Simple Summary
Glioblastoma (GBM) is the most common form of primary malignant brain tumor with a devastatingly poor prognosis. Tumor heterogeneity (cellular, molecular and immune) is the major obstacle to current treatment failure. We revisited the recent literature to understand the heterogeneous features of GBM and their potential role in treatment resistance. This review provides a comprehensive overview covering the GBM’s pathogenetic features, currently available treatment options and the treatments currently under development in the clinic.
Abstract
Glioblastoma (GBM) is the most common form of primary malignant brain tumor with a devastatingly poor prognosis. The disease does not discriminate, affecting adults and children of both sexes, and has an average overall survival of 12–15 months, despite advances in diagnosis and rigorous treatment with chemotherapy, radiation therapy, and surgical resection. In addition, most survivors will eventually experience tumor recurrence that only imparts survival of a few months. GBM is highly heterogenous, invasive, vascularized, and almost always inaccessible for treatment. Based on all these outstanding obstacles, there have been tremendous efforts to develop alternative treatment options that allow for more efficient targeting of the tumor including small molecule drugs and immunotherapies. A number of other strategies in development include therapies based on nanoparticles, light, extracellular vesicles, and micro-RNA, and vessel co-option. Advances in these potential approaches shed a promising outlook on the future of GBM treatment. In this review, we briefly discuss the current understanding of adult GBM’s pathogenetic features that promote treatment resistance. We also outline novel and promising targeted agents currently under development for GBM patients during the last few years with their current clinical status.
1. Introduction
Glioblastoma (GBM) is one of the most common forms of primary malignant brain tumor and has a very poor prognosis with an average patient survival lasting only 12–15 months [1,2]. This bleak outlook is due in part to the challenges that are presented by the anatomical location of the tumor as well as the heterogeneity of GBM cells and their rapid growth rate [3,4]. Although GBM is known to affect both adults and children, the incidence of GBM increases with age peaking in the 1970s [5]. Cancer incidence is roughly 2-3 individuals per 100,000 cases each year in the United States, with rates increasing slightly based on patient age [6,7,8]. There is also a slightly higher rate of incidence in men versus women, with men being 1.6 times more likely to develop GBM [9]. GBM accounts for approximately 46% of all diagnosed brain tumors and causes around 2.7% of all cancer-related deaths [3]. In fact, it is ranked as the third most common cause of death from cancer in patients between 15 and 34 years [7]. There are currently four grades of gliomas classified by the World Health Organization (grades I-IV) [7,10]. Grade IV gliomas are the most aggressive and invasive forms and are responsible for the poorest prognoses [10,11]. GBM typically refers to these grade IV gliomas and can be subdivided into primary and secondary types [12,13]. Although primary and secondary gliomas share similar histological characteristics, they have very different genetic profiles [14]. Primary GBM constitutes approximately 90% of GBM cases and is considered a de novo pathway of multistep tumorigenesis from glial cells while secondary GBM develops from lower-grade and pre-existing tumors such as diffuse astrocytomas [15]. Of the two, primary GBM is generally found to be more malignant than secondary GBM [16], and men are somewhat more likely to present with primary GBM while women are more likely to be diagnosed with secondary GBM [17].
The standard treatment options for GBM include surgery, chemotherapy, and radiation. However, even with these interventions, GBM still carries a dismal prognosis [18,19]. Diverse pathogenetic features and immunosuppression are two major contributors of current treatment failure. Although many studies have attempted to design effective treatments around these challenges, none have been developed that are capable of achieving long-term patient survival without causing unwanted damage to the delicate cells and neuronal tissues of the brain [4]. Over the past several years, targeted therapies and immunotherapies have shown great achievement in GBM management with promising results in clinical trials [18,19,20,21]. Other therapies in development include nanotechnology-based innovations, photodynamic strategies, gene therapy, and local destruction of the tumor via genetically modified bacteria or controlled hyperthermia. In this review, we discuss the current understanding of GBM’s pathogenetic features (i.e., cellular, molecular, and immunosuppressive properties) that contribute to treatment resistance. We also outline novel targeted therapies, different immunotherapeutic approaches, and a number of other promising/emerging treatment strategies for adult GBM that are currently under development.
2. Pathogenetic Features
2.1. Cellular Heterogeneity, Tumor Vascularity, and Extracellular Matrix
GBM is highly heterogeneous, both intrinsically and intratumorally, with multiple factors driving its development and growth [22,23]. The tumor grows in an infiltrative manner, making it difficult to distinguish and surgically remove from the normal brain [24]. Over half of primary GBMs occur in the four lobes of the brain, with smaller parts also found in the brain stem and spinal cord [7,16,25]. Among those occurring in the brain, approximately 25% are located in the frontal lobe, 20% in the temporal lobe, 13% in the parietal lobe, and 3% in the occipital lobe [25]. However, while primary GBM can develop in any number of locations in the brain, secondary GBM is found primarily in the frontal lobe [12]. GBMs were once thought to be derived from neural stem cells (NSCs), NSC-derived astrocytes, and oligodendrocyte precursor cells (OPCs) [26]. However, new evidence suggests that astrocyte-like NSCs in the subventricular zone (SVZ) are the cell of origin for GBM [27,28,29,30]. Lee and colleagues established that astrocyte-like NSCs harboring low mutational levels can migrate from the SVZ to a distinct site and develop high-grade gliomas [29]. In addition, NSCs in the SVZ were found to carry limited self-renewal abilities, thus, being able to escape replicative senescence and acquire driver mutations over time that play a role in GBM recurrence [29]. The cells that make up GBM are small, polymorphic, and anaplastic. In appearance, they are usually polygonal or spindle-shaped, with indistinct borders and acidophilic cytoplasm [28,31]. These cells are known to have clumped chromatin with distinct nucleoli, oval or elongated nuclei of varying sizes, and an increased nuclear to cytoplasmic ratio. In addition, some cells may be binuclear or multinucleated and may have large lipomatous vacuoles [28,31]. The endothelial cells of GBM are also unique in that they overlap focally and are heterogeneous in appearance. While normal brain endothelial cells lack Weibel–Palade bodies, which are the storage granules of endothelial cells, GBM endothelial cells (GECs) in new vasculature contain many of these granules [31]. Weibel–Palade bodies in GECs secrete von Willebrand Factor (VWF)—a pro-angiogenic factor that is associated with a three-fold higher risk of death in GBM patients [32]. GBM also contains a sub-population of cancer cells that display stem-cell qualities designated GBM stem-like cells (GSCs) [27,33,34]. Differentiation of these GSCs leads to an incredibly diverse population of cell types such as microglia-like cells [35,36,37,38,39] and oligodendrocyte-like cells within the tumor [40,41]. GSCs possess a number of other characteristics including self-renewal, increased proliferation and migration, suppression of immune responses, support of angiogenesis, and increased radio- and chemoresistance [27,33,34]. Because of these abilities and characteristics, GBMs often recur from GSCs and are often distinctly unique from the original glioma with a developed resistance to previously applied treatments [33].
GBM is a highly vascularized tumor [42] that occurs via multiple mechanisms such as vessel co-option, angiogenesis, vasculogenesis, endothelial cell trans-differentiation, and vascular mimicry [43]. Vessel co-option is a non-oncogenic process where tumor cells use nutrients from the pre-existing vasculature of normal/healthy tissues to support tumor growth and tumor cell survival [44]. This process involves the movement of tumor cells along the vasculature and contributes greatly to the infiltrative growth of gliomas [45,46]. Some pre-clinical studies show that vessel co-option is a preferred mechanism of vascularization in early-stage gliomas, which later may or may not switch to the process of angiogenesis (i.e., development of new blood vessels) in GBM [47,48,49]. Angiogenesis in GBM can be induced by different factors such as basic fibroblast growth factor (bFGF), platelet-derived growth factor (PDGF), transforming growth factor-beta (TGF-β), and angiopoietins [50]. However, tumor angiogenesis is oftentimes nonproductive due to abnormal vessels that lead to vascular occlusion [51,52] that causes a hypoxic environment surrounding the failed vessel and ensuring central necrotic tissue [31]. Tumor cells then actively migrate away from this central hypoxia, resulting in the formation of a zone of hypercellular tissue surrounding an area of necrosis (defined as a pseudopalisade). Pseudopalisades secrete hypoxia-inducible factor (HIF-1), vascular endothelial growth factor (VEGF), hepatocyte growth factor (HGF), matrix metalloproteases (MMPs), and interleukin-8 (IL-8) that promote microvascular proliferation, angiogenesis, and tumor expansion [51]. Clinically, pseudopalisades are thought to be a major contributor to the rapid disease progression and poor survival rate and response to treatment [53]. GBM endothelial cells can be derived from bone marrow (BM)-derived endothelial progenitor cells (EPCs) or GSCs through vasculogenesis [54] and endothelial cell trans-differentiation, respectively [37,55]. GSCs also possess an ability to form a non-endothelial tube-like structure that mimics the tumor vasculature and supports tumor growth—a process otherwise known as vascular mimicry that is associated with poor prognosis in GBM patients [56,57]. Importantly, the steps of GSC-induced tumor vascularization can also be potentiated by GBM standard-of-care interventions [58,59]. For instance, traditional temozolomide (TMZ) chemotherapy can promote neovascularization by inducing chemotherapeutic stress, which helps transdifferentiate GSCs into endothelial cells and vascular mimicry [58]. Similar to TMZ, ionizing radiation also promotes GSCs to transdifferentiate themselves into endothelial cells through the Tie2 signaling pathway [59]. GSCs may also survive or be enriched by anti-angiogenic therapy, leading to tumor recurrence and anti-angiogenic therapy (AAT)-resistant tumors [60,61].
The relative volume of extracellular space in the normal brain is about 24%, which can be increased by up to 48% during the progression from lower to higher-grade gliomas [62]. The expansion of this extracellular space is thought to create an ideal environment for tumor migration and invasion [62]. GBM cells produce different ECM components such as hyaluronic acid (HA), matrix metalloproteinase-2 (MMP2), matrix metalloproteinase-9 (MMP9), integrins, tenascin-C [63], and fibronectin [64] to degrade and remodel the ECM for tumor invasion [65,66,67]. Adhesion of tumor cells to ECM proteins is regulated by transmembrane receptors known as integrins like αvβ3 and αvβ5. Both receptors are highly expressed on GBM cells and GBM endothelial cells [68], and their overexpression is associated with poor overall survival (OS) in GBM patients solely treated with standard chemotherapy [69]. Inhibiting integrins blocks angiogenesis, tumor invasion, stemness, and immunosuppression [68]. In addition, integrins such as α5β1 are overexpressed in GBM tumors that have reduced p53 activity due to TMZ treatment. The negative cross-talk between α5β1 and p53 attributes to TMZ resistance [70]. GBM cells communicate with surrounding tumor and non-tumor cells by secreting special structures known as extracellular vesicles (EVs) that are often encapsulated with different factors to promote tumor growth [71,72]. For example, EVs derived from GBM cells stimulate normal astrocytes to be converted into a tumor-supportive phenotype via p53 and MYC signaling pathways, leading to ECM destruction [71]. GSCs secrete different pro-angiogenic factors such as VEGF or miRNAs such as miR-21 or miR-26a in EVs to promote angiogenesis [73,74,75] and facilitate tumor growth under hypoxic conditions [76]. The mRNA and protein expression of numerous invasion-associated ECM molecules such as Fms-related tyrosine kinase 4 (FLT4), mouse double minute 2 homolog (MDM2), and MMP-2 vary between different GBM subtypes, and such ECM compositions can be used as prognostic indicators for patients with GBM [77].
2.2. Molecular Heterogeneity
The Cancer Genome Atlas (TCGA) Project created a GBM classification system based on 600 genes that were sequenced from 200 human tumor samples [78]. This system has shed new light on the complexity of the genetic profile of GBM and led to the discovery that molecular alterations can be used to distinguish primary and secondary gliomas. Primary and secondary GBM are histologically similar but genetically and epigenetically different. Primary GBMs, which are also designated as isocitrate dehydrogenase wild-type GBMs (IDH-WT), are characterized by (i) overexpression of epidermal growth factor receptor (EGFR), (ii) mutated telomerase reverse transcriptase promoter, p53, and phosphate and tensin homologue (PTEN) genes, and (iii) loss of chromosome 10q and cyclin-dependent kinase inhibitor 2A (CDKN2A) gene [11,79]. Secondary GBMs (also referred to as IDH-mutant GBMs) typically harbor mutations in p53 and isocitrate dehydrogenase 1 (IDH1) genes alongside the loss of chromosome 19q [79]. IDH-WT GBMs are more common, respond poorly to treatment, and have an overall lower survival rate than IDH-mutant GBMs [79,80].
GBM can also be categorized into four tumor subtypes: proneural, mesenchymal, classical, and neural [81]. The signature gene alternations for each subtype are listed in Table 1. Briefly, proneural GBM is characterized by the alteration of platelet-derived growth factor receptor alpha (PDGFRA) and mutated IDH1 along with higher expression of certain proneural markers (e.g., SRY-related HMG-box genes [SOX], doublecortin [DCX], delta-like canonical Notch ligand 3 [DLL3], achaete-scute family BHLH transcription factor 1 [ASCL1], transcription factor 4 [TCF4] and oligodendrocytic development genes (e.g., PDGFRA, NK2 homeobox 2 [NKX2-2], oligodendrocyte transcription factor 2 [OLIG2] [81,82,83]. Mesenchymal GBM is featured by focal hemizygous deletions of a chromosomal region at 17q11.2, co-mutations in neurofibromin 1 (NF1) and PTEN genes, and enrichment of tumor necrosis factor (TNF) super family pathway and nuclear factor kappa B (NF-κB) pathway genes (e.g., TNFRSF1A associated via death domain [TRADD], v-rel avian reticuloendotheliosis viral oncogene homolog B [RELB], TNF receptor superfamily member 1A [TNFRSF1A]) [81]. Mesenchymal GBM is also linked to higher expression of mesenchymal and astrocytic markers such as CD44 or c-mer proto-oncogene tyrosine kinase (MERTK), which promote epithelial–mesenchymal (EMT) transition [81,84]. Classical GBM is characterized by amplification of chromosome 7 and loss of chromosome 10 [81]. Overexpression of EGFR, absence of TP53 mutations and focal 9p21.3 homozygous deletion that targets cyclin-dependent kinase inhibitor 2A (CDKN2A) are also predominant features in classical GBM [81]. The common markers for classical GBM include the neural precursor and stem cell markers nestin, notch (neurogen locus notch homolog protein 3 [NOTCH3], jagged canonical notch ligand 1 [JAG1], O-Fucosylpeptide 3-Beta-N-Acetylglucosaminyltransferase [LFNG] and sonic hedgehog (smoothened, frizzled class receptor [SMO], growth arrest-specific 1 [GAS1], GLI family zinc finger 2 [GLI2])) [81]. Lastly, the major properties of neural GBM include neuron projection, axon and synaptic transmission, and expression of neuronal markers such as neurofilament light (NEFL), gamma-aminobutyric acid type A receptor subunit alpha1 (GABRA1), synaptotagmin 1 (SYT1), and solute carrier family 12 member 5 (SLC12A5) [81].

Table 1.
Glioblastoma (GBM) molecular subtypes.
The distinction of the four GBM subtypes by the TCGA also revealed common themes in altered-signaling pathways that dictate cell growth and regulation, DNA repair, and apoptosis (Figure 1) [78,79,80]. In a TCGA-based study, GBM specimens routinely contained aberrations in the following signaling pathways: p53 (87%), RB (78%), and RTK/Ras/PI3K (88%) [78,79,85]. Disruptions in p53 signaling are associated with increased cell migration, invasion, and survival [86]. The Rb pathway is controlled by phosphorylation of Rb by cyclin D, cyclin-dependent kinase 4 (CDK4), or CDK6 [87]. CDK4 and CDK6 are further regulated by CDK inhibitors (CDKN2A, CDKN2B, CDKN2C), and mutations to CDKN2A or Rb1 cause cell cycle disruption, allowing uncontrolled cell proliferation and apoptosis evasion [87]. RTKs (once activated) regulate cell proliferation, differentiation, angiogenesis, and survival through either downstream PI3K/AKT/mTOR or Ras/MAPK/ERK signaling [88]. Mutations or alterations in genes that encode RTKs (e.g., epidermal growth factor receptor [EGFR], vascular endothelial growth factor [VEGF], or insulin-like growth factor 1 receptor [IGF-1R]), lead to unregulated cell proliferation and survival [80,88].
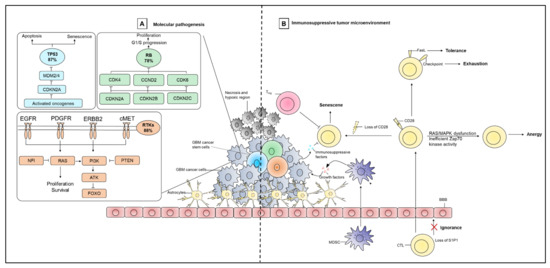
Figure 1.
Characteristics of the GBM tumor microenvironment. (A) Three dominant molecular alterations in GBM include P53, retinoblastoma (Rb), and receptor tyrosine kinase (RTK) signaling pathways and their corresponding frequencies in GBM. (B) The GBM tumor microenvironment (TME) is immunosuppressed mainly due to presence of dysfunctional T cells and myeloid-derived suppressor cells (MDSCs). T cell dysfunction results from different mechanisms that involve senescence, tolerance, anergy, exhaustion, and ignorance. T cells senescence is associated with loss of the co-stimulator CD28, which may occur as a result of telomere damage, presence of Tregs, or TME metabolic stress. Tolerance of T cells is mediated by FasL-induced apoptosis, recruitment of Tregs, and upregulation of factors that limit T cell effector functions. T cell anergy occurs due to RAS/MAPK dysfunction or inefficient Zap70 kinase activity. Co-expression of immune checkpoint molecules such as PD-1, LAG-3 and TIM-3 are also important contributors to T cell exhaustion in GBM. The loss of sphingsosine-1-phosphate receptor 1 (S1P1) is associated with T cell ignorance. MDSCs release immunosuppressive growth factors and other mediators to support growth of cancer cells. Cancer cells can also secret a number of immunosuppressive factors to maintain their overall survival.
2.3. Immunosuppressive Features
The CNS is separated from the circulatory system by the blood–brain barrier (BBB) that effectively prevents the passive diffusion of molecules. The CNS was previously viewed as an immune-privileged site [89]. However, recent discoveries have identified functional lymphatic vessels in the brain’s dura matter that drain cerebrospinal fluid into cervical lymph nodes [90,91]. In this way, tumor-derived antigens can concentrate within lymph nodes to stimulate immune cell responses involving T cells [90]. However, after infiltrating into the GBM tumor, T cells become dysfunctional via different mechanisms that could include senescence, tolerance, anergy, exhaustion, or ignorance that ultimately leads to poor GBM prognosis (Figure 1) [92]. T cell senescence is associated with the loss of the co-stimulatory molecule CD28, which could occur as a result of telomere damage, regulatory T cell (Treg) interaction, or metabolic competition in the tumor microenvironment (TME) [93,94]. T cell tolerance is mediated by Fas ligand (FasL)-induced apoptosis, recruitment of Tregs, and upregulation of factors that dampen T cell effector function such as cytotoxic T-lymphocyte antigen-4 (CTLA-4), indoleamine 2,3-dioxygenase 1 (IDO-1), and signal transducer and activator of transcription 3 (STAT3) [92,95,96,97]. Anergy is mainly observed in infiltrating CD4+ T cells and is due in part to RAS/MAPK dysfunction or inefficient Zap70 kinase activity that impairs interleukin-2 (IL-2) production (and, hence, T cell activation/proliferation) [92]. The co-expression of immune checkpoint molecules such as programmed cell death 1 (PD-1), lymphocyte-activation gene 3 (LAG3), and T-cell immunoglobulin mucin-3 (TIM-3) are also important contributors to T cell exhaustion in GBM [92]. For example, GBM may express PD-L1 and induce T cell exhaustion through interaction with its cognate receptor PD-1 expressed by T cells [98,99]. T cell ignorance as a result of sphingsosine-1-phosphate receptor 1 (S1P1) loss that prevents T cells from trafficking to the tumor site, disrupting T cell-mediated anti-tumor immunity and contributing to tumor progression [92]. Additionally, T cell function can be further impaired through the GBM vasculature instituting physically-induced constraints (e.g., hypoxia) and promoting infiltration of immunosuppressive immune cells (such as macrophages, neutrophils, and myeloid-derived suppressor cells [MDSCs]) [100]. Such immune cell subsets release pro-inflammatory mediators and cytotoxic cytokines, growth factors, bioactive lipids, hydrolytic enzymes, matrix metalloproteinases, reactive oxygen intermediates, and nitric oxide to support the continued growth of cancer cells [100]. GBM cells also release a number of immunosuppressive factors, including TGF-β, prostaglandin E (PGE), interleukin-1 (IL-1), interleukin-10 (IL-10), and fibrinogen-like protein 2 (FGL2) that suppress DC priming/activation of immune effector cells [101]. Tumor-derived immunosuppressive factors also help recruit pro-tumoral M2 macrophages and Tregs, which further secrete TGF-β1 and IL-10 and eventually suppress T cell effector functions [102,103,104,105]. In a recent study, an extensive level of tumor immunosuppression in GBM (excluding grade II and III gliomas), is found due to the presence of blood-derived macrophages, tumoral expression of programmed cell death ligand 1 (PD-L1), and T cell expression of PD-1 [106]. The same study reported that bone marrow-derived macrophages migrate to the tumor site and accumulate centrally in GBM lesions, exerting strong immune suppression by releasing iron that is necessary to maintain tumor cell survival and tumor progression. In contrast, resident microglia exert little to no immunosuppressive function [106].
3. Current Treatment
3.1. Standard of Care and Other FDA Approved Treatments
The current standards of care for GBM include maximal resection surgery, radiation, and temozolomide (TMZ) therapy—with TMZ and radiation being commenced within 30 days post-surgery [107,108,109]. Unfortunately, GBM response to TMZ varies between patients, and many types of GBM carry resistance to the compound. Treatment resistance to standard therapies is likely due to a combination of upregulated DNA repair mechanisms and the presence of GSCs that maintain an ability to self-renew and differentiate [110]. TMZ resistance also appears to be driven by the DNA repair enzyme O6-methylguanine-DNA methyltransferase (MGMT) that repairs DNA alkylation since patients bearing MGMT genes with methylated promoters seem to be more responsive to TMZ treatment [110]. However, TMZ damages both tumor and normal cells and does not eliminate GBM, so, options for alternative treatments are desperately needed [4].
Other treatments approved by the FDA for use in GBM therapy include bevacizumab and tumor-treating fields (TTFs). Bevacizumab is a humanized monoclonal antibody (mAb) that targets the angiogenic factor VEGF and was the first anti-angiogenic drug approved for patient use after showing increased overall survival in colorectal and non-small-cell lung cancers when combined with chemotherapy [111,112]. In addition, the antibody was observed to be safe in patients and mediate effective anti-tumor responses in Phase II clinical trial for recurrent GBM when combined with the chemotherapeutic drug irinotecan [113]. However, in two Phase III trials conducted in newly diagnosed GBM patients, treatment with bevacizumab in addition to either radiation or radiation plus TMZ showed no significant difference in overall survival compared to the placebo [114,115]. Although progression-free survival was better with treatment, patients suffered a higher frequency of adverse events and poorer quality of life [115].
TTFs are a non-invasive and anti-mitotic FDA-approved strategy for newly diagnosed cases of GBM (i.e., as adjuvant therapy) or recurring disease [116,117]. It involves using alternating electrical fields with a frequency range of 100-300 kHz and an intensity of 1 to 3 V/cm to interfere with the functions of rapidly dividing cancer cells, causing cessation of cell division and ultimately leading to cell death [116,118]. The theory behind this treatment is that the electrical fields create space between the growing ends of microtubules and tubulin dimers, thus, interfering with microtubule polymerization of the mitotic spindle [117]. Recently, TTF has been tested in combination with current standard-of-care in newly diagnosed GBM patients. Concurrent administration of TTF/radiation/TMZ followed by adjuvant TMZ/TTF demonstrated safety and promising preliminary efficacy [119], which warrants further clinical investigation in a larger patient cohort. In fact, a clinical study is currently ongoing utilizing this triple combination in 60 newly diagnosed GBM patients (NCT03869242).
3.2. Hurdles with Current Treatments
There are many aspects that make GBM difficult to effectively treat [120]. One complication is enabling the treatment drug to cross the BBB and reach the tumor [108]. It was previously thought that the BBB was uniformly disrupted in cases of GBM, and was, therefore, not an issue when designing treatment plans [108]. However, recent evidence suggests that a large portion of the BBB remains intact, presenting a challenge to many drug therapies [108]. Considering that drug molecules are unable to reach the tumor to potentiate effects, BBB transporters often remove most of the molecules that do manage to pass through [4].
The infiltrative and invasive growth of GBM also impedes complete surgical resection of tumor cells. Thus, secondary treatment is usually needed following surgery [121,122]. In addition, most GBMs that initially respond well to treatment recur after a period of a few months. Relapsed tumors generally have an even poorer overall survival and do not respond well to previously used treatments [123] as they acquire new mutations and evasive properties [124]. Tremendous efforts have been made to target those mutations by targeted therapies (discussed in more detail below).
Other major reasons for treatment failure can include: (i) GBM is extremely immunosuppressive [125,126,127], (ii) tumor cells contain a low somatic mutational load [78,128], which could explain poor responses to immune checkpoint blockade to the anti-PD-1 antibody (CheckMate-143), and (iii) presence of GSCs that help drive resistance to radiotherapy [129,130], chemotherapy [131], and anti-VEGF therapy [132]. Although TMZ is effective against MGMT-negative GSCs [133], the drug is incapable of eliminating MGMT-positive GSCs [134]. Resistance to TMZ in particular and other therapies mediated by GSCs also rely on an ability to regulate various miRNA molecules that can remodel different signaling pathways in response to treatment [135,136]. GSC plasticity also allows differentiation into a slow-cycling and persistent cellular state that can escape cytotoxicity from different targeted therapies [137]. Treatment-resistant GSCs further induce immunosuppression by recruiting M2-like tumor-associated macrophages (TAMs) and Tregs into the TME [138,139]. Lastly (iv), while effective anti-tumor immunity in GBM is profoundly inhibited, possibly by promoting subsets of dysfunctional T cells through various mechanisms [92], it is important to note that current standard therapies (including TMZ and high-dose corticosteroids) might worsen GBM’s immunosuppressive status [140,141,142]. Thus, there is a need to develop newer forms of immunotherapy that overcome immunosuppression and boost the host’s anti-tumor immune responses [121].
4. Treatments in Development
4.1. Targeted Therapies
Based on dysregulated signaling in GBM, targeted therapies are mainly categorized to ablate: the p53, RB, and receptor tyrosine kinase (RTK) signaling pathways (Figure 2). In general, intra- and inter-tumoral heterogeneity of mutated signaling pathways in GBM incite resistant mechanisms to monotherapeutic treatment with targeted agents. While combination therapies to target multiple pathways is one potential route to overcoming resistance, developing improved better strategies to impact each individual mutational alteration in GBM are gaining interest [121].
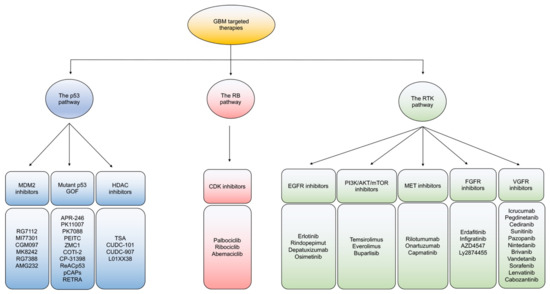
Figure 2.
An overview of targeted therapies in GBM. Classification of current targeted therapies in GBM according to the three main signaling pathway alterations of the P53, Rb, and RTK pathways.
4.1.1. Targeting the p53 Pathway
The P53 pathway (including CDKN2A, MDM2 and TP53) is dysregulated in 85% of GBM tumors [78], with prevalence varying between different GBM molecular subtypes (proneural 54%, mesenchymal 32%, neural 21%, and classical 0%) [81]. Current options to target the p53 pathway in GBM include inhibiting the MDM2/p53 complex, restoring wt-p53 conformation and gain of function (GOF), and degrading Mut-p53 [86].
Inhibiting MDM2/p53 interaction to reactivate the anti-tumor function of p53 offers a promising approach for patients, with many MDM2 inhibitors currently in development for GBM preclinically (such as RG7112, MI77301, CGM097, and MK8242) and clinically (such as RG-7388 and AMG-232). AMG-232 treatment, either as a monotherapy or in combination with other chemotherapies, markedly increases activation of p53 signaling in tumors, making the drug the most potent MDM2 inhibitor to date [143]. Clinical trials for MDM2 inhibitors are currently running in both newly diagnosed and recurrent GBM patients (Table 2).
Table 2.
Current on-going clinical trials of targeted therapies in GBM within the last 5 years.
Cancer-associated p53 mutation is often a single amino acid substitution that favors cancer cell survival and tumor progression [144]. Since mutant p53 is associated with malignant progression, chemoresistance, invasion, cancer maintenance, and metastasis, GOF p53 mutations represent promising targets for novel cancer therapy development [145]. PRIMA-1 and its structural analogue PRIMA-1MET (APR-246) are the most extensively studied compounds that restore wt-p53 conformation and p53 function [86]. Both drugs induce p21 expression, cell cycle arrest and apoptosis, and inhibit GBM stemness and growth [146,147,148,149]. While PRIMA-1 and APR-246 have been successfully tested in different cancer types, there is no clinical trial of APR-246 reported so far in GBM. Other drugs in this class include PK11007, PK7088, PEITC, ZMC1, COTI-2, CP-31398, small peptides (ReACp53 and pCAPs), and RETRA [86,145].
Another p53 targeting approach involves degrading mutant p53 by inhibiting interactions with the heat shock proteins (Hsp) Hsp70 and Hsp90. Histone deacetylase 6 (HDAC6) is required to form the mutant p53-Hsp70/Hsp90 complex. Thus, histone deacetylase inhibitors (HDACi) (such as trichostatin A, CUDC-101, CUDC-907, and vorinostat) used either alone or in combination with other drugs can disrupt this mutant p53-Hsp complex, resulting in the degradation of mutant p53 in GBM [150,151,152,153]. Detailed mechanisms and clinical significance of HDACi in GBM have been previously discussed [154]. Current clinical studies are focused on exploring the synergistic combination of HDACi and chemotherapy and/or radiotherapy in GBM patients as listed in Table 2.
4.1.2. Targeting the Rb Pathway
The genes that are mostly altered in the RB pathway in GBM include CDK4, CDK6, CCND2, CDKN2A/B, and Rb1 [78]. The tumor suppressor pRb plays a crucial role in cell cycle progression through the regulation of cyclin-dependent kinases (CDKs) at the G1/S phase. Although mutation of cycD1-CDK4/6-Rb1 occurs in approximately 80% of GBM cases and is one of the top three most altered pathways [78,155], the essential function of this pathway in normal healthy cells has limited its potential as a target for GBM treatment. CDK inhibitor such as palbociclib directly suppresses phosphorylated Rb1 and induces cell cycle arrest and apoptosis [156] and include drugs such as palbociclib, ribociclib or abemaciclib that are FDA approved for other cancer types and are currently being clinically evaluated in GBM (Table 2) [157,158]. Palbociclib alone has not been effective in patients with recurrent GBM [157], indicating combination with other therapies is likely required to improve therapeutic outcomes [156,159]. In fact, combined CDK4/6 inhibition with radiotherapy resulted in an improved survival advantage over monotherapies in a mouse model of GBM, which was associated with a significant increase in γH2AX (a marker for DNA damage) and cleaved poly (ADP-ribose) polymerase (PARP) (a measure for apoptosis) [156]. Combined CDK4/6 and mTOR inhibition has also resulted in disruption of GBM metabolic pathways, leading to significant induction of apoptosis compared to single treatments [159].
4.1.3. Targeting the RTK Pathway
Epidermal Growth Factor Receptor (EGFR) Inhibitors
EGFR mutations occur at high frequencies (57.4%) in GBM [78] and indicate poor prognosis [160]. The bulk of EGFR mutations in GBM are largely classified as EGFRvI (N-terminal deletion), vII (deletion of exons 14–15), vIII (deletion of exons 2–7), vIV (deletion of exons 25–27), and vV (deletion of exons 25–28) [161]. Among these mutants, vII/vIII are oncogenic [161], and vIII is the most common variant in GBM [78,162]. Unfortunately, previous attempts targeting EGFR in GBM have not been successful and may be a result of the diverse molecular features of the molecule [163]. In addition, Kwatra and colleagues [164] outlined the major flaws of failed clinical trials with first- and second-generation EGFR tyrosine kinase inhibitors (EGFR-TKIs): (i) EGFR-TKI gefitinib blocks the cell-surface receptor but does not abrogate downstream signaling, promoting alternate tumor growth signaling pathways; (ii) Failed clinical trials included both wild-type and EGFR-activated GBM patients instead of patients with activated EGFR or EGFRvIII; and (iii) Most EGFR-TKIs (e.g., erlotinib, gefitinib, afatinib, and lapatinib) are poorly penetrant to the brain [165]. However, a third-generation EGFR-TKI, AZD9291, has recently been approved for the treatment of non-small cell lung cancer, irreversibly binds with high affinity to EGFRvIII, and is ~10 times more efficient than first-generation EGFR inhibitors in inhibiting tumor cell proliferation in an orthotopic GBM model [166]. These findings suggest AZD9291 efficiently crosses the BBB and could be a useful EGFR-TKI for GBM patients.
Overall, current clinical approaches to specifically targeting EGFR and EGFRvIII include EGFR-TKIs, unmodified anti-EGFR antibodies (e.g., cetuximab, panitumumab, and nimotuzumab), engineered anti-EGFR antibodies (e.g., conjugated with a radioactive isotope [125I mAb 425] [167], antibody–drug conjugate (e.g., depatuxizumab mafodotin) [168], bispecific format [bscEGFRvIIIxCD3]), anti-EGFRvIII vaccines (CDX-110), EGFRvIII-specific CAR-T cells, and RNA-based formulations that have been described elsewhere [162]. In general, strategies directed against EGFR/EGFRvIII have not yet gained clinical benefits in a majority of patients and would likely benefit from a clearer understanding of the molecular dynamics of EGFR/EGFRvIII cancer cell signaling [162].
Phosphatidylinositol-3-Kinase/AKT/Mammalian Target of Rapamycin (PI3K/AKT/mTOR) Inhibitors
PI3K/AKT/mTOR is an important signaling pathway to regulate cell growth, motility, survival, metabolism, and angiogenesis [169,170]. Impairment in PI3K/AKT/mTOR pathway in GBM can be caused by different mechanisms such as loss/inactivation of PTEN, mutation/amplification of PIK3CA, and activation of RTKs or oncogenes upstream of PI3K [78,171]. Although more than 50 PI3K inhibitors have been tested in various types of cancer, a limited number of drugs have entered into clinical trials for GBM patients [172]. Development of resistance to PI3K inhibition, reduced BBB permeability, and poor safety profile of PI3K inhibitors are major contributors to a less than pronounced clinical translation [171].
AKT stands as a midpoint between upstream and downstream regulation of cell growth and survival signals—the main mechanism of resistance to chemo- and radiotherapy, making its inhibition an attractive target for GBM [173]. AKT inhibitors can be sub-divided into lipid-based phosphatidyl-inositol analogues, ATP-competitive inhibitors, and allosteric inhibitors [173]. However, the AKT pathway is complex and promiscuous and contains different AKT isozymes that differ in function and tissue distribution, making any promise of selective AKT drugs difficult to realize. For example, inhibiting AKTs as a single agent has shown marginal effects in a phase II trial (NCT00590954). Disrupting mTOR, which controls downstream targets of phosphorylated Akt that regulate protein synthesis, cancer cell survival, invasiveness and GSC maintenance, chemotherapy resistance, and angiogenesis, is another attractive approach for GBM [174]. In fact, combining mTOR inhibitors with other therapies such as CDK4/6 [159] or MDM2 blockade [175] has provided some promise in GBM treatment. Along with instigating direct anti-proliferative activities following mTOR inhibition, mTOR disruption offers another potential anti-tumor mechanism [176]. For instance, mTOR is activated in 39% of tumor-associated microglial cells (as tested in 42 human GBM tumor specimens) but is downregulated following treatment with an mTOR inhibitor, which suggests mTOR disbarment in GBM patients might reduce the frequency of pro-tumorigenic M2-type macrophages [176]. However, the direct role of mTOR inhibitors on the immune system is controversial, since mTOR blockade can lead to either immunosuppressive or immunomodulatory effects depending on the cell types and nature of stimuli involved [177,178]. Further research is obviously warranted to possibly exploit mTOR inhibition for the improvement of GBM immunity.
Hepatocyte Growth Factor Receptor (HGFR/c-MET) Inhibitors
Interaction of hepatocyte growth factor (HGF) to MET triggers several downstream signaling pathways and promotes carcinogenesis [179]. MET amplification is detected in 1.6–4% of GBM patients [78,155] and its expression is associated with poor prognosis [180,181]. MET targeted therapies can either be mAb or small molecule inhibitors. Despite a better understanding of MET signaling and its associated resistance mechanisms, the clinical benefit of anti-MET therapies remains minimal. Results from a recent randomized, double-blinded, placebo-controlled, multicentered phase II study in patients with recurrent GBM showed that the anti-MET mAb onartuzumab failed to provide additional clinical benefit when added alongside bevacizumab [182].
Fibroblast Growth Factor Receptor (FGFR) Inhibitors
The FGF-FGFR signaling pathway regulates many biological functions, including cell proliferation, survival, and cytoskeletal regulation [183]. FGFR mutations are found in 3% of GBM cases and associated with poor overall survival in GBM patients treated with chemoradiation [184]. The heterogeneous expression of different FGFRs (type 1-4) in GBM, along with a lack of understanding of FGFR’s contribution to GBM progression make this target’s outlook for GBM less clear. Interestingly, recent research finds that GBM upregulates the FGFR pathway in response to dual blockade of MET and EGFR [185], suggesting that a more comprehensive drug cocktail (i.e., inhibiting FGFR, MET, and EGFR) might provide patients added benefit.
Vascular Endothelial Growth Factor Receptor (VEGFR) Inhibitors
Since GBM is a highly vascularized tumor, disrupting the tumor vasculature by targeting the VEGF/VEGFR pathway represents an attractive approach to control GBM progression. The VEGF gene family consists of six secreted ligands, including VEGF-A, -B, -C, -D, and placental growth factors (PIGF) 1 and 2 [186]. These ligands bind their corresponding receptors (i.e., VEGFR-1, -2, and -3) and trigger angiogenesis, vasculogenesis, or lymphangiogenesis [186]. VEGF can be blocked using the anti-VEGF mAb bevacizumab or trapped by a soluble decoy receptor such as aflibercept. VEGFR signaling could also be inactivated by an interfering interaction with its ligand by icrucumab [VEGFR-1 inhibitor], ramucirumab [VEGFR-2 inhibitor] or tyrosine kinase activation (e.g., cediranib, sunitinib, pazopanib) [50]. The current clinical trials employing angiogenic VEGFR inhibitors in GBM are listed in Table 2. Although VEGFR inhibitors are being extensively studied, a recent meta-analysis on approximately 2000 GBM patients revealed that an anti-VEGF mAb in combination with standard therapy did not improve the OS of GBM patients compared to standard therapy alone [187]. GBM often develops resistance to vascular inhibitor drugs [188]. Essentially, a better understanding of acquired GBM resistance to VEGF/VEGFR targeted therapies is needed to select an optimal combined therapy to improve therapeutic outcomes.
Platelet-Derived Growth Factor Receptor (PDGFR) Inhibitors
PDGF amplifications are observed in 13% of GBM cases, which makes them the second most frequent somatic alteration in GBM after EGFR [78]. GBM expresses all isoforms of PDGF ligands including PDGF-A, -B, -C, and -D. These growth factors bind their transmembrane receptors PDGFR-α or -β and initiate downstream autophosphorylation events [189] that contribute to various physiological mechanisms of GBM progression including the transformation of glial cells into stem cells, angiogenesis, lymphangiogenesis, and immunosuppression [189,190]. Overexpression of PDGFR in GBM is oftentimes associated with a poor prognosis [191]. Although pre-clinical efficacy of PDGFR inhibitors is compelling, PDGFR inhibitors are facing clinical hurdles in patients since GBM cells often induce a number of signaling pathways such as receptor tyrosine-protein kinase erbB-3 (ERBB3), insulin-like growth factor 1 receptor (IGF1R), and transforming growth factor-beta receptor II (TGFBR2) that provoke resistance to PDGFR abrogation [192,193].
4.1.4. Targeting Other Pathways
Transforming Growth Factor-Beta (TGF-β) Inhibitors
TGF-β is a pleiotropic cytokine involved in GBM angiogenesis, proliferation, progression, and invasion [194]. High expression of TGF-β is associated with poor prognosis in newly diagnosed GBM patients rather than individuals with recurrent disease [194]. Due to TGF-β′s complex cross-talk with other signaling pathways such as Wnt, Notch, Hippo, MAPK, PI3K-Akt and NF-κB/IKK [195], TGF-β inhibition has not yet gained clinical benefits. In a recent phase Ib/IIa trial, the addition of galunisertib (a small molecule TGF-β inhibitor) with TMZ and radiotherapy in newly diagnosed GBM patients did not improve progression-free survival (PFS) or OS [196].
Proteasome Inhibitors
Proteasomal inhibition is another approach for GBM treatment intervention [197,198]. The over-expression and enhanced activity of the proteasome are observed in GBM cells following radiotherapy and subsequent proteasome inhibition prevents GBM recurrence [199]. Proteasome inhibitors that have gained access in GBM clinical studies include bortezomib, ixazomib, and marizomib [200]. However, results from bortezomib clinical trial are disappointing as the drug does not cross the BBB [200].
DNA Damage Response (DDR) Inhibitors
GSCs upregulate various DNA repair proteins such as ataxia telangiectasia mutated (ATM), ataxia telangiectasia and Rad3-related (ATR), or poly (ADP-ribose) polymerase 1 (PARP-1) that facilitate GBM resistance to chemotherapy and/or radiotherapy [201,202]. Therefore, inhibition of these DDR proteins offers an attractive therapeutic approach for GBM patients to overcome resistance to the current standard-of-care. In fact, ATM inhibition defeats DDR-mediated resistance and significantly radio-sensitizes GBM preclinically [203]. The current number of clinical trials with ATM/ATR inhibition in GBM is limited (Table 2). The PARP inhibitors (PARPi) such as olaparib, veliparib, pamiparib are also currently being tested in a number of clinical trials either as single agents or in combination with other therapies (Table 2).
4.2. Immunotherapy
4.2.1. Immune Checkpoint Inhibitors (ICIs)
Anti-PD-1/PD-L1 Antibodies
A number of newer therapies in development constitute a major area in immunotherapy (Figure 3) [20,21,204,205,206,207]. GBM is an immunosuppressive tumor, with varying degrees of PD-L1 expression in tumor cells in GBM patients ranging from 61% to 88% [208]. Blocking PD-1/PD-L1 interactions unleash anti-tumor immune responses in various cancers such as melanoma. In GBM, although preclinical experience with PD-1/PD-L1 inhibition is quite promising [209,210,211,212], the clinical outcome of PD-1 blockade has been disappointing. For instance, the first large-scale randomized trial in recurrent GBM (CheckMate-143) revealed no significant difference in overall survival between patients receiving bevacizumab or nivolumab (an anti-PD-1 mAb), leading to premature termination of the nivolumab arm [213]. The inability of ICIs to cross the BBB, reduced frequency of immune infiltrates in the GBM TME, and a high level of GBM immunosuppression are considered major contributors of treatment failure for this approach in general [213]. In addition, the TME of PD-1 blockade non-responders is enriched with PTEN mutations regardless of GBM subtype, suggesting that combined targeting of PTEN and PD-1 could provide additive treatment benefits for this lethal disease [214]. Although adjuvant monotherapy of anti-PD-1 mAb failed to generate effective anti-tumor immunity, neoadjuvant PD-1 blockade led to the activation of GBM-specific T cells and downregulation of genes associated with the tumor cell-cycle. Therefore, timing of anti-PD-1/PD-L1 interventions in patients is probably crucial for mediating objective response rates and managing GBM [215].
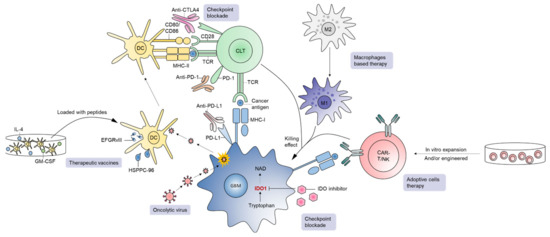
Figure 3.
A brief overview of immunotherapies in GBM. Current immunotherapeutic approaches in GBM include checkpoint blockade, oncolytic virus, therapeutic vaccines, adoptive cell therapy, and macrophage-based strategies.
Anti-CTLA-4 Antibody
Cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) is found on antigen-presenting cells (APCs) and Tregs, although the prognostic role of CTLA-4 expression in cancer patients is controversial [216]. A recent study demonstrated that increased expression of CTLA-4 in the GBM TME positively correlated with elevated expression of specific gene signatures in immune cells such as CD8+ T cells, Tregs, and macrophages, suggesting a greater immune cell infiltration in tumors with higher CTLA-4 expression [97,217]. Patients with increased levels of CTLA-4 are also likely to beneficially respond to CTLA-4 blockade. The same study concluded that glioma patients with lower CTLA-4 expression have significantly longer OS rates [97]. Relatedly, the safety and efficacy of anti-CTLA4 mAb therapy (e.g., ipilimumab) have been demonstrated in melanoma patients harboring brain metastasis, suggesting promise for individuals with GBM [217,218,219]. Consistent with convincing reports from preclinical studies [217,219], anti-CTLA4 treatment is currently showing encouraging results in clinical trials. In one study in recurrent GBM, ipilimumab in combination with GM-CSF and bevacizumab resulted in partial responses (31%) and stable disease (31%) in select patients [220]. Currently, several clinical trials are running in GBM to determine the safety and efficacy of ipilimumab in combination with drugs such as TMZ, bevacizumab, and other ICIs (Table 3).
Table 3.
Clinical status of immunotherapies in GBM within the last 5 years (since 2015).
IDO Inhibition
The less pronounced efficacy of various ICIs (such as anti-PD-1, PD-L1 or CTLA-4 mAbs) in GBM can be attributed, in part, to immunosuppression mediated by alternative pathways such as indoleamine 2,3 dioxygenase (IDO) [221,222]. Indoleamine 2,3 dioxygenase 1 (IDO1) is a rate-limiting enzyme in the kynurenine pathway of tryptophan metabolism [223,224]. Cancer cells activate IDO as a mechanism to limit the bioavailability of tryptophan and, thereby, limit the function of CD8+ T and NK cells while also inducing the differentiation of CD4+ Tregs [223,225]. The kynurenine pathway is considered a primary resistance mechanism of GBM to chemotherapy and radiation therapy [222], and increased expression of IDO1 in tumor-infiltrating T cells is usually linked to poor overall survival in brain tumor patients [226,227], while IDO loss is associated with reduced recruitment of Tregs in the brain and significantly better patient prognosis [95]. Since the IDO pathway plays a critical role in GBM immunosuppression, interest in IDO pathway inhibition is rapidly increasing. IDO-deficient gliomas recruit antigen-specific CD4+ T cells, leading to a significant extension of survival compared to IDO-competent tumors [228]. Anti-tumor efficacy of IDO blockade in vivo is further enhanced by anti-PD-1 and/or radiation therapy [229]. In addition, the combination of IDO inhibition and TMZ/radiation therapy increased tumor cell destruction and survival in mouse GBM models [107,230]. The simultaneous use of IDO inhibition and anti-PD-L1/anti-CTLA4 treatment also led to 100% long-term survivors of mice bearing intracranial gliomas [228]. Currently, several IDO inhibitors are being tested clinically in combination with standard therapies and/or ICIs in GBM (Table 3).
4.2.2. Oncolytic Viruses (OVs)
Oncolytic viruses (OVs) are a major developmental therapy of interest and have gained success in different cancer types, including GBM [139,205,231,232,233,234,235,236,237,238,239,240,241,242,243]. The recent FDA approval of an oncolytic herpes simplex virus (oHSV) (designated talimogene laherparepvec) has further fueled the field of oncolytic virotherapy. Tumor cells are noticeably distinct from normal cells by adopting behaviors such as increased proliferation and vascularization. OVs selectively infect tumor cells, killing them following replication, while leaving normal cells unscathed. At the same time, OVs trigger a cascade of anti-tumor immune responses [244], including increased tumor infiltration of immune cells [238,245]. OVs including oHSVs have shown promising efficacy in preclinical models of GBM [238,246,247] as well as in GBM patients [248]. In a recently published case study, four previously treated GBM patients received individualized treatment regimens comprised of three OVs (wild-type Newcastle disease virus [NDV], wild-type parvovirus [PV], and wild-type vaccinia virus [VV]). OVs were sequentially administered using the same catheter with a dose of 109 TCID50 for each virus in a volume of 10 mL and demonstrated impressive clinical and radiological responses with long-term survival up to 14 years [249]. OV-induced anti-tumor immunity can be further enhanced through OV-mediated expression of various cytokines/chemokines and immunomodulatory molecules [250]. OVs can also be used in combination with standard of care TMZ that produces synergistic anti-tumor effects in various preclinical cancer models including GBM [251,252,253,254,255,256]. However, a recent preclinical combination study (OV+TMZ) in GBM demonstrated conflicting results. Concurrent OV and TMZ therapy antagonized the anti-tumor properties of oncolytic virotherapy [239], indicating that co-applied administration of OV and TMZ represent a failed synergistic strategy as opposed to the pre-clinical benefits observed when TMZ was administered either before or after OV treatment [254,256,257]. Altogether, OVs serve to beneficially alter the TME to increase tumor immunogenicity, and synergize with ICIs [205,258]. Newer clinical studies are aiming to combine OVs and ICIs in order to improve patient outcomes as listed in Table 3 [258].
4.2.3. Therapeutic Vaccines
Vaccines are an active form of immunotherapy that has recently gained interest for GBM treatment [259,260]. The antigens such as EGFRvIII, heat shock protein (HSP), and any tumor-derived antigens can be loaded to DCs to incite immune responses against GBM [260]. In a randomized Phase II clinical trial in patients with relapsed EGFRvIII+ GBM, the EGFRvIII vaccine (designated Rindopepimut or CDX-110) delivered intradermally with GM-CSF (NCT01498328) resulted in the induction of EGFRvIII-specific immune responses, encouraging PFS and OS, and a significant extension of survival when the vaccine was administered in combination with bevacizumab [261]. The promising results of this trial led to a Phase III trial with Rindopepimut/GM-CSF in patients with newly diagnosed GBM, where all patients received standard-of-care TMZ (NCT01480479). Unfortunately, this Phase III trial was discontinued in early 2016 since Rindopepimut failed to significantly improve survival [262] and emphasizes the importance of identifying alternate and newer vaccine-based strategies to tackle GBM [263].
In contrast to EGFRvIII immunization that elicits immune responses to pre-defined tumor target, HSP vaccines offer immunity against a broad range of antigens. Induction of anti-tumor immunity against various antigenic targets is important to help minimize the outgrowth of target null variants, especially for cancer types that have high intra-tumoral heterogeneity like GBM [264]. The most well-known HSP vaccine is heat-shock protein peptide complex-96 (HSPPC-96) [265]. The safety and immunogenicity of HSPPC-96 monotherapy were demonstrated in a Phase I clinical trial in newly diagnosed GBM Patients [265]. HSPPC-96 is currently being tested in two separate Phase II clinical trials; one in combination with TMZ in patients with newly diagnosed GBM (NCT00905060) and the other in combination with bevacizumab in surgically resectable recurrent GBMs (NCT01814813) (Table 3).
DCs play a central role in linking innate and adaptive anti-tumor immune responses [266]. The principle of dendritic cell vaccines (DCV) is based on the ability of primed DCs to process/present tumor antigens and activate cytotoxic lymphocytes [267]. DCVs are prepared by isolating CD14+ monocytes from patient peripheral blood and further culturing cells ex vivo with granulocyte-macrophage colony-stimulating factor (GM-CSF), interleukin 4 (IL-4), and tumor antigens, prior to injecting the cells back into patients [268]. Although interest in DCV is further compelled by an FDA-approved DCV for the treatment of prostate cancer (Sipuleucel-T), most DCV-based clinical trials in GBM are still under phase I and II evaluations. For example, DCVax, an approved DCV for treatment of GBM in Switzerland, is currently being assessed in the US in patients with newly diagnosed GBM (NCT00045968) [269]. In a recent phase III study, DCVax was used alongside standard options and resulted in the extended survival of patients by 8 months compared to the control cohort [269,270]. Personalized neoantigen vaccine has also recently been tested in GBM clinical trials. For instance, in a Phase I/Ib study in newly diagnosed MGMT-unmethylated GBM, patients who did not receive dexamethasone had better neoantigen-specific CD4+ and CD8+ T cell responses with a higher number of TILs [271].
4.2.4. Adoptive Cell Therapies (ACTs)
In contrast to active immunotherapies such as OVs or anti-cancer vaccines that induce anti-tumor immunity within the host, ACTs are considered a passive form of immunotherapy where T cells are harvested from patients, expanded and activated ex vivo under appropriate conditions, and reinfused back into patients [272]. There are several types of ACT currently under development for GBM treatment, such as lymphokine-activated killer cells, allogenic donor lymphocyte infusion, autologous lymphocytes, tumor-infiltrating lymphocytes (TILs), transgenic T-cell receptor (TCR) T cells, antibody-armed T cells, and chimeric antigen receptor (CAR) T cells [272]. ACTs using autologous TILs and CAR-T cells are the most prevalent strategy being explored in GBM, as evidenced by the number of completed or running clinical trials [272]. ACT by autologous lymphocytes relies on MHC-restricted tumor antigen recognition via T cell receptors (TCRs) [273]. It has also been demonstrated that TILs harvested from GBM patients can recognize and kill autologous tumor cells [274], but recent studies reveal that not all TILs are tumor-specific. Ultimately, TILs that express CD39 (CD39+CD8+ T cells) are predominantly tumor-reactive [275,276] and these reports suggest that ACT strategies should incorporate CD39+CD8+ cells over bulk TILs also containing CD39-CD8+ cells.
Among ACT-based treatment strategies, CAR-T cell therapy is the furthest along in the clinic [277]. CAR-T cells are engineered to directly recognize tumor-specific antigens (e.g., EGFRvIII, HER2, IL-13Rα2, EphA2.) in an MHC-independent manner [273]. As an example, single intravenous infusion of CAR-T cells redirected to EGFRvIII antigen in patients with recurrent GBM lead to the loss of EGFRvIII in 5/7 patients [278]. Yet, CART-EGFRvIII therapy also induced the overexpression of inhibitory molecules such as immune checkpoints in the TME and increased tumor infiltration of immunosuppressive Tregs [278]. Recently, PD1-TILs have been engineered to overcome the inhibitory functions of the PD-1 molecule. PD1-TILs express full-length PD-1 antibody and are currently being tested in GBM patients in an early Phase I clinical trial (NCT03347097). In another study, a recurrent GBM patient received multiple intracranial infusions of IL13Rα2-specific CAR-T cells directly into the resected tumor cavity followed by infusions into the ventricular system over a period of 220 days. This strategy resulted in the regression of all intracranial and spinal tumors [279].
4.2.5. Macrophage and NK Cell-Based Immunotherapy
TAMs are another interesting therapeutic target in GBM since these cells are the most abundant infiltrating immune cells in GBM [280] and their immunosuppressive functions promote tumor progression [281]. TAMs can be divided into two major phenotypes: (i) M1-polarized cells that are generally considered anti-tumoral and (ii) M2-like cells that contribute to pro-tumoral activities [282]. The strategies to target TAMs in GBM include: (i) Inhibiting TAM recruitment by preventing interactions between C-C motif chemokine ligand 2 (CCL2) and C-C motif chemokine receptor 2 (CCR2) [283]; (ii) Increasing M1 polarization by disrupting the CD47-SIRPα-SHP-1 signaling pathway [284] or activating CD40 or toll-like receptors (TLRs) [285,286]; and (iii) Depleting TAMs using CSF1R inhibitors [287]. Overall, these aforementioned approaches have demonstrated promise in preclinical GBM studies and are well-tolerated among cancer patients [288]. Nevertheless, the safety and efficacy of macrophage-based immunotherapies need to be confirmed in GBM patients.
In contrast to macrophages, NK cells are the least abundant infiltrating immune cell type in GBM. Interestingly, NK cells contribute to immune surveillance by preventing spontaneous metastasis in GBM [289,290]. Therapeutic approaches that deploy NK cells in GBM are still in early developmental stages, with only one phase I clinical trial exploring the safety and objective response rates following NK cell-based immunotherapy in recurrent GBM patients (NCT04489420). In this study, CYNK-001 cells (i.e., NK cells derived from human placental CD34+ cells) will be intravenously infused after a lymphodepleting cyclophosphamide-based chemotherapy or provided intratumorally prior to surgery without lymphodepletion. Although no NK cell-based product has yet received FDA approval, numerous CAR-NK cells have been engineered to target tumor antigens such as EGFRvIII, HER2, IL-13Rα2, EphA2, CSPG4, CD133, or CD70 preclinically in GBM [289].
4.3. Nanomedicine
The neoplastic vasculature network is typically defective and leaky, which enhances the permeability and retention of nanoparticles in the TME [291,292]. Nanomedicine has been validated to enhance the efficacy of chemotherapies [293] and radiotherapy [294], but the safety and delivery of this approach have always been a major concern since nanoparticles preferentially deposit in the reticuloendothelial tissues of the kidneys, liver, and spleen [295,296]. Despite these issues, various anti-cancer nanoparticle therapies can produce superior efficacy versus non-nanoparticle formulation. For example, paclitaxel (PTX) or doxorubicin when administered as nanoparticles potentiate improved cytotoxicity against GBM compared to their parental compounds [297]. Several clinical trials are evaluating the therapeutic properties of different nanomedicine formulations such as nanoliposome (NCT00734682, NCT00944801, NCT01906385), Spherical Nucleic Acid (SNA) gold nanoparticles (NCT03020017), and nanocells (NCT02766699). To further advance this field in GBM and improve safety, an improved understanding of the long-term stability, biodistribution, and clearance mechanisms of nanoparticles is required.
4.4. Photodynamic Therapy (PDT)
Photodynamic therapy (PDT) is a form of light therapy that preferentially damages residual tumor cells following surgical resection [298]. PDT requires molecular oxygen (a photosensitizing [PS] agent) and a light source [299]. Based on their chemical structure, PS agents can be classified as derivatives of chlorin, porphyrin, bacteriochlorin, or phthalocyanine [300]. Ultimately, the PS agent is localized in tumor cells and remains non-toxic until activated by a light source. As the PS agent absorbs photons at a specific wavelength, the transfer of energy converts the PS agent to an excited state and promotes two types of photochemical reactions [301]: (i) Direct interaction between excited PS agents and biochemical molecules within target cells eventually generate cell-destroying free radicles; (ii) The radical anion of an excited PS indirectly interacts with oxygen, leading to the formation of single oxygen molecules that damage mitochondrial DNA and promote mitotic arrest and apoptosis [302,303]. Interestingly, the formation of these oxygen molecules is short-lived (4 micro-seconds) and limited to a maximum 1 μm migration path, which confer minimal harm to surrounding normal cells [304]. A meta-analysis of more than 1,000 GBM patients from several observational studies and three randomized clinical trials (RCTs) [305,306] (NCT01966809) indicate that PDT is safe and significantly improved the quality of life, survival, and delayed tumor relapse in patients (p < 0.001) [307]. The combination of PDT with standard therapies (i.e., maximum resection surgery followed by concomitant radio-chemotherapy and adjuvant chemotherapy) is also found to be safe and well-tolerated [308]. A comprehensive review of PDT (including its immunological effects and translational feasibility) has been previously discussed [303]. Although PDT shows favorable outcomes in GBM patients, the field still requires a greater number of pre-clinical studies and RCTs to better confirm safety and efficacy for GBM management.
4.5. Inhibition of Extracellular Vesicles (EVs) and Micro RNA (miRNA)-Based Therapies
Cells in general communicate and sense their environment by employing EVs loaded with nucleic acids, lipids, and proteins [309]. EVs can include exosomes, micro-vesicles, apoptotic bodies, and oncosomes, and are involved in directing cell metabolism and movement [309]. Within the realm of GBM, EVs can be utilized as biomarkers for diagnosis, prognosis and GBM recurrence [310,311]. More specifically, an increase in plasma EV concentration is associated with GBM recurrence [310] while EVs contribute to GBM invasion and recurrence after treatment by transferring therapy-resistant epigenetic materials between GBM cells [312]. Additionally, GBM cells can internalize bevacizumab and release it back into the TME where EVs neutralize the antibody to prevent VEGF-A binding, leading to drug resistance [313]. Therefore, EV inhibition is expected to enhance GBM treatment outcomes. EV production can be prevented by either preventing EV trafficking [312] or lipid metabolism [313]. CCR8 is usually required for EV fusion to cells [313]. As such, the small molecule CCR8 inhibitor R243 (when used in combination with TMZ) demonstrated a significant delay of tumor recurrence [313]. A separate lipid metabolism inhibitor (designated GW4869) also enhanced the cytotoxic effect of bevacizumab in GBM [313].
miRNA is the distinct cellular product transferred by EVs between GBM cells [309]. miRNAs are non-coding RNAs that consist of about 22 base pairs and regulate gene expression by binding complementary mRNA sequences to silence translation. miRNA is a major cellular communication scheme in GBM [314] and abnormal miRNA regulation favors tumor growth and invasion [315]. GBM patients have distinct miRNA expression profiles over healthy individuals—with at least 30 miRNAs that can be used as biomarkers [315]. The most frequently up-regulated miRNAs in GBM include miR-21, miR-10b, and miR-25, whereas miR-139, miR-218, and miR-124 are commonly down-regulated in GBM patients [315]. Emerging approaches to target miRNAs include inhibiting their occurrence by administering anti-sense oligonucleotides or therapeutics that silence miRNA expression. For example, inhibition of miR-21 suppresses GBM proliferation by inhibiting EGFR signaling pathway [316], increasing PTEN expression [317], and enhancing chemosensitivity [136,318]. Systemic administration of miR-10b antisense oligonucleotide inhibitors (ASO) also significantly prolonged survival in a xenograft model of mouse GBM [319]. Expression of miRNA-10b and its relation to OS and PFS in glioma patients is likewise currently under investigation (NCT01849952). The exogenous delivery of miR-124 decreases tumor growth and invasion and sensitizes GBM cells to chemotherapy [320]. Lastly, GSCs express different miRNAs over non-stem GBM cells that correlates with patient survival [321]. Some clinical trials consist of miRNA-based therapies and EV inhibitors are currently underway for a variety of cancer types (NCT02580552, NCT03713320, NCT03608631, NCT04167722), but these strategies have not yet been evaluated in GBM patients.
4.6. Targeting Vessel Co-Option and Vascular Mimicry
As discussed above, neovascularization of GBM comprises vessel co-option, angiogenesis, vasculogenesis, endothelial cell trans-differentiation, and vascular mimicry [43]. Among therapies that target GBM tumor vasculature, AAT is the most studied in the clinic. However, GBM treatment with AAT oftentimes increases vascular co-option [322] and results in resistance to AAT [323]. Mechanisms that drive vessel co-option in GBM and non-GBM cancers are poorly understood, although various tumor cell invasion/adhesion pathways are known to be involved as drivers of vessel co-option in glioma [45], such as bradykinin, CXC-chemokine receptor-4 (CXCR4)-binding cytokines, stromal cell-derived factor-1α (SDF1α), interleukin 8 (IL-8), angiopoietin 2 (Ang-2), cell division control protein 42 (CDC42), EGFRvIII, mammary-derived growth inhibitor/fatty acid-binding protein 3 (MDGI/FABP3), inositol-requiring enzyme-1α (IRE1α), homologous wingless and Int-1 (Wnt), and oligodendrocyte transcription factor (Olig2) [45,324,325,326]. It has been proposed that sequential treatment of vessel co-option inhibition by LGK974 (a porcupine inhibitor that blocks Wnt secretion) followed by anti-angiogenic therapy could produce synergistic effects that are superior to single treatments [323]. Vascular mimicry is a separate mechanism observed in GBM following AAT-induced resistance [61]. Treatment with AAT enhances a population of CXCR2-positive GBM-stem cells with endothelial-like phenotypes that promote tumor growth. Targeting vascular mimicry in this scenario by blocking the expression of CXCR2 with the compound SB225002 demonstrated significant reduction of tumor burden [61].
5. Conclusions
GBM is a devastating disease with an exceedingly poor prognosis and has an expected survival of only 12–15 months [1,2]. Currently approved treatments only manage to increase overall survival by a few months and further research is desperately needed in order to make a significant difference in the progression of the disease. Increasing our understanding of GBM pathogenesis is vital toward developing efficacious and long-lasting therapies. Heterogeneity of GBM is observed at both intra-and inter-tumoral levels, making targeted approaches (including small molecule drugs and immunotherapies) difficult to produce substantial clinical benefits. Therefore, a better understanding of the molecular heterogeneity and immunosuppressive profile of GBM would provide a more comprehensive insight into strategies that can overcome resistance acquired by this lethal disease. While combination therapies in development with the current standard of care options are gaining more attention, it is crucial to assess whether antagonizing properties develop. For example, while the addition of a histone deacetylase inhibitor to TMZ ensures GBM eradication [327], anti-EGFRvIII or anti-MAPK strategies potentially abrogate TMZ efficacy by interfering with the regulation of DNA mismatch repair in GBM [328]. Similarly, the combination of OV and TMZ shows a reduced efficacy against GBM compared to either agent alone [239]. It is also needed to investigate sequential administration of different therapies to potentially gain synergistic effects. For example, drugs that target EVs could be administered prior to AATs. Another consideration for improved GBM prognosis and treatment is the timing and method of diagnosis. The BBB is another major obstacle that should be addressed to achieve suitable therapeutic responses in individuals with GBM, particularly for emerging treatment approaches such as OVs, small molecule inhibitors, therapeutic vaccines, miRNA-based treatments, and EVs. It also remains vital to find new therapeutic targets in GBM. Since GSCs play an important role in GBM pathogenesis and therapy resistance, targeting GSCs would offer a unique way to eradicate the disease. Recently, ADAM Like Decysin 1 (ADAMDEC1) was discovered as a novel target in GBM and is overexpressed by GSCs, which regulate stem cell proliferation and sphere formation, and promote tumor growth through an ADAMDEC1-FGFR1-ZEB1 signaling loop [329]. Overall, the rapid growth and aggressive nature of GBM likely demand the necessary focus on developing effective treatments that will work alongside current mainstays of treatment.
Author Contributions
H.-M.N. wrote/edited the manuscript, prepared figures and tables, K.G.-M. wrote initial draft and edited the manuscript, D.B.L. reviewed/edited the manuscript, D.S. wrote/edited the manuscript, study supervision, and funding acquisition. All authors have read and agreed to the published version of the manuscript.
Funding
D.S. is supported in part by funds from the DOD (W81XWH-20-1-0702) and Dodge Jones Foundation-Abilene. D.B.L. is supported in part by funds from the NIH (R15 CA215874), DOD (W81XWH-18-1-0293), and Dodge Jones Foundation-Abilene.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Delgado-Lopez, P.D.; Corrales-Garcia, E.M. Survival in glioblastoma: A review on the impact of treatment modalities. Clin. Transl. Oncol. 2016, 18, 1062–1071. [Google Scholar] [CrossRef] [PubMed]
- Witthayanuwat, S.; Pesee, M.; Supaadirek, C.; Supakalin, N.; Thamronganantasakul, K.; Krusun, S. Survival Analysis of Glioblastoma Multiforme. Asian Pac. J. Cancer Prev. 2018, 19, 2613–2617. [Google Scholar] [CrossRef]
- Lyon, J.G.; Mokarram, N.; Saxena, T.; Carroll, S.L.; Bellamkonda, R.V. Engineering challenges for brain tumor immunotherapy. Adv. Drug Deliv. Rev. 2017, 114, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Shergalis, A.; Bankhead, A., 3rd; Luesakul, U.; Muangsin, N.; Neamati, N. Current Challenges and Opportunities in Treating Glioblastoma. Pharmacol. Rev. 2018, 70, 412–445. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Cioffi, G.; Gittleman, H.; Patil, N.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012–2016. Neuro-Oncology 2019, 21, v1–v100. [Google Scholar] [CrossRef]
- Lieberman, F. Glioblastoma update: Molecular biology, diagnosis, treatment, response assessment, and translational clinical trials. F1000Research 2017, 6, 1892. [Google Scholar] [CrossRef]
- Hanif, F.; Muzaffar, K.; Perveen, K.; Malhi, S.M.; Simjee, S.U. Glioblastoma Multiforme: A Review of its Epidemiology and Pathogenesis through Clinical Presentation and Treatment. Asian Pac. J. Cancer Prev. 2017, 18, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Melin, B.S.; Barnholtz-Sloan, J.S.; Wrensch, M.R.; Johansen, C.; Il’yasova, D.; Kinnersley, B.; Ostrom, Q.T.; Labreche, K.; Chen, Y.; Armstrong, G.; et al. Genome-wide association study of glioma subtypes identifies specific differences in genetic susceptibility to glioblastoma and non-glioblastoma tumors. Nat. Genet. 2017, 49, 789–794. [Google Scholar] [CrossRef]
- Yang, W.; Warrington, N.M.; Taylor, S.J.; Whitmire, P.; Carrasco, E.; Singleton, K.W.; Wu, N.; Lathia, J.D.; Berens, M.E.; Kim, A.H.; et al. Sex differences in GBM revealed by analysis of patient imaging, transcriptome, and survival data. Sci. Transl. Med. 2019, 11, eaao5253. [Google Scholar] [CrossRef]
- Vigneswaran, K.; Neill, S.; Hadjipanayis, C.G. Beyond the World Health Organization grading of infiltrating gliomas: Advances in the molecular genetics of glioma classification. Ann. Transl. Med. 2015, 3, 95. [Google Scholar] [CrossRef]
- Delgado-Martin, B.; Medina, M.A. Advances in the Knowledge of the Molecular Biology of Glioblastoma and Its Impact in Patient Diagnosis, Stratification, and Treatment. Adv. Sci (Weinh) 2020, 7, 1902971. [Google Scholar] [CrossRef]
- Li, R.; Li, H.; Yan, W.; Yang, P.; Bao, Z.; Zhang, C.; Jiang, T.; You, Y. Genetic and clinical characteristics of primary and secondary glioblastoma is associated with differential molecular subtype distribution. Oncotarget 2015, 6, 7318–7324. [Google Scholar] [CrossRef] [PubMed]
- Ohgaki, H.; Kleihues, P. The definition of primary and secondary glioblastoma. Clin. Cancer Res. 2013, 19, 764–772. [Google Scholar] [CrossRef]
- Ohgaki, H.; Kleihues, P. Genetic pathways to primary and secondary glioblastoma. Am. J. Pathol. 2007, 170, 1445–1453. [Google Scholar] [CrossRef]
- Lee, D.H.; Ryu, H.W.; Won, H.R.; Kwon, S.H. Advances in epigenetic glioblastoma therapy. Oncotarget 2017, 8, 18577–18589. [Google Scholar] [CrossRef]
- Davis, M.E. Glioblastoma: Overview of Disease and Treatment. Clin. J. Oncol. Nurs. 2016, 20, S2–S8. [Google Scholar] [CrossRef] [PubMed]
- Thakkar, J.P.; Dolecek, T.A.; Horbinski, C.; Ostrom, Q.T.; Lightner, D.D.; Barnholtz-Sloan, J.S.; Villano, J.L. Epidemiologic and molecular prognostic review of glioblastoma. Cancer Epidemiol. Biomark. Prev. 2014, 23, 1985–1996. [Google Scholar] [CrossRef] [PubMed]
- Jain, K.K. A Critical Overview of Targeted Therapies for Glioblastoma. Front. Oncol. 2018, 8, 419. [Google Scholar] [CrossRef] [PubMed]
- Touat, M.; Idbaih, A.; Sanson, M.; Ligon, K.L. Glioblastoma targeted therapy: Updated approaches from recent biological insights. Ann. Oncol. 2017, 28, 1457–1472. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Zhang, H.; Gu, L.; Ye, B.; Jian, Z.; Stary, C.; Xiong, X. Advances in Immunotherapy for Glioblastoma Multiforme. J. Immunol. Res. 2017, 2017, 3597613. [Google Scholar] [CrossRef]
- Tivnan, A.; Heilinger, T.; Lavelle, E.C.; Prehn, J.H. Advances in immunotherapy for the treatment of glioblastoma. J. Neurooncol. 2017, 131, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Friedmann-Morvinski, D. Glioblastoma heterogeneity and cancer cell plasticity. Crit. Rev. Oncog. 2014, 19, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Dirkse, A.; Golebiewska, A.; Buder, T.; Nazarov, P.V.; Muller, A.; Poovathingal, S.; Brons, N.H.C.; Leite, S.; Sauvageot, N.; Sarkisjan, D.; et al. Stem cell-associated heterogeneity in Glioblastoma results from intrinsic tumor plasticity shaped by the microenvironment. Nat. Commun. 2019, 10, 1787. [Google Scholar] [CrossRef]
- Lara-Velazquez, M.; Al-Kharboosh, R.; Jeanneret, S.; Vazquez-Ramos, C.; Mahato, D.; Tavanaiepour, D.; Rahmathulla, G.; Quinones-Hinojosa, A. Advances in Brain Tumor Surgery for Glioblastoma in Adults. Brain Sci. 2017, 7, 166. [Google Scholar] [CrossRef]
- Blissitt, P.A.; American Association of Neuroscience Nurses. Clinical practice guideline series update: Care of the adult patient with a brain tumor. J. Neurosci. Nurs. 2014, 46, 367–368. [Google Scholar] [CrossRef]
- Yao, M.; Li, S.; Wu, X.; Diao, S.; Zhang, G.; He, H.; Bian, L.; Lu, Y. Cellular origin of glioblastoma and its implication in precision therapy. Cell Mol. Immunol. 2018, 15, 737–739. [Google Scholar] [CrossRef]
- Matarredona, E.R.; Pastor, A.M. Neural Stem Cells of the Subventricular Zone as the Origin of Human Glioblastoma Stem Cells. Therapeutic Implications. Front. Oncol. 2019, 9, 779. [Google Scholar] [CrossRef] [PubMed]
- Alcantara Llaguno, S.; Chen, J.; Kwon, C.H.; Jackson, E.L.; Li, Y.; Burns, D.K.; Alvarez-Buylla, A.; Parada, L.F. Malignant astrocytomas originate from neural stem/progenitor cells in a somatic tumor suppressor mouse model. Cancer Cell 2009, 15, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Lee, J.E.; Kahng, J.Y.; Kim, S.H.; Park, J.S.; Yoon, S.J.; Um, J.Y.; Kim, W.K.; Lee, J.K.; Park, J.; et al. Human glioblastoma arises from subventricular zone cells with low-level driver mutations. Nature 2018, 560, 243–247. [Google Scholar] [CrossRef]
- Alcantara Llaguno, S.; Sun, D.; Pedraza, A.M.; Vera, E.; Wang, Z.; Burns, D.K.; Parada, L.F. Cell-of-origin susceptibility to glioblastoma formation declines with neural lineage restriction. Nat. Neurosci. 2019, 22, 545–555. [Google Scholar] [CrossRef]
- Urbanska, K.; Sokolowska, J.; Szmidt, M.; Sysa, P. Glioblastoma multiforme—An overview. Contemp. Oncol. (Pozn) 2014, 18, 307–312. [Google Scholar] [CrossRef]
- Marfia, G.; Navone, S.E.; Fanizzi, C.; Tabano, S.; Pesenti, C.; Abdel Hadi, L.; Franzini, A.; Caroli, M.; Miozzo, M.; Riboni, L.; et al. Prognostic value of preoperative von Willebrand factor plasma levels in patients with Glioblastoma. Cancer Med. 2016, 5, 1783–1790. [Google Scholar] [CrossRef]
- Gimple, R.C.; Bhargava, S.; Dixit, D.; Rich, J.N. Glioblastoma stem cells: Lessons from the tumor hierarchy in a lethal cancer. Genes Dev. 2019, 33, 591–609. [Google Scholar] [CrossRef]
- Wang, X.; Prager, B.C.; Wu, Q.; Kim, L.J.Y.; Gimple, R.C.; Shi, Y.; Yang, K.; Morton, A.R.; Zhou, W.; Zhu, Z.; et al. Reciprocal Signaling between Glioblastoma Stem Cells and Differentiated Tumor Cells Promotes Malignant Progression. Cell Stem. Cell 2018, 22, 514–528. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Huang, Z.; Zhou, W.; Wu, Q.; Donnola, S.; Liu, J.K.; Fang, X.; Sloan, A.E.; Mao, Y.; Lathia, J.D.; et al. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell 2013, 153, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Gong, T.; Heng, B.C.; Zhang, C.F. A systematic review: Differentiation of stem cells into functional pericytes. FASEB J. 2017, 31, 1775–1786. [Google Scholar] [CrossRef]
- Mei, X.; Chen, Y.-S.; Chen, F.-R.; Xi, S.-Y.; Chen, Z.-P. Glioblastoma stem cell differentiation into endothelial cells evidenced through live-cell imaging. Neuro-Oncology 2017, 19, 1109–1118. [Google Scholar] [CrossRef]
- Beier, C.P.; Rasmussen, T.; Dahlrot, R.H.; Tenstad, H.B.; Aarø, J.S.; Sørensen, M.F.; Heimisdóttir, S.B.; Sørensen, M.D.; Svenningsen, P.; Riemenschneider, M.J.; et al. Aberrant neuronal differentiation is common in glioma but is associated neither with epileptic seizures nor with better survival. Sci. Rep. 2018, 8, 14965. [Google Scholar] [CrossRef] [PubMed]
- Bien-Moller, S.; Balz, E.; Herzog, S.; Plantera, L.; Vogelgesang, S.; Weitmann, K.; Seifert, C.; Fink, M.A.; Marx, S.; Bialke, A.; et al. Association of Glioblastoma Multiforme Stem Cell Characteristics, Differentiation, and Microglia Marker Genes with Patient Survival. Stem. Cells Int. 2018, 2018, 9628289. [Google Scholar] [CrossRef] [PubMed]
- Guelfi, S.; Duffau, H.; Bauchet, L.; Rothhut, B.; Hugnot, J.P. Vascular Transdifferentiation in the CNS: A Focus on Neural and Glioblastoma Stem-Like Cells. Stem. Cells Int. 2016, 2016, 2759403. [Google Scholar] [CrossRef] [PubMed]
- Garnier, D.; Renoult, O.; Alves-Guerra, M.C.; Paris, F.; Pecqueur, C. Glioblastoma Stem-Like Cells, Metabolic Strategy to Kill a Challenging Target. Front. Oncol. 2019, 9, 118. [Google Scholar] [CrossRef]
- Das, S.; Marsden, P.A. Angiogenesis in glioblastoma. N. Engl. J. Med. 2013, 369, 1561–1563. [Google Scholar] [CrossRef]
- Kane, J.R. The Role of Brain Vasculature in Glioblastoma. Mol. Neurobiol. 2019, 56, 6645–6653. [Google Scholar] [CrossRef]
- Kuczynski, E.A.; Vermeulen, P.B.; Pezzella, F.; Kerbel, R.S.; Reynolds, A.R. Vessel co-option in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 469–493. [Google Scholar] [CrossRef]
- Seano, G.; Jain, R.K. Vessel co-option in glioblastoma: Emerging insights and opportunities. Angiogenesis 2020, 23, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Donnem, T.; Hu, J.; Ferguson, M.; Adighibe, O.; Snell, C.; Harris, A.L.; Gatter, K.C.; Pezzella, F. Vessel co-option in primary human tumors and metastases: An obstacle to effective anti-angiogenic treatment? Cancer Med. 2013, 2, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Baker, G.J.; Yadav, V.N.; Motsch, S.; Koschmann, C.; Calinescu, A.A.; Mineharu, Y.; Camelo-Piragua, S.I.; Orringer, D.; Bannykh, S.; Nichols, W.S.; et al. Mechanisms of glioma formation: Iterative perivascular glioma growth and invasion leads to tumor progression, VEGF-independent vascularization, and resistance to antiangiogenic therapy. Neoplasia 2014, 16, 543–561. [Google Scholar] [CrossRef] [PubMed]
- Holash, J.; Maisonpierre, P.C.; Compton, D.; Boland, P.; Alexander, C.R.; Zagzag, D.; Yancopoulos, G.D.; Wiegand, S.J. Vessel cooption, regression, and growth in tumors mediated by angiopoietins and VEGF. Science 1999, 284, 1994–1998. [Google Scholar] [CrossRef]
- Sakariassen, P.; Prestegarden, L.; Wang, J.; Skaftnesmo, K.O.; Mahesparan, R.; Molthoff, C.; Sminia, P.; Sundlisaeter, E.; Misra, A.; Tysnes, B.B.; et al. Angiogenesis-independent tumor growth mediated by stem-like cancer cells. Proc. Natl. Acad. Sci. USA 2006, 103, 16466–16471. [Google Scholar] [CrossRef]
- Ahir, B.K.; Engelhard, H.H.; Lakka, S.S. Tumor Development and Angiogenesis in Adult Brain Tumor: Glioblastoma. Mol. Neurobiol. 2020, 57, 2461–2478. [Google Scholar] [CrossRef]
- Ayuso, J.M.; Monge, R.; Martinez-Gonzalez, A.; Virumbrales-Munoz, M.; Llamazares, G.A.; Berganzo, J.; Hernandez-Lain, A.; Santolaria, J.; Doblare, M.; Hubert, C.; et al. Glioblastoma on a microfluidic chip: Generating pseudopalisades and enhancing aggressiveness through blood vessel obstruction events. Neuro-Oncology 2017, 19, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Rong, Y.; Durden, D.L.; Van Meir, E.G.; Brat, D.J. ‘Pseudopalisading’ necrosis in glioblastoma: A familiar morphologic feature that links vascular pathology, hypoxia, and angiogenesis. J. Neuropathol. Exp. Neurol. 2006, 65, 529–539. [Google Scholar] [CrossRef]
- Brat, D.J.; Van Meir, E.G. Vaso-occlusive and prothrombotic mechanisms associated with tumor hypoxia, necrosis, and accelerated growth in glioblastoma. Lab. Investig. 2004, 84, 397–405. [Google Scholar] [CrossRef]
- Machein, M.R.; Renninger, S.; de Lima-Hahn, E.; Plate, K.H. Minor contribution of bone marrow-derived endothelial progenitors to the vascularization of murine gliomas. Brain Pathol. 2003, 13, 582–597. [Google Scholar] [CrossRef] [PubMed]
- Ricci-Vitiani, L.; Pallini, R.; Biffoni, M.; Todaro, M.; Invernici, G.; Cenci, T.; Maira, G.; Parati, E.A.; Stassi, G.; Larocca, L.M.; et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature 2010, 468, 824–828. [Google Scholar] [CrossRef]
- Liu, X.M.; Zhang, Q.P.; Mu, Y.G.; Zhang, X.H.; Sai, K.; Pang, J.C.; Ng, H.K.; Chen, Z.P. Clinical significance of vasculogenic mimicry in human gliomas. J. Neurooncol. 2011, 105, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.Y.; Ke, Y.Q.; Lu, G.H.; Song, Z.H.; Yu, L.; Xiao, S.; Sun, X.L.; Jiang, X.D.; Yang, Z.L.; Hu, C.C. Vasculogenic mimicry is a prognostic factor for postoperative survival in patients with glioblastoma. J. Neurooncol. 2013, 112, 339–345. [Google Scholar] [CrossRef]
- Baisiwala, S.; Auffinger, B.; Caragher, S.P.; Shireman, J.M.; Ahsan, R.; Lee, G.; Hasan, T.; Park, C.; Saathoff, M.R.; Christensen, A.C.; et al. Chemotherapeutic Stress Induces Transdifferentiation of Glioblastoma Cells to Endothelial Cells and Promotes Vascular Mimicry. Stem. Cells Int. 2019, 2019, 6107456. [Google Scholar] [CrossRef]
- Deshors, P.; Toulas, C.; Arnauduc, F.; Malric, L.; Siegfried, A.; Nicaise, Y.; Lemarie, A.; Larrieu, D.; Tosolini, M.; Cohen-Jonathan Moyal, E.; et al. Ionizing radiation induces endothelial transdifferentiation of glioblastoma stem-like cells through the Tie2 signaling pathway. Cell Death Dis. 2019, 10, 816. [Google Scholar] [CrossRef]
- Lu, K.V.; Bergers, G. Mechanisms of evasive resistance to anti-VEGF therapy in glioblastoma. CNS Oncol. 2013, 2, 49–65. [Google Scholar] [CrossRef] [PubMed]
- Angara, K.; Borin, T.F.; Rashid, M.H.; Lebedyeva, I.; Ara, R.; Lin, P.C.; Iskander, A.; Bollag, R.J.; Achyut, B.R.; Arbab, A.S. CXCR2-Expressing Tumor Cells Drive Vascular Mimicry in Antiangiogenic Therapy-Resistant Glioblastoma. Neoplasia 2018, 20, 1070–1082. [Google Scholar] [CrossRef]
- Zamecnik, J. The extracellular space and matrix of gliomas. Acta Neuropathol. 2005, 110, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Lal, B.; Tung, B.; Wang, S.; Goodwin, C.R.; Laterra, J. Tumor microenvironment tenascin-C promotes glioblastoma invasion and negatively regulates tumor proliferation. Neuro-Oncology 2016, 18, 507–517. [Google Scholar] [CrossRef]
- Serres, E.; Debarbieux, F.; Stanchi, F.; Maggiorella, L.; Grall, D.; Turchi, L.; Burel-Vandenbos, F.; Figarella-Branger, D.; Virolle, T.; Rougon, G.; et al. Fibronectin expression in glioblastomas promotes cell cohesion, collective invasion of basement membrane in vitro and orthotopic tumor growth in mice. Oncogene 2014, 33, 3451–3462. [Google Scholar] [CrossRef]
- Chen, J.E.; Pedron, S.; Shyu, P.; Hu, Y.; Sarkaria, J.N.; Harley, B.A.C. Influence of Hyaluronic Acid Transitions in Tumor Microenvironment on Glioblastoma Malignancy and Invasive Behavior. Front. Mater. 2018, 5, 39. [Google Scholar] [CrossRef] [PubMed]
- Schittenhelm, J.; Schwab, E.I.; Sperveslage, J.; Tatagiba, M.; Meyermann, R.; Fend, F.; Goodman, S.L.; Sipos, B. Longitudinal expression analysis of alphav integrins in human gliomas reveals upregulation of integrin alphavbeta3 as a negative prognostic factor. J. Neuropathol. Exp. Neurol. 2013, 72, 194–210. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wang, T.; Liu, S.; Yoshida, D.; Teramoto, A. The expression of matrix metalloproteinase-2 and -9 in human gliomas of different pathological grades. Brain Tumor. Pathol. 2003, 20, 65–72. [Google Scholar] [CrossRef]
- Roth, P.; Silginer, M.; Goodman, S.L.; Hasenbach, K.; Thies, S.; Maurer, G.; Schraml, P.; Tabatabai, G.; Moch, H.; Tritschler, I.; et al. Integrin control of the transforming growth factor-beta pathway in glioblastoma. Brain 2013, 136, 564–576. [Google Scholar] [CrossRef]
- Malric, L.; Monferran, S.; Gilhodes, J.; Boyrie, S.; Dahan, P.; Skuli, N.; Sesen, J.; Filleron, T.; Kowalski-Chauvel, A.; Cohen-Jonathan Moyal, E.; et al. Interest of integrins targeting in glioblastoma according to tumor heterogeneity and cancer stem cell paradigm: An update. Oncotarget 2017, 8, 86947–86968. [Google Scholar] [CrossRef] [PubMed]
- Janouskova, H.; Maglott, A.; Leger, D.Y.; Bossert, C.; Noulet, F.; Guerin, E.; Guenot, D.; Pinel, S.; Chastagner, P.; Plenat, F.; et al. Integrin alpha5beta1 plays a critical role in resistance to temozolomide by interfering with the p53 pathway in high-grade glioma. Cancer Res. 2012, 72, 3463–3470. [Google Scholar] [CrossRef]
- Hallal, S.; Mallawaaratchy, D.M.; Wei, H.; Ebrahimkhani, S.; Stringer, B.W.; Day, B.W.; Boyd, A.W.; Guillemin, G.J.; Buckland, M.E.; Kaufman, K.L. Extracellular Vesicles Released by Glioblastoma Cells Stimulate Normal Astrocytes to Acquire a Tumor-Supportive Phenotype Via p53 and MYC Signaling Pathways. Mol. Neurobiol. 2019, 56, 4566–4581. [Google Scholar] [CrossRef]
- Abels, E.R.; Maas, S.L.N.; Nieland, L.; Wei, Z.; Cheah, P.S.; Tai, E.; Kolsteeg, C.J.; Dusoswa, S.A.; Ting, D.T.; Hickman, S.; et al. Glioblastoma-Associated Microglia Reprogramming Is Mediated by Functional Transfer of Extracellular miR-21. Cell Rep. 2019, 28, 3105–3119. [Google Scholar] [CrossRef]
- Treps, L.; Perret, R.; Edmond, S.; Ricard, D.; Gavard, J. Glioblastoma stem-like cells secrete the pro-angiogenic VEGF-A factor in extracellular vesicles. J. Extracell Vesicles 2017, 6, 1359479. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Ma, X.; Wang, J.; Zhao, Y.; Wang, Y.; Bihl, J.C.; Chen, Y.; Jiang, C. Glioma stem cells-derived exosomes promote the angiogenic ability of endothelial cells through miR-21/VEGF signal. Oncotarget 2017, 8, 36137–36148. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.F.; Liao, F.; Wu, H.; Dai, J. Glioma stem cells-derived exosomal miR-26a promotes angiogenesis of microvessel endothelial cells in glioma. J. Exp. Clin. Cancer Res. 2019, 38, 201. [Google Scholar] [CrossRef]
- Kucharzewska, P.; Christianson, H.C.; Welch, J.E.; Svensson, K.J.; Fredlund, E.; Ringner, M.; Morgelin, M.; Bourseau-Guilmain, E.; Bengzon, J.; Belting, M. Exosomes reflect the hypoxic status of glioma cells and mediate hypoxia-dependent activation of vascular cells during tumor development. Proc. Natl. Acad. Sci. USA 2013, 110, 7312–7317. [Google Scholar] [CrossRef]
- Virga, J.; Szivos, L.; Hortobagyi, T.; Chalsaraei, M.K.; Zahuczky, G.; Steiner, L.; Toth, J.; Remenyi-Puskar, J.; Bognar, L.; Klekner, A. Extracellular matrix differences in glioblastoma patients with different prognoses. Oncol. Lett. 2019, 17, 797–806. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef]
- Park, A.K.; Kim, P.; Ballester, L.Y.; Esquenazi, Y.; Zhao, Z. Subtype-specific signaling pathways and genomic aberrations associated with prognosis of glioblastoma. Neuro-Oncology 2019, 21, 59–70. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Noble, M.; Proschel, C.; Mayer-Proschel, M. Getting a GR(i)P on oligodendrocyte development. Dev. Biol. 2004, 265, 33–52. [Google Scholar] [CrossRef]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef]
- Thiery, J.P. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef]
- Behnan, J.; Finocchiaro, G.; Hanna, G. The landscape of the mesenchymal signature in brain tumours. Brain 2019, 142, 847–866. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Dube, C.; Gibert, M., Jr.; Cruickshanks, N.; Wang, B.; Coughlan, M.; Yang, Y.; Setiady, I.; Deveau, C.; Saoud, K.; et al. The p53 Pathway in Glioblastoma. Cancers 2018, 10, 297. [Google Scholar] [CrossRef]
- Dick, F.A.; Goodrich, D.W.; Sage, J.; Dyson, N.J. Non-canonical functions of the RB protein in cancer. Nat. Rev. Cancer 2018, 18, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Pearson, J.R.D.; Regad, T. Targeting cellular pathways in glioblastoma multiforme. Signal. Transduct Target. Ther. 2017, 2, 17040. [Google Scholar] [CrossRef] [PubMed]
- Louveau, A.; Harris, T.H.; Kipnis, J. Revisiting the Mechanisms of CNS Immune Privilege. Trends Immunol. 2015, 36, 569–577. [Google Scholar] [CrossRef]
- Aspelund, A.; Antila, S.; Proulx, S.T.; Karlsen, T.V.; Karaman, S.; Detmar, M.; Wiig, H.; Alitalo, K. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J. Exp. Med. 2015, 212, 991–999. [Google Scholar] [CrossRef]
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.D.; Rouhani, S.J.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015, 523, 337–341. [Google Scholar] [CrossRef]
- Woroniecka, K.I.; Rhodin, K.E.; Chongsathidkiet, P.; Keith, K.A.; Fecci, P.E. T-cell Dysfunction in Glioblastoma: Applying a New Framework. Clin. Cancer Res. 2018, 24, 3792–3802. [Google Scholar] [CrossRef] [PubMed]
- Huff, W.X.; Kwon, J.H.; Henriquez, M.; Fetcko, K.; Dey, M. The Evolving Role of CD8(+)CD28(-) Immunosenescent T Cells in Cancer Immunology. Int. J. Mol. Sci. 2019, 20, 2810. [Google Scholar] [CrossRef]
- Liu, X.; Mo, W.; Ye, J.; Li, L.; Zhang, Y.; Hsueh, E.C.; Hoft, D.F.; Peng, G. Regulatory T cells trigger effector T cell DNA damage and senescence caused by metabolic competition. Nat. Commun. 2018, 9, 249. [Google Scholar] [CrossRef]
- Wainwright, D.A.; Balyasnikova, I.V.; Chang, A.L.; Ahmed, A.U.; Moon, K.S.; Auffinger, B.; Tobias, A.L.; Han, Y.; Lesniak, M.S. IDO expression in brain tumors increases the recruitment of regulatory T cells and negatively impacts survival. Clin. Cancer Res. 2012, 18, 6110–6121. [Google Scholar] [CrossRef]
- Hussain, S.F.; Kong, L.Y.; Jordan, J.; Conrad, C.; Madden, T.; Fokt, I.; Priebe, W.; Heimberger, A.B. A novel small molecule inhibitor of signal transducers and activators of transcription 3 reverses immune tolerance in malignant glioma patients. Cancer Res. 2007, 67, 9630–9636. [Google Scholar] [CrossRef]
- Liu, F.; Huang, J.; Liu, X.; Cheng, Q.; Luo, C.; Liu, Z. CTLA-4 correlates with immune and clinical characteristics of glioma. Cancer Cell Int. 2020, 20, 7. [Google Scholar] [CrossRef]
- Davidson, T.B.; Lee, A.; Hsu, M.; Sedighim, S.; Orpilla, J.; Treger, J.; Mastall, M.; Roesch, S.; Rapp, C.; Galvez, M.; et al. Expression of PD-1 by T Cells in Malignant Glioma Patients Reflects Exhaustion and Activation. Clin. Cancer Res. 2019, 25, 1913–1922. [Google Scholar] [CrossRef]
- Litak, J.; Mazurek, M.; Grochowski, C.; Kamieniak, P.; Rolinski, J. PD-L1/PD-1 Axis in Glioblastoma Multiforme. Int. J. Mol. Sci. 2019, 20, 5347. [Google Scholar] [CrossRef] [PubMed]
- Mostofa, A.G.; Punganuru, S.R.; Madala, H.R.; Al-Obaide, M.; Srivenugopal, K.S. The Process and Regulatory Components of Inflammation in Brain Oncogenesis. Biomolecules 2017, 7, 34. [Google Scholar] [CrossRef] [PubMed]
- Nduom, E.K.; Weller, M.; Heimberger, A.B. Immunosuppressive mechanisms in glioblastoma. Neuro-Oncology 2015, 17 (Suppl. 7), vii9–vii14. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Ke, S.Q.; Huang, Z.; Flavahan, W.; Fang, X.; Paul, J.; Wu, L.; Sloan, A.E.; McLendon, R.E.; Li, X.; et al. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat. Cell Biol. 2015, 17, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Crane, C.A.; Ahn, B.J.; Han, S.J.; Parsa, A.T. Soluble factors secreted by glioblastoma cell lines facilitate recruitment, survival, and expansion of regulatory T cells: Implications for immunotherapy. Neuro-Oncology 2012, 14, 584–595. [Google Scholar] [CrossRef]
- Qi, L.; Yu, H.; Zhang, Y.; Zhao, D.; Lv, P.; Zhong, Y.; Xu, Y. IL-10 secreted by M2 macrophage promoted tumorigenesis through interaction with JAK2 in glioma. Oncotarget 2016, 7, 71673–71685. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Kuang, W.; Zhou, Q.; Zhang, Y. TGF-beta1 secreted by M2 phenotype macrophages enhances the stemness and migration of glioma cells via the SMAD2/3 signalling pathway. Int. J. Mol. Med. 2018, 42, 3395–3403. [Google Scholar] [CrossRef] [PubMed]
- Pinton, L.; Masetto, E.; Vettore, M.; Solito, S.; Magri, S.; D’Andolfi, M.; Del Bianco, P.; Lollo, G.; Benoit, J.-P.; Okada, H.; et al. The immune suppressive microenvironment of human gliomas depends on the accumulation of bone marrow-derived macrophages in the center of the lesion. J. Immun.Ther. Cancer 2019, 7, 58. [Google Scholar] [CrossRef]
- Xu, S.; Tang, L.; Li, X.; Fan, F.; Liu, Z. Immunotherapy for glioma: Current management and future application. Cancer Lett. 2020, 476, 1–12. [Google Scholar] [CrossRef]
- Sarkaria, J.N.; Hu, L.S.; Parney, I.F.; Pafundi, D.H.; Brinkmann, D.H.; Laack, N.N.; Giannini, C.; Burns, T.C.; Kizilbash, S.H.; Laramy, J.K.; et al. Is the blood-brain barrier really disrupted in all glioblastomas? A critical assessment of existing clinical data. Neuro-Oncology 2018, 20, 184–191. [Google Scholar] [CrossRef]
- Paolillo, M.; Boselli, C.; Schinelli, S. Glioblastoma under Siege: An Overview of Current Therapeutic Strategies. Brain Sci. 2018, 8, 15. [Google Scholar] [CrossRef]
- Jiapaer, S.; Furuta, T.; Tanaka, S.; Kitabayashi, T.; Nakada, M. Potential Strategies Overcoming the Temozolomide Resistance for Glioblastoma. Neurol. Med. Chir. (Tokyo) 2018, 58, 405–421. [Google Scholar] [CrossRef]
- Rosen, L.S.; Jacobs, I.A.; Burkes, R.L. Bevacizumab in Colorectal Cancer: Current Role in Treatment and the Potential of Biosimilars. Target. Oncol. 2017, 12, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Russo, A.E.; Priolo, D.; Antonelli, G.; Libra, M.; McCubrey, J.A.; Ferrau, F. Bevacizumab in the treatment of NSCLC: Patient selection and perspectives. Lung Cancer (Auckl.) 2017, 8, 259–269. [Google Scholar] [CrossRef]
- Friedman, H.S.; Prados, M.D.; Wen, P.Y.; Mikkelsen, T.; Schiff, D.; Abrey, L.E.; Yung, W.K.; Paleologos, N.; Nicholas, M.K.; Jensen, R.; et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J. Clin. Oncol. 2009, 27, 4733–4740. [Google Scholar] [CrossRef]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Chinot, O.L.; Wick, W.; Mason, W.; Henriksson, R.; Saran, F.; Nishikawa, R.; Carpentier, A.F.; Hoang-Xuan, K.; Kavan, P.; Cernea, D.; et al. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 709–722. [Google Scholar] [CrossRef] [PubMed]
- Kinzel, A.; Ambrogi, M.; Varshaver, M.; Kirson, E.D. Tumor Treating Fields for Glioblastoma Treatment: Patient Satisfaction and Compliance With the Second-Generation Optune((R)) System. Clin. Med. Insights Oncol. 2019, 13. [Google Scholar] [CrossRef]
- Fabian, D.; Guillermo Prieto Eibl, M.D.P.; Alnahhas, I.; Sebastian, N.; Giglio, P.; Puduvalli, V.; Gonzalez, J.; Palmer, J.D. Treatment of Glioblastoma (GBM) with the Addition of Tumor-Treating Fields (TTF): A Review. Cancers 2019, 11, 174. [Google Scholar] [CrossRef]
- Burri, S.H.; Gondi, V.; Brown, P.D.; Mehta, M.P. The Evolving Role of Tumor Treating Fields in Managing Glioblastoma: Guide for Oncologists. Am. J. Clin. Oncol. 2018, 41, 191–196. [Google Scholar] [CrossRef]
- Bokstein, F.; Blumenthal, D.; Limon, D.; Harosh, C.B.; Ram, Z.; Grossman, R. Concurrent Tumor Treating Fields (TTFields) and Radiation Therapy for Newly Diagnosed Glioblastoma: A Prospective Safety and Feasibility Study. Front. Oncol. 2020, 10, 411. [Google Scholar] [CrossRef]
- Noch, E.K.; Ramakrishna, R.; Magge, R. Challenges in the Treatment of Glioblastoma: Multisystem Mechanisms of Therapeutic Resistance. World Neurosurg. 2018, 116, 505–517. [Google Scholar] [CrossRef] [PubMed]
- Osuka, S.; Van Meir, E.G. Overcoming therapeutic resistance in glioblastoma: The way forward. J. Clin. Investig. 2017, 127, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, J.P.; Nahed, B.V.; Madden, M.W.; Oliveira, S.M.; Springer, S.; Bhere, D.; Chi, A.S.; Wakimoto, H.; Rothenberg, S.M.; Sequist, L.V.; et al. Brain tumor cells in circulation are enriched for mesenchymal gene expression. Cancer Discov. 2014, 4, 1299–1309. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.D.; Chow, D.S.; Yang, P.H.; Filippi, C.G.; Lignelli, A. Predicting Glioblastoma Recurrence by Early Changes in the Apparent Diffusion Coefficient Value and Signal Intensity on FLAIR Images. AJR Am. J. Roentgenol. 2017, 208, 57–65. [Google Scholar] [CrossRef]
- Muscat, A.M.; Wong, N.C.; Drummond, K.J.; Algar, E.M.; Khasraw, M.; Verhaak, R.; Field, K.; Rosenthal, M.A.; Ashley, D.M. The evolutionary pattern of mutations in glioblastoma reveals therapy-mediated selection. Oncotarget 2018, 9, 7844–7858. [Google Scholar] [CrossRef]
- Razavi, S.M.; Lee, K.E.; Jin, B.E.; Aujla, P.S.; Gholamin, S.; Li, G. Immune Evasion Strategies of Glioblastoma. Front. Surg. 2016, 3, 11. [Google Scholar] [CrossRef] [PubMed]
- Perng, P.; Lim, M. Immunosuppressive Mechanisms of Malignant Gliomas: Parallels at Non-CNS Sites. Front. Oncol. 2015, 5, 153. [Google Scholar] [CrossRef]
- Preusser, M.; Lim, M.; Hafler, D.A.; Reardon, D.A.; Sampson, J.H. Prospects of immune checkpoint modulators in the treatment of glioblastoma. Nat. Rev. Neurol. 2015, 11, 504–514. [Google Scholar] [CrossRef]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Goffart, N.; Lombard, A.; Lallemand, F.; Kroonen, J.; Nassen, J.; Di Valentin, E.; Berendsen, S.; Dedobbeleer, M.; Willems, E.; Robe, P.; et al. CXCL12 mediates glioblastoma resistance to radiotherapy in the subventricular zone. Neuro-Oncology 2017, 19, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Ventero, M.P.; Fuentes-Baile, M.; Quereda, C.; Perez-Valeciano, E.; Alenda, C.; Garcia-Morales, P.; Esposito, D.; Dorado, P.; Manuel Barbera, V.; Saceda, M. Radiotherapy resistance acquisition in Glioblastoma. Role of SOCS1 and SOCS3. PLoS ONE 2019, 14, e0212581. [Google Scholar] [CrossRef]
- Chen, J.; Li, Y.; Yu, T.S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef]
- Piao, Y.; Liang, J.; Holmes, L.; Zurita, A.J.; Henry, V.; Heymach, J.V.; de Groot, J.F. Glioblastoma resistance to anti-VEGF therapy is associated with myeloid cell infiltration, stem cell accumulation, and a mesenchymal phenotype. Neuro-Oncology 2012, 14, 1379–1392. [Google Scholar] [CrossRef] [PubMed]
- Beier, D.; Rohrl, S.; Pillai, D.R.; Schwarz, S.; Kunz-Schughart, L.A.; Leukel, P.; Proescholdt, M.; Brawanski, A.; Bogdahn, U.; Trampe-Kieslich, A.; et al. Temozolomide preferentially depletes cancer stem cells in glioblastoma. Cancer Res. 2008, 68, 5706–5715. [Google Scholar] [CrossRef] [PubMed]
- Clement, V.; Sanchez, P.; de Tribolet, N.; Radovanovic, I.; Ruiz i Altaba, A. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr. Biol. 2007, 17, 165–172. [Google Scholar] [CrossRef]
- Tezcan, G.; Tunca, B.; Bekar, A.; Preusser, M.; Berghoff, A.S.; Egeli, U.; Cecener, G.; Ricken, G.; Budak, F.; Taskapilioglu, M.O.; et al. microRNA expression pattern modulates temozolomide response in GBM tumors with cancer stem cells. Cell Mol. Neurobiol. 2014, 34, 679–692. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.T.; Zhang, X.Q.; Zhuang, J.T.; Chan, H.L.; Li, C.H.; Leung, G.K. MicroRNA-21 inhibition enhances in vitro chemosensitivity of temozolomide-resistant glioblastoma cells. Anticancer Res. 2012, 32, 2835–2841. [Google Scholar]
- Liau, B.B.; Sievers, C.; Donohue, L.K.; Gillespie, S.M.; Flavahan, W.A.; Miller, T.E.; Venteicher, A.S.; Hebert, C.H.; Carey, C.D.; Rodig, S.J.; et al. Adaptive Chromatin Remodeling Drives Glioblastoma Stem Cell Plasticity and Drug Tolerance. Cell Stem Cell 2017, 20, 233–246. [Google Scholar] [CrossRef] [PubMed]
- Codony-Servat, J.; Rosell, R. Cancer stem cells and immunoresistance: Clinical implications and solutions. Transl. Lung Cancer Res. 2015, 4, 689–703. [Google Scholar] [CrossRef]
- Saha, D.; Martuza, R.L.; Rabkin, S.D. Curing glioblastoma: Oncolytic HSV-IL12 and checkpoint blockade. Oncoscience 2017, 4, 67–69. [Google Scholar] [CrossRef]
- Fadul, C.E.; Fisher, J.L.; Gui, J.; Hampton, T.H.; Cote, A.L.; Ernstoff, M.S. Immune modulation effects of concomitant temozolomide and radiation therapy on peripheral blood mononuclear cells in patients with glioblastoma multiforme. Neuro-Oncology 2011, 13, 393–400. [Google Scholar] [CrossRef]
- Wang, S.; Yao, F.; Lu, X.; Li, Q.; Su, Z.; Lee, J.H.; Wang, C.; Du, L. Temozolomide promotes immune escape of GBM cells via upregulating PD-L1. Am. J. Cancer Res. 2019, 9, 1161–1171. [Google Scholar] [PubMed]
- Cenciarini, M.; Valentino, M.; Belia, S.; Sforna, L.; Rosa, P.; Ronchetti, S.; D’Adamo, M.C.; Pessia, M. Dexamethasone in Glioblastoma Multiforme Therapy: Mechanisms and Controversies. Front. Mol. Neurosci. 2019, 12, 65. [Google Scholar] [CrossRef]
- Canon, J.; Osgood, T.; Olson, S.H.; Saiki, A.Y.; Robertson, R.; Yu, D.; Eksterowicz, J.; Ye, Q.; Jin, L.; Chen, A.; et al. The MDM2 Inhibitor AMG 232 Demonstrates Robust Antitumor Efficacy and Potentiates the Activity of p53-Inducing Cytotoxic Agents. Mol. Cancer Ther. 2015, 14, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, F.; Collavin, L.; Del Sal, G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 2019, 26, 199–212. [Google Scholar] [CrossRef]
- Schulz-Heddergott, R.; Moll, U.M. Gain-of-Function (GOF) Mutant p53 as Actionable Therapeutic Target. Cancers 2018, 10, 188. [Google Scholar] [CrossRef]
- Walerych, D.; Lisek, K.; Del Sal, G. Mutant p53: One, No One, and One Hundred Thousand. Front. Oncol 2015, 5, 289. [Google Scholar] [CrossRef] [PubMed]
- Patyka, M.; Sharifi, Z.; Petrecca, K.; Mansure, J.; Jean-Claude, B.; Sabri, S. Sensitivity to PRIMA-1MET is associated with decreased MGMT in human glioblastoma cells and glioblastoma stem cells irrespective of p53 status. Oncotarget 2016, 7, 60245–60269. [Google Scholar] [CrossRef]
- Perdrix, A.; Najem, A.; Saussez, S.; Awada, A.; Journe, F.; Ghanem, G.; Krayem, M. PRIMA-1 and PRIMA-1(Met) (APR-246): From Mutant/Wild Type p53 Reactivation to Unexpected Mechanisms Underlying Their Potent Anti-Tumor Effect in Combinatorial Therapies. Cancers 2017, 9, 172. [Google Scholar] [CrossRef]
- Zache, N.; Lambert, J.M.; Wiman, K.G.; Bykov, V.J. PRIMA-1MET inhibits growth of mouse tumors carrying mutant p53. Cell. Oncol. 2008, 30, 411–418. [Google Scholar] [CrossRef]
- Pal, S.; Kozono, D.; Yang, X.; Fendler, W.; Fitts, W.; Ni, J.; Alberta, J.A.; Zhao, J.; Liu, K.X.; Bian, J.; et al. Dual HDAC and PI3K Inhibition Abrogates NFkappaB- and FOXM1-Mediated DNA Damage Response to Radiosensitize Pediatric High-Grade Gliomas. Cancer Res. 2018, 78, 4007–4021. [Google Scholar] [CrossRef]
- Staberg, M.; Michaelsen, S.R.; Rasmussen, R.D.; Villingshoj, M.; Poulsen, H.S.; Hamerlik, P. Inhibition of histone deacetylases sensitizes glioblastoma cells to lomustine. Cell Oncol. (Dordr.) 2017, 40, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Liffers, K.; Kolbe, K.; Westphal, M.; Lamszus, K.; Schulte, A. Histone Deacetylase Inhibitors Resensitize EGFR/EGFRvIII-Overexpressing, Erlotinib-Resistant Glioblastoma Cells to Tyrosine Kinase Inhibition. Target. Oncol. 2016, 11, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.M.; Johnson, B.; Venkatarayan, A.; Flores, E.R.; Zhang, J.; Su, X.; Barton, M.; Lang, F.; Chandra, J. Preclinical activity of combined HDAC and KDM1A inhibition in glioblastoma. Neuro-Oncology 2015, 17, 1463–1473. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.; Murphy, B.; Miller, R.; Menon, V.; Banik, N.L.; Giglio, P.; Lindhorst, S.M.; Varma, A.K.; Vandergrift, W.A., 3rd; Patel, S.J.; et al. Mechanisms and clinical significance of histone deacetylase inhibitors: Epigenetic glioblastoma therapy. Anticancer Res. 2015, 35, 615–625. [Google Scholar] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Whittaker, S.; Madani, D.; Joshi, S.; Chung, S.A.; Johns, T.; Day, B.; Khasraw, M.; McDonald, K.L. Combination of palbociclib and radiotherapy for glioblastoma. Cell Death Discov. 2017, 3, 17033. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.W.; Parikh, M.; Phillips, J.J.; James, C.D.; Molinaro, A.M.; Butowski, N.A.; Clarke, J.L.; Oberheim-Bush, N.A.; Chang, S.M.; Berger, M.S.; et al. Phase-2 trial of palbociclib in adult patients with recurrent RB1-positive glioblastoma. J. Neurooncol. 2018, 140, 477–483. [Google Scholar] [CrossRef]
- Wen, P.Y.; Weller, M.; Lee, E.Q.; Alexander, B.M.; Barnholtz-Sloan, J.S.; Barthel, F.P.; Batchelor, T.T.; Bindra, R.S.; Chang, S.M.; Chiocca, E.A.; et al. Glioblastoma in adults: A Society for Neuro-Oncology (SNO) and European Society of Neuro-Oncology (EANO) consensus review on current management and future directions. Neuro-Oncology 2020, 22, 1073–1113. [Google Scholar] [CrossRef]
- Olmez, I.; Brenneman, B.; Xiao, A.; Serbulea, V.; Benamar, M.; Zhang, Y.; Manigat, L.; Abbas, T.; Lee, J.; Nakano, I.; et al. Combined CDK4/6 and mTOR Inhibition Is Synergistic against Glioblastoma via Multiple Mechanisms. Clin. Cancer Res. 2017, 23, 6958–6968. [Google Scholar] [CrossRef]
- Li, J.; Liang, R.; Song, C.; Xiang, Y.; Liu, Y. Prognostic significance of epidermal growth factor receptor expression in glioma patients. Onco Targets Ther. 2018, 11, 731–742. [Google Scholar] [CrossRef]
- Furnari, F.B.; Cloughesy, T.F.; Cavenee, W.K.; Mischel, P.S. Heterogeneity of epidermal growth factor receptor signalling networks in glioblastoma. Nat. Rev. Cancer 2015, 15, 302–310. [Google Scholar] [CrossRef]
- An, Z.; Aksoy, O.; Zheng, T.; Fan, Q.W.; Weiss, W.A. Epidermal growth factor receptor and EGFRvIII in glioblastoma: Signaling pathways and targeted therapies. Oncogene 2018, 37, 1561–1575. [Google Scholar] [CrossRef]
- Eskilsson, E.; Rosland, G.V.; Solecki, G.; Wang, Q.; Harter, P.N.; Graziani, G.; Verhaak, R.G.W.; Winkler, F.; Bjerkvig, R.; Miletic, H. EGFR heterogeneity and implications for therapeutic intervention in glioblastoma. Neuro-Oncology 2018, 20, 743–752. [Google Scholar] [CrossRef]
- Kwatra, M.M. A Rational Approach to Target the Epidermal Growth Factor Receptor in Glioblastoma. Curr. Cancer Drug Targets 2017, 17, 290–296. [Google Scholar] [CrossRef]
- Kim, M.; Laramy, J.K.; Mohammad, A.S.; Talele, S.; Fisher, J.; Sarkaria, J.N.; Elmquist, W.F. Brain Distribution of a Panel of Epidermal Growth Factor Receptor Inhibitors Using Cassette Dosing in Wild-Type and Abcb1/Abcg2-Deficient Mice. Drug Metab. Dispos. 2019, 47, 393–404. [Google Scholar] [CrossRef]
- Liu, X.; Chen, X.; Shi, L.; Shan, Q.; Cao, Q.; Yue, C.; Li, H.; Li, S.; Wang, J.; Gao, S.; et al. The third-generation EGFR inhibitor AZD9291 overcomes primary resistance by continuously blocking ERK signaling in glioblastoma. J. Exp. Clin. Cancer Res. 2019, 38, 219. [Google Scholar] [CrossRef]
- Li, L.; Quang, T.S.; Gracely, E.J.; Kim, J.H.; Emrich, J.G.; Yaeger, T.E.; Jenrette, J.M.; Cohen, S.C.; Black, P.; Brady, L.W. A Phase II study of anti-epidermal growth factor receptor radioimmunotherapy in the treatment of glioblastoma multiforme. J. Neurosurg. 2010, 113, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Von Achenbach, C.; Silginer, M.; Blot, V.; Weiss, W.A.; Weller, M. Depatuxizumab Mafodotin (ABT-414)-induced Glioblastoma Cell Death Requires EGFR Overexpression, but not EGFR(Y1068) Phosphorylation. Mol. Cancer Ther. 2020, 19, 1328–1339. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Luo, J.; Cantley, L.C. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 2006, 7, 606–619. [Google Scholar] [CrossRef] [PubMed]
- Katso, R.; Okkenhaug, K.; Ahmadi, K.; White, S.; Timms, J.; Waterfield, M.D. Cellular function of phosphoinositide 3-kinases: Implications for development, homeostasis, and cancer. Annu. Rev. Cell Dev. Biol. 2001, 17, 615–675. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in cancer: Mechanisms and advances in clinical trials. Mol. Cancer 2019, 18, 26. [Google Scholar] [CrossRef]
- Zhao, H.F.; Wang, J.; Shao, W.; Wu, C.P.; Chen, Z.P.; To, S.T.; Li, W.P. Recent advances in the use of PI3K inhibitors for glioblastoma multiforme: Current preclinical and clinical development. Mol. Cancer 2017, 16, 100. [Google Scholar] [CrossRef]
- Djuzenova, C.S.; Fiedler, V.; Memmel, S.; Katzer, A.; Sisario, D.; Brosch, P.K.; Gohrung, A.; Frister, S.; Zimmermann, H.; Flentje, M.; et al. Differential effects of the Akt inhibitor MK-2206 on migration and radiation sensitivity of glioblastoma cells. BMC Cancer 2019, 19, 299. [Google Scholar] [CrossRef] [PubMed]
- Mecca, C.; Giambanco, I.; Donato, R.; Arcuri, C. Targeting mTOR in Glioblastoma: Rationale and Preclinical/Clinical Evidence. Dis. Markers 2018, 2018, 9230479. [Google Scholar] [CrossRef]
- Daniele, S.; Costa, B.; Zappelli, E.; Da Pozzo, E.; Sestito, S.; Nesi, G.; Campiglia, P.; Marinelli, L.; Novellino, E.; Rapposelli, S.; et al. Combined inhibition of AKT/mTOR and MDM2 enhances Glioblastoma Multiforme cell apoptosis and differentiation of cancer stem cells. Sci. Rep. 2015, 5, 9956. [Google Scholar] [CrossRef] [PubMed]
- Lisi, L.; Ciotti, G.M.P.; Chiavari, M.; Pizzoferrato, M.; Mangiola, A.; Kalinin, S.; Feinstein, D.L.; Navarra, P. Phospho-mTOR expression in human glioblastoma microglia-macrophage cells. Neurochem. Int. 2019, 129, 104485. [Google Scholar] [CrossRef]
- Geissler, E.K. The influence of mTOR inhibitors on immunity and the relationship to post-transplant malignancy. Transplant. Res. 2013, 2, S2. [Google Scholar] [CrossRef] [PubMed]
- Bak, S.; Tischer, S.; Dragon, A.; Ravens, S.; Pape, L.; Koenecke, C.; Oelke, M.; Blasczyk, R.; Maecker-Kolhoff, B.; Eiz-Vesper, B. Selective Effects of mTOR Inhibitor Sirolimus on Naive and CMV-Specific T Cells Extending Its Applicable Range Beyond Immunosuppression. Front. Immunol. 2018, 9, 2953. [Google Scholar] [CrossRef]
- Zhang, Y.; Xia, M.; Jin, K.; Wang, S.; Wei, H.; Fan, C.; Wu, Y.; Li, X.; Li, X.; Li, G.; et al. Function of the c-Met receptor tyrosine kinase in carcinogenesis and associated therapeutic opportunities. Mol. Cancer 2018, 17, 45. [Google Scholar] [CrossRef]
- Petterson, S.A.; Dahlrot, R.H.; Hermansen, S.K.; Sune, K.A.M.; Gundesen, M.T.; Wohlleben, H.; Rasmussen, T.; Beier, C.P.; Hansen, S.; Kristensen, B.W. High levels of c-Met is associated with poor prognosis in glioblastoma. J. Neurooncol. 2015, 122, 517–527. [Google Scholar] [CrossRef]
- Kong, D.S.; Song, S.Y.; Kim, D.H.; Joo, K.M.; Yoo, J.S.; Koh, J.S.; Dong, S.M.; Suh, Y.L.; Lee, J.I.; Park, K.; et al. Prognostic significance of c-Met expression in glioblastomas. Cancer 2009, 115, 140–148. [Google Scholar] [CrossRef]
- Cloughesy, T.; Finocchiaro, G.; Belda-Iniesta, C.; Recht, L.; Brandes, A.A.; Pineda, E.; Mikkelsen, T.; Chinot, O.L.; Balana, C.; Macdonald, D.R.; et al. Randomized, Double-Blind, Placebo-Controlled, Multicenter Phase II Study of Onartuzumab Plus Bevacizumab Versus Placebo Plus Bevacizumab in Patients With Recurrent Glioblastoma: Efficacy, Safety, and Hepatocyte Growth Factor and O(6)-Methylguanine-DNA Methyltransferase Biomarker Analyses. J. Clin. Oncol. 2017, 35, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Beenken, A.; Mohammadi, M. The FGF family: Biology, pathophysiology and therapy. Nat. Rev. Drug Discov. 2009, 8, 235–253. [Google Scholar] [CrossRef] [PubMed]
- Ducassou, A.; Uro-Coste, E.; Verrelle, P.; Filleron, T.; Benouaich-Amiel, A.; Lubrano, V.; Sol, J.C.; Delisle, M.B.; Favre, G.; Ken, S.; et al. alphavbeta3 Integrin and Fibroblast growth factor receptor 1 (FGFR1): Prognostic factors in a phase I-II clinical trial associating continuous administration of Tipifarnib with radiotherapy for patients with newly diagnosed glioblastoma. Eur. J. Cancer 2013, 49, 2161–2169. [Google Scholar] [CrossRef]
- Day, E.K.; Sosale, N.G.; Xiao, A.; Zhong, Q.; Purow, B.; Lazzara, M.J. Glioblastoma Cell Resistance to EGFR and MET Inhibition Can Be Overcome via Blockade of FGFR-SPRY2 Bypass Signaling. Cell Rep. 2020, 30, 3383–3396. [Google Scholar] [CrossRef]
- Lange, C.; Storkebaum, E.; de Almodovar, C.R.; Dewerchin, M.; Carmeliet, P. Vascular endothelial growth factor: A neurovascular target in neurological diseases. Nat. Rev. Neurol. 2016, 12, 439–454. [Google Scholar] [CrossRef]
- Xiao, Q.; Yang, S.; Ding, G.; Luo, M. Anti-vascular endothelial growth factor in glioblastoma: A systematic review and meta-analysis. Neurol. Sci. 2018, 39, 2021–2031. [Google Scholar] [CrossRef]
- Haibe, Y.; Kreidieh, M.; El Hajj, H.; Khalifeh, I.; Mukherji, D.; Temraz, S.; Shamseddine, A. Resistance Mechanisms to Anti-angiogenic Therapies in Cancer. Front. Oncol. 2020, 10, 221. [Google Scholar] [CrossRef]
- Nazarenko, I.; Hede, S.M.; He, X.; Hedren, A.; Thompson, J.; Lindstrom, M.S.; Nister, M. PDGF and PDGF receptors in glioma. Ups. J. Med. Sci. 2012, 117, 99–112. [Google Scholar] [CrossRef]
- Cao, Y. Multifarious functions of PDGFs and PDGFRs in tumor growth and metastasis. Trends Mol. Med. 2013, 19, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Cantanhede, I.G.; de Oliveira, J.R.M. PDGF Family Expression in Glioblastoma Multiforme: Data Compilation from Ivy Glioblastoma Atlas Project Database. Sci. Rep. 2017, 7, 15271. [Google Scholar] [CrossRef]
- Song, K.; Yuan, Y.; Lin, Y.; Wang, Y.X.; Zhou, J.; Gai, Q.J.; Zhang, L.; Mao, M.; Yao, X.X.; Qin, Y.; et al. ERBB3, IGF1R, and TGFBR2 expression correlate with PDGFR expression in glioblastoma and participate in PDGFR inhibitor resistance of glioblastoma cells. Am. J. Cancer Res. 2018, 8, 792–809. [Google Scholar] [PubMed]
- Pastorino, S.; Langley, E.J.; Chao, Y.; Jiang, P.; Mukthavaram, R.; Pingle, S.C.; Kim, P.S.; Singh, S.; Kesari, S. Mechanisms of resistance to PDGFR inhibition in glioblastoma. J. Clin. Oncol. 2014, 32, e13030. [Google Scholar] [CrossRef]
- Roy, L.O.; Poirier, M.B.; Fortin, D. Differential Expression and Clinical Significance of Transforming Growth Factor-Beta Isoforms in GBM Tumors. Int. J. Mol. Sci. 2018, 19, 1113. [Google Scholar] [CrossRef] [PubMed]
- Luo, K. Signaling Cross Talk between TGF-beta/Smad and Other Signaling Pathways. Cold Spring Harb. Perspect Biol. 2017, 9, a022137. [Google Scholar] [CrossRef]
- Wick, A.; Desjardins, A.; Suarez, C.; Forsyth, P.; Gueorguieva, I.; Burkholder, T.; Cleverly, A.L.; Estrem, S.T.; Wang, S.; Lahn, M.M.; et al. Phase 1b/2a study of galunisertib, a small molecule inhibitor of transforming growth factor-beta receptor I, in combination with standard temozolomide-based radiochemotherapy in patients with newly diagnosed malignant glioma. Investig. New Drugs 2020. [Google Scholar] [CrossRef]
- Areeb, Z.; Stylli, S.S.; Ware, T.M.; Harris, N.C.; Shukla, L.; Shayan, R.; Paradiso, L.; Li, B.; Morokoff, A.P.; Kaye, A.H.; et al. Inhibition of glioblastoma cell proliferation, migration and invasion by the proteasome antagonist carfilzomib. Med. Oncol. 2016, 33, 53. [Google Scholar] [CrossRef]
- Vlachostergios, P.J.; Voutsadakis, I.A.; Papandreou, C.N. The shaping of invasive glioma phenotype by the ubiquitin-proteasome system. Cell Commun. Adhes. 2013, 20, 87–92. [Google Scholar] [CrossRef][Green Version]
- Rajendra, J.; Datta, K.K.; Ud Din Farooqee, S.B.; Thorat, R.; Kumar, K.; Gardi, N.; Kaur, E.; Nair, J.; Salunkhe, S.; Patkar, K.; et al. Enhanced proteasomal activity is essential for long term survival and recurrence of innately radiation resistant residual glioblastoma cells. Oncotarget 2018, 9, 27667–27681. [Google Scholar] [CrossRef] [PubMed]
- Roth, P.; Mason, W.P.; Richardson, P.G.; Weller, M. Proteasome inhibition for the treatment of glioblastoma. Expert Opin. Investig Drugs 2020, 29, 1133–1141. [Google Scholar] [CrossRef] [PubMed]
- Lesueur, P.; Lequesne, J.; Grellard, J.M.; Dugue, A.; Coquan, E.; Brachet, P.E.; Geffrelot, J.; Kao, W.; Emery, E.; Berro, D.H.; et al. Phase I/IIa study of concomitant radiotherapy with olaparib and temozolomide in unresectable or partially resectable glioblastoma: OLA-TMZ-RTE-01 trial protocol. BMC Cancer 2019, 19, 198. [Google Scholar] [CrossRef] [PubMed]
- Ferri, A.; Stagni, V.; Barila, D. Targeting the DNA Damage Response to Overcome Cancer Drug Resistance in Glioblastoma. Int. J. Mol. Sci. 2020, 21, 4910. [Google Scholar] [CrossRef]
- Frosina, G.; Marubbi, D.; Marcello, D.; Vecchio, D.; Daga, A. The efficacy and toxicity of ATM inhibition in glioblastoma initiating cells-driven tumor models. Crit. Rev. Oncol. Hematol. 2019, 138, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Fisusi, F.A.; Schatzlein, A.G.; Uchegbu, I.F. Nanomedicines in the treatment of brain tumors. Nanomedicine (Lond.) 2018, 13, 579–583. [Google Scholar] [CrossRef]
- Saha, D.; Martuza, R.L.; Rabkin, S.D. Oncolytic herpes simplex virus immunovirotherapy in combination with immune checkpoint blockade to treat glioblastoma. Immunotherapy 2018, 10, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Migliorini, D.; Dietrich, P.Y.; Stupp, R.; Linette, G.P.; Posey, A.D., Jr.; June, C.H. CAR T-Cell Therapies in Glioblastoma: A First Look. Clin. Cancer Res. 2018, 24, 535–540. [Google Scholar] [CrossRef]
- Kong, Z.; Wang, Y.; Ma, W. Vaccination in the immunotherapy of glioblastoma. Hum. Vaccin. Immunother. 2018, 14, 255–268. [Google Scholar] [CrossRef]
- Caccese, M.; Indraccolo, S.; Zagonel, V.; Lombardi, G. PD-1/PD-L1 immune-checkpoint inhibitors in glioblastoma: A concise review. Crit. Rev. Oncol. Hematol. 2019, 135, 128–134. [Google Scholar] [CrossRef]
- Zeng, J.; See, A.P.; Phallen, J.; Jackson, C.M.; Belcaid, Z.; Ruzevick, J.; Durham, N.; Meyer, C.; Harris, T.J.; Albesiano, E.; et al. Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. Int. J. Radiat. Oncol. Biol. Phys. 2013, 86, 343–349. [Google Scholar] [CrossRef]
- Kim, J.E.; Patel, M.A.; Mangraviti, A.; Kim, E.S.; Theodros, D.; Velarde, E.; Liu, A.; Sankey, E.W.; Tam, A.; Xu, H.; et al. Combination Therapy with Anti-PD-1, Anti-TIM-3, and Focal Radiation Results in Regression of Murine Gliomas. Clin. Cancer Res. 2017, 23, 124–136. [Google Scholar] [CrossRef]
- Huang, B.Y.; Zhan, Y.P.; Zong, W.J.; Yu, C.J.; Li, J.F.; Qu, Y.M.; Han, S. The PD-1/B7-H1 pathway modulates the natural killer cells versus mouse glioma stem cells. PLoS ONE 2015, 10, e0134715. [Google Scholar] [CrossRef] [PubMed]
- Jahan, N.; Talat, H.; Alonso, A.; Saha, D.; Curry, W.T. Triple combination immunotherapy with GVAX, anti-PD-1 monoclonal antibody, and agonist anti-OX40 monoclonal antibody is highly effective against murine intracranial glioma. Oncoimmunology 2019, 8, e1577108. [Google Scholar] [CrossRef] [PubMed]
- Filley, A.C.; Henriquez, M.; Dey, M. Recurrent glioma clinical trial, CheckMate-143: The game is not over yet. Oncotarget 2017, 8, 91779–91794. [Google Scholar] [CrossRef]
- Zhao, J.; Chen, A.X.; Gartrell, R.D.; Silverman, A.M.; Aparicio, L.; Chu, T.; Bordbar, D.; Shan, D.; Samanamud, J.; Mahajan, A.; et al. Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat. Med. 2019, 25, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.R.; Hugo, W.; Lee, A.H.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M.; et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat. Med. 2019, 25, 477–486. [Google Scholar] [CrossRef]
- Hu, P.; Liu, Q.; Deng, G.; Zhang, J.; Liang, N.; Xie, J.; Zhang, J. The prognostic value of cytotoxic T-lymphocyte antigen 4 in cancers: A systematic review and meta-analysis. Sci. Rep. 2017, 7, 42913. [Google Scholar] [CrossRef]
- Grauer, O.M.; Nierkens, S.; Bennink, E.; Toonen, L.W.; Boon, L.; Wesseling, P.; Sutmuller, R.P.; Adema, G.J. CD4+FoxP3+ regulatory T cells gradually accumulate in gliomas during tumor growth and efficiently suppress antiglioma immune responses in vivo. Int. J. Cancer 2007, 121, 95–105. [Google Scholar] [CrossRef]
- Margolin, K.; Ernstoff, M.S.; Hamid, O.; Lawrence, D.; McDermott, D.; Puzanov, I.; Wolchok, J.D.; Clark, J.I.; Sznol, M.; Logan, T.F.; et al. Ipilimumab in patients with melanoma and brain metastases: An open-label, phase 2 trial. Lancet Oncol. 2012, 13, 459–465. [Google Scholar] [CrossRef]
- Vom Berg, J.; Vrohlings, M.; Haller, S.; Haimovici, A.; Kulig, P.; Sledzinska, A.; Weller, M.; Becher, B. Intratumoral IL-12 combined with CTLA-4 blockade elicits T cell-mediated glioma rejection. J. Exp. Med. 2013, 210, 2803–2811. [Google Scholar] [CrossRef]
- Carter, T.; Shaw, H.; Cohn-Brown, D.; Chester, K.; Mulholland, P. Ipilimumab and Bevacizumab in Glioblastoma. Clin. Oncol. (R. Coll. Radiol.) 2016, 28, 622–626. [Google Scholar] [CrossRef]
- Wainwright, D.A.; Dey, M.; Chang, A.; Lesniak, M.S. Targeting Tregs in Malignant Brain Cancer: Overcoming IDO. Front. Immunol. 2013, 4, 116. [Google Scholar] [CrossRef] [PubMed]
- Sordillo, P.P.; Sordillo, L.A.; Helson, L. The Kynurenine Pathway: A Primary Resistance Mechanism in Patients with Glioblastoma. Anticancer Res. 2017, 37, 2159–2171. [Google Scholar] [CrossRef] [PubMed]
- Munn, D.H.; Mellor, A.L. Indoleamine 2,3 dioxygenase and metabolic control of immune responses. Trends Immunol. 2013, 34, 137–143. [Google Scholar] [CrossRef]
- Zhai, L.; Lauing, K.L.; Chang, A.L.; Dey, M.; Qian, J.; Cheng, Y.; Lesniak, M.S.; Wainwright, D.A. The role of IDO in brain tumor immunotherapy. J. Neurooncol. 2015, 123, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Abedalthagafi, M.; Barakeh, D.; Foshay, K.M. Immunogenetics of glioblastoma: The future of personalized patient management. NPJ Precis Oncol. 2018, 2, 27. [Google Scholar] [CrossRef] [PubMed]
- Zhai, L.; Ladomersky, E.; Lauing, K.L.; Wu, M.; Genet, M.; Gritsina, G.; Gyorffy, B.; Brastianos, P.K.; Binder, D.C.; Sosman, J.A.; et al. Infiltrating T Cells Increase IDO1 Expression in Glioblastoma and Contribute to Decreased Patient Survival. Clin. Cancer Res. 2017, 23, 6650–6660. [Google Scholar] [CrossRef]
- Mitsuka, K.; Kawataki, T.; Satoh, E.; Asahara, T.; Horikoshi, T.; Kinouchi, H. Expression of indoleamine 2,3-dioxygenase and correlation with pathological malignancy in gliomas. Neurosurgery 2013, 72, 1031–1038. [Google Scholar] [CrossRef] [PubMed]
- Wainwright, D.A.; Chang, A.L.; Dey, M.; Balyasnikova, I.V.; Kim, C.K.; Tobias, A.; Cheng, Y.; Kim, J.W.; Qiao, J.; Zhang, L.; et al. Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA-4, and PD-L1 in mice with brain tumors. Clin. Cancer Res. 2014, 20, 5290–5301. [Google Scholar] [CrossRef]
- Ladomersky, E.; Zhai, L.; Lenzen, A.; Lauing, K.L.; Qian, J.; Scholtens, D.M.; Gritsina, G.; Sun, X.; Liu, Y.; Yu, F.; et al. IDO1 Inhibition Synergizes with Radiation and PD-1 Blockade to Durably Increase Survival Against Advanced Glioblastoma. Clin. Cancer Res. 2018, 24, 2559–2573. [Google Scholar] [CrossRef]
- Li, M.; Bolduc, A.R.; Hoda, M.N.; Gamble, D.N.; Dolisca, S.B.; Bolduc, A.K.; Hoang, K.; Ashley, C.; McCall, D.; Rojiani, A.M.; et al. The indoleamine 2,3-dioxygenase pathway controls complement-dependent enhancement of chemo-radiation therapy against murine glioblastoma. J. Immunother. Cancer 2014, 2, 21. [Google Scholar] [CrossRef]
- Eissa, I.R.; Bustos-Villalobos, I.; Ichinose, T.; Matsumura, S.; Naoe, Y.; Miyajima, N.; Morimoto, D.; Mukoyama, N.; Zhiwen, W.; Tanaka, M.; et al. The Current Status and Future Prospects of Oncolytic Viruses in Clinical Trials against Melanoma, Glioma, Pancreatic, and Breast Cancers. Cancers 2018, 10, 356. [Google Scholar] [CrossRef] [PubMed]
- O’bryan, S.M.; Mathis, J.M. Oncolytic Virotherapy for Breast Cancer Treatment. Curr. Gene Ther. 2018, 18, 192–205. [Google Scholar] [CrossRef]
- Ghouse, S.M.; Nguyen, H.M.; Bommareddy, P.K.; Guz-Montgomery, K.; Saha, D. Oncolytic Herpes Simplex Virus Encoding IL12 Controls Triple-Negative Breast Cancer Growth and Metastasis. Front. Oncol. 2020, 10, 384. [Google Scholar] [CrossRef] [PubMed]
- Bommareddy, P.K.; Peters, C.; Saha, D.; Rabkin, S.D.; Kaufman, H.L. Oncolytic Herpes Simplex Viruses as a Paradigm for the Treatment of Cancer. Annu. Rev. Cancer Biol. 2018, 2, 155–173. [Google Scholar] [CrossRef]
- Peters, C.; Paget, M.; Tshilenge, K.T.; Saha, D.; Antoszczyk, S.; Baars, A.; Frost, T.; Martuza, R.L.; Wakimoto, H.; Rabkin, S.D. Restriction of Replication of Oncolytic Herpes Simplex Virus with a Deletion of gamma34.5 in Glioblastoma Stem-Like Cells. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- Saha, D.; Ahmed, S.S.; Rabkin, S.D. Exploring the Antitumor Effect of Virus in Malignant Glioma. Drugs Future 2015, 40, 739–749. [Google Scholar] [CrossRef]
- Saha, D.; Martuza, R.L.; Curry, W.T. Viral oncolysis of glioblastoma. In Neurotropic Viral Infections, 2nd ed.; Reiss, C.S., Ed.; Springer: New York, NY, USA, 2016; Volume 2, pp. 481–517. [Google Scholar]
- Saha, D.; Martuza, R.L.; Rabkin, S.D. Macrophage Polarization Contributes to Glioblastoma Eradication by Combination Immunovirotherapy and Immune Checkpoint Blockade. Cancer Cell 2017, 32, 253–267. [Google Scholar] [CrossRef]
- Saha, D.; Rabkin, S.D.; Martuza, R.L. Temozolomide antagonizes oncolytic immunovirotherapy in glioblastoma. J. Immunother. Cancer 2020, 8, e000345. [Google Scholar] [CrossRef] [PubMed]
- Saha, D.; Wakimoto, H.; Peters, C.W.; Antoszczyk, S.J.; Rabkin, S.D.; Martuza, R.L. Combinatorial Effects of VEGFR Kinase Inhibitor Axitinib and Oncolytic Virotherapy in Mouse and Human Glioblastoma Stem-Like Cell Models. Clin. Cancer Res. 2018, 24, 3409–3422. [Google Scholar] [CrossRef]
- Siurala, M.; Havunen, R.; Saha, D.; Lumen, D.; Airaksinen, A.J.; Tahtinen, S.; Cervera-Carrascon, V.; Bramante, S.; Parviainen, S.; Vaha-Koskela, M.; et al. Adenoviral Delivery of Tumor Necrosis Factor-alpha and Interleukin-2 Enables Successful Adoptive Cell Therapy of Immunosuppressive Melanoma. Mol. Ther. 2016, 24, 1435–1443. [Google Scholar] [CrossRef] [PubMed]
- Tahtinen, S.; Blattner, C.; Vaha-Koskela, M.; Saha, D.; Siurala, M.; Parviainen, S.; Utikal, J.; Kanerva, A.; Umansky, V.; Hemminki, A. T-Cell Therapy Enabling Adenoviruses Coding for IL2 and TNFalpha Induce Systemic Immunomodulation in Mice With Spontaneous Melanoma. J. Immunother. 2016, 39, 343–354. [Google Scholar] [CrossRef]
- Vaha-Koskela, M.; Tahtinen, S.; Gronberg-Vaha-Koskela, S.; Taipale, K.; Saha, D.; Merisalo-Soikkeli, M.; Ahonen, M.; Rouvinen-Lagerstrom, N.; Hirvinen, M.; Veckman, V.; et al. Overcoming tumor resistance by heterologous adeno-poxvirus combination therapy. Mol. Ther. Oncol. 2015, 1, 14006. [Google Scholar] [CrossRef] [PubMed]
- Saha, D.; Wakimoto, H.; Rabkin, S.D. Oncolytic herpes simplex virus interactions with the host immune system. Curr. Opin. Virol. 2016, 21, 26–34. [Google Scholar] [CrossRef]
- Saha, D.; Rabkin, S.D. Immunohistochemistry for Tumor-Infiltrating Immune Cells After Oncolytic Virotherapy. Methods Mol. Biol. 2020, 2058, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Alessandrini, F.; Menotti, L.; Avitabile, E.; Appolloni, I.; Ceresa, D.; Marubbi, D.; Campadelli-Fiume, G.; Malatesta, P. Eradication of glioblastoma by immuno-virotherapy with a retargeted oncolytic HSV in a preclinical model. Oncogene 2019, 38, 4467–4479. [Google Scholar] [CrossRef] [PubMed]
- Alayo, Q.A.; Ito, H.; Passaro, C.; Zdioruk, M.; Mahmoud, A.B.; Grauwet, K.; Zhang, X.; Lawler, S.E.; Reardon, D.A.; Goins, W.F.; et al. Glioblastoma infiltration of both tumor- and virus-antigen specific cytotoxic T cells correlates with experimental virotherapy responses. Sci. Rep. 2020, 10, 5095. [Google Scholar] [CrossRef] [PubMed]
- Todo, T. ATIM-14. RESULTS OF PHASE II CLINICAL TRIAL OF ONCOLYTIC HERPES VIRUS G47Δ IN PATIENTS WITH GLIOBLASTOMA. Neuro-Oncology 2019, 21, vi4. [Google Scholar] [CrossRef]
- Gesundheit, B.; Ben-David, E.; Posen, Y.; Ellis, R.; Wollmann, G.; Schneider, E.M.; Aigner, K.; Brauns, L.; Nesselhut, T.; Ackva, I.; et al. Effective Treatment of Glioblastoma Multiforme With Oncolytic Virotherapy: A Case-Series. Front. Oncol. 2020, 10, 702. [Google Scholar] [CrossRef]
- Nguyen, H.M.; Guz-Montgomery, K.; Saha, D. Oncolytic Virus Encoding a Master Pro-Inflammatory Cytokine Interleukin 12 in Cancer Immunotherapy. Cells 2020, 9, 400. [Google Scholar] [CrossRef]
- Alonso, M.M.; Gomez-Manzano, C.; Jiang, H.; Bekele, N.B.; Piao, Y.; Yung, W.K.; Alemany, R.; Fueyo, J. Combination of the oncolytic adenovirus ICOVIR-5 with chemotherapy provides enhanced anti-glioma effect in vivo. Cancer Gene Ther. 2007, 14, 756–761. [Google Scholar] [CrossRef]
- Gomez-Gutierrez, J.G.; Nitz, J.; Sharma, R.; Wechman, S.L.; Riedinger, E.; Martinez-Jaramillo, E.; Sam Zhou, H.; McMasters, K.M. Combined therapy of oncolytic adenovirus and temozolomide enhances lung cancer virotherapy in vitro and in vivo. Virology 2016, 487, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Kanai, R.; Rabkin, S.D.; Yip, S.; Sgubin, D.; Zaupa, C.M.; Hirose, Y.; Louis, D.N.; Wakimoto, H.; Martuza, R.L. Oncolytic virus-mediated manipulation of DNA damage responses: Synergy with chemotherapy in killing glioblastoma stem cells. J. Natl. Cancer Inst. 2012, 104, 42–55. [Google Scholar] [CrossRef]
- Kleijn, A.; van den Bossche, W.; Haefner, E.S.; Belcaid, Z.; Burghoorn-Maas, C.; Kloezeman, J.J.; Pas, S.D.; Leenstra, S.; Debets, R.; de Vrij, J.; et al. The Sequence of Delta24-RGD and TMZ Administration in Malignant Glioma Affects the Role of CD8(+)T Cell Anti-tumor Activity. Mol. Ther. Oncol. 2017, 5, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Garza-Morales, R.; Gonzalez-Ramos, R.; Chiba, A.; Montes de Oca-Luna, R.; McNally, L.R.; McMasters, K.M.; Gomez-Gutierrez, J.G. Temozolomide Enhances Triple-Negative Breast Cancer Virotherapy In Vitro. Cancers 2018, 10, 144. [Google Scholar] [CrossRef]
- Pisklakova, A.; McKenzie, B.; Zemp, F.; Lun, X.; Kenchappa, R.S.; Etame, A.B.; Rahman, M.M.; Reilly, K.; Pilon-Thomas, S.; McFadden, G.; et al. M011L-deficient oncolytic myxoma virus induces apoptosis in brain tumor-initiating cells and enhances survival in a novel immunocompetent mouse model of glioblastoma. Neuro-Oncology 2016, 18, 1088–1098. [Google Scholar] [CrossRef]
- Bai, Y.; Chen, Y.; Hong, X.; Liu, X.; Su, X.; Li, S.; Dong, X.; Zhao, G.; Li, Y. Newcastle disease virus enhances the growth-inhibiting and proapoptotic effects of temozolomide on glioblastoma cells in vitro and in vivo. Sci. Rep. 2018, 8, 11470. [Google Scholar] [CrossRef]
- Sivanandam, V.; LaRocca, C.J.; Chen, N.G.; Fong, Y.; Warner, S.G. Oncolytic Viruses and Immune Checkpoint Inhibition: The Best of Both Worlds. Mol. Ther. Oncol. 2019, 13, 93–106. [Google Scholar] [CrossRef]
- Dejaegher, J.; Van Gool, S.; De Vleeschouwer, S. Dendritic cell vaccination for glioblastoma multiforme: Review with focus on predictive factors for treatment response. Immunotargets Ther. 2014, 3, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Oh, T.; Sayegh, E.T.; Fakurnejad, S.; Oyon, D.; Lamano, J.B.; DiDomenico, J.D.; Bloch, O.; Parsa, A.T. Vaccine therapies in malignant glioma. Curr. Neurol. Neurosci. Rep. 2015, 15, 508. [Google Scholar] [CrossRef]
- Reardon, D.A.; Schuster, J.; Tran, D.D.; Fink, K.L.; Nabors, L.B.; Li, G.; Bota, D.A.; Lukas, R.V.; Desjardins, A.; Ashby, L.S.; et al. ReACT: Overall survival from a randomized phase II study of rindopepimut (CDX-110) plus bevacizumab in relapsed glioblastoma. J. Clin. Oncol. 2015, 33, 2009. [Google Scholar] [CrossRef]
- Weller, M.; Butowski, N.; Tran, D.D.; Recht, L.D.; Lim, M.; Hirte, H.; Ashby, L.; Mechtler, L.; Goldlust, S.A.; Iwamoto, F.; et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): A randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017, 18, 1373–1385. [Google Scholar] [CrossRef]
- Binder, D.C.; Ladomersky, E.; Lenzen, A.; Zhai, L.; Lauing, K.L.; Otto-Meyer, S.D.; Lukas, R.V.; Wainwright, D.A. Lessons learned from rindopepimut treatment in patients with EGFRvIII-expressing glioblastoma. Transl. Cancer Res. 2018, 7, S510–S513. [Google Scholar] [CrossRef]
- Khansur, E.M.; Shah, A.H.; Lacy, K.; Kuchakulla, M.; Komotar, R.J. Novel Immunotherapeutics for the Treatment of Glioblastoma: The Last Decade of Research. Cureus 2018, 10, e2130. [Google Scholar] [CrossRef] [PubMed]
- Ji, N.; Zhang, Y.; Liu, Y.; Xie, J.; Wang, Y.; Hao, S.; Gao, Z. Heat shock protein peptide complex-96 vaccination for newly diagnosed glioblastoma: A phase I, single-arm trial. JCI Insight 2018, 3, e99145. [Google Scholar] [CrossRef]
- Palucka, K.; Banchereau, J. Cancer immunotherapy via dendritic cells. Nat. Rev. Cancer 2012, 12, 265–277. [Google Scholar] [CrossRef]
- Perez, C.R.; De Palma, M. Engineering dendritic cell vaccines to improve cancer immunotherapy. Nat. Commun. 2019, 10, 5408. [Google Scholar] [CrossRef] [PubMed]
- Nava, S.; Dossena, M.; Pogliani, S.; Pellegatta, S.; Antozzi, C.; Baggi, F.; Gellera, C.; Pollo, B.; Parati, E.A.; Finocchiaro, G.; et al. An optimized method for manufacturing a clinical scale dendritic cell-based vaccine for the treatment of glioblastoma. PLoS ONE 2012, 7, e52301. [Google Scholar] [CrossRef]
- Liau, L.M.; Ashkan, K.; Tran, D.D.; Campian, J.L.; Trusheim, J.E.; Cobbs, C.S.; Heth, J.A.; Salacz, M.; Taylor, S.; D’Andre, S.D.; et al. First results on survival from a large Phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J. Transl. Med. 2018, 16, 142. [Google Scholar] [CrossRef] [PubMed]
- Weenink, B.; French, P.J.; Sillevis Smitt, P.A.E.; Debets, R.; Geurts, M. Immunotherapy in Glioblastoma: Current Shortcomings and Future Perspectives. Cancers 2020, 12, 751. [Google Scholar] [CrossRef] [PubMed]
- Keskin, D.B.; Anandappa, A.J.; Sun, J.; Tirosh, I.; Mathewson, N.D.; Li, S.; Oliveira, G.; Giobbie-Hurder, A.; Felt, K.; Gjini, E.; et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 2019, 565, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.D.; Maus, M.V.; June, C.H.; Sampson, J.H. Immunotherapy for Glioblastoma: Adoptive T-cell Strategies. Clin. Cancer Res. 2019, 25, 2042–2048. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Shen, F.; Yao, Y.; Wang, L.L.; Zhu, Y.; Hu, J. Adoptive Cell Therapy: A Novel and Potential Immunotherapy for Glioblastoma. Front. Oncol. 2020, 10, 59. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Meng, Q.; Bartek, J., Jr.; Poiret, T.; Persson, O.; Rane, L.; Rangelova, E.; Illies, C.; Peredo, I.H.; Luo, X.; et al. Tumor-infiltrating lymphocytes (TILs) from patients with glioma. Oncoimmunology 2017, 6, e1252894. [Google Scholar] [CrossRef]
- Duhen, T.; Duhen, R.; Montler, R.; Moses, J.; Moudgil, T.; de Miranda, N.F.; Goodall, C.P.; Blair, T.C.; Fox, B.A.; McDermott, J.E.; et al. Co-expression of CD39 and CD103 identifies tumor-reactive CD8 T cells in human solid tumors. Nat. Commun. 2018, 9, 2724. [Google Scholar] [CrossRef]
- Simoni, Y.; Becht, E.; Fehlings, M.; Loh, C.Y.; Koo, S.L.; Teng, K.W.W.; Yeong, J.P.S.; Nahar, R.; Zhang, T.; Kared, H.; et al. Bystander CD8(+) T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature 2018, 557, 575–579. [Google Scholar] [CrossRef] [PubMed]
- Salinas, R.D.; Durgin, J.S.; O’Rourke, D.M. Potential of Glioblastoma-Targeted Chimeric Antigen Receptor (CAR) T-Cell Therapy. CNS Drugs 2020, 34, 127–145. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9, eaaa0984. [Google Scholar] [CrossRef]
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N. Engl. J. Med. 2016, 375, 2561–2569. [Google Scholar] [CrossRef] [PubMed]
- Orrego, E.; Castaneda, C.A.; Castillo, M.; Bernabe, L.A.; Casavilca, S.; Chakravarti, A.; Meng, W.; Garcia-Corrochano, P.; Villa-Robles, M.R.; Zevallos, R.; et al. Distribution of tumor-infiltrating immune cells in glioblastoma. CNS Oncol. 2018, 7, CNS21. [Google Scholar] [CrossRef]
- Morisse, M.C.; Jouannet, S.; Dominguez-Villar, M.; Sanson, M.; Idbaih, A. Interactions between tumor-associated macrophages and tumor cells in glioblastoma: Unraveling promising targeted therapies. Expert Rev. Neurother. 2018, 18, 729–737. [Google Scholar] [CrossRef]
- Di Martile, M.; Farini, V.; Consonni, F.M.; Trisciuoglio, D.; Desideri, M.; Valentini, E.; D’Aguanno, S.; Tupone, M.G.; Buglioni, S.; Ercolani, C.; et al. Melanoma-specific bcl-2 promotes a protumoral M2-like phenotype by tumor-associated macrophages. J. Immunother. Cancer 2020, 8, e000489. [Google Scholar] [CrossRef]
- Cho, H.R.; Kumari, N.; Thi Vu, H.; Kim, H.; Park, C.K.; Choi, S.H. Increased Antiangiogenic Effect by Blocking CCL2-dependent Macrophages in a Rodent Glioblastoma Model: Correlation Study with Dynamic Susceptibility Contrast Perfusion MRI. Sci. Rep. 2019, 9, 11085. [Google Scholar] [CrossRef]
- Shi, L.; Bian, Z.; Liu, Y. Dual role of SIRPα in macrophage activation: Inhibiting M1 while promoting M2 polarization via selectively activating SHP-1 and SHP-2 signal. J. Immunol. 2017, 198, 67.12. [Google Scholar]
- Khalil, D.N.; Suek, N.; Campesato, L.F.; Budhu, S.; Redmond, D.; Samstein, R.M.; Krishna, C.; Panageas, K.S.; Capanu, M.; Houghton, S.; et al. In situ vaccination with defined factors overcomes T cell exhaustion in distant tumors. J. Clin. Investig. 2019, 129, 3435–3447. [Google Scholar] [CrossRef]
- Suek, N.; Campesato, L.F.; Merghoub, T.; Khalil, D.N. Targeted APC Activation in Cancer Immunotherapy to Enhance the Abscopal Effect. Front. Immunol. 2019, 10, 604. [Google Scholar] [CrossRef]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef] [PubMed]
- Gregoire, H.; Roncali, L.; Rousseau, A.; Cherel, M.; Delneste, Y.; Jeannin, P.; Hindre, F.; Garcion, E. Targeting Tumor Associated Macrophages to Overcome Conventional Treatment Resistance in Glioblastoma. Front. Pharmacol. 2020, 11, 368. [Google Scholar] [CrossRef] [PubMed]
- Burger, M.C.; Zhang, C.; Harter, P.N.; Romanski, A.; Strassheimer, F.; Senft, C.; Tonn, T.; Steinbach, J.P.; Wels, W.S. CAR-Engineered NK Cells for the Treatment of Glioblastoma: Turning Innate Effectors Into Precision Tools for Cancer Immunotherapy. Front. Immunol. 2019, 10, 2683. [Google Scholar] [CrossRef]
- Lee, S.J.; Kang, W.Y.; Yoon, Y.; Jin, J.Y.; Song, H.J.; Her, J.H.; Kang, S.M.; Hwang, Y.K.; Kang, K.J.; Joo, K.M.; et al. Natural killer (NK) cells inhibit systemic metastasis of glioblastoma cells and have therapeutic effects against glioblastomas in the brain. BMC Cancer 2015, 15, 1011. [Google Scholar] [CrossRef]
- Matsumura, Y.; Maeda, H. A new concept for macromolecular therapeutics in cancer chemotherapy: Mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986, 46, 6387–6392. [Google Scholar]
- Karathanasis, E.; Ghaghada, K.B. Crossing the barrier: Treatment of brain tumors using nanochain particles. WIREs Nanomed. Nanobiotechnol. 2016, 8, 678–695. [Google Scholar] [CrossRef] [PubMed]
- Lam, F.C.; Morton, S.W.; Wyckoff, J.; Vu Han, T.L.; Hwang, M.K.; Maffa, A.; Balkanska-Sinclair, E.; Yaffe, M.B.; Floyd, S.R.; Hammond, P.T. Enhanced efficacy of combined temozolomide and bromodomain inhibitor therapy for gliomas using targeted nanoparticles. Nat. Commun. 2018, 9, 1991. [Google Scholar] [CrossRef]
- Hainfeld, J.F.; Ridwan, S.M.; Stanishevskiy, Y.; Panchal, R.; Slatkin, D.N.; Smilowitz, H.M. Iodine nanoparticles enhance radiotherapy of intracerebral human glioma in mice and increase efficacy of chemotherapy. Sci. Rep. 2019, 9, 4505. [Google Scholar] [CrossRef]
- Samec, N.; Zottel, A.; Videtic Paska, A.; Jovcevska, I. Nanomedicine and Immunotherapy: A Step Further towards Precision Medicine for Glioblastoma. Molecules 2020, 25, 490. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; van Straten, D.; Broekman, M.L.D.; Preat, V.; Schiffelers, R.M. Nanocarrier-based drug combination therapy for glioblastoma. Theranostics 2020, 10, 1355–1372. [Google Scholar] [CrossRef]
- Walter, K.A.; Cahan, M.A.; Gur, A.; Tyler, B.; Hilton, J.; Colvin, O.M.; Burger, P.C.; Domb, A.; Brem, H. Interstitial taxol delivered from a biodegradable polymer implant against experimental malignant glioma. Cancer Res. 1994, 54, 2207–2212. [Google Scholar] [PubMed]
- Perria, C.; Capuzzo, T.; Cavagnaro, G.; Datti, R.; Francaviglia, N.; Rivano, C.; Tercero, V.E. Fast attempts at the photodynamic treatment of human gliomas. J. Neurosurg. Sci. 1980, 24, 119–129. [Google Scholar]
- Castano, A.P.; Demidova, T.N.; Hamblin, M.R. Mechanisms in photodynamic therapy: Part three-Photosensitizer pharmacokinetics, biodistribution, tumor localization and modes of tumor destruction. Photodiagnosis Photodyn Ther. 2005, 2, 91–106. [Google Scholar] [CrossRef]
- Abrahamse, H.; Hamblin, M.R. New photosensitizers for photodynamic therapy. Biochem. J. 2016, 473, 347–364. [Google Scholar] [CrossRef]
- Baptista, M.S.; Cadet, J.; Di Mascio, P.; Ghogare, A.A.; Greer, A.; Hamblin, M.R.; Lorente, C.; Nunez, S.C.; Ribeiro, M.S.; Thomas, A.H.; et al. Type I and Type II Photosensitized Oxidation Reactions: Guidelines and Mechanistic Pathways. Photochem. Photobiol. 2017, 93, 912–919. [Google Scholar] [CrossRef]
- De Groof, T.W.M.; Mashayekhi, V.; Fan, T.S.; Bergkamp, N.D.; Sastre Torano, J.; van Senten, J.R.; Heukers, R.; Smit, M.J.; Oliveira, S. Nanobody-Targeted Photodynamic Therapy Selectively Kills Viral GPCR-Expressing Glioblastoma Cells. Mol. Pharm. 2019, 16, 3145–3156. [Google Scholar] [CrossRef]
- Cramer, S.W.; Chen, C.C. Photodynamic Therapy for the Treatment of Glioblastoma. Front. Surg. 2019, 6, 81. [Google Scholar] [CrossRef]
- Vasilev, A.; Sofi, R.; Rahman, R.; Smith, S.J.; Teschemacher, A.G.; Kasparov, S. Using Light for Therapy of Glioblastoma Multiforme (GBM). Brain Sci. 2020, 10, 75. [Google Scholar] [CrossRef]
- Eljamel, M.S.; Goodman, C.; Moseley, H. ALA and Photofrin fluorescence-guided resection and repetitive PDT in glioblastoma multiforme: A single centre Phase III randomised controlled trial. Lasers Med. Sci. 2008, 23, 361–367. [Google Scholar] [CrossRef]
- Stepp, H.; Beck, T.; Pongratz, T.; Meinel, T.; Kreth, F.W.; Tonn, J.; Stummer, W. ALA and malignant glioma: Fluorescence-guided resection and photodynamic treatment. J. Environ. Pathol. Toxicol. Oncol. 2007, 26, 157–164. [Google Scholar] [CrossRef]
- Eljamel, S. Photodynamic applications in brain tumors: A comprehensive review of the literature. Photodiagnosis Photodyn Ther. 2010, 7, 76–85. [Google Scholar] [CrossRef]
- Dupont, C.; Vermandel, M.; Leroy, H.A.; Quidet, M.; Lecomte, F.; Delhem, N.; Mordon, S.; Reyns, N. INtraoperative photoDYnamic Therapy for GliOblastomas (INDYGO): Study Protocol for a Phase I Clinical Trial. Neurosurgery 2019, 84, E414–E419. [Google Scholar] [CrossRef]
- Yekula, A.; Yekula, A.; Muralidharan, K.; Kang, K.; Carter, B.S.; Balaj, L. Extracellular Vesicles in Glioblastoma Tumor Microenvironment. Front. Immunol. 2019, 10, 3137. [Google Scholar] [CrossRef]
- Osti, D.; Del Bene, M.; Rappa, G.; Santos, M.; Matafora, V.; Richichi, C.; Faletti, S.; Beznoussenko, G.V.; Mironov, A.; Bachi, A.; et al. Clinical Significance of Extracellular Vesicles in Plasma from Glioblastoma Patients. Clin. Cancer Res. 2019, 25, 266–276. [Google Scholar] [CrossRef]
- Simon, T.; Jackson, E.; Giamas, G. Breaking through the glioblastoma micro-environment via extracellular vesicles. Oncogene 2020, 39, 4477–4490. [Google Scholar] [CrossRef]
- Berenguer, J.; Lagerweij, T.; Zhao, X.W.; Dusoswa, S.; van der Stoop, P.; Westerman, B.; de Gooijer, M.C.; Zoetemelk, M.; Zomer, A.; Crommentuijn, M.H.W.; et al. Glycosylated extracellular vesicles released by glioblastoma cells are decorated by CCL18 allowing for cellular uptake via chemokine receptor CCR8. J. Extracell. Vesicles 2018, 7, 1446660. [Google Scholar] [CrossRef] [PubMed]
- Simon, T.; Pinioti, S.; Schellenberger, P.; Rajeeve, V.; Wendler, F.; Cutillas, P.R.; King, A.; Stebbing, J.; Giamas, G. Shedding of bevacizumab in tumour cells-derived extracellular vesicles as a new therapeutic escape mechanism in glioblastoma. Mol. Cancer 2018, 17, 132. [Google Scholar] [CrossRef]
- Buruiana, A.; Florian, S.I.; Florian, A.I.; Timis, T.L.; Mihu, C.M.; Miclaus, M.; Osan, S.; Hrapsa, I.; Cataniciu, R.C.; Farcas, M.; et al. The Roles of miRNA in Glioblastoma Tumor Cell Communication: Diplomatic and Aggressive Negotiations. Int. J. Mol. Sci. 2020, 21, 1950. [Google Scholar] [CrossRef] [PubMed]
- Piwecka, M.; Rolle, K.; Belter, A.; Barciszewska, A.M.; Zywicki, M.; Michalak, M.; Nowak, S.; Naskret-Barciszewska, M.Z.; Barciszewski, J. Comprehensive analysis of microRNA expression profile in malignant glioma tissues. Mol. Oncol. 2015, 9, 1324–1340. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Ren, Y.; Moore, L.; Mei, M.; You, Y.; Xu, P.; Wang, B.; Wang, G.; Jia, Z.; Pu, P.; et al. Downregulation of miR-21 inhibits EGFR pathway and suppresses the growth of human glioblastoma cells independent of PTEN status. Lab. Investig. 2010, 90, 144–155. [Google Scholar] [CrossRef]
- Belter, A.; Rolle, K.; Piwecka, M.; Fedoruk-Wyszomirska, A.; Naskret-Barciszewska, M.Z.; Barciszewski, J. Inhibition of miR-21 in glioma cells using catalytic nucleic acids. Sci. Rep. 2016, 6, 24516. [Google Scholar] [CrossRef]
- Ananta, J.S.; Paulmurugan, R.; Massoud, T.F. Nanoparticle-Delivered Antisense MicroRNA-21 Enhances the Effects of Temozolomide on Glioblastoma Cells. Mol. Pharm. 2015, 12, 4509–4517. [Google Scholar] [CrossRef]
- Teplyuk, N.M.; Uhlmann, E.J.; Gabriely, G.; Volfovsky, N.; Wang, Y.; Teng, J.; Karmali, P.; Marcusson, E.; Peter, M.; Mohan, A.; et al. Therapeutic potential of targeting microRNA-10b in established intracranial glioblastoma: First steps toward the clinic. EMBO Mol. Med. 2016, 8, 268–287. [Google Scholar] [CrossRef]
- Sharif, S.; Ghahremani, M.H.; Soleimani, M. Delivery of Exogenous miR-124 to Glioblastoma Multiform Cells by Wharton’s Jelly Mesenchymal Stem Cells Decreases Cell Proliferation and Migration, and Confers Chemosensitivity. Stem. Cell Rev. Rep. 2018, 14, 236–246. [Google Scholar] [CrossRef] [PubMed]
- Sana, J.; Busek, P.; Fadrus, P.; Besse, A.; Radova, L.; Vecera, M.; Reguli, S.; Stollinova Sromova, L.; Hilser, M.; Lipina, R.; et al. Identification of microRNAs differentially expressed in glioblastoma stem-like cells and their association with patient survival. Sci. Rep. 2018, 8, 2836. [Google Scholar] [CrossRef]
- Rubenstein, J.L.; Kim, J.; Ozawa, T.; Zhang, M.; Westphal, M.; Deen, D.F.; Shuman, M.A. Anti-VEGF antibody treatment of glioblastoma prolongs survival but results in increased vascular cooption. Neoplasia 2000, 2, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Voutouri, C.; Kirkpatrick, N.D.; Chung, E.; Mpekris, F.; Baish, J.W.; Munn, L.L.; Fukumura, D.; Stylianopoulos, T.; Jain, R.K. Experimental and computational analyses reveal dynamics of tumor vessel cooption and optimal treatment strategies. Proc. Natl. Acad. Sci. USA 2019, 116, 2662–2671. [Google Scholar] [CrossRef] [PubMed]
- Montana, V.; Sontheimer, H. Bradykinin promotes the chemotactic invasion of primary brain tumors. J. Neurosci. 2011, 31, 4858–4867. [Google Scholar] [CrossRef] [PubMed]
- Yadav, V.N.; Zamler, D.; Baker, G.J.; Kadiyala, P.; Erdreich-Epstein, A.; DeCarvalho, A.C.; Mikkelsen, T.; Castro, M.G.; Lowenstein, P.R. CXCR4 increases in-vivo glioma perivascular invasion, and reduces radiation induced apoptosis: A genetic knockdown study. Oncotarget 2016, 7, 83701–83719. [Google Scholar] [CrossRef]
- Griveau, A.; Seano, G.; Shelton, S.J.; Kupp, R.; Jahangiri, A.; Obernier, K.; Krishnan, S.; Lindberg, O.R.; Yuen, T.J.; Tien, A.C.; et al. A Glial Signature and Wnt7 Signaling Regulate Glioma-Vascular Interactions and Tumor Microenvironment. Cancer Cell 2018, 33, 874–889. [Google Scholar] [CrossRef]
- Rabe, M.; Dumont, S.; Alvarez-Arenas, A.; Janati, H.; Belmonte-Beitia, J.; Calvo, G.F.; Thibault-Carpentier, C.; Sery, Q.; Chauvin, C.; Joalland, N.; et al. Identification of a transient state during the acquisition of temozolomide resistance in glioblastoma. Cell Death Dis. 2020, 11, 19. [Google Scholar] [CrossRef] [PubMed]
- Struve, N.; Binder, Z.A.; Stead, L.F.; Brend, T.; Bagley, S.J.; Faulkner, C.; Ott, L.; Muller-Goebel, J.; Weik, A.S.; Hoffer, K.; et al. EGFRvIII upregulates DNA mismatch repair resulting in increased temozolomide sensitivity of MGMT promoter methylated glioblastoma. Oncogene 2020, 39, 3041–3055. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Pascual, A.; Hale, J.S.; Kordowski, A.; Pugh, J.; Silver, D.J.; Bayik, D.; Roversi, G.; Alban, T.J.; Rao, S.; Chen, R.; et al. ADAMDEC1 Maintains a Growth Factor Signaling Loop in Cancer Stem Cells. Cancer Discov. 2019, 9, 1574–1589. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).