
Mouse Preclinical Cancer Immunotherapy Modeling Involving Anti-PD-1 Therapies Reveals the Need to Use Mouse Reagents to Mirror Clinical Paradigms


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
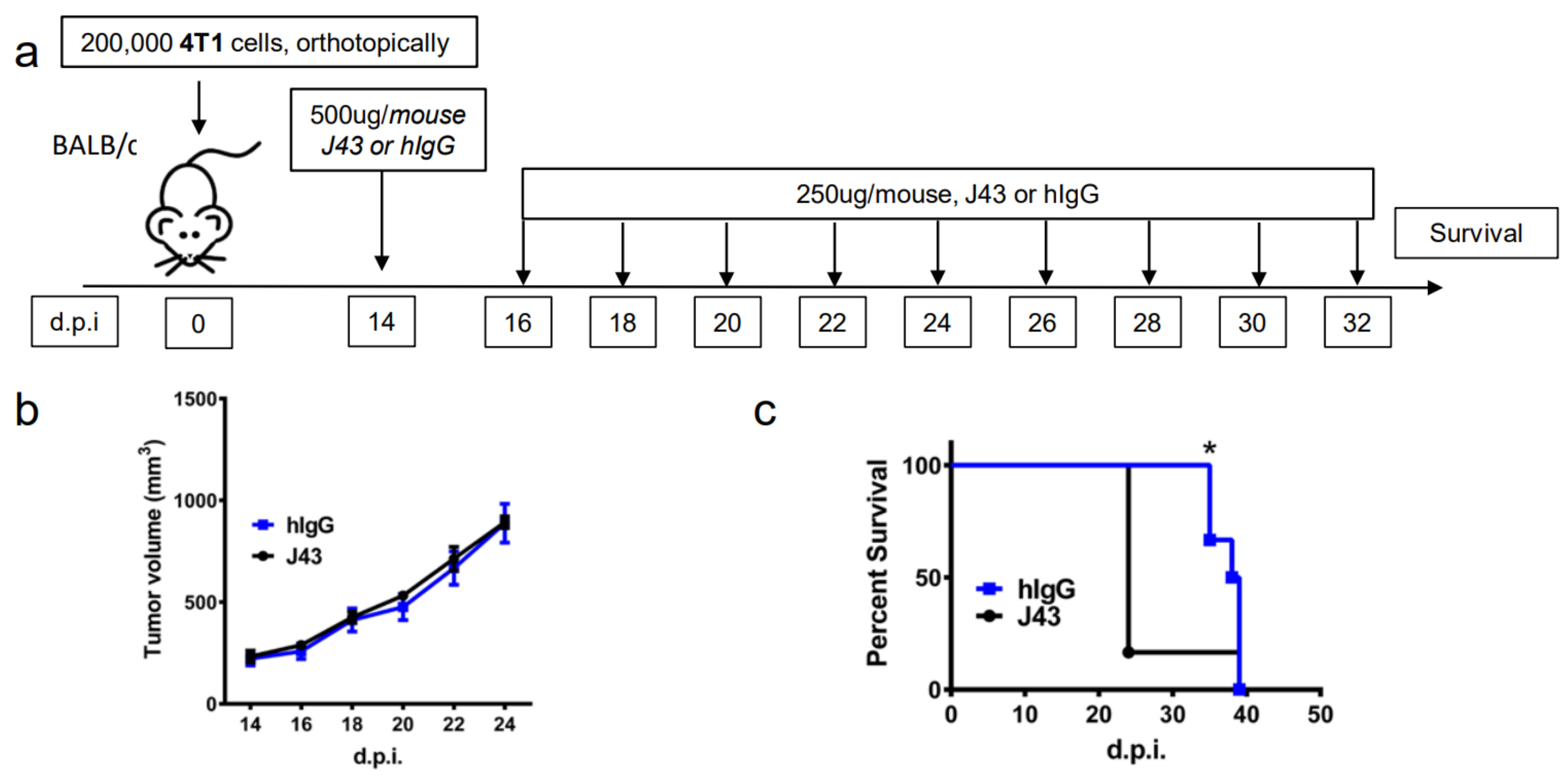
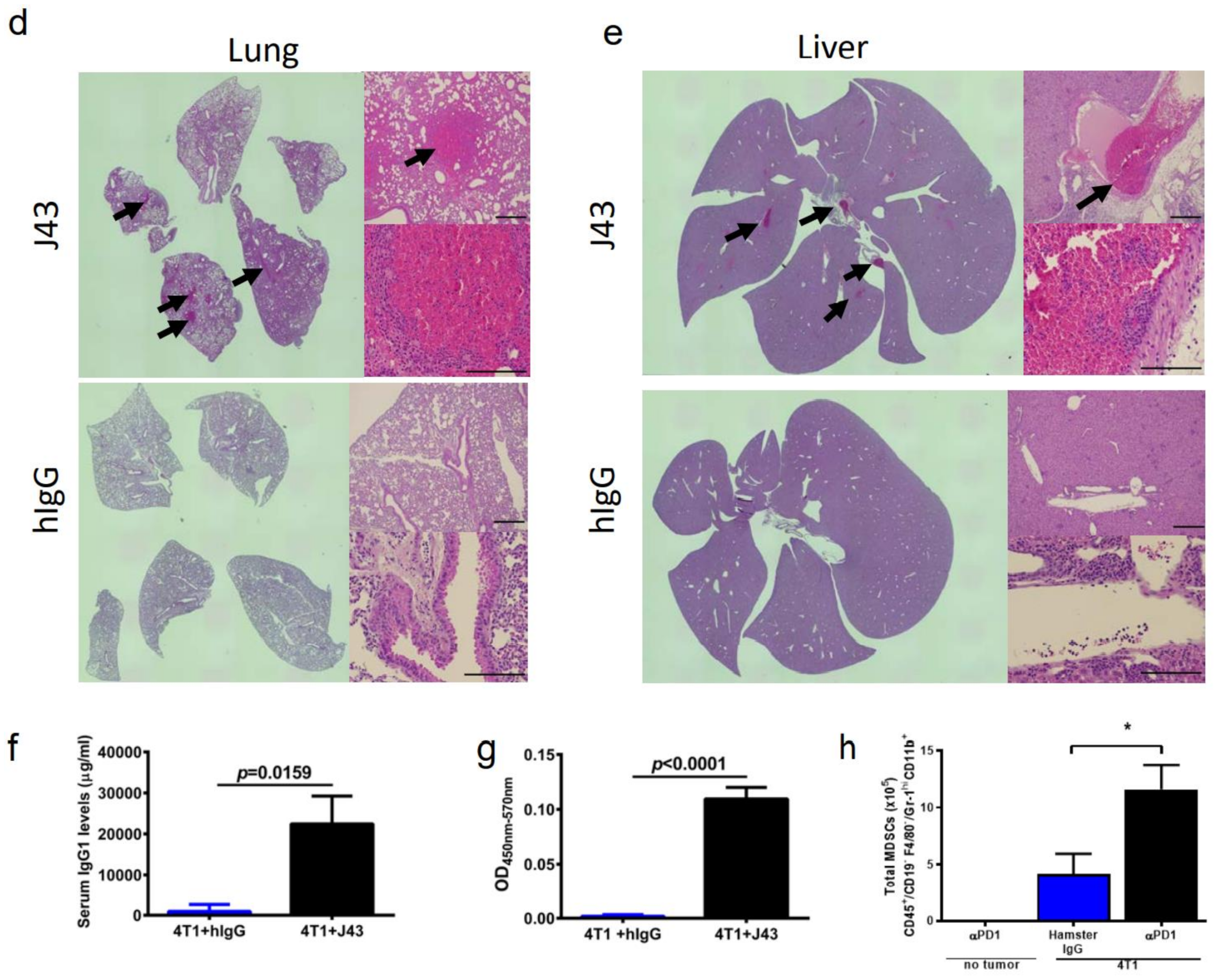
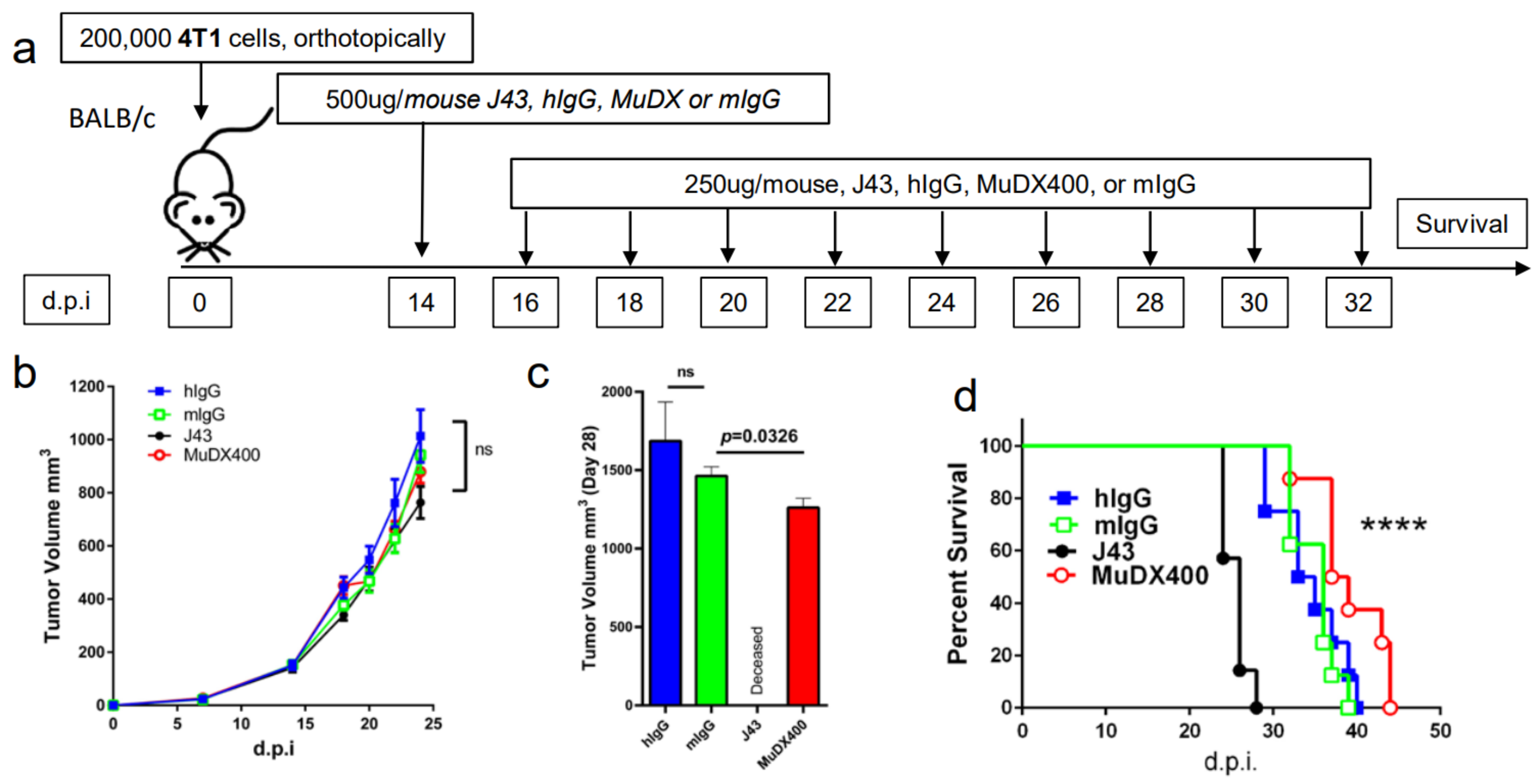
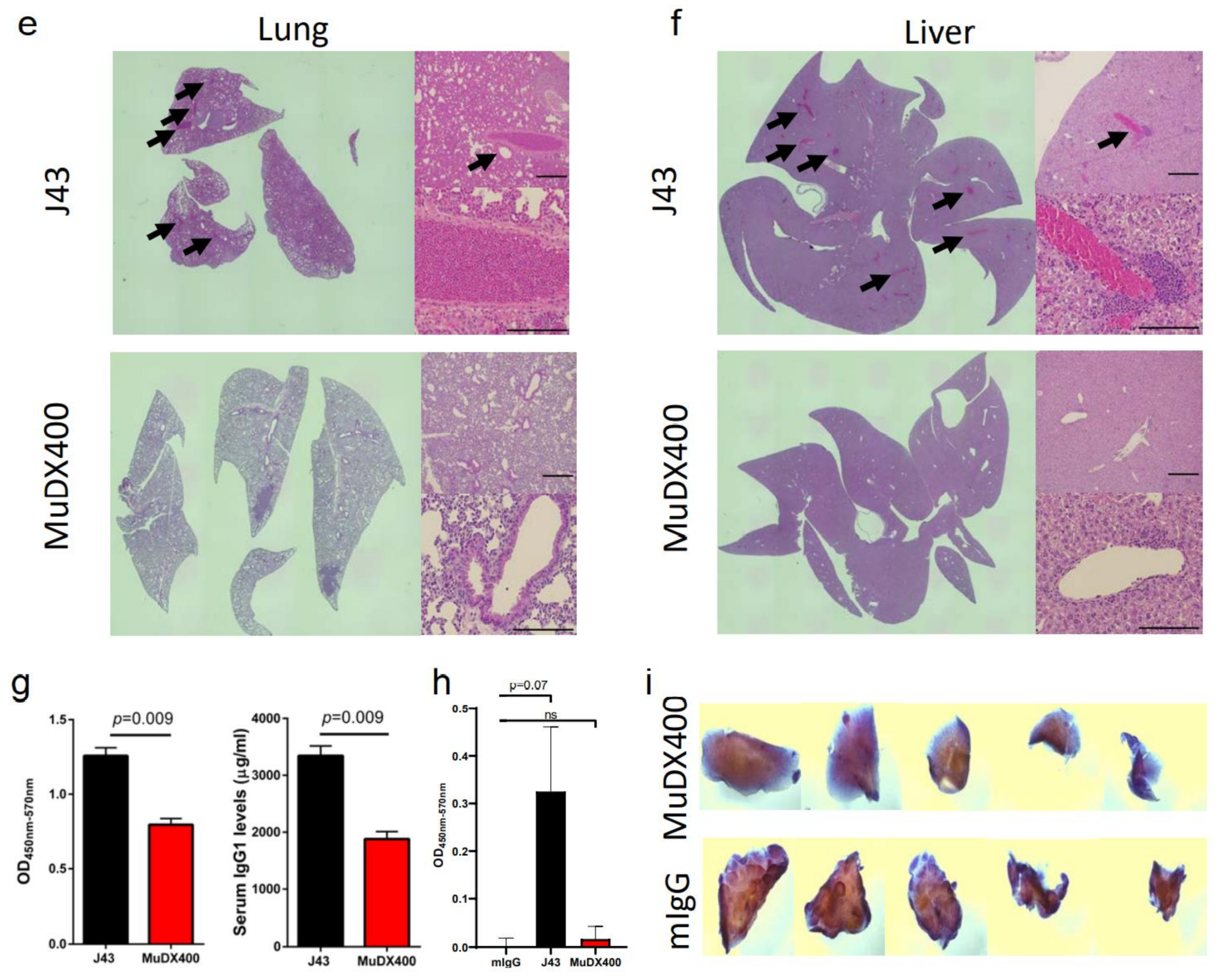
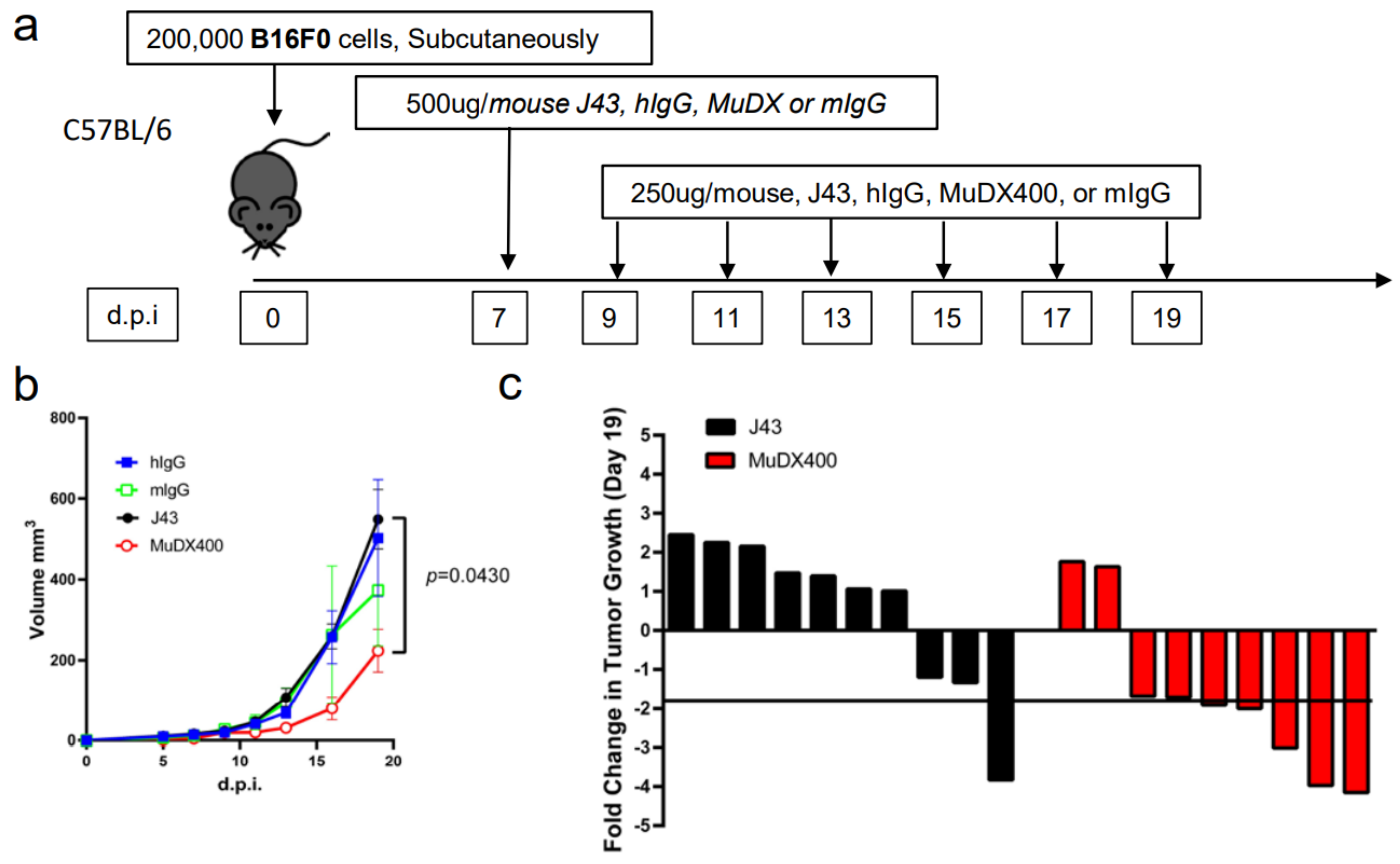
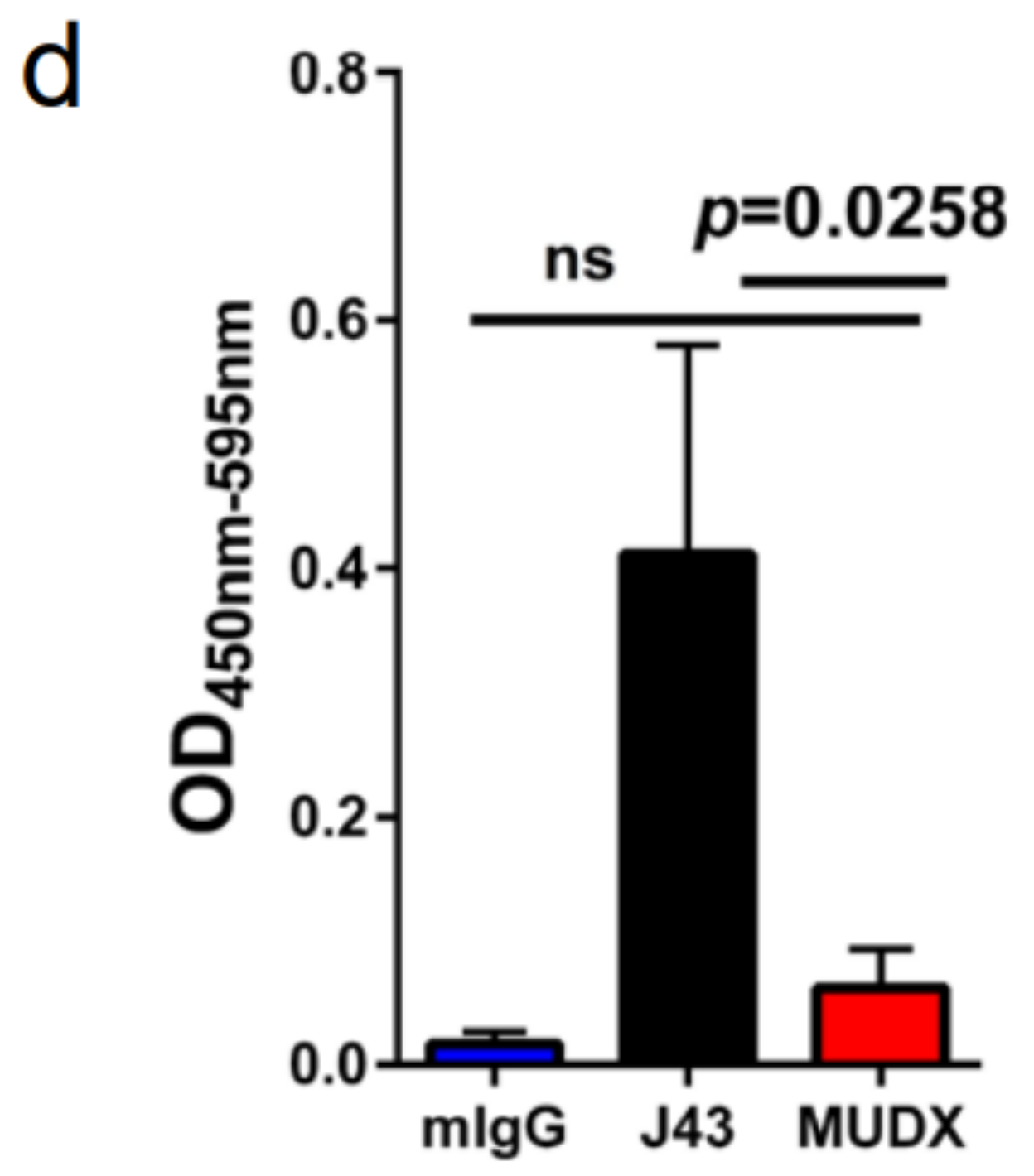
2. Results
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Tumor Cell Line and Treatment
4.3. Mouse Lung Whole-Mount Preparation
4.4. H&E Staining
4.5. ELISA
4.6. Statistics
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sanmamed, F.M.; Chen, A.L. Paradigm shift in cancer immunotherapy: From enhancement to normalization. Cell 2018, 175, 313–326. [Google Scholar] [CrossRef]
- Sharpe, A.H.; Wherry, E.J.; Ahmed, R.; Freeman, G.J. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat. Immunol. 2007, 8, 239. [Google Scholar] [CrossRef] [PubMed]
- Baumeister, H.S.; Freeman, J.G.; Dranoff, G.; Sharpe, H.A. Coinhibitory pathways in immunotherapy for cancer. Annu. Rev. Immunol. 2016, 34, 539–573. [Google Scholar] [CrossRef] [PubMed]
- Chen, L. Co-inhibitory molecules of the B7–CD28 family in the control of T-cell immunity. Nat. Rev. Immunol. 2004, 4, 336. [Google Scholar] [CrossRef] [PubMed]
- Greenwald, R.J.; Freeman, G.J.; Sharpe, A.H. The B7 family revisited. Annu Rev. Immunol. 2005, 23, 515–548. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, T.; Honjo, T. The PD-1–PD-L pathway in immunological tolerance. Trends Immunol. 2006, 27, 195–201. [Google Scholar] [CrossRef]
- Nakae, S.; Suto, H.; Iikura, M.; Kakurai, M.; Sedgwick, J.D.; Tsai, M.; Galli, S.J. Mast cells enhance T cell activation: Importance of mast cell costimulatory molecules and secreted TNF. J. Immunol. 2006, 176, 2238–2248. [Google Scholar] [CrossRef]
- Latchman, Y.; Wood, C.R.; Chernova, T.; Chaudhary, D.; Borde, M.; Chernova, I.; Freeman, G. J PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2001, 2, 261. [Google Scholar] [CrossRef]
- Brown, J.A.; Dorfman, D.M.; Ma, F.R.; Sullivan, E.L.; Munoz, O.; Wood, C.R.; Freeman, G.J. Blockade of programmed death-1 ligands on dendritic cells enhances T cell activation and cytokine production. J. Immunol. 2003, 170, 1257–1266. [Google Scholar] [CrossRef]
- LaFleur, M.W.; Muroyama, Y.; Drake, C.G.; Sharpe, A.H. Inhibitors of the PD-1 pathway in tumor therapy. J. Immunol. 2018, 200, 375–383. [Google Scholar] [CrossRef]
- Twyman-Saint Victor, C.; Rech, A.J.; Maity, A.; Rengan, R.; Pauken, K.E.; Stelekati, E.; Minn, A.J. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 2015, 520, 373. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Ribas, A. Pembrolizumab versus ipilimumab in advanced melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Gandhi, L. Pembrolizumab for the treatment of non–small-cell lung cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef] [PubMed]
- Powles, T.; Fine, G.D.; Braiteh, F.S.; Loriot, Y.; Cruz, C.; Vogelzang, N.J. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature 2014, 515, 558–562. [Google Scholar] [CrossRef] [PubMed]
- Rangarajan, A.; Weinberg, R.A. Comparative biology of mouse versus human cells: Modelling human cancer in mice. Nat. Rev. Cancer 2003, 3, 952. [Google Scholar] [CrossRef] [PubMed]
- Masopust, D.; Sivula, C.P.; Jameson, S.C. Of mice, dirty mice, and men: Using mice to understand human immunology. J. Immunol. 2017, 199, 383–388. [Google Scholar] [CrossRef]
- Moore, K.J. Utilization of mouse models in the discovery of human disease genes. Drug Discov. Today 1999, 4, 123–128. [Google Scholar] [CrossRef]
- Mall, C.; Sckisel, G.D.; Proia, D.A.; Mirsoian, A.; Grossenbacher, S.K.; Pai, C.C.S.; Murphy, W.J. Repeated PD-1/PD-L1 monoclonal antibody administration induces fatal xenogeneic hypersensitivity reactions in a murine model of breast cancer. Oncoimmunology 2016, 5, e1075114. [Google Scholar] [CrossRef]
- Brüggemann, M.; Winter, G.; Waldmann, H.; Neuberger, M.S. The immunogenicity of chimeric antibodies. J. Exp. Med. 1989, 170, 2153–2157. [Google Scholar] [CrossRef] [PubMed]
- Hunter, K.W., Jr. Murine mammary carcinoma 4T1 induces a leukemoid reaction with splenomegaly: Association with tumor-derived growth factors. Exp. Mol. Pathol. 2007, 82, 12–24. [Google Scholar]
- Heppner, G.H.; Miller, F.R.; Shekhar, P.M. Nontransgenic models of breast cancer. Breast Cancer Res. 2000, 2, 331. [Google Scholar] [CrossRef] [PubMed]
- Lelekakis, M.; Moseley, J.M.; Martin, T.J.; Hards, D.; Williams, E.; Ho, P.; Anderson, R.L. A novel orthotopic model of breast cancer metastasis to bone. Clin. Exp. Metastasis 1999, 17, 163–170. [Google Scholar] [CrossRef]
- De Souza Garcia, C.M.; de Araújo, M.R.; Lopes, M.T.P.; Ferreira, M.; Cassali, G.D. Morphological and immunophenotipical characterization of murine mammary carcinoma 4t1. Braz. J. Vet. Pathol. 2014, 7, 158–165. [Google Scholar]
- McMaster, P.D.; Kruse, H. Peripheral vascular reactions in anaphylaxis of the mouse. J. Exp. Med. 1949, 89, 583–596. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Weiser, R.S.; Golub, O.J.; Hamre, D.M. Studies on anaphylaxis in the mouse. J. Infect. Dis. 1941, 68, 97–112. [Google Scholar] [CrossRef]
- Cameron, J. The effect of aspirin, cortisone and other compounds on susceptibility to anaphylactic shock in mice. Br. J. Exp. Pathol. 1957, 38, 512. [Google Scholar]
- Presta, L.G. Engineering of therapeutic antibodies to minimize immunogenicity and optimize function. Adv. Drug Deliv. Rev. 2006, 58, 640–656. [Google Scholar] [CrossRef] [PubMed]
- Khodoun, M.V.; Strait, R.; Armstrong, L.; Yanase, N.; Finkelman, F.D. Identification of markers that distinguish IgE-from IgG-mediated anaphylaxis. Proc. Natl. Acad. Sci. USA 2011, 108, 12413–12418. [Google Scholar] [CrossRef]
- Jönsson, F.; Mancardi, D.A.; Kita, Y.; Karasuyama, H.; Iannascoli, B.; Van Rooijen, N.; Bruhns, P. Mouse and human neutrophils induce anaphylaxis. J. Clin. Invest. 2011, 121, 1484–1496. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monjazeb, A.M.; Wang, Z.; Vick, L.V.; Dunai, C.; Minnar, C.; Khuat, L.T.; Murphy, W.J. Mouse Preclinical Cancer Immunotherapy Modeling Involving Anti-PD-1 Therapies Reveals the Need to Use Mouse Reagents to Mirror Clinical Paradigms. Cancers 2021, 13, 729. https://doi.org/10.3390/cancers13040729
Monjazeb AM, Wang Z, Vick LV, Dunai C, Minnar C, Khuat LT, Murphy WJ. Mouse Preclinical Cancer Immunotherapy Modeling Involving Anti-PD-1 Therapies Reveals the Need to Use Mouse Reagents to Mirror Clinical Paradigms. Cancers. 2021; 13(4):729. https://doi.org/10.3390/cancers13040729
Chicago/Turabian StyleMonjazeb, Arta M., Ziming Wang, Logan V. Vick, Cordelia Dunai, Christine Minnar, Lam T. Khuat, and William J. Murphy. 2021. "Mouse Preclinical Cancer Immunotherapy Modeling Involving Anti-PD-1 Therapies Reveals the Need to Use Mouse Reagents to Mirror Clinical Paradigms" Cancers 13, no. 4: 729. https://doi.org/10.3390/cancers13040729
APA StyleMonjazeb, A. M., Wang, Z., Vick, L. V., Dunai, C., Minnar, C., Khuat, L. T., & Murphy, W. J. (2021). Mouse Preclinical Cancer Immunotherapy Modeling Involving Anti-PD-1 Therapies Reveals the Need to Use Mouse Reagents to Mirror Clinical Paradigms. Cancers, 13(4), 729. https://doi.org/10.3390/cancers13040729