
CAR-T Therapy, the End of a Chapter or the Beginning of a New One?


{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Mechanism of Relapse to CAR-T Therapy
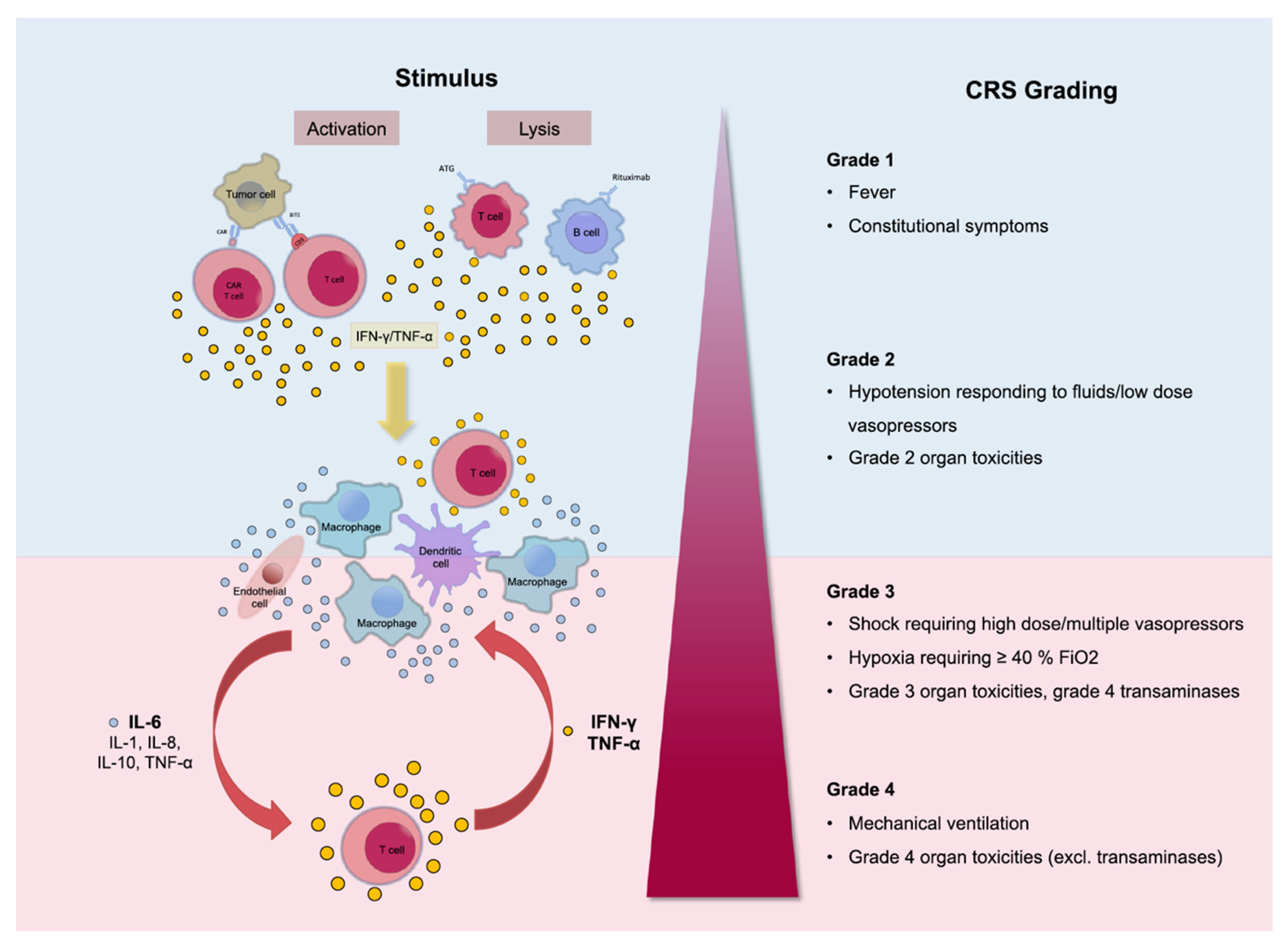
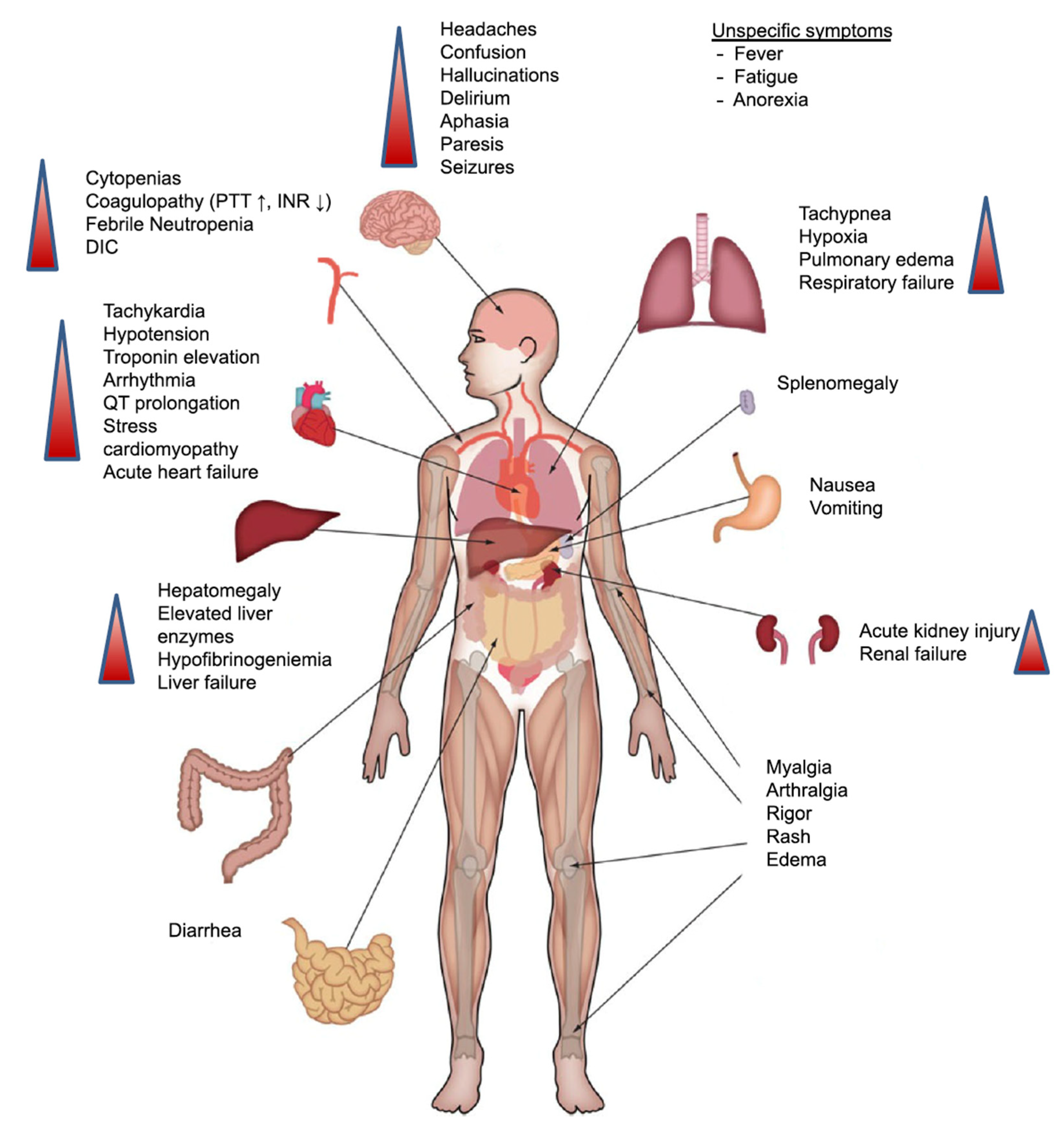
3. CRS: A Limit to the Full Therapeutic Potential of CAR-T Therapy?
4. CAR-T Therapy in Other Haematologic Malignancies: Is There a Role?
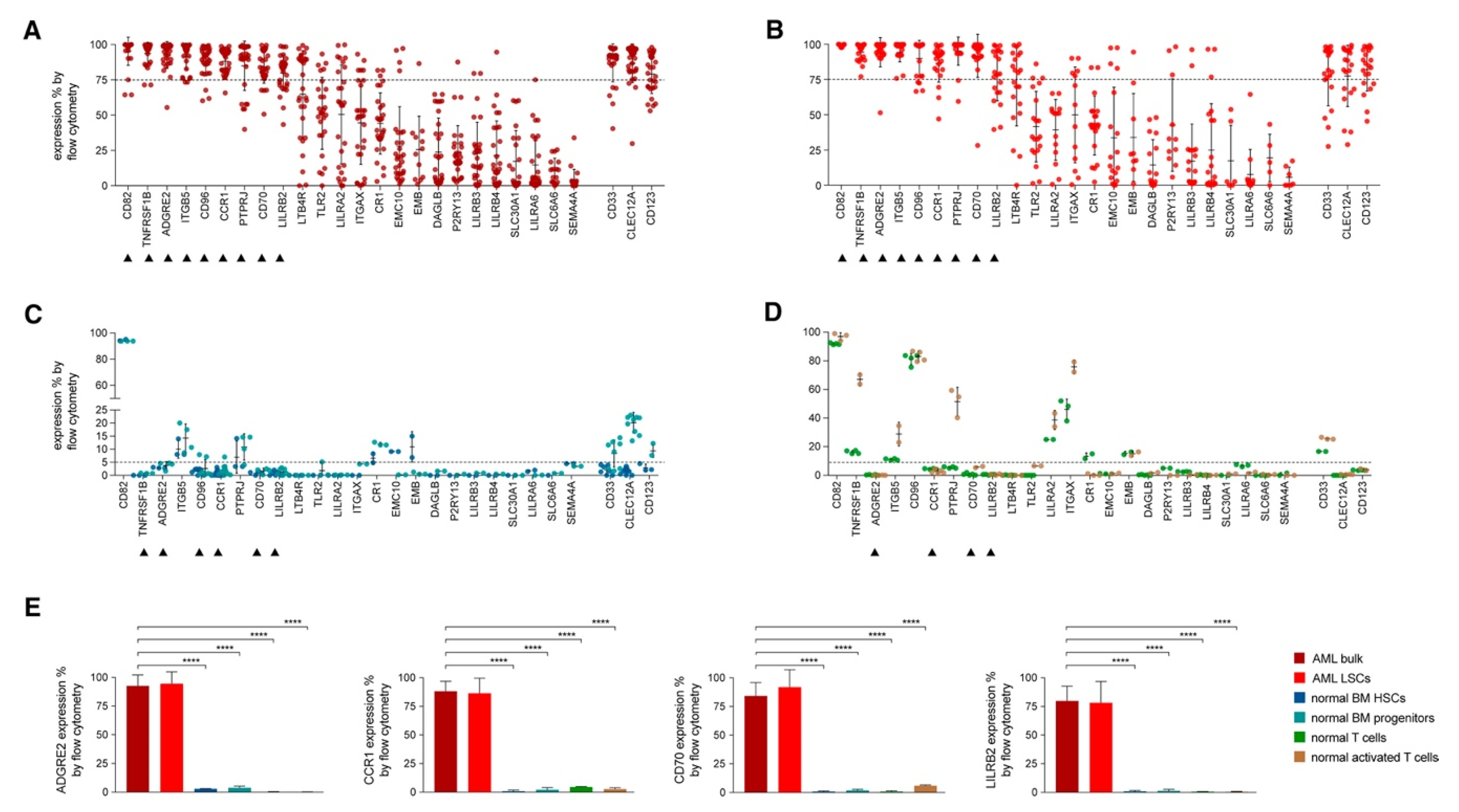
4.1. CAR-T Therapy in AML
4.2. CAR-T Therapy in CLL
4.3. CAR-T Therapy in Hodgkin Lymphoma (HL)
4.4. CAR-T Therapy in MM
5. What Is Next?
6. Conclusions
Funding
Data Availability Statement
Conflicts of Interest
References
- Yescarta Package Insert. Available online: https://www.fda.gov/media/108377/download (accessed on 28 January 2021).
- Kymeriah Package Insert. Available online: https://www.fda.gov/files/vaccines%2C%20blood%20%26%20biologics/published/Package-Insert---KYMRIAH.pdf/ (accessed on 28 January 2021).
- Srour, S.A.; Singh, H.; McCarty, J.; de Groot, E.; Huls, H.; Rondon, G.; Qazilbash, M.; Ciurea, S.; Bardelli, G.; Buck, J.; et al. Long-term outcomes of Sleeping Beauty–generated CD19-specific CAR T-cell therapy for relapsed-refractory B-cell lymphomas. Blood 2020, 135, 862–865. [Google Scholar] [CrossRef]
- June, C.H.; Sadelain, M. Chimeric antigen receptor therapy. N. Engl. J. Med. 2018, 379, 64–73. [Google Scholar] [CrossRef]
- Grupp, S.A.; Maude, S.L.; Rives, S.; Baruchel, A.; Boyer, M.W.; Bittencourt, H.; Bader, P.; Büchner, J.; Laetsch, T.W.; Stefanski, H.; et al. Updated analysis of the efficacy and safety of tisagenlecleucel in pediatric and young adult patients with relapsed/refractory (r/r) acute lymphoblastic leukemia. Blood 2018, 132 (Suppl. S1), 895. [Google Scholar] [CrossRef]
- Xu, X.; Sun, Q.; Liang, X.; Chen, Z.; Zhang, X.; Zhou, X.; Li, M.; Tu, H.; Liu, Y.; Tu, S.; et al. Mechanisms of relapse after CD19 CAR T-cell therapy for acute lymphoblastic leukemia and its prevention and treatment strategies. Front. Immunol. 2019, 10, 2664. [Google Scholar] [CrossRef] [PubMed]
- Gardner, R.; Finney, O.; Brakke, H.; Rhea, S.; Hicks, R.; Doolittle, D.; Lopez, M.; Orentas, R.J.; Li, D.; Jensen, M.C. Starting T cell and cell product phenotype are associated with durable remission of leukemia following CD19 CAR-T cell immunotherapy. Blood 2018, 132 (Suppl. S1), 4022. [Google Scholar] [CrossRef]
- Kotani, H.; Li, G.; Yao, J.; Mesa, T.E.; Chen, J.; Boucher, J.C.; Yoder, S.J.; Zhou, J.; Davila, M.L. Aged CAR T cells exhibit enhanced cytotoxicity and effector function but shorter persistence and less memory-like phenotypes. Blood 2018, 132 (Suppl. S1), 2047. [Google Scholar] [CrossRef]
- Guha, P.; Cunetta, M.; Somasundar, P.; Espat, N.J.; Junghans, R.P.; Katz, S.C. Frontline Science: Functionally impaired geriatric CAR-T cells rescued by increased α5β1 integrin expression. J. Leukoc. Biol. 2017, 102, 201–208. [Google Scholar] [CrossRef]
- Sommermeyer, D.; Hill, T.; Shamah, S.M.; Salter, A.I.; Chen, Y.; Mohler, K.M.; Riddell, S.R. Fully human CD19-specific chimeric antigen receptors for T-cell therapy. Leukemia 2017, 31, 2191–2199. [Google Scholar] [CrossRef] [PubMed]
- Orlando, E.J.; Han, X.; Tribouley, C.; Wood, P.A.; Leary, R.J.; Riester, M.; Levine, J.E.; Qayed, M.; Grupp, S.A.; Boyer, M.; et al. Genetic mechanisms of target antigen loss in CAR19 therapy of acute lymphoblastic leukemia. Nat. Med. 2018, 24, 1504–1506. [Google Scholar] [CrossRef]
- Fischer, J.; Paret, C.; El Malki, K.; Alt, F.; Wingerter, A.; Neu, M.A.; Kron, B.; Russo, A.; Lehmann, N.; Roth, L.; et al. CD19 isoforms enabling resistance to CART-19 immunotherapy are expressed in B-ALL patients at initial diagnosis. J. Immunother. (Hagerstown, Md.: 1997) 2017, 40, 187. [Google Scholar] [CrossRef]
- Inoue, D.; Bradley, R.K.; Abdel-Wahab, O. Spliceosomal gene mutations in myelodysplasia: Molecular links to clonal abnormalities of hematopoiesis. Genes. Dev. 2016, 30, 989–1001. [Google Scholar] [CrossRef]
- Frey, N.V.; Porter, D.L. Cytokine release syndrome with novel therapeutics for acute lymphoblastic leukemia. Hematology 2016, 2016, 567–572. [Google Scholar] [CrossRef] [PubMed]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [PubMed]
- Shimabukuro-Vornhagen, A.; Gödel, P.; Subklewe, M.; Stemmler, H.J.; Schlößer, H.A.; Schlaak, M.; Kochanek, M.; Böll, B.; von Bergwelt-Baildon, M.S. Cytokine release syndrome. J. Immunother. Cancer 2018, 6, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Teachey, D.T.; Rheingold, S.R.; Maude, S.L.; Zugmaier, G.; Barrett, D.M.; Seif, A.E.; Nichols, K.E.; Suppa, E.K.; Kalos, M.; Berg, R.A.; et al. Cytokine release syndrome after blinatumomab treatment related to abnormal macrophage activation and ameliorated with cytokine-directed therapy. Blood 2013, 121, 5154–5157. [Google Scholar] [CrossRef]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Brudno, J.N.; Kochenderfer, J.N. Toxicities of chimeric antigen receptor T cells: Recognition and management. Blood 2016, 127, 3321–3330. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Tummala, S.; Kebriaei, P.; Wierda, W.; Gutierrez, C.; Locke, F.L.; Komanduri, K.V.; Lin, Y.; Jain, N.; Daver, N.; et al. Chimeric antigen receptor T-cell therapy—assessment and management of toxicities. Nat. Rev. Clin. Oncol. 2018, 15, 47. [Google Scholar] [CrossRef]
- Lee, D.W.; Gardner, R.; Porter, D.L.; Louis, C.U.; Ahmed, N.; Jensen, M.; Grupp, S.A.; Mackall, C.L. Current concepts in the diagnosis and management of cytokine release syndrome. Blood 2014, 124, 188–195. [Google Scholar] [CrossRef]
- Lee, D.W.; Santomasso, B.D.; Locke, F.L.; Ghobadi, A.; Turtle, C.J.; Brudno, J.N.; Maus, M.V.; Park, J.H.; Mead, E.; Pavletic, S.; et al. ASTCT consensus grading for cytokine release syndrome and neurologic toxicity associated with immune effector cells. Biol. Blood Marrow Transplant. 2019, 25, 625–638. [Google Scholar] [CrossRef] [PubMed]
- Davila, M.L.; Riviere, I.; Wang, X.; Bartido, S.; Park, J.; Curran, K.; Chung, S.S.; Stefanski, J.; Borquez-Ojeda, O.; Olszewska, M.; et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci. Transl. Med. 2014, 6, 224ra25. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Tummala, S.; Kebriaei, P.; Wierda, W.; Locke, F.L.; Lin, Y.; Jain, N.; Daver, N.; Gulbis, A.M.; Adkins, S.; et al. Toxicity management after chimeric antigen receptor T cell therapy: One size does not fit ‘ALL’. Nat. Rev. Clin. Oncol. 2018, 15, 218. [Google Scholar] [CrossRef]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Nassif Kerbauy, L.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef]
- Frey, N.V.; Shaw, P.A.; Hexner, E.O.; Pequignot, E.; Gill, S.; Luger, S.M.; Mangan, J.K.; Loren, A.W.; Perl, A.E.; Maude, S.L.; et al. Optimizing chimeric antigen receptor T-cell therapy for adults with acute lymphoblastic leukemia. J. Clin. Oncol. 2020, 38, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Gardner, R.A.; Ceppi, F.; Rivers, J.; Annesley, C.; Summers, C.; Taraseviciute, A.; Gust, J.; Leger, K.J.; Tarlock, K.; Cooper, T.M.; et al. Preemptive mitigation of CD19 CAR T-cell cytokine release syndrome without attenuation of antileukemic efficacy. Blood 2019, 134, 2149–2158. [Google Scholar] [CrossRef]
- Gardner, R.A.; Finney, O.; Annesley, C.; Brakke, H.; Summers, C.; Leger, K.; Bleakley, M.; Brown, C.; Mgebroff, S.; Kelly-Spratt, K.S.; et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood 2017, 129, 3322–3331. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Key Statistics for Acute Myeloid Leukemia (AML). Available online: https://www.cancer.org/cancer/acute-myeloid-leukemia/about/key-statistics.html (accessed on 28 January 2021).
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Estey, E.H.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Burnett, A.K.; Dombret, H.; Fenaux, P.; Grimwade, D.; Larson, R.A. Diagnosis and management of acute myeloid leukemia in adults: Recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood 2010, 115, 453–474. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute myeloid leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef]
- Tasian, S.K. Acute myeloid leukemia chimeric antigen receptor T-cell immunotherapy: How far up the road have we traveled? Ther. Adv. Hematol. 2018, 9, 135–148. [Google Scholar] [CrossRef] [PubMed]
- Gross, G.; Eshhar, Z. Therapeutic potential of T cell chimeric antigen receptors (CARs) in cancer treatment: Counteracting off-tumor toxicities for safe CAR T cell therapy. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 59–83. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.M.; DeAngelo, D.J.; Stelljes, M.; Martinelli, G.; Liedtke, M.; Stock, W.; Gökbuget, N.; O’Brien, S.; Wang, K.; Wang, T. Inotuzumab ozogamicin versus standard therapy for acute lymphoblastic leukemia. N. Engl. J. Med. 2016, 375, 740–753. [Google Scholar] [CrossRef] [PubMed]
- Petrov, J.C.; Wada, M.; Pinz, K.G.; Yan, L.E.; Chen, K.H.; Shuai, X.; Liu, H.; Chen, X.; Leung, L.H.; Salman, H. Compound CAR T-cells as a double-pronged approach for treating acute myeloid leukemia. Leukemia 2018, 32, 1317–1326. [Google Scholar] [CrossRef] [PubMed]
- Larson, R.A.; Sievers, E.L.; Stadtmauer, E.A.; Löwenberg, B.; Estey, E.H.; Dombret, H.; Theobald, M.; Voliotis, D.; Bennett, J.M.; Richie, M. Final report of the efficacy and safety of gemtuzumab ozogamicin (Mylotarg) in patients with CD33-positive acute myeloid leukemia in first recurrence. Cancer 2005, 104, 1442–1452. [Google Scholar] [CrossRef] [PubMed]
- Gamis, A.S.; Alonzo, T.A.; Meshinchi, S.; Sung, L.; Gerbing, R.B.; Raimondi, S.C.; Hirsch, B.A.; Kahwash, S.B.; Heerema-McKenney, A.; Winter, L. Gemtuzumab ozogamicin in children and adolescents with de novo acute myeloid leukemia improves event-free survival by reducing relapse risk: Results from the randomized phase III Children’s Oncology Group trial AAML0531. J. Clin. Oncol. 2014, 32, 3021. [Google Scholar] [CrossRef]
- Hills, R.K.; Castaigne, S.; Appelbaum, F.R.; Delaunay, J.; Petersdorf, S.; Othus, M.; Estey, E.H.; Dombret, H.; Chevret, S.; Ifrah, N. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: A meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol. 2014, 15, 986–996. [Google Scholar] [CrossRef]
- Mylotarg. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2006/021174s020lbl.pdf (accessed on 28 January 2021).
- Perna, F.; Berman, S.H.; Soni, R.K.; Mansilla-Soto, J.; Eyquem, J.; Hamieh, M.; Hendrickson, R.C.; Brennan, C.W.; Sadelain, M. Integrating proteomics and transcriptomics for systematic combinatorial chimeric antigen receptor therapy of AML. Cancer Cell 2017, 32, 506–519. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A. Tissue-based map of the human proteome. Science 2015, 347. [Google Scholar] [CrossRef]
- Kim, M.S.; Pinto, S.M.; Getnet, D.; Nirujogi, R.S.; Manda, S.S.; Chaerkady, R.; Madugundu, A.K.; Kelkar, D.S.; Isserlin, R.; Jain, S. A draft map of the human proteome. Nature 2014, 509, 575–581. [Google Scholar] [CrossRef]
- Wilhelm, M.; Schlegl, J.; Hahne, H.; Gholami, A.M.; Lieberenz, M.; Savitski, M.M.; Ziegler, E.; Butzmann, L.; Gessulat, S.; Marx, H. Mass-spectrometry-based draft of the human proteome. Nature 2014, 509, 582–587. [Google Scholar] [CrossRef] [PubMed]
- Cummins, K.D.; Frey, N.; Nelson, A.M.; Schmidt, A.; Luger, S.; Isaacs, R.E.; Lacey, S.F.; Hexner, E.; Melenhorst, J.J.; June, C.H. Treating relapsed/refractory (RR) AML with biodegradable anti-CD123 CAR modified T cells. Blood 2017, 130 (Suppl. S1), 1359. [Google Scholar]
- Ritchie, D.S.; Neeson, P.J.; Khot, A.; Peinert, S.; Tai, T.; Tainton, K.; Chen, K.; Shin, M.; Wall, D.M.; Hönemann, D. Persistence and efficacy of second generation CAR T cell against the LeY antigen in acute myeloid leukemia. Mol. Ther. 2013, 21, 2122–2129. [Google Scholar] [CrossRef] [PubMed]
- Nikiforow, S.; Murad, J.; Daley, H.; Negren, H.; Reder, J.; Sentman, C. A first-in-human phase I trial of NKG2D chimeric antigen receptor-T cells in AML/MDS and multiple myeloma. J. Clin. Oncol. 2016, 34 (Suppl. S15), TPS3102. [Google Scholar] [CrossRef]
- Verma, B.; Aftimos, P.G.; Awada, A.; Machiels, J.P.H.; Brayer, J.B.; Sallman, D.A.; Kerre, T.; Odunsi, K.; Lonez, C.; Gilham, D.E. A NKG2D-based CAR-T therapy in a multinational phase I dose escalation and expansion study targeting multiple solid and hematologic tumor types. J. Clin. Oncol. 2017, 35 (Suppl. S15), TPS3093. [Google Scholar] [CrossRef]
- Kochenderfer, J.N.; Feldman, S.A.; Zhao, Y.; Xu, H.; Black, M.A.; Morgan, R.A.; Wilson, W.H.; Rosenberg, S.A. Construction and pre-clinical evaluation of an anti-CD19 chimeric antigen receptor. J. Immunother. (Hagerstown, Md.: 1997) 2009, 32, 689. [Google Scholar] [CrossRef]
- Zou, Y.; Xu, W.; Li, J. Chimeric antigen receptor-modified T cell therapy in chronic lymphocytic leukemia. J. Hematol. Oncol. 2018, 11, 1–12. [Google Scholar] [CrossRef]
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric antigen receptor–modified T cells in chronic lymphoid leukemia. N. Engl. J. Med. 2011, 365, 725–733. [Google Scholar] [CrossRef]
- Cruz, C.R.Y.; Micklethwaite, K.P.; Savoldo, B.; Ramos, C.A.; Lam, S.; Ku, S.; Diouf, O.; Liu, E.; Barrett, A.J.; Ito, S.; et al. Infusion of donor-derived CD19-redirected virus-specific T cells for B-cell malignancies relapsed after allogeneic stem cell transplant: A phase 1 study. Blood 2013, 122, 2965–2973. [Google Scholar] [CrossRef]
- Kochenderfer, J.N.; Dudley, M.E.; Kassim, S.H.; Somerville, R.P.; Carpenter, R.O.; Stetler-Stevenson, M.; Yang, J.C.; Phan, G.Q.; Hughes, M.S.; Sherry, R.M. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J. Clin. Oncol. 2015, 33, 540. [Google Scholar] [CrossRef] [PubMed]
- Kalos, M.; Levine, B.L.; Porter, D.L.; Katz, S.; Grupp, S.A.; Bagg, A.; June, C.H. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci. Transl. Med. 2011, 3, 95ra73. [Google Scholar] [CrossRef]
- Kochenderfer, J.N.; Dudley, M.E.; Feldman, S.A.; Wilson, W.H.; Spaner, D.E.; Maric, I.; Stetler-Stevenson, M.; Phan, G.Q.; Hughes, M.S.; Sherry, R.M. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor–transduced T cells. Blood 2012, 119, 2709–2720. [Google Scholar] [CrossRef] [PubMed]
- Fraietta, J.A.; Beckwith, K.A.; Patel, P.R.; Ruella, M.; Zheng, Z.; Barrett, D.M.; Lacey, S.F.; Melenhorst, J.J.; McGettigan, S.E.; Cook, D.R. Ibrutinib enhances chimeric antigen receptor T-cell engraftment and efficacy in leukemia. Blood 2016, 127, 1117–1127. [Google Scholar] [CrossRef]
- Gauthier, J.; Hirayama, A.V.; Hay, K.A.; Li, D.; Lymp, J.; Sheih, A.; Purushe, J.; Pender, B.S.; Hawkins, R.M.; Vakil, A. Efficacy and toxicity of CD19-specific chimeric antigen receptor T cells alone or in combination with ibrutinib for relapsed and/or refractory CLL. Biol. Blood Marrow Transplant. 2019, 25, S9–S10. [Google Scholar] [CrossRef][Green Version]
- Gill, S.I.; Vides, V.; Frey, N.V.; Metzger, S.; O’Brien, M.; Hexner, E.; Mato, A.R.; Lacey, S.F.; Melenhorst, J.J.; Pequignot, E. Prospective clinical trial of anti-CD19 CAR T cells in combination with ibrutinib for the treatment of chronic lymphocytic leukemia shows a high response rate. Blood 2018, 132 (Suppl. S1), 298. [Google Scholar] [CrossRef]
- Lemal, R.; Tournilhac, O. State-of-the-art for CAR T-cell therapy for chronic lymphocytic leukemia in 2019. J. Immunother. Cancer 2019, 7, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Ramos, C.A.; Bilgi, M.; Gerken, C.P.; Dakhova, O.; Mei, Z.; Grilley, B.J.; Gee, A.P.; Rooney, C.M.; Dotti, G.; Savoldo, B. CD30-chimeric antigen receptor (CAR) T cells for therapy of Hodgkin lymphoma (HL). Blood 2018, 132 (Suppl. S1), 680. [Google Scholar] [CrossRef]
- Grover, N.S.; Park, S.I.; Ivanova, A.; Eldridge, P.; McKay, K.; Cheng, C.; Laing, S.; Covington, D.; West, J.; Sharf, E. A phase Ib/II study of anti-CD30 chimeric antigen receptor T cells for relapsed/refractory CD30+ lymphomas. Biol. Blood Marrow Transplant. 2019, 25, S66. [Google Scholar] [CrossRef]
- Goldschmidt, H.; Ashcroft, J.; Szabo, Z.; Garderet, L. Navigating the treatment landscape in multiple myeloma: Which combinations to use and when? Ann. Hematol. 2019, 98, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Chim, C.S.; Kumar, S.K.; Orlowski, R.Z.; Cook, G.; Richardson, P.G.; Gertz, M.A.; Giralt, S.; Mateos, M.V.; Leleu, X.; Anderson, K.C. Management of relapsed and refractory multiple myeloma: Novel agents, antibodies, immunotherapies and beyond. Leukemia 2018, 32, 252–262. [Google Scholar] [CrossRef]
- Sonneveld, P. Management of multiple myeloma in the relapsed/refractory patient. Hematology 2017, 2017, 508–517. [Google Scholar] [CrossRef]
- Nijhof, I.S.; van de Donk, N.W.; Zweegman, S.; Lokhorst, H.M. Current and new therapeutic strategies for relapsed and refractory multiple myeloma: An update. Drugs 2018, 78, 19–37. [Google Scholar] [CrossRef]
- Tai, Y.T.; Anderson, K.C. Targeting B-cell maturation antigen in multiple myeloma. Immunotherapy 2015, 7, 1187–1199. [Google Scholar] [CrossRef]
- Novak, A.J.; Darce, J.R.; Arendt, B.K.; Harder, B.; Henderson, K.; Kindsvogel, W.; Gross, J.A.; Greipp, P.R.; Jelinek, D.F. Expression of BCMA, TACI, and BAFF-R in multiple myeloma: A mechanism for growth and survival. Blood 2004, 103, 689–694. [Google Scholar] [CrossRef]
- Brudno, J.N.; Maric, I.; Hartman, S.D.; Rose, J.J.; Wang, M.; Lam, N.; Stetler-Stevenson, M.; Salem, D.; Yuan, C.; Pavletic, S.; et al. T cells genetically modified to express an anti–B-cell maturation antigen chimeric antigen receptor cause remissions of poor-prognosis relapsed multiple myeloma. J. Clin. Oncol. 2018, 36, 2267. [Google Scholar] [CrossRef] [PubMed]
- Friedman, K.M.; Garrett, T.E.; Evans, J.W.; Horton, H.M.; Latimer, H.J.; Seidel, S.L.; Horvath, C.J.; Morgan, R.A. Effective targeting of multiple B-cell maturation antigen–expressing hematological malignances by anti-B-cell maturation antigen chimeric antigen receptor T cells. Hum. Gene Ther. 2018, 29, 585–601. [Google Scholar] [CrossRef]
- Raje, N.; Berdeja, J.; Lin, Y.I.; Siegel, D.; Jagannath, S.; Madduri, D.; Liedtke, M.; Rosenblatt, J.; Maus, M.V.; Turka, A.; et al. Anti-BCMA CAR T-cell therapy bb2121 in relapsed or refractory multiple myeloma. N. Engl. J. Med. 2019, 380, 1726–1737. [Google Scholar] [CrossRef] [PubMed]
- Topp, M.S.; Duell, J.; Zugmaier, G.; Attal, M.; Moreau, P.; Langer, C.; Krönke, J.; Facon, T.; Salnikov, A.V.; Lesley, R. Anti–B-cell maturation antigen BiTE molecule AMG 420 induces responses in multiple myeloma. J. Clin. Oncol. 2020, 38, 775–783. [Google Scholar] [CrossRef]
- Madduri, D.; Berdeja, J.G.; Usmani, S.Z.; Jakubowiak, A.; Agha, M.; Cohen, A.D.; Stewart, A.K.; Hari, P.; Htut, M.; O’Donnell, E.; et al. CARTITUDE-1: Phase 1b/2 study of ciltacabtagene autoleucel, a B-cell maturation antigen–directed chimeric antigen receptor T cell therapy, in relapsed/refractory multiple myeloma. In Proceedings of the 2020 American Society of Hematology Annual Meeting, San Diego, CA, USA, 5 December 2020. [Google Scholar]
- Munshi, N.; Anderson, L.; Shah, N.; Jagannath, S.; Berdeja, J.G.; Lonial, S.; Raje, N.S.; Siegel, D.S.D.; Lin, Y.; Oriol, A.; et al. Idecabtagene vicleucel (ide-cel; bb2121), a BCMA-targeted CAR T-cell therapy, in patients with relapsed and refractory multiple myeloma (RRMM): Initial KarMMa results. J. Clin. Oncol. 2020, 38, 8503. [Google Scholar] [CrossRef]
- Britten, O.; Ragusa, D.; Tosi, S.; Mostafa Kamel, Y. MLL-Rearranged Acute Leukemia with t (4; 11)(q21; q23)—Current Treatment Options. Is There a Role for CAR-T Cell Therapy? Cells 2019, 8, 1341. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mostafa Kamel, Y. CAR-T Therapy, the End of a Chapter or the Beginning of a New One? Cancers 2021, 13, 853. https://doi.org/10.3390/cancers13040853
Mostafa Kamel Y. CAR-T Therapy, the End of a Chapter or the Beginning of a New One? Cancers. 2021; 13(4):853. https://doi.org/10.3390/cancers13040853
Chicago/Turabian StyleMostafa Kamel, Yasser. 2021. "CAR-T Therapy, the End of a Chapter or the Beginning of a New One?" Cancers 13, no. 4: 853. https://doi.org/10.3390/cancers13040853
APA StyleMostafa Kamel, Y. (2021). CAR-T Therapy, the End of a Chapter or the Beginning of a New One? Cancers, 13(4), 853. https://doi.org/10.3390/cancers13040853