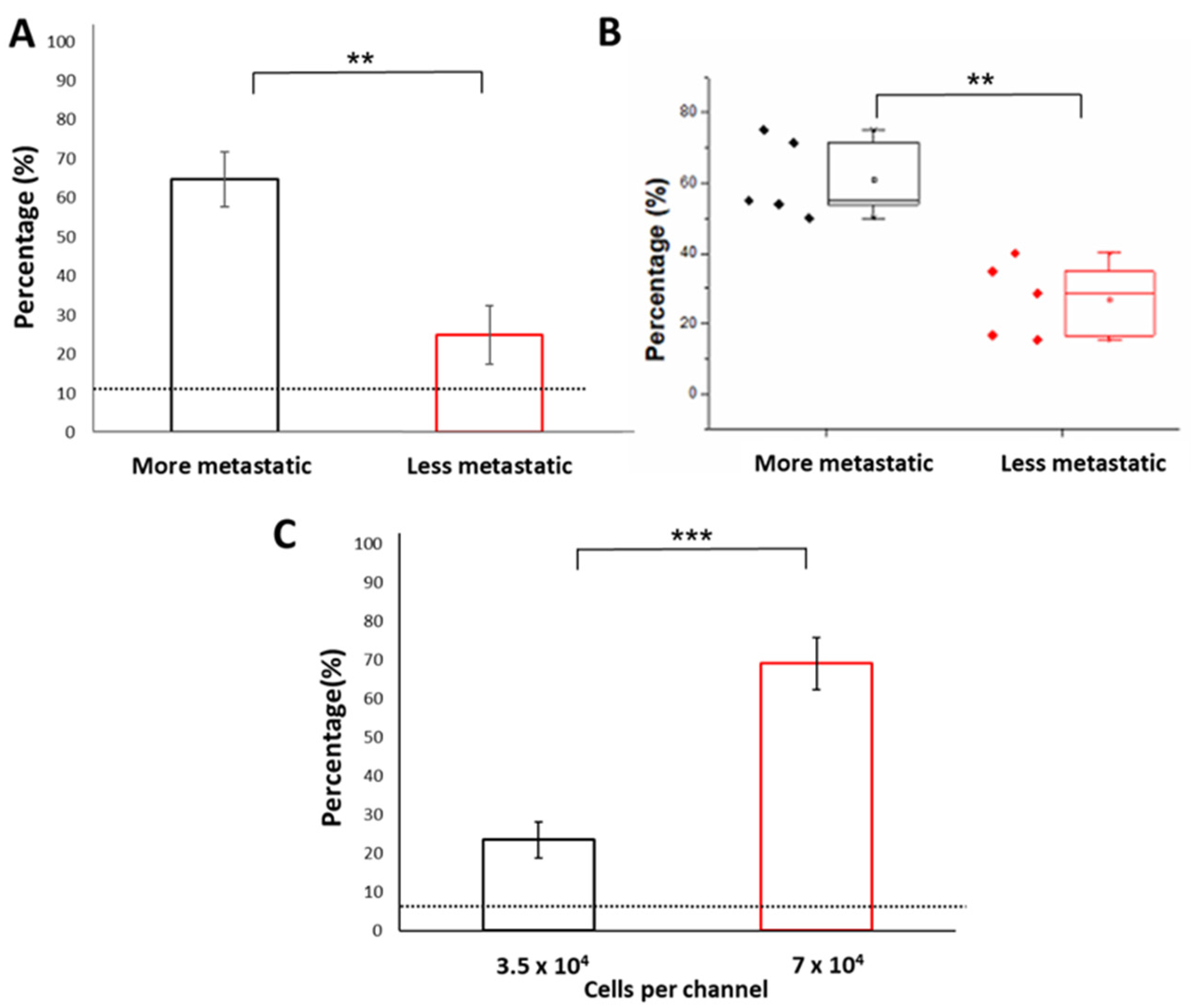
3.2. Characterization of Parameters for the WB Biosensor
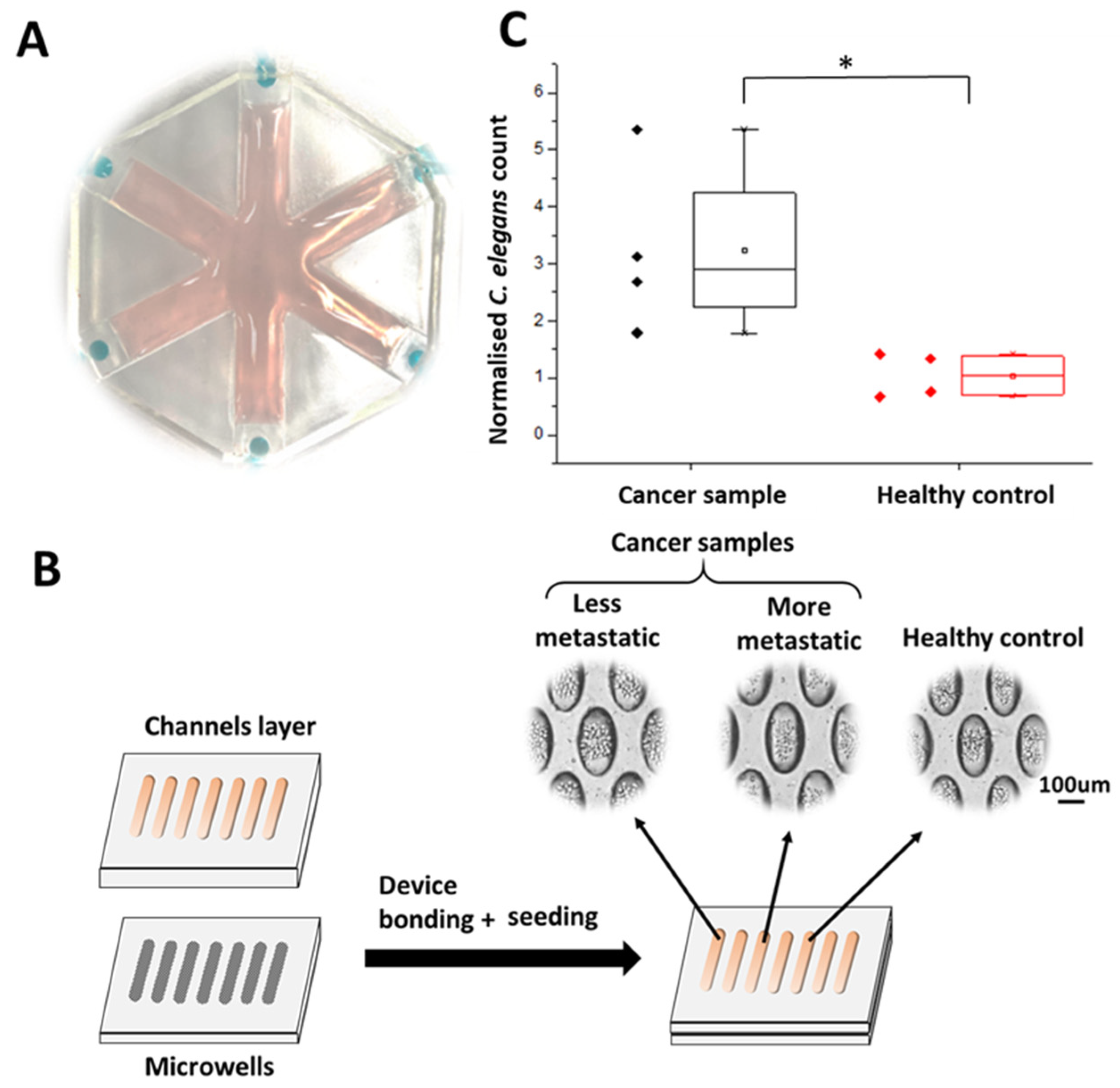
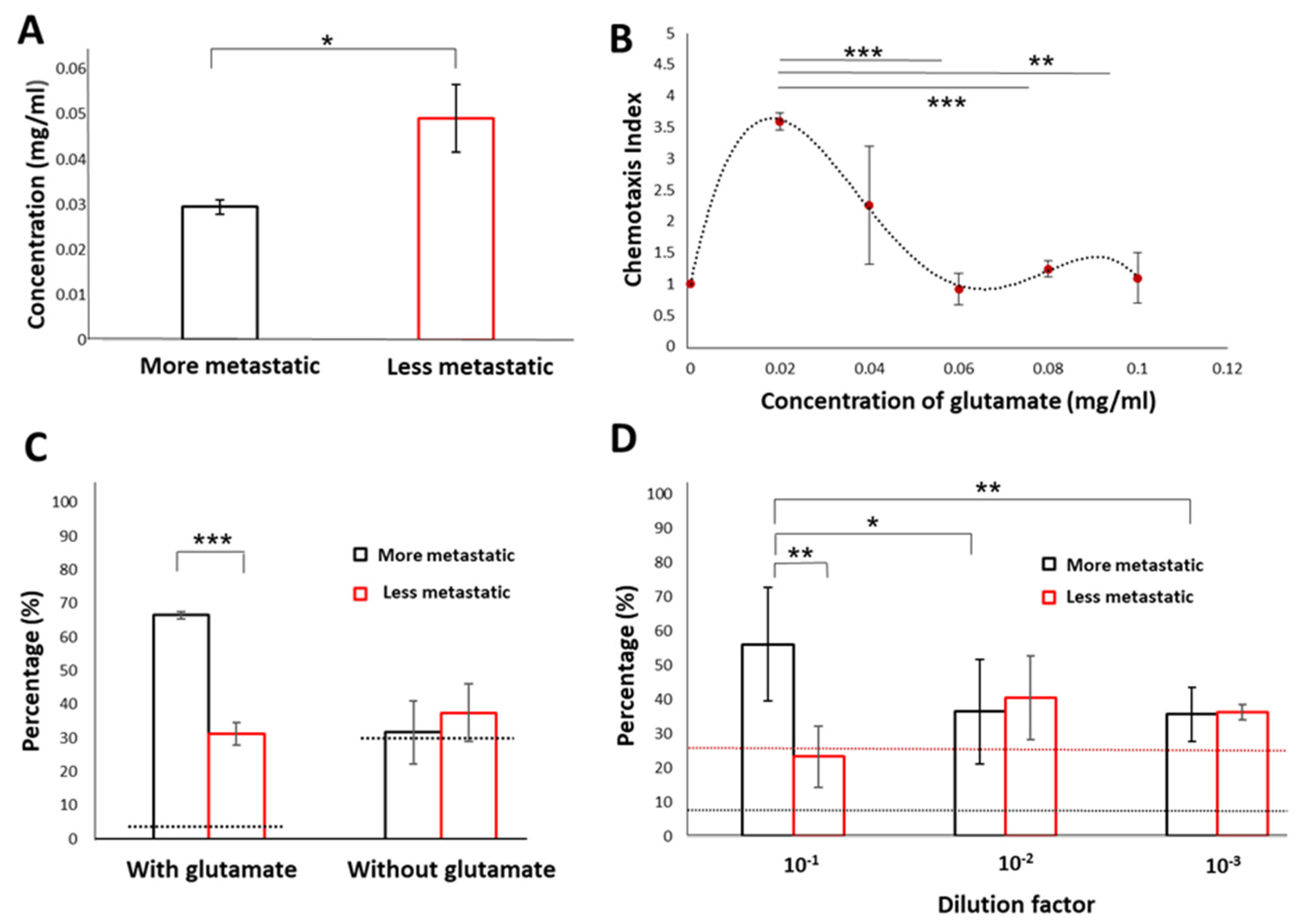
3.3. Detection of Samples Associated with Higher Metastatic Potential Using the WB Biosensor
3.4. Low Glutamate Levels Reflect the Presence of Metastasis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| T | Tumor |
| N | Node |
| M | Metastasis |
| WB | Worm-based |
| C. elegans | Caenorhabditis elegans |
| CI | Chemotaxis index |
| PDMS | Polydimethylsiloxane |
| PET | Positron emission tomography |
| SPECT | Single-photon emission computer tomography |
| CTCs | Circulating tumor cells |
| MRI | Magnetic resonance imaging |
| CT scan | Computed tomography scan |
| T | Time |
| RBC | Red blood cells |
References
- Lefebvre, C.; Bachelot, T.; Filleron, T.; Pedrero, M.; Campone, M.; Soria, J.-C.; Massard, C.; Levy, C.; Arnedos, M.; Lacroix-Triki, M. Mutational profile of metastatic breast cancers: A retrospective analysis. PLoS Med. 2016, 13, e1002201. [Google Scholar] [CrossRef]
- Redig, A.J.; McAllister, S.S. Breast cancer as a systemic disease: A view of metastasis. J. Intern. Med. 2013, 274, 113–126. [Google Scholar] [CrossRef]
- Reiter, W.; Stieber, P.; Reuter, C.; Nagel, D.; Lau-Werner, U.; Lamerz, R. Multivariate analysis of the prognostic value of CEA and CA 19-9 serum levels in colorectal cancer. Anticancer Res. 2000, 20, 5195–5198. [Google Scholar] [PubMed]
- Mitchell, P.S.; Parkin, R.K.; Kroh, E.M.; Fritz, B.R.; Wyman, S.K.; Pogosova-Agadjanyan, E.L.; Peterson, A.; Noteboom, J.; O’Briant, K.C.; Allen, A.; et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc. Natl. Acad. Sci. USA 2008, 105, 10513–10518. [Google Scholar] [CrossRef] [PubMed]
- Ng, E.K.; Chong, W.W.; Jin, H.; Lam, E.K.; Shin, V.Y.; Yu, J.; Poon, T.C.; Ng, S.S.; Sung, J.J. Differential expression of microRNAs in plasma of patients with colorectal cancer: A potential marker for colorectal cancer screening. Gut 2009, 58, 1375–1381. [Google Scholar] [CrossRef]
- Tan, E.; Gouvas, N.; Nicholls, R.J.; Ziprin, P.; Xynos, E.; Tekkis, P.P. Diagnostic precision of carcinoembryonic antigen in the detection of recurrence of colorectal cancer. Surg. Oncol. 2009, 18, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Luo, D.; Cai, S.; Li, Q.; Li, X. Real-World Implications of Nonbiological Factors with Staging, Prognosis and Clinical Management in Colon Cancer. Cancers 2018, 10, 263. [Google Scholar] [CrossRef] [PubMed]
- Wood, W.B. (Ed.) The nematode Caenorhabditis elegans. Cold Spring Harb. Lab. 1988, 1, 988. [Google Scholar]
- Sudhaus, W.; Kiontke, K. Phylogeny of Rhabditis subgenus Caenorhabditis (Rhabditidae, Nematoda). J. Zool. Syst. Evol. Res. 1996, 34, 217–233. [Google Scholar] [CrossRef]
- Chronis, N.; Zimmer, M.; Bargmann, C.I. Microfluidics for in vivo imaging of neuronal and behavioral activity in Caenorhabditis elegans. Nat. Methods 2007, 4, 727–731. [Google Scholar] [CrossRef]
- Chokshi, T.V.; Bazopoulou, D.; Chronis, N. An automated microfluidic platform for calcium imaging of chemosensory neurons in Caenorhabditis elegans. Lab Chip 2010, 10, 2758–2763. [Google Scholar] [CrossRef]
- Gokce, S.K.; Hegarty, E.M.; Mondal, S.; Zhao, P.; Ghorashian, N.; Hilliard, M.A.; Ben-Yakar, A. A multi-trap microfluidic chip enabling longitudinal studies of nerve regeneration in Caenorhabditis elegans. Sci. Rep. 2017, 7, 9837. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, D.L.; Badhiwala, K.N.; Vercosa, D.G.; Avants, B.W.; Liu, Z.; Zhong, W.; Robinson, J.T. Scalable electrophysiology in intact small animals with nanoscale suspended electrode arrays. Nat. Nanotechnol. 2017, 12, 684–691. [Google Scholar] [CrossRef] [PubMed]
- McFadden, B.; Patel, T. Axenic and synchronous cultures of Caenorhabditis elegans. Nematologica 1978, 24, 51–62. [Google Scholar] [CrossRef]
- Bargmann, C.I. Chemosensation in C. elegans. WormBook 2006, 25, 1–29. [Google Scholar] [CrossRef]
- Hirotsu, T.; Sonoda, H.; Uozumi, T.; Shinden, Y.; Mimori, K.; Maehara, Y.; Ueda, N.; Hamakawa, M. A highly accurate inclusive cancer screening test using Caenorhabditis elegans scent detection. PLoS ONE 2015, 10, e0118699. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Yang, W.; Ren, J.; Hall, Q.; Zhang, Y.; Kaplan, J.M. Thioredoxin shapes the C. elegans sensory response to Pseudomonas produced nitric oxide. Elife 2018, 7, e36833. [Google Scholar] [CrossRef]
- Collins, C.L.; Wasa, M.; Souba, W.W.; Abcouwer, S.F. Determinants of glutamine dependence and utilization by normal and tumor-derived breast cell lines. J. Cell. Physiol. 1998, 176, 166–178. [Google Scholar] [CrossRef]
- Takano, T.; Lin, J.H.-C.; Arcuino, G.; Gao, Q.; Yang, J.; Nedergaard, M. Glutamate release promotes growth of malignant gliomas. Nat. Med. 2001, 7, 1010–1015. [Google Scholar] [CrossRef]
- Seidlitz, E.P.; Sharma, M.K.; Saikali, Z.; Ghert, M.; Singh, G. Cancer cell lines release glutamate into the extracellular environment. Clin. Exp. Metastasis 2009, 26, 781. [Google Scholar] [CrossRef]
- Khoo, B.L.; Grenci, G.; Lim, J.S.Y.; Lim, Y.P.; Fong, J.; Yeap, W.H.; Bin Lim, S.; Chua, S.L.; Wong, S.C.; Yap, Y.S.; et al. Low-dose anti-inflammatory combinatorial therapy reduced cancer stem cell formation in patient-derived preclinical models for tumour relapse prevention. Br. J. Cancer 2019, 120, 407–423. [Google Scholar] [CrossRef]
- Carretta, A. Clinical value of nodal micrometastases in patients with non-small cell lung cancer: Time for reconsideration? J. Thorac. Dis. 2016, 8, E1755. [Google Scholar] [CrossRef] [PubMed]
- Bedard, P.L.; Hansen, A.R.; Ratain, M.J.; Siu, L.L. Tumour heterogeneity in the clinic. Nature 2013, 501, 355–364. [Google Scholar] [CrossRef]
- Meacham, C.E.; Morrison, S.J. Tumour heterogeneity and cancer cell plasticity. Nature 2013, 501, 328–337. [Google Scholar] [CrossRef]
- Liu, X.; Feng, R. Inhibition of epithelial to mesenchymal transition in metastatic breast carcinoma cells by c-Src suppression. Acta. Biochim. Biophys Sin. 2010, 42, 496–501. [Google Scholar] [CrossRef]
- Asgeirsson, K.; McCulley, S.; Pinder, S.; Macmillan, R. Size of invasive breast cancer and risk of local recurrence after breast-conservation therapy. Eur. J. Cancer 2003, 39, 2462–2469. [Google Scholar] [CrossRef]
- Tot, T. Clinical relevance of the distribution of the lesions in 500 consecutive breast cancer cases documented in large-format histologic sections. Cancer: Interdiscip. Int. J. Am. Cancer Soc. 2007, 110, 2551–2560. [Google Scholar] [CrossRef]
- Tot, T. DCIS, cytokeratins, and the theory of the sick lobe. Virchows Arch. 2005, 447, 1–8. [Google Scholar] [CrossRef]
- Andea, A.A.; Wallis, T.; Newman, L.A.; Bouwman, D.; Dey, J.; Visscher, D.W. Pathologic analysis of tumor size and lymph node status in multifocal/multicentric breast carcinoma. Cancer 2002, 94, 1383–1390. [Google Scholar] [CrossRef] [PubMed]
- Foschini, M.P.; Flamminio, F.; Miglio, R.; Calò, D.G.; Cucchi, M.C.; Masetti, R.; Eusebi, V. The impact of large sections on the study of in situ and invasive duct carcinoma of the breast. Hum. Pathol. 2007, 38, 1736–1743. [Google Scholar] [CrossRef]
- Fowble, B.; Yeh, I.-T.; Schultz, D.J.; Solin, L.J.; Rosato, E.F.; Jardines, L.; Hoffman, J.; Eisenberg, B.; Weiss, M.C.; Hanks, G. The role of mastectomy in patients with stage I-II breast cancer presenting with gross multifocal or multicentric disease or diffuse microcalcifications. Int. J. Radiat. Oncol. Biol. Phys. 1993, 27, 567–573. [Google Scholar] [CrossRef]
- Rosenberg, J.; Chia, Y.L.; Plevritis, S. The effect of age, race, tumor size, tumor grade, and disease stage on invasive ductal breast cancer survival in the US SEER database. Breast Cancer Res. Treat. 2005, 89, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Winnike, J.H.; Stewart, D.A.; Pathmasiri, W.W.; McRitchie, S.L.; Sumner, S.J. Stable isotope-resolved metabolomic differences between hormone-responsive and triple-negative breast cancer cell lines. Int. J. Breast Cancer 2018, 2018. [Google Scholar] [CrossRef]
- Ueda, Y.; Kawamoto, K.; Konno, M.; Noguchi, K.; Kaifuchi, S.; Satoh, T.; Eguchi, H.; Doki, Y.; Hirotsu, T.; Mori, M. Application of C. elegans cancer screening test for the detection of pancreatic tumor in genetically engineered mice. Oncotarget 2019, 10, 5412. [Google Scholar] [CrossRef]
- Menezes, M.; Das, S.; Minn, I.; Emdad, L.; Wang, X.-Y.; Sarkar, D.; Pomper, M.G.; Fisher, P. Detecting tumor metastases: The road to therapy starts here. In Advances in Cancer Research; Elsevier: Amsterdam, The Netherlands, 2016; Volume 132, pp. 1–44. [Google Scholar]
- Taube, S.E.; Jacobson, J.W.; Lively, T.G. Cancer Diagnostics. Am. J. Pharm. 2005, 5, 357–364. [Google Scholar] [CrossRef]
- Mordente, A.; Meucci, E.; Martorana, G.E.; Silvestrini, A. Cancer biomarkers discovery and validation: State of the art, problems and future perspectives. In Advances in Cancer Biomarkers; Springer: Berlin, Germany, 2015; pp. 9–26. [Google Scholar]
- Hoda, S.A.; Cheng, E. Robbins Basic Pathology; Oxford University Press US: Oxford, UK, 2017. [Google Scholar]
- Gauvain, K.M.; Garbow, J.R.; Song, S.-K.; Hirbe, A.C.; Weilbaecher, K. MRI detection of early bone metastases in B16 mouse melanoma models. Clin. Exp. Metastasis 2005, 22, 403–411. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pomper, M.; Fisher, P.B. Emerging Applications of Molecular Imaging to Oncology; Elsevier: Amsterdam, The Netherlands, 2014. [Google Scholar]
- Engelhard, K.; Hollenbach, H.; Wohlfart, K.; Von Imhoff, E.; Fellner, F. Comparison of whole-body MRI with automatic moving table technique and bone scintigraphy for screening for bone metastases in patients with breast cancer. Eur. Radiol. 2004, 14, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, G.; Dinter, D.; Reiser, M.F.; Schoenberg, S.O. The uses and limitations of whole-body magnetic resonance imaging. Dtsch. Deutsches Ärzteblatt Int. 2010, 107, 383. [Google Scholar] [CrossRef]
- Fass, L. Imaging and cancer: A review. Mol. Oncol. 2008, 2, 115–152. [Google Scholar] [CrossRef] [PubMed]
- Lind, P.; Igerc, I.; Beyer, T.; Reinprecht, P.; Hausegger, K. Advantages and limitations of FDG PET in the follow-up of breast cancer. Eur. J. Nucl. Med. Mol. Imaging 2004, 31, S125–S134. [Google Scholar]
- Kato, T.; Tsukamoto, E.; Kuge, Y.; Takei, T.; Shiga, T.; Shinohara, N.; Katoh, C.; Nakada, K.; Tamaki, N. Accumulation of [11 C] acetate in normal prostate and benign prostatic hyperplasia: Comparison with prostate cancer. Eur. J. Nucl. Med. Mol. Imaging 2002, 29, 1492–1495. [Google Scholar] [CrossRef] [PubMed]
- Salmon, E.; Ir, C.B.; Hustinx, R. Pitfalls and limitations of PET/CT in brain imaging. Semin. Nucl. Med. 2015, 45, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Halldorsson, S.; Lucumi, E.; Gómez-Sjöberg, R.; Fleming, R.M. Advantages and challenges of microfluidic cell culture in polydimethylsiloxane devices. Biosens. Bioelectron. 2015, 63, 218–231. [Google Scholar] [CrossRef] [PubMed]
- Henry, N.L.; Hayes, D.F. Cancer biomarkers. Mol. Oncol. 2012, 6, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Ellis, I.; Galea, M.; Broughton, N.; Locker, A.; Blamey, R.; Elston, C. Pathological prognostic factors in breast cancer. II. Histological type. Relationship with survival in a large study with long-term follow-up. Histopathology 1992, 20, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Cristofanilli, M.; Budd, G.T.; Ellis, M.J.; Stopeck, A.; Matera, J.; Miller, M.C.; Reuben, J.M.; Doyle, G.V.; Allard, W.J.; Terstappen, L.W. Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N. Engl. J. Med. 2004, 351, 781–791. [Google Scholar] [CrossRef]
- Yu, M.; Bardia, A.; Aceto, N.; Bersani, F.; Madden, M.W.; Donaldson, M.C.; Desai, R.; Zhu, H.; Comaills, V.; Zheng, Z.; et al. Cancer therapy. Ex vivo culture of circulating breast tumor cells for individualized testing of drug susceptibility. Science 2014, 345, 216–220. [Google Scholar] [CrossRef]
- Zhang, L.; Ridgway, L.D.; Wetzel, M.D.; Ngo, J.; Yin, W.; Kumar, D.; Goodman, J.C.; Groves, M.D.; Marchetti, D. The identification and characterization of breast cancer CTCs competent for brain metastasis. Sci. Transl. Med. 2013, 5, 180ra148. [Google Scholar] [CrossRef]
- Fu, Y.; Zhang, Y.; Khoo, B.L. Liquid biopsy technologies for hematological diseases. Med. Res. Rev. 2020. [Google Scholar] [CrossRef]
- Khoo, B.L.; Grenci, G.; Lim, Y.B.; Lee, S.C.; Han, J.; Lim, C.T. Expansion of patient-derived circulating tumor cells from liquid biopsies using a CTC microfluidic culture device. Nat. Protoc. 2018, 13, 34–58. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Chua, S.L.; Khoo, B.L. Worm-Based Microfluidic Biosensor for Real-Time Assessment of the Metastatic Status. Cancers 2021, 13, 873. https://doi.org/10.3390/cancers13040873
Zhang J, Chua SL, Khoo BL. Worm-Based Microfluidic Biosensor for Real-Time Assessment of the Metastatic Status. Cancers. 2021; 13(4):873. https://doi.org/10.3390/cancers13040873
Chicago/Turabian StyleZhang, Jing, Song Lin Chua, and Bee Luan Khoo. 2021. "Worm-Based Microfluidic Biosensor for Real-Time Assessment of the Metastatic Status" Cancers 13, no. 4: 873. https://doi.org/10.3390/cancers13040873
APA StyleZhang, J., Chua, S. L., & Khoo, B. L. (2021). Worm-Based Microfluidic Biosensor for Real-Time Assessment of the Metastatic Status. Cancers, 13(4), 873. https://doi.org/10.3390/cancers13040873