
Antigenic Essence: Upgrade of Cellular Cancer Vaccines



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
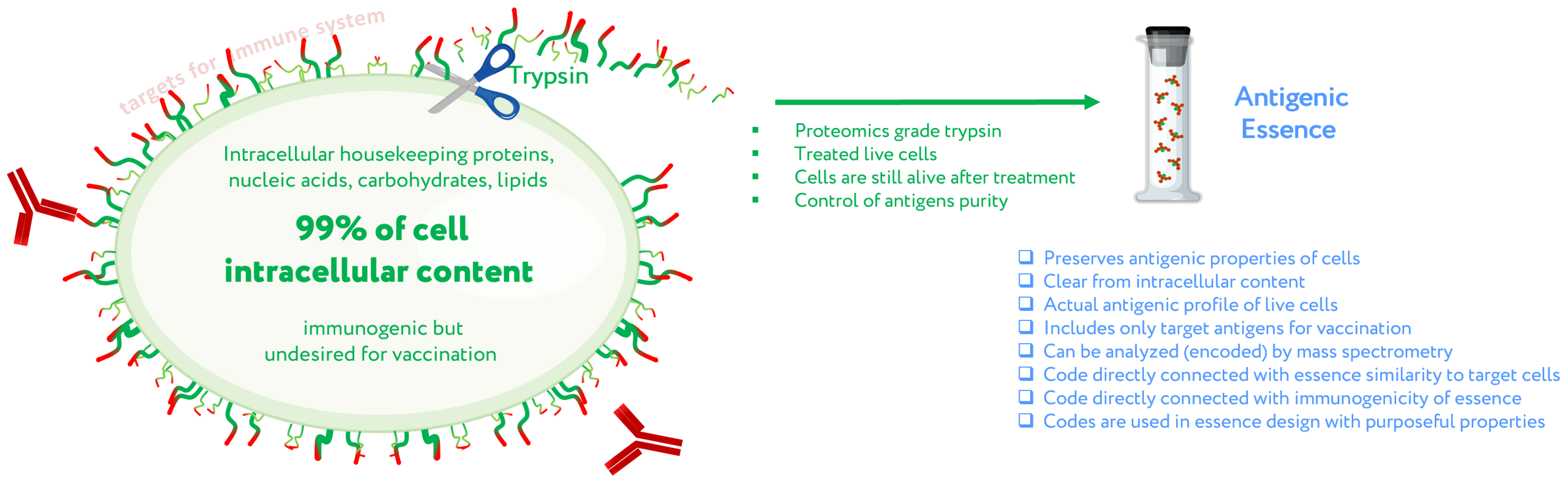
2. Antigenic Essence Concept
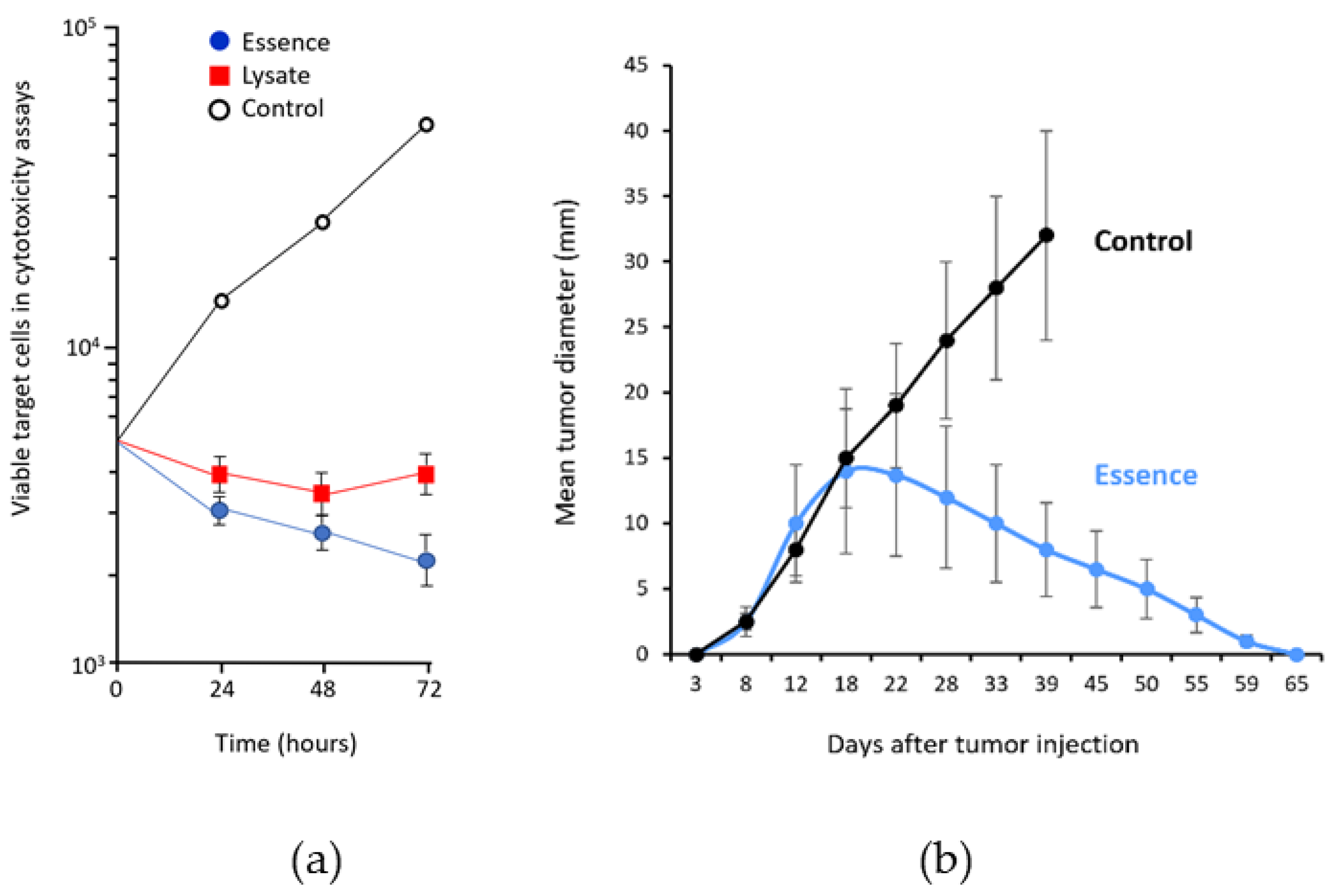
3. Antigenic Essence Equivalence to Cell Antigens
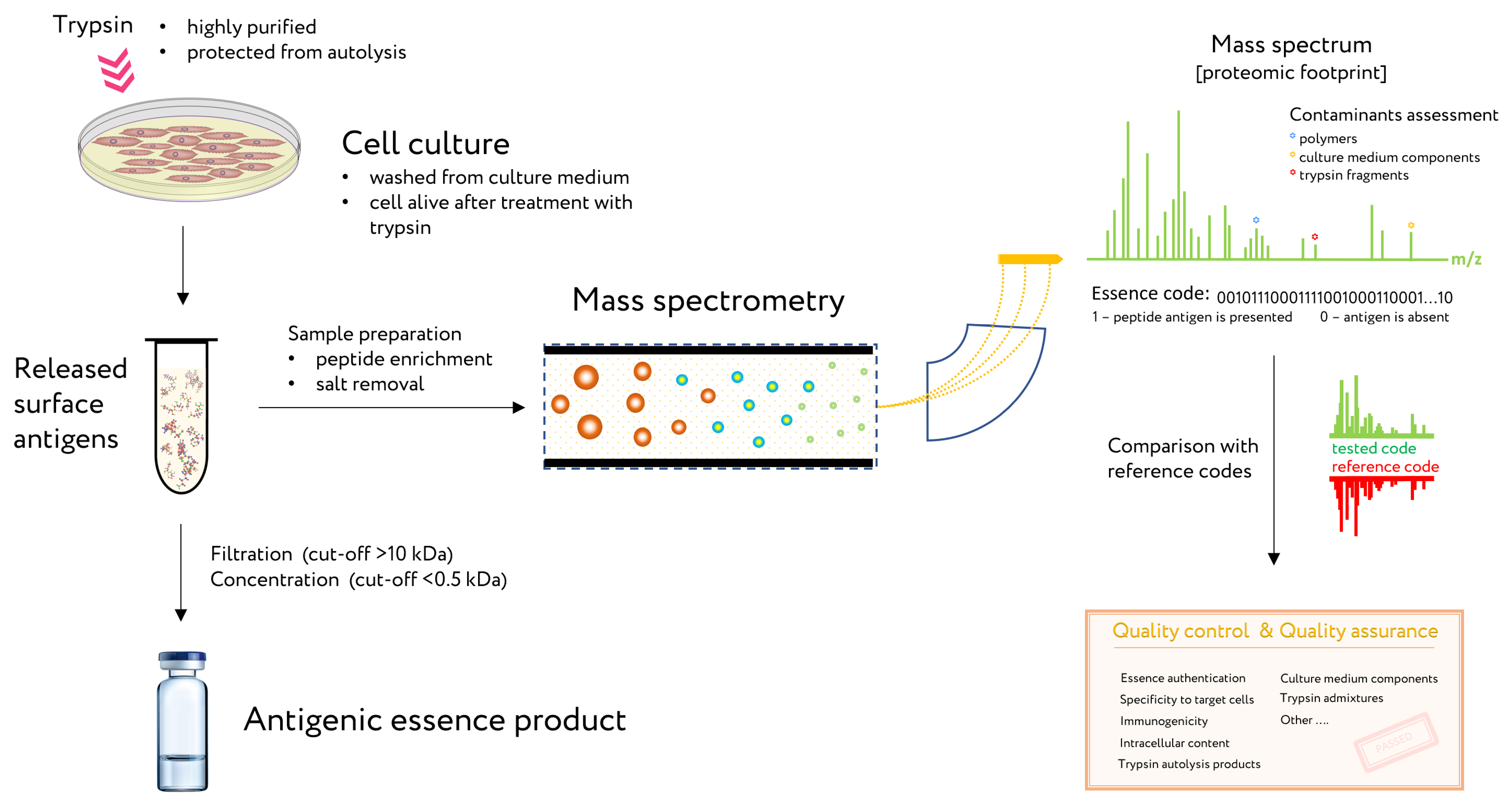
4. Control of Antigenic Essence Composition
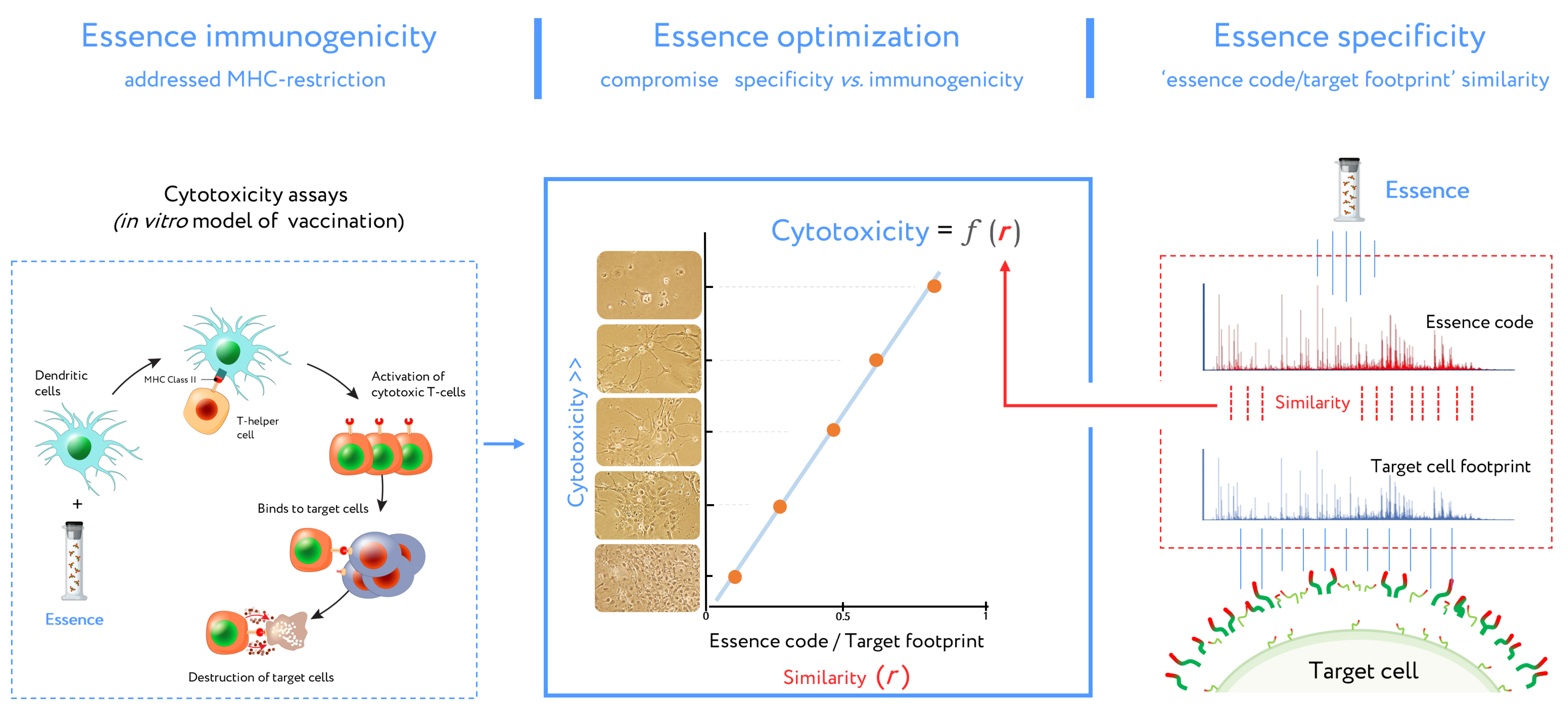
5. Antigenic Essence and MHC Restriction
5.1. Antigens Size in Essence and MHC
5.2. Essence Composition and MHC Mediated Immunogenicity
5.3. Antigenic Essence and Immunopeptidome
- Antigenic essences are composed of peptides of a size ideal for presentation by MHC.
- Antigenic essence compositions strike an optimal balance between similarity of essence code with the surface of target cells and enrichment of MHC-restricted peptide antigens.
- Antigenic essences are probable to contain the intracellular antigens present in the immunopeptidome relevant for vaccination.
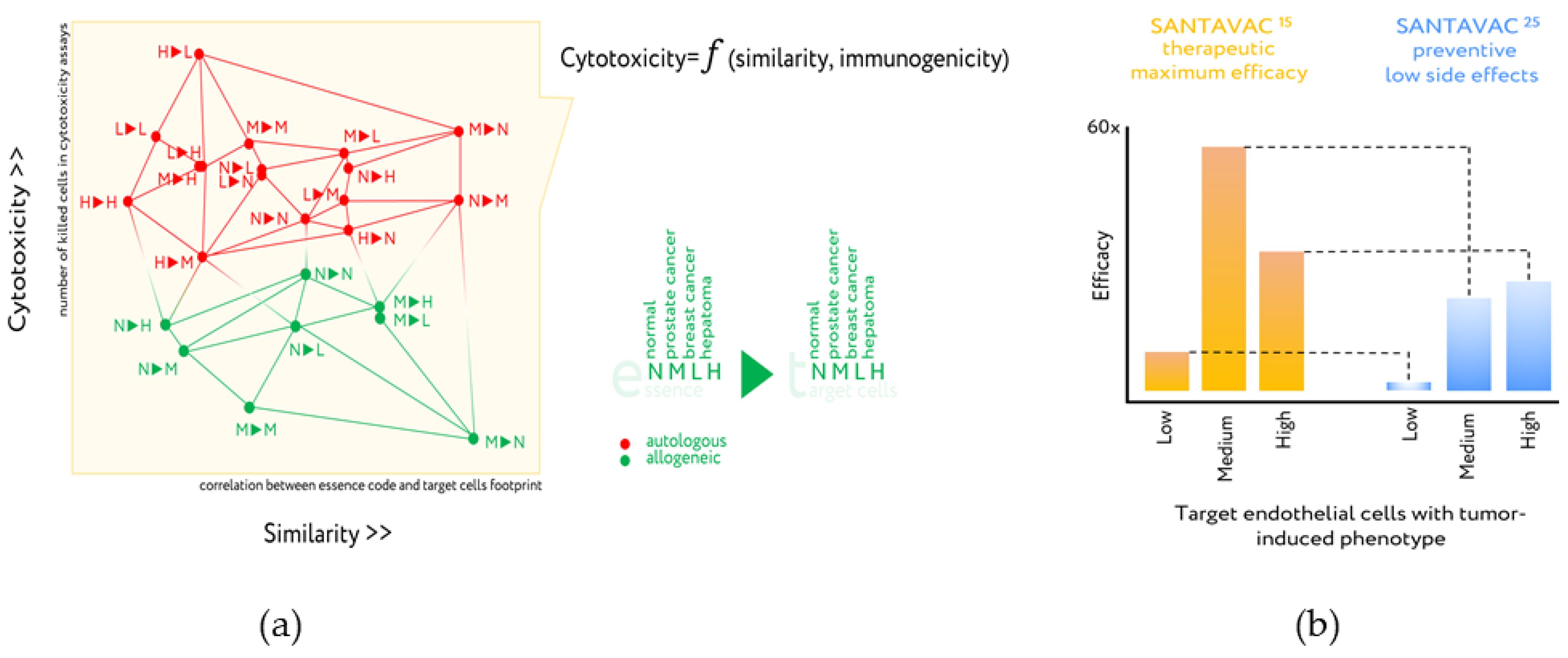
6. Antigenic Essence Products
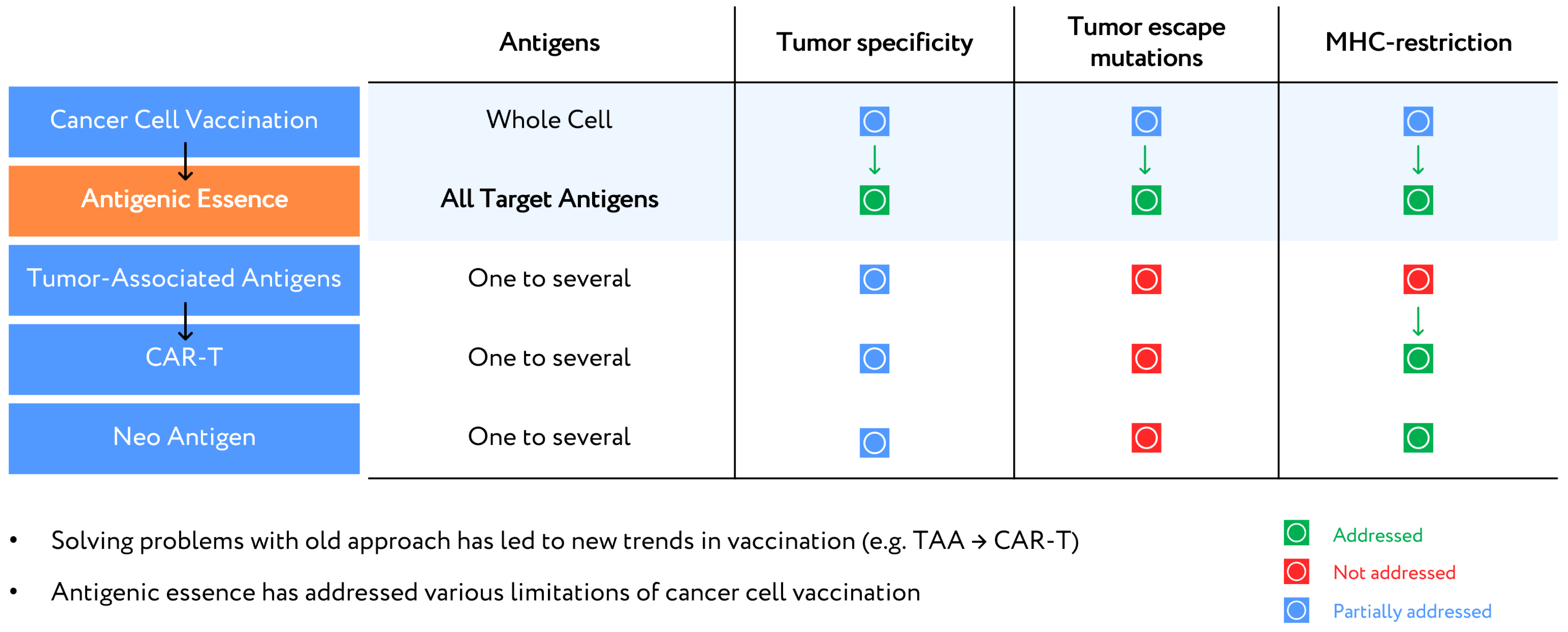
7. Upgrade of Developed Cellular Cancer Vaccines
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hanna, M.G.; Peters, L.C. Specific immunotherapy of established visceral micrometastases by BCG-tumor cell vaccine alone or as an adjunct to surgery. Cancer 1978, 42, 2613–2625. [Google Scholar] [CrossRef]
- Berger, M.; Kreutz, F.T.; Horst, J.L.; Baldi, A.C.; Koff, W.J. Phase I study with an autologous tumor cell vaccine for locally advanced or metastatic prostate cancer. J. Pharm. Pharm. Sci. 2007, 10, 144–152. [Google Scholar]
- Harris, J.E.; Ryan, L.; Hoover, H.C.; Stuart, R.K.; Oken, M.M.; Benson, A.B.; Mansour, E.; Haller, D.G.; Manola, J.; Hanna, M.G. Adjuvant active specific immunotherapy for stage II and III colon cancer with an autologous tumor cell vaccine: Eastern Cooperative Oncology Group study E5283. J. Clin. Oncol. 2000, 18, 148–157. [Google Scholar] [CrossRef]
- Maver, С.; McKneally, M. Preparation of autologous tumor cell vaccine from human lung cancer. Cancer Res. 1979, 39, 3276. [Google Scholar]
- Schulof, R.S.; Mai, D.; Nelson, M.A.; Paxton, H.M.; Cox, J.W.; Turner, M.L., Jr.; Mills, M.; Hix, W.R.; Nochomovitz, L.E.; Peters, L.C.; et al. Active specific immunotherapy with an autologous tumor cell vaccine in patients with resected non-small cell lung cancer. Mol. Biother. 1988, 1, 30–36. [Google Scholar] [PubMed]
- Nemunaitis, J.; Sterman, D.; Jablons, D.; Smith, J.W.; Fox, B.; Maples, P.; Hamilton, S.; Borellini, F.; Lin, A.; Morali, S.; et al. Granulocyte-macrophage colony-stimulating factor gene-modified autologous tumor vaccines in non-small-cell lung cancer. J. Natl. Cancer Inst. 2004, 96, 326–331. [Google Scholar] [CrossRef] [PubMed]
- Rüttinger, D.; van den Engel, N.K.; Winter, H.; Schlemmer, M.; Pohla, H.; Grützner, S.; Wagner, B.; Schendel, D.J.; Fox, B.A.; Jauch, K.W.; et al. Adjuvant therapeutic vaccination in patients with non-small cell lung cancer made lymphopenic and reconstituted with autologous PBMC: First clinical experience and evidence of an immune response. J. Transl. Med. 2007, 5, 43. [Google Scholar] [CrossRef]
- de Weger, V.A.; Turksma, A.W.; Voorham, Q.J.M.; Euler, Z.; Bril, H.; van den Eertwegh, A.J.; Bloemena, E.; Pinedo, H.M.; Vermorken, J.B.; van Tinteren, H.; et al. Clinical effects of adjuvant active specific immunotherapy differ between patients with microsatellite-stable and microsatellite-instable colon cancer. Clin. Cancer Res. 2012, 18, 882–889. [Google Scholar] [CrossRef] [PubMed]
- Hanna, M.G.; Hoover, H.C.; Vermorken, J.B.; Harris, J.E.; Pinedo, H.M. Adjuvant active specific immunotherapy of stage II and stage III colon cancer with an autologous tumor cell vaccine: First randomized phase III trials show promise. Vaccine 2001, 19, 2576–2582. [Google Scholar] [CrossRef]
- Ockert, D.; Schirrmacher, V.; Beck, N.; Stoelben, E.; Ahlert, T.; Flechtenmacher, J.; Hagmüller, E.; Buchcik, R.; Nagel, M.; Saeger, H.D. Newcastle disease virus-infected intact autologous tumor cell vaccine for adjuvant active specific immunotherapy of resected colorectal carcinoma. Clin. Cancer Res. 1996, 2, 21–28. [Google Scholar]
- Baars, A.; van Riel, J.M.G.H.; Cuesta, M.A.; Jaspars, E.H.; Pinedo, H.M.; van den Eertwegh, A.J.M. Metastasectomy and active specific immunotherapy for a large single melanoma metastasis. Hepatogastroenterology 2002, 49, 691–693. [Google Scholar]
- Berd, D.; Maguire, H.C.; McCue, P.; Mastrangelo, M.J. Treatment of metastatic melanoma with an autologous tumor-cell vaccine: Clinical and immunologic results in 64 patients. J. Clin. Oncol. 1990, 8, 1858–1867. [Google Scholar] [CrossRef]
- Méndez, R.; Ruiz-Cabello, F.; Rodríguez, T.; del Campo, A.; Paschen, A.; Schadendorf, D.; Garrido, F. Identification of different tumor escape mechanisms in several metastases from a melanoma patient undergoing immunotherapy. Cancer Immunol. Immunother. 2007, 56, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Antonia, S.J.; Seigne, J.; Diaz, J.; Muro-Cacho, C.; Extermann, M.; Farmelo, M.J.; Friberg, M.; Alsarraj, M.; Mahany, J.J.; Pow-Sang, J.; et al. Phase I trial of a B7-1 (CD80) gene modified autologous tumor cell vaccine in combination with systemic interleukin-2 in patients with metastatic renal cell carcinoma. J. Urol. 2002, 167, 1995–2000. [Google Scholar] [CrossRef]
- Fishman, M.; Hunter, T.B.; Soliman, H.; Thompson, P.; Dunn, M.; Smilee, R.; Farmelo, M.J.; Noyes, D.R.; Mahany, J.J.; Lee, J.H.; et al. Phase II trial of B7-1 (CD-86) transduced, cultured autologous tumor cell vaccine plus subcutaneous interleukin-2 for treatment of stage IV renal cell carcinoma. J. Immunother. 2008, 31, 72–80. [Google Scholar] [CrossRef]
- Kinoshita, Y.; Kono, T.; Yasumoto, R.; Kishimoto, T.; Wang, C.Y.; Haas, G.P.; Nishisaka, N. Antitumor effect on murine renal cell carcinoma by autologous tumor vaccines genetically modified with granulocyte-macrophage colony-stimulating factor and interleukin-6 cells. J. Immunother. 2001, 24, 205–211. [Google Scholar] [CrossRef]
- Buonaguro, L.; Petrizzo, A.; Tornesello, M.L.; Buonaguro, F.M. Translating tumor antigens into cancer vaccines. Clin. Vaccine Immunol. 2011, 18, 23–34. [Google Scholar] [CrossRef]
- Hollingsworth, R.E.; Jansen, K. Turning the corner on therapeutic cancer vaccines. npj Vaccines 2019, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef]
- Srivastava, S.; Riddell, S.R. Engineering CAR-T cells: Design concepts. Trends Immunol. 2015, 36, 494–502. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Keenan, B.P.; Jaffee, E.M. Whole cell vaccines—Past progress and future strategies. Semin. Oncol. 2012, 39, 276–286. [Google Scholar] [CrossRef]
- Chiang, C.L.L.; Benencia, F.; Coukos, G. Whole tumor antigen vaccines. Semin. Immunol. 2010, 22, 132–143. [Google Scholar] [CrossRef]
- Khong, H.T.; Restifo, N.P. Natural selection of tumor variants in the generation of “tumor escape” phenotypes. Nat. Immunol. 2002, 3, 999–1005. [Google Scholar] [CrossRef] [PubMed]
- Balashova, E.E.; Dashtiev, M.I.; Lokhov, P.G. Proteomic Footprinting of Drug-Treated Cancer Cells as a Measure of Cellular Vaccine Efficacy for the Prevention of Cancer Recurrence. Mol. Cell. Proteom. 2012, 11, M111.014480. [Google Scholar] [CrossRef]
- Caron, E.; Kowalewski, D.J.; Koh, C.C.; Sturm, T.; Schuster, H.; Aebersold, R. Analysis of MHC immuM111.014480. eptidomes using mass spectrometry. Mol. Cell. Proteom. 2015, 14, 3105–3117. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.C.L.; Goubier, A.; Kohrt, H.E. A quantitative analysis of therapeutic cancer vaccines in phase 2 or phase 3 trial. J. Immunother. Cancer 2015, 3, 48. [Google Scholar] [CrossRef]
- Lokhov, P.G.; Balashova, E.E. Cellular cancer vaccines: An update on the development of vaccines generated from cell surface antigens. J. Cancer 2010, 1, 230–241. [Google Scholar] [CrossRef]
- Lokhov, P.; Balashova, E.; Dashtiev, M. Cell proteomic footprint. Rapid Commun. Mass Spectrom. 2009, 23, 680–682. [Google Scholar] [CrossRef]
- Balashova, E.E.; Lokhov, P.G. Proteolytically-cleaved fragments of cell-surface proteins from live tumor cells stimulate anti-tumor immune response in vitro. J. Carcinog. Mutagen. 2010, 1, 1–3. [Google Scholar] [CrossRef]
- Balashova, E.E.; Lokhov, P.G. Proteolytically-cleaved fragments of cell surface proteins stimulate a cytotoxic immune response against tumor-activated endothelial cells in vitro. J. Cancer Sci. Ther. 2010, 2, 126–131. [Google Scholar] [CrossRef]
- Lokhov, P.G. Method for Producing An Antitumoral Vaccine Based on Surface Endothelial Cell Antigens. U.S. Patent Application No. 9844586, 19 December 2007. [Google Scholar]
- Stacey, G.N.; Masters, J.R.W.; Hay, R.J.; Drexler, H.G.; MacLeod, R.A.F.; Freshney, R.I. Cell contamination leads to inaccurate data: We must take action now. Nature 2000, 403, 356. [Google Scholar] [CrossRef] [PubMed]
- MacLeod, R.A.F.; Dirks, W.G.; Matsuo, Y.; Kaufmann, M.; Milch, H.; Drexler, H.G. Widespread intraspecies cross-contamination of human tumor cell lines arising at source. Int. J. Cancer 1999, 83, 555–563. [Google Scholar] [CrossRef]
- Harris, J.E.; Ryan, L.; Hoover, H.C., Jr.; Stuart, R.K.; Oken, M.M.; Mansour, A.B.B.I.; Haller , D.G.; Manola, J.; Hanna, M.G., Jr. Identity crisis. Nature 2009, 457, 935–936. [Google Scholar] [CrossRef]
- Cabrera, C.M.; Cobo, F.; Nieto, A.; Cortés, J.L.; Montes, R.M.; Catalina, P.; Concha, A. Identity tests: Determination of cell line cross-contamination. Cytotechnology 2006, 51, 45–50. [Google Scholar] [CrossRef]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Wright, W.E.; Shay, J.W. Historical claims and current interpretations of replicative aging. Nat. Biotechnol. 2002, 20, 682–688. [Google Scholar] [CrossRef]
- Huggins, J.W.; Chestnut, R.W.; Durham, N.N.; Carraway, K.L. Molecular changes in cell surface membranes resulting from trypsinization of sarcoma 180 tumor cells. BBA Biomembr. 1976, 426, 630–637. [Google Scholar] [CrossRef]
- Angus, R.; Collins, C.M.P.; Symes, M.O. Expression of major histocompatibility complex (MHC) antigens and their loss on culture in renal carcinoma. Eur. J. Cancer 1993, 29, 2158–2160. [Google Scholar] [CrossRef]
- Nabholz, M.; MacDonald, H.R. Cytolytic T Lymphocytes. Annu. Rev. Immunol. 1983, 1, 273. [Google Scholar] [CrossRef]
- Lanzavecchia, A. Identifying strategies for immune intervention. Science 1993, 260, 937. [Google Scholar] [CrossRef]
- Berke, G. The Binding and Lysis of Target Cells by Cytotoxic Lymphocytes: Molecular and Cellular Aspects. Annu. Rev. Immunol. 1994, 12, 735. [Google Scholar] [CrossRef]
- Malonis, R.J.; Lai, J.R.; Vergnolle, O. Peptide-Based Vaccines: Current Progress and Future Challenges. Chem. Rev. 2020, 120, 3210–3229. [Google Scholar] [CrossRef] [PubMed]
- van den Eynde, B.J.; Boon, T. Tumor antigens recognized by T lymphocytes. Int. J. Clin. Lab. Res. 1997, 18, 267–268. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A. Cancer vaccines based on the identification of genes encoding cancer regression antigens. Immunol. Today 1997, 18, 175. [Google Scholar] [CrossRef]
- Fisk, B.; Blevins, T.L.; Wharton, J.T.; Ioannides, C.G. Identification of an immunodominant peptide of HER-2/neu protooncogene recognized by ovarian tumor-specific cytotoxic t lymphocyte lines. J. Exp. Med. 1995, 181, 2109. [Google Scholar] [CrossRef]
- Peoples, G.E.; Goedegebuure, P.S.; Smith, R.; Linehan, D.C.; Yoshino, I.; Eberlein, T.J. Breast and ovarian cancer-specific cytotoxic T lymphocytes recognize the same HER2/neu-derived peptide. Proc. Natl. Acad. Sci. USA 1995, 92, 432. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.L.; Skipper, J.; Chen, Y.; Henderson, R.A.; Darrow, T.L.; Shabanowitz, J.; Engelhard, V.H.; Hunt, D.F.; Slingluff, C.L. Identification of a peptide recognized by five melanoma-specific human cytotoxic T cell lines. Science 1994, 264, 716. [Google Scholar] [CrossRef]
- Coulie, P.G.; van den Eynde, B.J.; van der Bruggen, P.; Boon, T. Tumour antigens recognized by T lymphocytes: At the core of cancer immunotherapy. Nat. Rev. Cancer 2014, 14, 135–146. [Google Scholar] [CrossRef]
- Celis, E.; Sette, A.; Grey, H.M. Epitope selection and development of peptide based vaccines to treat cancer. Semin. Cancer Biol. 1995, 6, 329. [Google Scholar] [CrossRef]
- Appella, E.; Loftus, D.J.; Sakaguchi, K.; Sette, A.; Celis, E. Synthetic antigenic peptides as a new strategy for immunotherapy of cancer. Biomed. Pept. Proteins Nucleic Acids 1995, 1, 177. [Google Scholar] [PubMed]
- Celis, E.; Fikes, J.; Wentworth, P.; Sidney, J.; Southwood, S.; Maewal, A.; del Guercio, M.F.; Sette, A.; Livingston, B. Identification of potential CTL epitopes of tumor-associated antigen MAGE-1 for five common HLA-A alleles. Mol. Immunol. 1994, 31, 1423. [Google Scholar] [CrossRef]
- Tsai, V.; Southwood, S.; Sidney, J.; Sakaguchi, K.; Kawakami, Y.; Appella, E.; Sette, A.; Celis, E. Identification of subdominant CTL epitopes of the GP100 melanoma-associated tumor antigen by primary in vitro immunization with peptide-pulsed dendritic cells. J. Immunol. 1997, 158, 1796. [Google Scholar] [PubMed]
- Celis, E.; Tsai, V.; Crimi, C.; Demars, R.; Wentworth, P.A.; Chesnut, R.W.; Grey, H.M.; Sette, A.; Serra, H.M. Induction of anti-tumor cytotoxic T lymphocytes in normal humans using primary cultures and synthetic peptide epitopes. Proc. Natl. Acad. Sci. USA 1994, 91, 2105. [Google Scholar] [CrossRef] [PubMed]
- Weynants, P.; Lethé, B.; Brasseur, F.; Marchand, M.; Boon, T. Expression of MAGE genes by non-small-cell lung carcinomas. Int. J. Cancer 1994, 56, 826. [Google Scholar] [CrossRef] [PubMed]
- Vincent, R.G.; Chu, T.M. Carcinoembryonic antigen in patients with carcinoma of the lung. J. Thorac. Cardiovasc. Surg. 1973, 66, 320. [Google Scholar] [CrossRef]
- Slamon, D.J.; Godolphin, W.; Jones, L.A.; Holt, J.A.; Wong, S.G.; Keith, D.E.; Levin, W.J.; Stuart, S.G.; Udove, J.; Ullrich, A.; et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science 1989, 244, 707. [Google Scholar] [CrossRef]
- Kawashima, I.; Hudson, S.J.; Tsai, V.; Southwood, S.; Takesako, K.; Appella, E.; Sette, A.; Celis, E. The multi-epitope approach for immunotherapy for cancer: Identification of several CTL epitopes from various tumor-associated antigens expressed on solid epithelial tumors. Hum. Immunol. 1998, 59, 1–14. [Google Scholar] [CrossRef]
- Sugawara, S.; Abo, T.; Kumagai, K. A simple method to eliminate the antigenicity of surface class I MHC molecules from the membrane of viable cells by acid treatment at pH 3. J. Immunol. Methods 1987, 100, 83–90. [Google Scholar] [CrossRef]
- Bassani-Sternberg, M.; Barnea, E.; Beer, I.; Avivi, I.; Katz, T.; Admon, A. Soluble plasma HLA peptidome as a potential source for cancer biomarkers. Proc. Natl. Acad. Sci. USA 2010, 107, 18769–18776. [Google Scholar] [CrossRef]
- Demaria, S.; Schwab, R.; Gottesman, S.R.S.; Bushkin, Y. Soluble β2-microglobulin-free class I heavy chains are released from the surface of activated and leukemia cells by a metalloprotease. J. Biol. Chem. 1994, 269, 6689–6694. [Google Scholar] [CrossRef]
- Demaria, S.; Bushkin, Y. Soluble HLA proteins with bound peptides are released from the cell surface by the membrane metalloproteinase. Hum. Immunol. 2000, 61, 1332–1338. [Google Scholar] [CrossRef]
- Galati, G.; Arcelloni, C.; Paroni, R.; Heltai, S.; Rovere, P.; Rugarli, C.; Manfredi, A.A. Quantitative cytometry of MHC class I digestion from living cells. Cytometry 1997, 27, 77–83. [Google Scholar] [CrossRef]
- Antwi, K.; Hanavan, P.D.; Myers, C.E.; Ruiz, Y.W.; Thompson, E.J.; Lake, D.F. Proteomic identification of an MHC-binding peptidome from pancreas and breast cancer cell lines. Mol. Immunol. 2009, 46, 2931–2937. [Google Scholar] [CrossRef]
- Muenst, S.; Läubli, H.; Soysal, S.D.; Zippelius, A.; Tzankov, A.; Hoeller, S. The immune system and cancer evasion strategies: Therapeutic concepts. J. Intern. Med. 2016, 279, 541–562. [Google Scholar] [CrossRef]
- Boehm, T.; Folkman, J.; Browder, T.; O’Reilly, M.S. Antiangiogenic therapy of experimental cancer does not induce acquired drug resistance. Nature 1997, 390, 404–407. [Google Scholar] [CrossRef]
- Uldry, E.; Faes, S.; Demartines, N.; Dormond, O. Fine-tuning tumor endothelial cells to selectively kill cancer. Int. J. Mol. Sci. 2017, 18, 1401. [Google Scholar] [CrossRef] [PubMed]
- Algire, G.H.; Chalkley, H.W.; Legallais, F.Y.; Park, H.D. Vasculae reactions of normal and malignant tissues in vivo. i. vascular reactions of mice to wounds and to normal and neoplastic transplants. J. Natl. Cancer Inst. 1945, 6, 73–85. [Google Scholar] [CrossRef]
- Wei, Y.Q.; Wang, Q.R.; Zhao, X.; Yang, L.; Tian, L.; Lu, Y.; Kang, B.; Lu, C.J.; Huang, M.J.; Lou, Y.Y.; et al. Immunotherapy of tumors with xenogeneic endothelial cells as a vaccine. Nat. Med. 2000, 6, 1160–1166. [Google Scholar] [CrossRef]
- Okaji, Y.; Tsuno, N.H.; Kitayama, J.; Saito, S.; Takahashi, T.; Kawai, K.; Yazawa, K.; Asakage, M.; Hori, N.; Watanabe, T.; et al. Vaccination with autologous endothelium inhibits angiogenesis and metastasis of colon cancer through autoimmunity. Cancer Sci. 2004, 95, 85–90. [Google Scholar] [CrossRef]
- Chen, X.-Y.; Zhang, W.; Zhang, W.; Wu, S.; Bi, F.; Su, Y.-J.; Tan, X.-Y.; Liu, J.-N.; Zhang, J. Vaccination with viable human umbilical vein endothelial cells prevents metastatic tumors by attack on tumor vasculature with both cellular and humoral immunity. Clin. Cancer Res. 2006, 12, 5834–5840. [Google Scholar] [CrossRef][Green Version]
- Okaji, Y.; Tsuno, N.H.; Tanaka, M.; Yoneyama, S.; Matsuhashi, M.; Kitayama, J.; Saito, S.; Nagura, Y.; Tsuchiya, T.; Yamada, J.; et al. Pilot study of anti-angiogenic vaccine using fixed whole endothelium in patients with progressive malignancy after failure of conventional therapy. Eur. J. Cancer 2008, 44, 383–390. [Google Scholar] [CrossRef]
- Lokhov, P.G.; Balashova, E.E. Design of universal cancer vaccines using natural tumor vessel-specific antigens (SANTAVAC). Hum. Vaccin. Immunother. 2015, 11, 689–698. [Google Scholar] [CrossRef]
- Lokhov, P.G.; Balashova, E.E. Allogeneic antigen composition for preparing universal cancer vaccines. J. Immunol. Res. 2016, 2016, 5031529. [Google Scholar] [CrossRef]
- Lokhov, P.G.; Mkrtichyan, M.; Mamikonyan, G.; Balashova, E.E. SANTAVACTM: Summary of research and development. Vaccines 2019, 7, 186. [Google Scholar] [CrossRef]
- Lokhov, P.G.; Balashova, E.E. Tumor-induced endothelial cell surface heterogeneity directly affects endothelial cell escape from a cell-mediated immune response in vitro. Hum. Vaccin. Immunother. 2013, 9, 198–209. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Higano, C.S.; Corman, J.M.; Smith, D.C.; Centeno, A.S.; Steidle, C.P.; Gittleman, M.; Simons, J.W.; Sacks, N.; Aimi, J.; Small, E.J. Phase 1/2 dose-escalation study of a GM-CSF-secreting, allogeneic, cellular immunotherapy for metastatic hormone-refractory prostate cancer. Cancer 2008, 113, 975–984. [Google Scholar] [CrossRef]
- Moasser, M.M. The oncogene HER2: Its signaling and transforming functions and its role in human cancer pathogenesis. Oncogene 2007, 26, 6469–6487. [Google Scholar] [CrossRef] [PubMed]
- Kudrin, A. Overview of cancer vaccines: Considerations for development. Hum. Vaccines Immunother. 2012, 8, 1335–1353. [Google Scholar] [CrossRef] [PubMed]
- Guidance for Industry Clinical Considerations for Therapeutic Cancer Vaccines [excerpts]. Biotechnol. Law Rep. 2012, 31, 303. [CrossRef]
- Hoofnagle, A.N.; Whiteaker, J.R.; Carr, S.A.; Kuhn, E.; Liu, T.; Massoni, S.A.; Thomas, S.N.; Reid Townsend, R.; Zimmerman, L.J.; Boja, E.; et al. Recommendations for the generation, quantification, storage, and handling of peptides used for mass spectrometry-based assays. Clin. Chem. 2016, 62, 48–69. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.L.; Hsing, H.W.; Lai, T.C.; Chen, Y.W.; Lee, T.R.; Chan, H.T.; Lyu, P.C.; Wu, C.L.; Lu, Y.C.; Lin, S.T.; et al. Trypsin-induced proteome alteration during cell subculture in mammalian cells. J. Biomed. Sci. 2010, 17, 36. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lokhov, P.G.; Balashova, E.E. Antigenic Essence: Upgrade of Cellular Cancer Vaccines. Cancers 2021, 13, 774. https://doi.org/10.3390/cancers13040774
Lokhov PG, Balashova EE. Antigenic Essence: Upgrade of Cellular Cancer Vaccines. Cancers. 2021; 13(4):774. https://doi.org/10.3390/cancers13040774
Chicago/Turabian StyleLokhov, Petr G., and Elena E. Balashova. 2021. "Antigenic Essence: Upgrade of Cellular Cancer Vaccines" Cancers 13, no. 4: 774. https://doi.org/10.3390/cancers13040774
APA StyleLokhov, P. G., & Balashova, E. E. (2021). Antigenic Essence: Upgrade of Cellular Cancer Vaccines. Cancers, 13(4), 774. https://doi.org/10.3390/cancers13040774