
Natural Killer Cells and Anti-Cancer Therapies: Reciprocal Effects on Immune Function and Therapeutic Response



 ,
,  , ,
, , 
Simple Summary
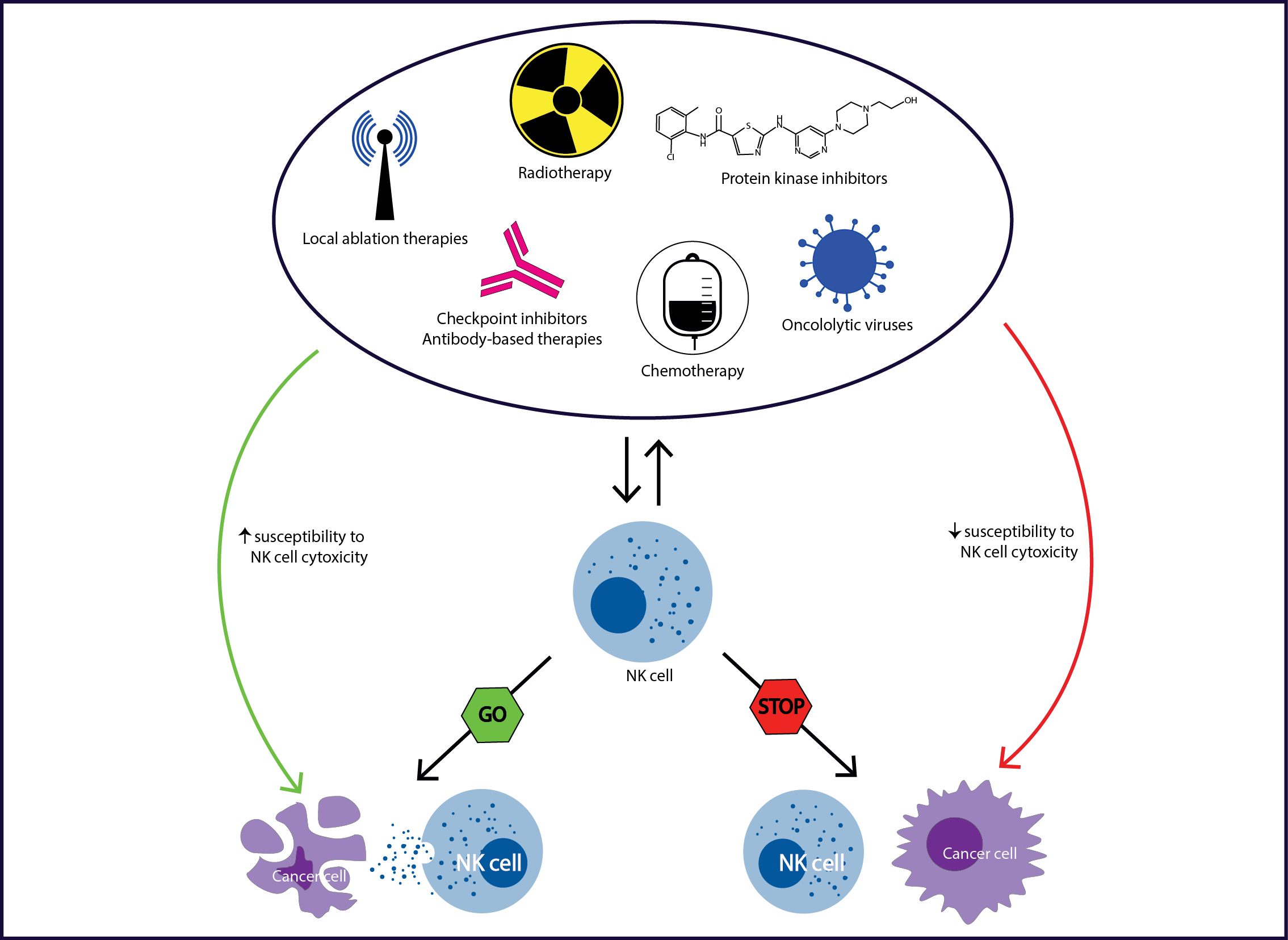
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Radiotherapy
3. Local Ablation Therapies
3.1. Radiofrequency Ablation
3.2. Microwave Ablation Therapy
3.3. High Intensity Focused Ultrasound Ablation
4. Checkpoint Inhibitors
4.1. PD-1-PD-L1 Axis
4.2. CTLA-4
4.3. TIM3
4.4. TIGIT-CD96
4.5. LAG3
5. Chemotherapy
5.1. Alkylating and Alkylating-Like Agents
5.2. Microtubule Targeting Agents
5.3. Antimetabolites
5.4. Anthracyclines
5.5. Other Anti-Cancer Agents
5.6. Combination Therapies
6. Protein Kinase Inhibitors
6.1. Src-Kinase Inhibitors
6.2. BRAF Inhibitors
6.3. GSK-3β Inhibitors
6.4. Other Protein Kinase Inhibitors
7. Oncolytic Viruses
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Campbell, K.S.; Hasegawa, J. Natural killer cell biology: An update and future directions. J. Allergy Clin. Immunol. 2013, 132, 536–544. [Google Scholar] [CrossRef]
- Vivier, E.; Tomasello, E.; Baratin, M.; Walzer, T.; Ugolini, S. Functions of natural killer cells. Nat. Immunol. 2008, 9, 503–510. [Google Scholar] [CrossRef]
- Cooper, M.A.; Fehniger, T.A.; Caligiuri, M.A. The biology of human natural killer-cell subsets. Trends Immunol. 2001, 22, 633–640. [Google Scholar] [CrossRef]
- Zamai, L.; Ahmad, M.; Bennett, I.M.; Azzoni, L.; Alnemri, E.S.; Perussia, B. Natural killer (NK) cell-mediated cytotoxicity: Differential use of TRAIL and Fas ligand by immature and mature primary human NK cells. J. Exp. Med. 1998, 188, 2375–2380. [Google Scholar] [CrossRef] [PubMed]
- Lanier, L.L. Up on the tightrope: Natural killer cell activation and inhibition. Nat. Immunol. 2008, 9, 495–502. [Google Scholar] [CrossRef]
- Veluchamy, J.P.; Kok, N.; van der Vliet, H.J.; Verheul, H.M.W.; de Gruijl, T.D.; Spanholtz, J. The rise of allogeneic Natural killer cells as a platform for cancer immunotherapy: Recent innovations and future developments. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- WHO Key Statistics. Available online: https://www.who.int/cancer/resources/keyfacts/en/ (accessed on 30 October 2020).
- WHO Europe Cancer. Available online: https://www.euro.who.int/en/health-topics/noncommunicable-diseases/cancer/cancer (accessed on 30 October 2020).
- Takeuchi, H.; Maehara, Y.; Tokunaga, E.; Koga, T.; Kakeji, Y.; Sugimachi, K. Prognostic significance of natural killer cell activity in patients with gastric carcinoma: A multivariate analysis. Am. J. Gastroenterol. 2001, 96, 574–578. [Google Scholar] [CrossRef] [PubMed]
- Tartter, P.I.; Steinberg, B.; Barron, D.M.; Martinelli, G. The Prognostic Significance of Natural Killer Cytotoxicity in Patients with Colorectal Cancer. Arch. Surg. 1987, 122, 1264–1268. [Google Scholar] [CrossRef]
- Luna, J.I.; Grossenbacher, S.K.; Murphy, W.J.; Canter, R.J. Natural Killer Cell Immunotherapy Targeting Cancer Stem Cells. Expert Opin. Biol. Ther. 2018, 17, 313–324. [Google Scholar] [CrossRef]
- Battella, S.; Cox, M.C.; Santoni, A.; Palmieri, G. Natural killer (NK) cells and anti-tumor therapeutic mAb: Unexplored interactions. J. Leukoc. Biol. 2016, 99, 87–96. [Google Scholar] [CrossRef]
- Baskar, R.; Lee, K.A.; Yeo, R.; Yeoh, K.W. Cancer and radiation therapy: Current advances and future directions. Int. J. Med. Sci. 2012, 9, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Delaney, G.; Jacob, S.; Featherstone, C.; Barton, M. The role of Radiother in cancer treatment: Estimating optimal utilization from a review of evidence-based clinical guidelines. Cancer 2005, 104, 1129–1137. [Google Scholar] [CrossRef]
- Hietanen, T.; Pitkänen, M.; Kapanen, M.; Kellokumpu-Lehtinen, P.L. Post-irradiation viability and cytotoxicity of natural killer cells isolated from human peripheral blood using different methods. Int. J. Radiat. Biol. 2016, 92, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Falcke, S.E.; Rühle, P.F.; Deloch, L.; Fietkau, R.; Frey, B.; Gaipl, U.S. Clinically relevant radiation exposure differentially impacts forms of cell death in human cells of the innate and adaptive immune system. Int. J. Mol. Sci. 2018, 19, 3574. [Google Scholar] [CrossRef]
- Hietanen, T.; Pitkänen, M.; Kapanen, M.; Kellokumpu-Lehtinen, P.L. Effects of single and fractionated irradiation on natural killer cell populations: Radiobiological characteristics of viability and cytotoxicity in vitro. Anticancer Res. 2015, 35, 5193–5200. [Google Scholar]
- Yang, G.; Kong, Q.; Wang, G.; Jin, H.; Zhou, L.; Yu, D.; Niu, C.; Han, W.; Li, W.; Cui, J. Low-dose ionizing radiation induces direct activation of natural killer cells and provides a novel approach for adoptive cellular immunotherapy. Cancer Biother. Radiopharm. 2014, 29. [Google Scholar] [CrossRef] [PubMed]
- Eric, A.; Juranic, Z.; Tisma, N.; Plesinac, V.; Borojevic, N.; Jovanovic, D.; Milovanovic, Z.; Gavrilovic, D.; Ilic, B. Radiotherapy-induced changes of peripheral blood lymphocyte subpopulations in cervical cancer patients: Relationship to clinical response. J. BUON 2009, 14, 79–83. [Google Scholar] [PubMed]
- Clave, E.; Socié, G.; Cosset, J.M.; Chaillet, M.P.; Tartour, E.; Girinsky, T.; Carosella, E.; Fridman, H.; Gluckman, E.; Mathiot, C. Multicolor flow cytometry analysis of blood cell subsets in patients given total body irradiation before bone marrow transplantation. Int. J. Radiat. Oncol. Biol. Phys. 1995. [Google Scholar] [CrossRef]
- Louagie, H.; van Eijkeren, M.; Philippe, J.; Thierens, H.; de Ridder, L. Changes in peripheral blood lymphocyte subsets in patients undergoing radiotherapy. Int. J. Radiat. Biol. 1999, 75, 767–771. [Google Scholar] [CrossRef]
- Mozaffari, F.; Lindemalm, C.; Choudhury, A.; Granstam-Björneklett, H.; Helander, I.; Lekander, M.; Mikaelsson, E.; Nilsson, B.; Ojutkangas, M.L.; Österborg, A.; et al. NK-cell and T-cell functions in patients with breast cancer: Effects of surgery and adjuvant chemo- and radiotherapy. Br. J. Cancer 2007, 97, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, Y.; Makino, S.; Fukuda, Y.; Ikemoto, T.; Shimizu, A. Varied Effects of Thoracic Irradiation on Peripheral Lymphocyte Subsets in Lung Cancer Patients. Intern. Med. 1995. [Google Scholar] [CrossRef] [PubMed]
- Belka, C.; Ottinger, H.; Kreuzfelder, E.; Weinmann, M.; Lindemann, M.; Lepple-Wienhues, A.; Budach, W.; Grosse-Wilde, H.; Bamberg, M. Impact of localized radiotherapy on blood immune cells counts and function in humans. Radiother. Oncol. 1999. [Google Scholar] [CrossRef]
- Domouchtsidou, A.; Barsegian, V.; Mueller, S.P.; Best, J.; Ertle, J.; Bedreli, S.; Horn, P.A.; Bockisch, A.; Lindemann, M. Impaired lymphocyte function in patients with hepatic malignancies after selective internal radiotherapy. Cancer Immunol. Immunother. 2018. [Google Scholar] [CrossRef]
- Yamaue, H.; Tanimura, H.; Aoki, Y.; Tsunoda, T.; Iwahashi, M.; Tani, M.; Tamai, M.; Noguchi, K.; Kashiwagi, H.; Sasaki, M.; et al. Clinical and immunological evaluation of intraoperative radiation therapy for patients with unresectable pancreatic cancer. J. Surg. Oncol. 1992. [Google Scholar] [CrossRef]
- Blomgren, H.; Baral, E.; Edsmyr, F.; Strender, L.E.; Petrini, B.; Wasserman, J. Natural killer activity in peripheral lymphocyte population following local radiation therapy. Acta Radiol. Oncol. Radiat. Phys. Biol. 1980, 19, 139–143. [Google Scholar] [CrossRef]
- Kim, J.Y.; Son, Y.O.; Park, S.W.; Bae, J.H.; Joo, S.C.; Hyung, H.K.; Chung, B.S.; Kim, S.H.; Kang, C.D. Increase of NKG2D ligands and sensitivity to NK cell-mediated cytotoxicity of tumor cells by heat shock and ionizing radiation. Exp. Mol. Med. 2006, 38, 474–484. [Google Scholar] [CrossRef]
- Fine, J.H.; Chen, P.; Mesci, A.; Allan, D.S.J.; Gasser, S.; Raulet, D.H.; Carlyle, J.R. Chemotherapy-induced genotoxic stress promotes sensitivity to natural killer cell cytotoxicity by enabling missing-self recognition. Cancer Res. 2010, 70, 7102–7113. [Google Scholar] [CrossRef]
- Balaji, G.R.; Aguilar, O.A.; Tanaka, M.; Shingu-Vazquez, M.A.; Fu, Z.; Gully, B.S.; Lanier, L.L.; Carlyle, J.R.; Rossjohn, J.; Berry, R. Recognition of host Clr-b by the inhibitory NKR-P1B receptor provides a basis for missing-self recognition. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]
- Heo, W.; Lee, Y.S.; Son, C.H.; Yang, K.; Park, Y.S.; Bae, J. Radiation-induced matrix metalloproteinases limit natural killer cell-mediated anticancer immunityin NCI-H23 lung cancer cells. Mol. Med. Rep. 2015, 1800–1806. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.W.; Kim, J.E.; Hwang, M.H.; Jeon, Y.H.; Lee, S.W.; Lee, J.; Zeon, S.K.; Ahn, B.C. Enhancement of Natural Killer Cell Cytotoxicity by Sodium/Iodide Symporter Gene-Mediated Radioiodine Pretreatment in Breast Cancer Cells. PLoS ONE 2013, 8, e70194. [Google Scholar] [CrossRef] [PubMed]
- Garnett, C.T.; Palena, C.; Chakarborty, M.; Tsang, K.Y.; Schlom, J.; Hodge, J.W. Sublethal irradiation of human tumor cells modulates phenotype resulting in enhanced killing by cytotoxic T lymphocytes. Cancer Res. 2004, 64, 7985–7994. [Google Scholar] [CrossRef]
- Michelin, S.; Gallegos, C.E.; Dubner, D.; Favier, B.; Carosella, E.D. Ionizing radiation modulates the surface expression of human leukocyte antigen-G in a human melanoma cell line. Hum. Immunol. 2009, 70, 1010–1015. [Google Scholar] [CrossRef]
- Urosevic, M.; Kempf, W.; Zagrodnik, B.; Panizzon, R.; Burg, G.; Dummer, R. HLA-G expression in basal cell carcinomas of the skin recurring after radiotherapy. Clin. Exp. Derm. 2005, 422–425. [Google Scholar] [CrossRef]
- Jeong, J.U.; Uong, T.N.T.; Chung, W.K.; Nam, T.K.; Ahn, S.J.; Song, J.Y.; Kim, S.K.; Shin, D.J.; Cho, E.; Kim, K.W.; et al. Effect of irradiation-induced intercellular adhesion molecule-1 expression on natural killer cell-mediated cytotoxicity toward human cancer cells. Cytotherapy 2018, 20, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Ames, E.; Canter, R.J.; Grossenbacher, S.K.; Mac, S.; Smith, R.C.; Monjazeb, A.M.; Chen, M.; Murphy, W.J. Enhanced targeting of stem-like solid tumor cells with radiation and natural killer cells. OncoImmunology 2015, 4, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.J.; Xu, L.J.; Yang, L.; Tsai, Y.; Keng, P.C.; Chen, Y.; Lee, S.O.; Chen, Y. Radiation alters PD-L1/NKG2D ligand levels in lung cancer cells and leads to immune escape from NK cell cytotoxicity via IL-6- MEK/Erk signaling pathway. Oncotarget 2017, 8, 80506–80520. [Google Scholar] [CrossRef] [PubMed]
- Multhoff, G.; Botzler, C.; Jennen, L.; Schmidt, J.; Ellwart, J.; Issels, R. Heat shock protein 72 on tumor cells: A recognition structure for natural killer cells. J. Immunol. 1997, 4341–4350. [Google Scholar]
- Multhoff, G.; Pockley, A.G.; Schmid, T.E.; Schilling, D. The role of heat shock protein 70 (Hsp70) in radiation-induced immunomodulation. Cancer Lett. 2015, 179–184. [Google Scholar] [CrossRef]
- Calini, V.; Urani, C.; Camatini, M. Overexpression of HSP70 is induced by ionizing radiation in C3H 10T1/2 cells and protects from DNA damage. Toxicol. In Vitro 2003, 17, 561–566. [Google Scholar] [CrossRef]
- Gastpar, R.; Gross, C.; Rossbacher, L.; Ellwart, J.; Riegger, J.; Multhoff, G. The Cell Surface-Localized Heat Shock Protein 70 Epitope TKD Induces Migration and Cytolytic Activity Selectively in Human NK Cells. J. Immunol. 2004. [Google Scholar] [CrossRef]
- Multhoff, G.; Mizzen, L.; Winchester, C.C.; Milner, C.M.; Wenk, S.; Eissner, G.; Kampinga, H.H.; Laumbacher, B.; Johnson, J. Heat shock protein 70 (Hsp70) stimulates proliferation and cytolytic activity of natural killer cells. Exp. Hematol. 1999, 27, 1627–1636. [Google Scholar] [CrossRef]
- Stangl, S.; Gross, C.; Pockley, A.G.; Asea, A.A.; Multhoff, G. Influence of Hsp70 and HLA-E on the killing of leukemic blasts by cytokine/Hsp70 peptide-activated human natural killer (NK) cells. Cell Stress Chaperones 2008, 13, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P.; Multhoff, G. Adenosine can thwart anti-tumor immune responses elicited by radiotherapy. Strahlenther. Und Onkol. 2016, 192, 279–287. [Google Scholar] [CrossRef]
- Young, A.; Ngiow, S.F.; Gao, Y.; Patch, A.M.; Barkauskas, D.S.; Messaoudene, M.; Lin, G.; Coudert, J.D.; Stannard, K.A.; Zitvogel, L.; et al. A2AR adenosine signaling suppresses natural killer cell maturation in the tumor microenvironment. Cancer Res. 2018, 78, 1003–1016. [Google Scholar] [CrossRef]
- Yoon, M.S.; Pham, C.T.; Phan, M.T.T.; Shin, D.J.; Jang, Y.Y.; Park, M.H.; Kim, S.K.; Kim, S.; Cho, D. Irradiation of breast cancer cells enhances CXCL16 ligand expression and induces the migration of natural killer cells expressing the CXCR6 receptor. Cytotherapy 2016, 18, 1532–1542. [Google Scholar] [CrossRef]
- Thandassery, R.B.; Goenka, U.; Goenka, M.K. Role of Local Ablative Therapy for hepatocellular carcinoma. J. Clin. Exp. Hepatol. 2014, 4, S104–S111. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.L.; Jennings, P.E. Lung radiofrequency and microwave ablation: A review of indications, techniques and post-procedural imaging appearances. Br. J. Radiol. 2015, 88. [Google Scholar] [CrossRef]
- Deng, Z.; Zhang, W.; Han, Y.; Zhang, S. Radiofrequency ablation inhibits lung metastasis ofbreast cancer in mice. Zhonghua Zhong Liu Za Zhi 2015, 37, 497–500. [Google Scholar] [PubMed]
- Mo, Z.; Lu, H.; Mo, S.; Fu, X.; Chang, S.; Yue, J. Ultrasound-guided radiofrequency ablation enhances natural killer-mediated antitumor immunity against liver cancer. Oncol. Lett. 2018, 15, 7014–7020. [Google Scholar] [CrossRef]
- Todorova, V.K.; Klimberg, V.S.; Hennings, L.; Kieber-Emmons, T.; Pashov, A. Immunomodulatory effects of radiofrequency ablation in a breast cancer model. Immunol. Investig. 2010, 39, 74–92. [Google Scholar] [CrossRef] [PubMed]
- Matuszewski, M.; Michajłowski, J.; Michajłowski, I.; Ruckermann-Dizurdzińska, K.; Witkowski, J.M.; Biernat, W.; Krajka, K. Impact of radiofrequency ablation on PBMC subpopulation in patients with renal cell carcinoma. Urol. Oncol. Semin. Orig. Investig. 2011, 29, 724–730. [Google Scholar] [CrossRef] [PubMed]
- Zerbini, A.; Pilli, M.; Laccabue, D.; Pelosi, G.; Molinari, A.; Negri, E.; Cerioni, S.; Fagnoni, F.; Soliani, P.; Ferrari, C.; et al. Radiofrequency Thermal Ablation for Hepatocellular Carcinoma Stimulates Autologous NK-Cell Response. Gastroenterology 2010, 138, 1931–1942.e2. [Google Scholar] [CrossRef] [PubMed]
- Guan, H.T.; Wang, J.; Yang, M.; Song, L.; Tong, X.Q.; Zou, Y.H. Changes in immunological function after treatment with transarterial chemoembolization plus radiofrequency ablation in hepatocellular carcinoma patients. Chin. Med. J. 2013, 126, 3651–3655. [Google Scholar] [CrossRef]
- Rochigneux, P.; Nault, J.C.; Mallet, F.; Chretien, A.S.; Barget, N.; Garcia, A.J.; del Pozo, L.; Bourcier, V.; Blaise, L.; Grando-Lemaire, V.; et al. Dynamic of systemic immunity and its impact on tumor recurrence after radiofrequency ablation of hepatocellular carcinoma. OncoImmunology 2019, 8, 1–11. [Google Scholar] [CrossRef]
- Lencioni, R. Loco-regional treatment of hepatocellular carcinoma. Hepatology 2010, 52, 762–773. [Google Scholar] [CrossRef]
- Yu, M.; Pan, H.; Che, N.; Li, L.; Wang, C.; Wang, Y.; Ma, G.; Qian, M.; Liu, J.; Zheng, M.; et al. Microwave ablation of primary breast cancer inhibits metastatic progression in model mice via activation of natural killer cells. Cell. Mol. Immunol. 2020, 1–12. [Google Scholar] [CrossRef]
- Dong, B.W.; Zhang, J.; Liang, P.; Yu, X.L.; Su, L.; Yu, D.J.; Ji, X.L.; Yu, G. Sequential pathological and immunologic analysis of percutaneous microwave coagulation therapy of hepatocellular carcinoma. Int. J. Hyperth. 2003, 19, 119–133. [Google Scholar] [CrossRef]
- Zhang, H.; Hou, X.; Cai, H.; Zhuang, X. Effects of microwave ablation on T-cell subsets and cytokines of patients with hepatocellular carcinoma. Minim. Invasive Allied Technol. 2017, 26, 207–211. [Google Scholar] [CrossRef]
- Szmigielski, S.; Sobczynski, J.; Sokolska, G.; Stawarz, B.; Zielinski, H.; Petrovich, Z. Effects of local prostatic hyperthermia on human NK and t cell function. Int. J. Hyperth. 1991, 7, 869–880. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, Z.B. High-intensity focused ultrasound tumor ablation: Review of ten years of clinical experience. Front. Med. China 2010, 4, 294–302. [Google Scholar] [CrossRef]
- Wu, F.; Wang, Z.B.; Lu, P.; Xu, Z.L.; Chen, W.Z.; Zhu, H.; Jin, C.B. Activated anti-tumor immunity in cancer patients after high intensity focused ultrasound ablation. Ultrasound Med. Biol. 2004, 30, 1217–1222. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Qin, J.; Chen, J.; Wang, L.; Chen, W.; Tang, L. The effect of high-intensity focused ultrasound treatment on immune function in patients with uterine fibroids. Int. J. Hyperth. 2013, 29, 225–233. [Google Scholar] [CrossRef]
- Ma, B.; Liu, X.; Yu, Z. The effect of high intensity focused ultrasound on the treatment of liver cancer and patients’ immunity. Cancer Biomark. 2019, 24, 85–90. [Google Scholar] [CrossRef]
- Lu, P.; Zhu, X.Q.; Xu, Z.L.; Zhou, Q.; Zhang, J.; Wu, F. Increased infiltration of activated tumor-infiltrating lymphocytes after high intensity focused ultrasound ablation of human breast cancer. Surgery 2009, 145, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, C.M.; White, M.J.; Goodier, M.R.; Riley, E.M. Functional significance of CD57 expression on human NK cells and relevance to disease. Front. Immunol. 2013, 4, 422. [Google Scholar] [CrossRef]
- Kim, N.; Kim, H.S. Targeting checkpoint receptors and molecules for therapeutic modulation of natural killer cells. Front. Immunol. 2018, 9, 2041. [Google Scholar] [CrossRef]
- Niu, C.; Li, M.; Zhu, S.; Chen, Y.; Zhou, L.; Xu, D.; Xu, J.; Li, Z.; Li, W.; Cui, J. Pd-1-positive natural killer cells have a weaker antitumor function than that of pd-1-negative natural killer cells in lung cancer. Int. J. Med. Sci. 2020, 17, 1964–1973. [Google Scholar] [CrossRef]
- Pesce, S.; Greppi, M.; Tabellini, G.; Rampinelli, F.; Parolini, S.; Olive, D.; Moretta, L.; Moretta, A.; Marcenaro, E. Identification of a subset of human natural killer cells expressing high levels of programmed death 1: A phenotypic and functional characterization. J. Allergy Clin. Immunol. 2017, 139, 335–346.e3. [Google Scholar] [CrossRef]
- Tumino, N.; Martini, S.; Munari, E.; Scordamaglia, F.; Besi, F.; Mariotti, F.R.; Bogina, G.; Mingari, M.C.; Vacca, P.; Moretta, L. Presence of innate lymphoid cells in pleural effusions of primary and metastatic tumors: Functional analysis and expression of PD-1 receptor. Int. J. Cancer 2019, 145, 1660–1668. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cheng, Y.; Xu, Y.; Wang, Z.; Du, X.; Li, C.; Peng, J.; Gao, L.; Liang, X.; Ma, C. Increased expression of programmed cell death protein 1 on NK cells inhibits NK-cell-mediated anti-tumor function and indicates poor prognosis in digestive cancers. Oncogene 2017, 36, 6143–6153. [Google Scholar] [CrossRef]
- Concha-Benavente, F.; Kansy, B.; Moskovitz, J.; Moy, J.; Chandran, U.; Ferris, R.L. PD-L1 Mediates Dysfunction in Activated PD-1+ NK Cells in Head and Neck Cancer Patients. Cancer Immunol. Res. 2018, 6, 1548–1560. [Google Scholar] [CrossRef]
- Trefny, M.P.; Kaiser, M.; Stanczak, M.A.; Herzig, P.; Savic, S.; Wiese, M.; Lardinois, D.; Läubli, H.; Uhlenbrock, F.; Zippelius, A. PD-1+ natural killer cells in human non-small cell lung cancer can be activated by PD-1/PD-L1 blockade. Cancer Immunol. Immunother. 2020, 69, 1505–1517. [Google Scholar] [CrossRef]
- Hsu, J.; Hodgins, J.J.; Marathe, M.; Nicolai, C.J.; Bourgeois-Daigneault, M.C.; Trevino, T.N.; Azimi, C.S.; Scheer, A.K.; Randolph, H.E.; Thompson, T.W.; et al. Contribution of NK cells to immunotherapy mediated by PD-1/PD-L1 blockade. J. Clin. Investig. 2018, 128, 4654–4668. [Google Scholar] [CrossRef] [PubMed]
- Oyer, J.L.; Gitto, S.B.; Altomare, D.A.; Copik, A.J. PD-L1 blockade enhances anti-tumor efficacy of NK cells. Oncoimmunology 2018, 7. [Google Scholar] [CrossRef]
- Juliá, E.P.; Amante, A.; Pampena, M.B.; Mordoh, J.; Levy, E.M. Avelumab, an IgG1 anti-PD-L1 immune checkpoint inhibitor, triggers NK cell-mediated cytotoxicity and cytokine production against triple negative breast cancer cells. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Boyerinas, B.; Jochems, C.; Fantini, M.; Heery, C.R.; Gulley, J.L.; Tsang, K.Y.; Schlom, J. Antibody-dependent cellular cytotoxicity activity of a Novel Anti-PD-L1 antibody avelumab (MSB0010718C) on human tumor cells. Cancer Immunol. Res. 2015, 3, 1148–1157. [Google Scholar] [CrossRef]
- Barry, K.C.; Hsu, J.; Broz, M.L.; Cueto, F.J.; Binnewies, M.; Combes, A.J.; Nelson, A.E.; Loo, K.; Kumar, R.; Rosenblum, M.D.; et al. A natural killer–dendritic cell axis defines checkpoint therapy–responsive tumor microenvironments. Nat. Med. 2018, 24, 1178–1191. [Google Scholar] [CrossRef]
- Deng, L.; Weichselbaum, R.R.; Fu, Y.; Deng, L.; Liang, H.; Burnette, B.; Beckett, M. Irradiation and anti—PD-L1 treatment synergistically promote antitumor immunity in mice Find the latest version: Irradiation and anti—PD-L1 treatment synergistically promote antitumor immunity in mice. J. Clin. Investig. 2014, 124, 687–695. [Google Scholar] [CrossRef]
- Makowska, A.; Meier, S.; Shen, L.; Busson, P.; Baloche, V.; Kontny, U. Anti-PD-1 antibody increases NK cell cytotoxicity towards nasopharyngeal carcinoma cells in the context of chemotherapy-induced upregulation of PD-1 and PD-L1. Cancer Immunol. Immunother. 2020. [Google Scholar] [CrossRef] [PubMed]
- Cameron, F.; Whiteside, G.; Perry, C. Ipilimumab: First global approval. Drugs 2011, 71, 1093–1104. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.; Gandhi, H.; Upaganlawar, A. Ipilimumab: Melanoma and beyond. J. Pharm. Bioallied Sci. 2011, 3, 546. [Google Scholar]
- Cabel, L.; Loir, E.; Gravis, G.; Lavaud, P.; Massard, C.; Albiges, L.; Baciarello, G.; Loriot, Y.; Fizazi, K. Long-term complete remission with Ipilimumab in metastatic castrate-resistant prostate cancer: Case report of two patients. J. Immunother. Cancer 2017, 5. [Google Scholar] [CrossRef]
- Stojanovic, A.; Fiegler, N.; Brunner-Weinzierl, M.; Cerwenka, A. CTLA-4 Is Expressed by Activated Mouse NK Cells and Inhibits NK Cell IFN-γ Production in Response to Mature Dendritic Cells. J. Immunol. 2014, 192, 4184–4191. [Google Scholar] [CrossRef]
- Lang, S.; Vujanovic, N.L.; Wollenberg, B.; Whiteside, T.L. Absence of B7.1-CD28/CTLA-4-mediated co-stimulation in human NK cells. Eur. J. Immunol. 1998, 28, 780–786. [Google Scholar] [CrossRef]
- Hutmacher, C.; Nuñez, N.G.; Liuzzi, A.R.; Becher, B.; Neri, D. Targeted delivery of IL2 to the tumor stroma potentiates the action of immune checkpoint inhibitors by preferential activation of NK and CD8þ T cells. Cancer Immunol. Res. 2019, 7, 572–583. [Google Scholar] [CrossRef]
- Kohlhapp, F.J.; Broucek, J.R.; Hughes, T.; Huelsmann, E.J.; Lusciks, J.; Zayas, J.P.; Dolubizno, H.; Fleetwood, V.A.; Grin, A.; Hill, G.E.; et al. NK cells and CD8+ T cells cooperate to improve therapeutic responses in melanoma treated with interleukin-2 (IL-2) and CTLA-4 blockade. J. Immunother. Cancer 2015, 3. [Google Scholar] [CrossRef]
- Sanseviero, E.; Karras, J.R.; Shabaneh, T.B.; Arman, B.; Xu, W.; Zheng, C.; Yin, X.; Xu, X.; Karakousis, G.; Nam, B.T.; et al. Anti-CTLA4 activates intratumoral NK cells and combination with IL15/IL15Rα complexes enhances tumor control. Cancer Immunol. Res. 2020, 7, 1371–1380. [Google Scholar] [CrossRef]
- Tallerico, R.; Cristiani, C.M.; Staaf, E.; Garofalo, C.; Sottile, R.; Capone, M.; de Coaña, Y.P.; Madonna, G.; Palella, E.; Wolodarski, M.; et al. IL-15, TIM-3 and NK cells subsets predict responsiveness to anti-CTLA-4 treatment in melanoma patients. OncoImmunology 2017, 6. [Google Scholar] [CrossRef]
- Sottile, R.; Tannazi, M.; Johansson, M.H.; Cristiani, C.M.; Calabró, L.; Ventura, V.; Cutaia, O.; Chiarucci, C.; Covre, A.; Garofalo, C.; et al. NK- and T-cell subsets in malignant mesothelioma patients: Baseline pattern and changes in the context of anti-CTLA-4 therapy. Int. J. Cancer 2019, 145, 2238–2248. [Google Scholar] [CrossRef]
- Tietze, J.K.; Angelova, D.; Heppt, M.V.; Ruzicka, T.; Berking, C. Low baseline levels of NK cells may predict a positive response to ipilimumab in melanoma therapy. Exp. Derm. 2017, 26, 622–629. [Google Scholar] [CrossRef] [PubMed]
- Jie, H.B.; Schuler, P.J.; Lee, S.C.; Srivastava, R.M.; Argiris, A.; Ferrone, S.; Whiteside, T.L.; Ferris, R.L. CTLA-4+ regulatory t cells increased in cetuximab-treated head and neck cancer patients suppress nk cell cytotoxicity and correlate with poor prognosis. Cancer Res. 2015, 75, 2200–2210. [Google Scholar] [CrossRef] [PubMed]
- Romano, E.; Kusio-Kobialka, M.; Foukas, P.G.; Baumgaertner, P.; Meyer, C.; Ballabeni, P.; Michielin, O.; Weide, B.; Romero, P.; Speiser, D.E. Ipilimumab-dependent cell-mediated cytotoxicity of regulatory T cells ex vivo by nonclassical monocytes in melanoma patients. Proc. Natl. Acad. Sci. USA 2015. [Google Scholar] [CrossRef]
- Simpson, T.R.; Li, F.; Montalvo-Ortiz, W.; Sepulveda, M.A.; Bergerhoff, K.; Arce, F.; Roddie, C.; Henry, J.Y.; Yagita, H.; Wolchok, J.D.; et al. Fc-dependent depletion of tumor-infiltrating regulatory t cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J. Exp. Med. 2013. [Google Scholar] [CrossRef]
- Vargas, F.A.; Furness, A.J.S.; Litchfield, K.; Joshi, K.; Rosenthal, R.; Ghorani, E.; Solomon, I.; Lesko, M.H.; Ruef, N.; Roddie, C.; et al. Fc Effector Function Contributes to the Activity of Human Anti-CTLA-4 Antibodies. Cancer Cell 2018, 33, 649–663.e4. [Google Scholar] [CrossRef]
- Laurent, S.; Queirolo, P.; Boero, S.; Salvi, S.; Piccioli, P.; Boccardo, S.; Minghelli, S.; Morabito, A.; Fontana, V.; Pietra, G.; et al. The engagement of CTLA-4 on primary melanoma cell lines induces antibody-dependent cellular cytotoxicity and TNF-α production. J. Transl. Med. 2013, 11, 1–13. [Google Scholar] [CrossRef]
- Da Silva, I.P.; Gallois, A.; Jimenez-Baranda, S.; Khan, S.; Anderson, A.C.; Kuchroo, V.K.; Osman, I.; Bhardwaj, N. Reversal of NK-cell exhaustion in advanced melanoma by Tim-3 blockade. Cancer Immunol. Res. 2014, 2, 410–422. [Google Scholar] [CrossRef]
- Komita, H.; Koido, S.; Hayashi, K.; Kan, S.; Ito, M.; Kamata, Y.; Suzuki, M.; Homma, S. Expression of immune checkpoint molecules of T cell immunoglobulin and mucin protein 3/galectin-9 for NK cell suppression in human gastrointestinal stromal tumors. Oncol. Rep. 2015, 34, 2099–2105. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Huang, Y.; Tan, L.; Yu, W.; Chen, D.; Lu, C.; He, J.; Wu, G.; Liu, X.; Zhang, Y. Increased Tim-3 expression in peripheral NK cells predicts a poorer prognosis and Tim-3 blockade improves NK cell-mediated cytotoxicity in human lung adenocarcinoma. Int. Immunopharmacol. 2015, 29, 635–641. [Google Scholar] [CrossRef]
- Datar, I.; Sanmamed, M.F.; Wang, J.; Henick, B.S.; Choi, J.; Badri, T.; Dong, W.; Mani, N.; Toki, M.; Mejías, L.D.; et al. Expression analysis and significance of PD-1, LAG-3, and TIM-3 in human non-small cell lung cancer using spatially resolved and multiparametric single-cell analysis. Clin. Cancer Res. 2019, 25, 4663–4673. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.; Xu, Y.; Wang, Z.; Wang, T.; Du, X.; Song, X.; Guo, X.; Peng, J.; Zhang, J.; Liang, Y.; et al. Tim-3 hampers tumor surveillance of liver-resident and conventional NK cells by disrupting PI3K signaling. Cancer Res. 2020, 80, 1130–1142. [Google Scholar] [CrossRef]
- Seo, H.; Kim, B.S.; Bae, E.A.; Min, B.S.; Han, Y.D.; Shin, S.J.; Kang, C.Y. IL21 therapy combined with PD-1 and Tim-3 blockade provides enhanced NK cell antitumor activity against MHC class I-deficient tumors. Cancer Immunol. Res. 2018, 6, 685–695. [Google Scholar] [CrossRef]
- Gleason, M.K.; Lenvik, T.R.; McCullar, V.; Felices, M.; O’Brien, M.S.; Cooley, S.A.; Verneris, M.R.; Cichocki, F.; Holman, C.J.; Panoskaltsis-Mortari, A.; et al. Tim-3 is an inducible human natural killer cell receptor that enhances interferon gamma production in response to galectin-9. Blood 2012, 119, 3064–3072. [Google Scholar] [CrossRef] [PubMed]
- So, E.C.; Khaladj-Ghom, A.; Ji, Y.; Amin, J.; Song, Y.; Burch, E.; Zhou, H.; Sun, H.; Chen, S.; Bentzen, S.; et al. NK cell expression of Tim-3: First impressions matter. Immunobiology 2019, 224, 362–370. [Google Scholar] [CrossRef] [PubMed]
- Sarhan, D.; Cichocki, F.; Zhang, B.; Yingst, A.; Spellman, S.R.; Cooley, S.; Verneris, M.R.; Blazar, B.R.; Miller, J.S. Adaptive NK cells with low TIGIT expression are inherently resistant to myeloid-derived suppressor cells. Cancer Res. 2016, 76, 5696–5706. [Google Scholar] [CrossRef] [PubMed]
- Blake, S.J.; Stannard, K.; Liu, J.; Allen, S.; Yong, M.C.R.; Mittal, D.; Aguilera, A.R.; Miles, J.J.; Lutzky, V.P.; de Andrade, L.F.; et al. Suppression of metastases using a new lymphocyte checkpoint target for cancer immunotherapy. Cancer Discov. 2016, 6, 446–459. [Google Scholar] [CrossRef]
- Wang, F.; Hou, H.; Wu, S.; Tang, Q.; Liu, W.; Huang, M.; Yin, B.; Huang, J.; Mao, L.; Lu, Y.; et al. TIGIT expression levels on human NK cells correlate with functional heterogeneity among healthy individuals. Eur. J. Immunol. 2015, 45, 2886–2897. [Google Scholar] [CrossRef]
- Peng, Y.P.; Xi, C.H.; Zhu, Y.; di Yin, L.; Wei, J.S.; Zhang, J.J.; Liu, X.C.; Guo, S.; Fu, Y.; Miao, Y. Altered expression of CD226 and CD96 on natural killer cells in patients with pancreatic cancer. Oncotarget 2016, 7, 66586–66594. [Google Scholar] [CrossRef]
- Zhang, Q.; Bi, J.; Zheng, X.; Chen, Y.; Wang, H.; Wu, W.; Wang, Z.; Wu, Q.; Peng, H.; Wei, H.; et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat. Immunol. 2018, 19, 723–732. [Google Scholar] [CrossRef]
- Stanietsky, N.; Simic, H.; Arapovic, J.; Toporik, A.; Levy, O.; Novik, A.; Levine, Z.; Beiman, M.; Dassa, L.; Achdout, H.; et al. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 17858–17863. [Google Scholar] [CrossRef]
- Sun, H.; Huang, Q.; Huang, M.; Wen, H.; Lin, R.; Zheng, M.; Qu, K.; Li, K.; Wei, H.; Xiao, W.; et al. Human CD96 Correlates to Natural Killer Cell Exhaustion and Predicts the Prognosis of Human Hepatocellular Carcinoma. Hepatology 2019, 70. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.J.; Martinet, L.; Gilfillan, S.; Souza-Fonseca-Guimaraes, F.; Chow, M.T.; Town, L.; Ritchie, D.S.; Colonna, M.; Andrews, D.M.; Smyth, M.J. The receptors CD96 and CD226 oppose each other in the regulation of natural killer cell functions. Nat. Immunol. 2014, 15, 431–438. [Google Scholar] [CrossRef]
- Brooks, J.; Fleischmann-Mundt, B.; Woller, N.; Niemann, J.; Ribback, S.; Peters, K.; Demir, I.E.; Armbrecht, N.; Ceyhan, G.O.; Manns, M.P.; et al. Perioperative, spatiotemporally coordinated activation of T and NK cells prevents recurrence of pancreatic cancer. Cancer Res. 2018, 78, 475–488. [Google Scholar] [CrossRef]
- Roman Aguilera, A.; Lutzky, V.P.; Mittal, D.; Li, X.Y.; Stannard, K.; Takeda, K.; Bernhardt, G.; Teng, M.W.L.; Dougall, W.C.; Smyth, M.J. CD96 targeted antibodies need not block CD96-CD155 interactions to promote NK cell anti-metastatic activity. OncoImmunology 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Baixeras, E.; Huard, B.; Miossec, C.; Jitsukawa, S.; Martin, M.; Hercend, T.; Auffray, C.; Triebel, F.; Piatier-Tonneau, D. Characterization of the Lymphocyte Activation Gene 3-Encoded Protein. A New Ligand for Human Leukocy Antigen Class H Antigens. J. Exp. Med. 1992, 176, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Huard, B.; Tournier, M.; Triebel, F. LAG-3 does not define a specific mode of natural killing in human. Immunol. Lett. 1998, 61, 109–112. [Google Scholar] [CrossRef]
- Zingoni, A.; Fionda, C.; Borrelli, C.; Cippitelli, M.; Santoni, A.; Soriani, A. Natural Killer Cell Response to Chemotherapy-Stressed Cancer Cells: Role in Tumor Immunosurveillance. Front. Immunol. 2017, 8, 1194. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Apetoh, L.; Ghiringhelli, F.; Kroemer, G. Immunological aspects of cancer chemotherapy. Nat. Rev. Immunol. 2008, 8, 59–73. [Google Scholar] [CrossRef] [PubMed]
- Carson, W.E., 3rd; Shapiro, C.L.; Crespin, T.R.; Thornton, L.M.; Andersen, B.L. Cellular immunity in breast cancer patients completing taxane treatment. Clin. Cancer Res. 2004, 10, 3401–3409. [Google Scholar] [CrossRef] [PubMed]
- Verma, C.; Kaewkangsadan, V.; Eremin, J.M.; Cowley, G.P.; Ilyas, M.; El-Sheemy, M.A.; Eremin, O. Natural killer (NK) cell profiles in blood and tumour in women with large and locally advanced breast cancer (LLABC) and their contribution to a pathological complete response (PCR) in the tumour following neoadjuvant chemotherapy (NAC): Differential rest. J. Transl. Med. 2015, 13, 180. [Google Scholar] [CrossRef]
- Krysko, D.V.; Garg, A.D.; Kaczmarek, A.; Krysko, O.; Agostinis, P.; Vandenabeele, P. Immunogenic cell death and DAMPs in cancer therapy. Nat. Rev. Cancer 2012, 12, 860–875. [Google Scholar] [CrossRef]
- Swift, L.H.; Golsteyn, R.M. Genotoxic Anti-Cancer Agents and Their Relationship to DNA Damage, Mitosis, and Checkpoint Adaptation in Proliferating Cancer Cells. Int. J. Mol. Sci. 2014, 15, 3403–3431. [Google Scholar] [CrossRef]
- Markasz, L.; Stuber, G.; Vanherberghen, B.; Flaberg, E.; Olah, E.; Carbone, E.; Eksborg, S.; Klein, E.; Skribek, H.; Szekely, L. Effect of frequently used chemotherapeutic drugs on the cytotoxic activity of human natural killer cells. Mol. Cancer 2007, 6, 644. [Google Scholar] [CrossRef]
- Multhoff, G.; Meier, T.; Botzler, C.; Wiesnet, M.; Allenbacher, A.; Wilmanns, W.; Issels, R.D. Differential effects of ifosfamide on the capacity of cytotoxic T lymphocytes and natural killer cells to lyse their target cells correlate with intracellular glutathione levels. Blood 1995, 85, 2124–2131. [Google Scholar] [CrossRef]
- Botzler, C.; Kis, K.; Issels, R.; Multhoff, G. A comparison of the effects of ifosfamide vs. mafosfamide treatment on intracellular glutathione levels and immunological functions of immunocompetent lymphocyte subsets. Exp. Hematol. 1997, 25, 338–344. [Google Scholar]
- Kuppner, M.C.; Bleifuß, E.; Noessner, E.; Mocikat, R.; von Hesler, C.; Mayerhofer, C.; Issels, R.D. Differential effects of ifosfamide on dendritic cell-mediated stimulation of T cell interleukin-2 production, natural killer cell cytotoxicity and interferon-γ production. Clin. Exp. Immunol. 2008, 153, 429–438. [Google Scholar] [CrossRef]
- Ghiringhelli, F.; Menard, C.; Puig, P.E.; Ladoire, S.; Roux, S.; Martin, F.; Solary, E.; le Cesne, A.; Zitvogel, L.; Chauffert, B. Metronomic cyclophosphamide regimen selectively depletes CD4+ CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer Immunol. Immunother. 2007, 56, 641–648. [Google Scholar] [CrossRef]
- Siew, Y.Y.; Neo, S.Y.; Yew, H.C.; Lim, S.W.; Ng, Y.C.; Lew, S.M.; Seetoh, W.G.; Seow, S.V.; Koh, H.L. Oxaliplatin regulates expression of stress ligands in ovarian cancer cells and modulates their susceptibility to natural killer cell-mediated cytotoxicity. Int. Immunol. 2015, 27, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Veneziani, I.; Brandetti, E.; Ognibene, M.; Pezzolo, A.; Pistoia, V.; Cifaldi, L. Neuroblastoma cell lines are refractory to genotoxic drug-mediated induction of ligands for NK cell-activating receptors. J. Immunol. Res. 2018, 2018. [Google Scholar] [CrossRef] [PubMed]
- Tesniere, A.; Schlemmer, F.; Boige, V.; Kepp, O.; Martins, I.; Ghiringhelli, F.; Aymeric, L.; Michaud, M.; Apetoh, L.; Barault, L.; et al. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene 2010, 29, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Okita, R.; Yukawa, T.; Nojima, Y.; Maeda, A.; Saisho, S.; Shimizu, K.; Nakata, M. MHC class I chain-related molecule A and B expression is upregulated by cisplatin and associated with good prognosis in patients with non-small cell lung cancer. Cancer Immunol. Immunother. 2016, 65, 499–509. [Google Scholar] [CrossRef]
- Okita, R.; Maeda, A.; Shimizu, K.; Nojima, Y.; Saisho, S.; Nakata, M. Effect of platinum-based chemotherapy on the expression of natural killer group 2 member D ligands, programmed cell death-1 ligand 1 and HLA class i in non-small cell lung cancer. Oncol. Rep. 2019, 42, 839–848. [Google Scholar] [CrossRef]
- Shi, L.; Lin, H.; Li, G.; Sun, Y.; Shen, J.; Xu, J.; Lin, C.; Yeh, S.; Cai, X.; Chang, C. Cisplatin enhances NK cells immunotherapy efficacy to suppress HCC progression via altering the androgen receptor (AR)-ULBP2 signals. Cancer Lett. 2016, 373, 45–56. [Google Scholar] [CrossRef]
- Cao, G.; Wang, J.; Zheng, X.; Wei, H.; Tian, Z.; Sun, R. Tumor Therapeutics Work as Stress Inducers to Enhance Tumor Sensitivity to Natural Killer (NK) Cell Cytolysis by Up-regulating NKp30 Ligand B7-H6. J. Biol. Chem. 2015, 290, 29964–29973. [Google Scholar] [CrossRef]
- Guerriero, J.L.; Ditsworth, D.; Catanzaro, J.M.; Sabino, G.; Furie, M.B.; Kew, R.R.; Crawford, H.C.; Zong, W.-X. DNA Alkylating Therapy Induces Tumor Regression through an HMGB1-Mediated Activation of Innate Immunity. J. Immunol. 2011, 186, 3517–3526. [Google Scholar] [CrossRef] [PubMed]
- Martins, I.; Tesniere, A.; Kepp, O.; Michaud, M.; Schlemmer, F.; Senovilla, L.; Séror, C.; Métivier, D.; Perfettini, J.-L.; Zitvogel, L.; et al. Chemotherapy induces ATP release from tumor cells. Cell Cycle 2009, 8, 3723–3728. [Google Scholar] [CrossRef] [PubMed]
- Čermák, V.; Dostál, V.; Jelínek, M.; Libusová, L.; Kovář, J.; Rösel, D.; Brábek, J. Microtubule-targeting agents and their impact on cancer treatment. Eur. J. Cell Biol. 2020, 99, 151075. [Google Scholar] [CrossRef]
- Sako, T.; Burioka, N.; Yasuda, K.; Tomita, K.; Miyata, M.; Kurai, J.; Chikumi, H.; Watanabe, M.; Suyama, H.; Fukuoka, Y.; et al. Cellular Immune Profile in Patients with Non-small Cell Lung Cancer after Weekly Paclitaxel Therapy. Acta Oncol. 2004, 43, 15–19. [Google Scholar] [CrossRef]
- Tong, A.W.; Seamour, B.; Lawson, J.M.; Ordonez, G.; Vukelja, S.; Hyman, W.; Richards, D.; Stein, L.; Maples, P.B.; Nemunaitis, J. Cellular immune profile of patients with advanced cancer before and after taxane treatment. Am. J. Clin. Oncol. Cancer Clin. Trials 2000, 23, 463–472. [Google Scholar] [CrossRef]
- Wu, X.; Feng, Q.M.; Wang, Y.; Shi, J.; Ge, H.L.; Di, W. The immunologic aspects in advanced ovarian cancer patients treated with paclitaxel and carboplatin chemotherapy. Cancer Immunol. Immunother. 2010, 59, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Acebes-Huerta, A.; Lorenzo-Herrero, S.; Folgueras, A.R.; Huergo-Zapico, L.; Lopez-Larrea, C.; López-Soto, A.; Gonzalez, S. Drug-induced hyperploidy stimulates an antitumor NK cell response mediated by NKG2D and DNAM-1 receptors. OncoImmunology 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- di Modica, M.; Sfondrini, L.; Regondi, V.; Varchetta, S.; Oliviero, B.; Mariani, G.; Bianchi, G.V.; Generali, D.; Balsari, A.; Triulzi, T.; et al. Taxanes enhance trastuzumab-mediated ADCC on tumor cells through NKG2D-mediated NK cell recognition. Oncotarget 2016, 7, 255–265. [Google Scholar] [CrossRef]
- Luengo, A.; Gui, D.Y.; van der Heiden, M.G. Targeting Metabolism for Cancer Therapy. Cell Chem. Biol. 2017, 24, 1161–1180. [Google Scholar] [CrossRef]
- Soriani, A.; Zingoni, A.; Cerboni, C.; Iannitto, M.L.; Ricciardi, M.R.; di Gialleonardo, V.; Cippitelli, M.; Fionda, C.; Petrucci, M.T.; Guarini, A.; et al. ATM-ATR–dependent up-regulation of DNAM-1 and NKG2D ligands on multiple myeloma cells by therapeutic agents results in enhanced NK-cell susceptibility and is associated with a senescent phenotype. Blood 2009, 113, 3503–3511. [Google Scholar] [CrossRef]
- Soriani, A.; Iannitto, M.L.; Ricci, B.; Fionda, C.; Malgarini, G.; Morrone, S.; Peruzzi, G.; Ricciardi, M.R.; Petrucci, M.T.; Cippitelli, M.; et al. Reactive oxygen species–and DNA damage response–dependent NK cell activating ligand upregulation occurs at transcriptional levels and requires the transcriptional factor E2F1. J. Immunol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Rayner, D.M.; Cutts, S.M. Anthracyclines. In Side Effects of Drugs Annual; Elsevier B.V.: Amsterdam, The Netherlands, 2014; Volume 36, pp. 683–694. [Google Scholar]
- Feng, H.; Dong, Y.; Wu, J.; Qiao, Y.; Zhu, G.; Jin, H.; Cui, J.; Li, W.; Liu, Y.J.; Chen, J.; et al. Epirubicin pretreatment enhances NK cell-mediated cytotoxicity against breast cancer cells in vitro. Am. J. Transl. Res. 2016, 8, 473–484. [Google Scholar]
- Wennerberg, E.; Sarhan, D.; Carlsten, M.; Kaminskyy, V.O.; D’Arcy, P.; Zhivotovsky, B.; Childs, R.; Lundqvist, A. Doxorubicin sensitizes human tumor cells to NK cell- and T-cell-mediated killing by augmented TRAIL receptor signaling. Int. J. Cancer 2013, 133, 1643–1652. [Google Scholar] [CrossRef] [PubMed]
- Hull, E.E.; Montgomery, M.R.; Leyva, K.J. HDAC Inhibitors as Epigenetic Regulators of the Immune System: Impacts on Cancer Therapy and Inflammatory Diseases. BioMed Res. Int. 2016, 2016, 1–15. [Google Scholar] [CrossRef]
- Ogbomo, H.; Michaelis, M.; Kreuter, J.; Doerr, H.W.; Cinatl, J. Histone deacetylase inhibitors suppress natural killer cell cytolytic activity. Febs Lett. 2007, 581, 1317–1322. [Google Scholar] [CrossRef]
- Ni, L.; Wang, L.; Yao, C.; Ni, Z.; Liu, F.; Gong, C.; Zhu, X.; Yan, X.; Watowich, S.S.; Lee, D.A.; et al. The histone deacetylase inhibitor valproic acid inhibits NKG2D expression in natural killer cells through suppression of STAT3 and HDAC3. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Kelly-Sell, M.J.; Kim, Y.H.; Straus, S.; Benoit, B.; Harrison, C.; Sutherland, K.; Armstrong, R.; Weng, W.-K.; Showe, L.C.; Wysocka, M.; et al. The histone deacetylase inhibitor, romidepsin, suppresses cellular immune functions of cutaneous T-cell lymphoma patients. Am. J. Hematol. 2012, 87, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Armeanu, S.; Bitzer, M.; Lauer, U.M.; Venturelli, S.; Pathil, A.; Krusch, M.; Kaiser, S.; Jobst, J.; Smirnow, I.; Wagner, A.; et al. Natural killer cell-mediated lysis of hepatoma cells via specific induction of NKG2D ligands by the histone deacetylase inhibitor sodium valproate. Cancer Res. 2005, 65, 6321–6329. [Google Scholar] [CrossRef]
- Shi, P.; Yin, T.; Zhou, F.; Cui, P.; Gou, S.; Wang, C. Valproic acid sensitizes pancreatic cancer cells to natural killer cell-mediated lysis by upregulating MICA and MICB via the PI3K/Akt signaling pathway. BMC Cancer 2014, 14. [Google Scholar] [CrossRef]
- Diermayr, S.; Himmelreich, H.; Durovic, B.; Mathys-Schneeberger, A.; Siegler, U.; Langenkamp, U.; Hofsteenge, J.; Gratwohl, A.; Tichelli, A.; Paluszewska, M.; et al. NKG2D ligand expression in AML increases in response to HDAC inhibitor valproic acid and contributes to allorecognition by NK-cell lines with single KIR-HLA class I specificities. Blood 2008, 111, 1428–1436. [Google Scholar] [CrossRef] [PubMed]
- Kato, N.; Tanaka, J.; Sugita, J.; Toubai, T.; Miura, Y.; Ibata, M.; Syono, Y.; Ota, S.; Kondo, T.; Asaka, M.; et al. Regulation of the expression of MHC class I-related chain A, B (MICA, MICB) via chromatin remodeling and its impact on the susceptibility of leukemic cells to the cytotoxicity of NKG2D-expressing cells. Leukemia 2007, 21, 2103–2108. [Google Scholar] [CrossRef]
- Fiegler, N.; Textor, S.; Arnold, A.; Rölle, A.; Oehme, I.; Breuhahn, K.; Moldenhauer, G.; Witzens-Harig, M.; Cerwenka, A. Downregulation of the activating NKp30 ligand B7-H6 by HDAC inhibitors impairs tumor cell recognition by NK cells. Blood 2013, 122, 684–693. [Google Scholar] [CrossRef]
- Nakata, S.; Yoshida, T.; Horinaka, M.; Shiraishi, T.; Wakada, M.; Sakai, T. Histone deacetylase inhibitors upregulate death receptor 5/TRAIL-R2 and sensitize apoptosis induced by TRAIL/APO2-L in human malignant tumor cells. Oncogene 2004, 23, 6261–6271. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-H. Sodium butyrate sensitizes TRAIL-mediated apoptosis by induction of transcription from the DR5 gene promoter through Sp1 sites in colon cancer cells. Carcinogenesis 2004, 25, 1813–1820. [Google Scholar] [CrossRef] [PubMed]
- Insinga, A.; Monestiroli, S.; Ronzoni, S.; Gelmetti, V.; Marchesi, F.; Viale, A.; Altucci, L.; Nervi, C.; Minucci, S.; Pelicci, P.G. Inhibitors of histone deacetylases induce tumor-selective apoptosis through activation of the death receptor pathway. Nat. Med. 2005, 11, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Manasanch, E.E.; Orlowski, R.Z. Proteasome Inhibitors in Cancer Therapy HHS Public Access. Nat. Rev. Clin. Oncol. 2017, 14, 417–433. [Google Scholar] [CrossRef]
- Armeanu, S.; Krusch, M.; Baltz, K.M.; Weiss, T.S.; Smirnow, I.; Steinle, A.; Lauer, U.M.; Bitzer, M.; Salih, H.R. Direct and natural killer cell-mediated antitumor effects of low-dose bortezomib in hepatocellular carcinoma. Clin. Cancer Res. 2008, 14, 3520–3528. [Google Scholar] [CrossRef]
- Niu, C.; Jin, H.; Li, M.; Zhu, S.; Zhou, L.; Jin, F.; Zhou, Y.; Xu, D.; Xu, J.; Zhao, L.; et al. Low-dose bortezomib increases the expression of NKG2D and DNAM-1 ligands and enhances induced NK and γδ T cellmediated lysis in multiple myeloma. Oncotarget 2017, 8, 5954–5964. [Google Scholar] [CrossRef]
- Shi, J.; Tricot, G.J.; Garg, T.K.; Malaviarachchi, P.A.; Szmania, S.M.; Kellum, R.E.; Storrie, B.; Mulder, A.; Shaughnessy, J.D.; Barlogie, B.; et al. Bortezomib down-regulates the cell-surface expression of HLA class I and enhances natural killer cell-mediated lysis of myeloma. Blood 2008, 111, 1309–1317. [Google Scholar] [CrossRef]
- Lundqvist, A.; Abrams, S.I.; Schrump, D.S.; Alvarez, G.; Suffredini, D.; Berg, M.; Childs, R. Bortezomib and depsipeptide sensitize tumors to tumor necrosis factor-related apoptosis-inducing ligand: A novel method to potentiate natural killer cell tumor cytotoxicity. Cancer Res. 2006, 66, 7317–7325. [Google Scholar] [CrossRef]
- Kabore, A.F.; Sun, J.; Hu, X.; McCrea, K.; Johnston, J.B.; Gibson, S.B. The TRAIL apoptotic pathway mediates proteasome inhibitor induced apoptosis in primary chronic lymphocytic leukemia cells. Apoptosis 2006, 11, 1175–1193. [Google Scholar] [CrossRef]
- Liu, X.; Yue, P.; Chen, S.; Hu, L.; Lonial, S.; Khuri, F.R.; Sun, S.Y. The proteasome inhibitor PS-341 (bortezomib) up-regulates DR5 expression leading to induction of apoptosis and enhancement of TRAIL-induced apoptosis despite up-regulation of c-FLIP and survivin expression in human NSCLC cells. Cancer Res. 2007, 67, 4981–4988. [Google Scholar] [CrossRef] [PubMed]
- Massa, C.; Karn, T.; Denkert, C.; Schneeweiss, A.; Hanusch, C.; Blohmer, J.U.; Zahm, D.M.; Jackisch, C.; van Mackelenbergh, M.; Thomalla, J.; et al. Differential effect on different immune subsets of neoadjuvant chemotherapy in patients with TNBC. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Aldarouish, M.; Su, X.; Qiao, J.; Gao, C.; Chen, Y.; Dai, A.; Zhang, T.; Shu, Y.; Wang, C. Immunomodulatory effects of chemotherapy on blood lymphocytes and survival of patients with advanced non-small cell lung cancer. Int. J. Immunopathol. Pharmacol. 2019, 33, 2058738419839592. [Google Scholar] [CrossRef] [PubMed]
- Shinko, D.; McGuire, H.M.; Diakos, C.I.; Pavlakis, N.; Clarke, S.J.; Byrne, S.N.; Charles, K.A. Mass Cytometry Reveals a Sustained Reduction in CD16+ Natural Killer Cells Following Chemotherapy in Colorectal Cancer Patients. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- Beitsch, P.; Lotzová, E.; Hortobagyi, G.; Pollock, R. Natural immunity in breast cancer patients during neoadjuvant chemotherapy and after surgery. Surg. Oncol. 1994, 3, 211–219. [Google Scholar] [CrossRef]
- Sewell, H.F.; Halbert, C.F.; Robins, R.A.; Galvin, A.; Chan, S.; Blamey, R.W. Chemotherapy-induced differential changes in lymphocyte subsets and natural-killer-cell function in patients with advanced breast cancer. Int. J. Cancer 1993, 55, 735–738. [Google Scholar] [CrossRef]
- Brenner, B.G.; Margolese, R.G. The relationship of chemotherapeutic and endocrine intervention on natural killer cell activity in human breast cancer. Cancer 1991, 68, 482–488. [Google Scholar] [CrossRef]
- Ogura, M.; Ishida, T.; Tsukasaki, K.; Takahashi, T.; Utsunomiya, A. Effects of first-line chemotherapy on natural killer cells in adult T-cell leukemia–lymphoma and peripheral T-cell lymphoma. Cancer Chemother. Pharm. 2016, 78, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR Signaling in Growth and Metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Bhullar, K.S.; Lagarón, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-targeted cancer therapies: Progress, challenges and future directions. Mol. Cancer 2018, 17, 48. [Google Scholar] [CrossRef]
- Adams, J.A. Kinetic and catalytic mechanisms of protein kinases. Chem. Rev. 2001, 101, 2271–2290. [Google Scholar] [CrossRef]
- Johnson, L.N.; Lewis, R.J. Structural basis for control by phosphorylation. Chem. Rev. 2001, 101, 2209–2242. [Google Scholar] [CrossRef] [PubMed]
- DiDonato, J.A.; Mercurio, F.; Karin, M. NF-κB and the link between inflammation and cancer. Immunol. Rev. 2012, 246, 379–400. [Google Scholar] [CrossRef]
- Sun, C.; Bernards, R. Feedback and redundancy in receptor tyrosine kinase signaling: Relevance to cancer therapies. Trends Biochem. Sci. 2014, 39, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Shen, A.; Ding, J.; Geng, M. Molecularly targeted cancer therapy: Some lessons from the past decade. Trends Pharm. Sci. 2014, 35, 41–50. [Google Scholar] [CrossRef]
- Kreutzman, A.; Porkka, K.; Mustjoki, S. Immunomodulatory effects of tyrosine kinase inhibitors. Int. Trends Immun. 2013, 1, 17–28. [Google Scholar]
- Blake, S.J.; Lyons, A.B.; Fraser, C.K.; Hayball, J.D.; Hughes, T.P. Dasatinib suppresses in vitro natural killer cell cytotoxicity. Blood 2008, 111, 4415–4416. [Google Scholar] [CrossRef]
- Mustjoki, S.; Auvinen, K.; Kreutzman, A.; Rousselot, P.; Hernesniemi, S.; Melo, T.; Lahesmaa-Korpinen, A.-M.; Hautaniemi, S.; Bouchet, S.; Molimard, M.; et al. Rapid mobilization of cytotoxic lymphocytes induced by dasatinib therapy. Leukemia 2013, 27, 914–924. [Google Scholar] [CrossRef]
- Hayashi, Y.; Nakamae, H.; Katayama, T.; Nakane, T.; Koh, H.; Nakamae, M.; Hirose, A.; Hagihara, K.; Terada, Y.; Nakao, Y.; et al. Different immunoprofiles in patients with chronic myeloid leukemia treated with imatinib, nilotinib or dasatinib. Leuk. Lymphoma 2012, 53, 1084–1089. [Google Scholar] [CrossRef]
- Kreutzman, A.; Yadav, B.; Brummendorf, T.H.; Gjertsen, B.T.; Lee, M.H.; Janssen, J.; Kasanen, T.; Koskenvesa, P.; Lotfi, K.; Markevärn, B.; et al. Immunological monitoring of newly diagnosed CML patients treated with bosutinib or imatinib first-line. Oncoimmunology 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Ilander, M.; Olsson-Strömberg, U.; Schlums, H.; Guilhot, J.; Brück, O.; Lähteenmäki, H.; Kasanen, T.; Koskenvesa, P.; Söderlund, S.; Höglund, M.; et al. Increased proportion of mature NK cells is associated with successful imatinib discontinuation in chronic myeloid leukemia. Leukemia 2017, 31, 1108–1116. [Google Scholar] [CrossRef]
- Rea, D.; Henry, G.; Khaznadar, Z.; Etienne, G.; Guilhot, F.; Nicolini, F.; Guilhot, J.; Rousselot, P.; Huguet, F.; Legros, L.; et al. Natural killer-cell counts are associated with molecular relapse-free survival after imatinib discontinuation in chronic myeloid leukemia: The IMMUNOSTIM study. Haematologica 2017, 102, 1368–1377. [Google Scholar] [CrossRef] [PubMed]
- Roberts, P.J.; Der, C.J. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 2007, 26, 3291–3310. [Google Scholar] [CrossRef]
- Subbiah, V.; Baik, C.; Kirkwood, J.M. Clinical Development of BRAF plus MEK Inhibitor Combinations. Trends Cancer 2020, 6, 797–810. [Google Scholar] [CrossRef]
- Kannaiyan, R.; Mahadevan, D. A comprehensive review of protein kinase inhibitors for cancer therapy HHS Public Access. Expert Rev. Anticancer 2018, 18, 1249–1270. [Google Scholar] [CrossRef] [PubMed]
- Shirley, M. Encorafenib and Binimetinib: First Global Approvals. Drugs 2018, 78, 1277–1284. [Google Scholar] [CrossRef] [PubMed]
- De Andrade, L.F.; Ngiow, S.F.; Stannard, K.; Rusakiewicz, S.; Kalimutho, M.; Khanna, K.K.; Tey, S.K.; Takeda, K.; Zitvogel, L.; Martinet, L.; et al. Natural killer cells are essential for the ability of BRAF inhibitors to control BRAFV600E-mutant metastatic melanoma. Cancer Res. 2014, 74, 7298–7308. [Google Scholar] [CrossRef]
- Frazao, A.; Colombo, M.; Fourmentraux-Neves, E.; Messaoudene, M.; Rusakiewicz, S.; Zitvogel, L.; Vivier, E.; Vély, F.; Faure, F.; Dréno, B.; et al. Shifting the Balance of Activating and Inhibitory Natural Killer Receptor Ligands on BRAF(V600E) Melanoma Lines with Vemurafenib. Cancer Immunol. Res. 2017, 5, 582–593. [Google Scholar] [CrossRef] [PubMed]
- López-Cobo, S.; Pieper, N.; Campos-Silva, C.; García-Cuesta, E.M.; Reyburn, H.T.; Paschen, A.; Valés-Gómez, M. Impaired NK cell recognition of vemurafenib-treated melanoma cells is overcome by simultaneous application of histone deacetylase inhibitors. OncoImmunology 2018, 7. [Google Scholar] [CrossRef]
- Frazao, A.; Rethacker, L.; Jeudy, G.; Colombo, M.; Pasmant, E.; Avril, M.-F.; Toubert, A.; Moins-Teisserenc, H.; Roelens, M.; Dalac, S.; et al. BRAF inhibitor resistance of melanoma cells triggers increased susceptibility to natural killer cell-mediated lysis. J. Immunother. Cancer 2020, 8, e000275. [Google Scholar] [CrossRef]
- Sottile, R.; Pangigadde, P.N.; Tan, T.; Anichini, A.; Sabbatino, F.; Trecroci, F.; Favoino, E.; Orgiano, L.; Roberts, J.; Ferrone, S.; et al. HLA class I downregulation is associated with enhanced NK-cell killing of melanoma cells with acquired drug resistance to BRAF inhibitors. Eur. J. Immunol. 2016, 46, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Augello, G.; Emma, M.R.; Cusimano, A.; Azzolina, A.; Montalto, G.; McCubrey, J.A.; Cervello, M. The Role of GSK-3 in Cancer Immunotherapy: GSK-3 Inhibitors as a New Frontier in Cancer Treatment. Cells 2020, 9, 1427. [Google Scholar] [CrossRef]
- Parameswaran, R.; Ramakrishnan, P.; Moreton, S.A.; Xia, Z.; Hou, Y.; Lee, D.A.; Gupta, K.; Delima, M.; Beck, R.C.; Wald, D.N. Repression of GSK3 restores NK cell cytotoxicity in AML patients. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Cichocki, F.; Valamehr, B.; Bjordahl, R.; Zhang, B.; Rezner, B.; Rogers, P.; Gaidarova, S.; Moreno, S.; Tuininga, K.; Dougherty, P.; et al. GSK3 inhibition drives maturation of NK cells and enhances their antitumor activity. Cancer Res. 2017, 77, 5664–5675. [Google Scholar] [CrossRef]
- Cosman, D.; Müllberg, J.; Sutherland, C.L.; Chin, W.; Armitage, R.; Fanslow, W.; Kubin, M.; Chalupny, N.J. ULBPs, novel MHC class I-related molecules, bind to CMV glycoprotein UL16 and stimulate NK cytotoxicity through the NKG2D receptor. Immunity 2001, 14, 123–133. [Google Scholar] [CrossRef]
- Fionda, C.; Malgarini, G.; Soriani, A.; Zingoni, A.; Cecere, F.; Iannitto, M.L.; Ricciardi, M.R.; Federico, V.; Petrucci, M.T.; Santoni, A.; et al. Inhibition of glycogen synthase kinase-3 increases NKG2D ligand MICA expression and sensitivity to NK cell-mediated cytotoxicity in multiple myeloma cells: Role of STAT3. J. Immunol. 2013, 190, 6662–6672. [Google Scholar] [CrossRef]
- Krusch, M.; Salih, J.; Schlicke, M.; Baessler, T.; Kampa, K.M.; Mayer, F.; Salih, H.R. The Kinase Inhibitors Sunitinib and Sorafenib Differentially Affect NK Cell Antitumor Reactivity In Vitro. J. Immunol. 2009, 183, 8286–8294. [Google Scholar] [CrossRef]
- Lohmeyer, J.; Nerreter, T.; Dotterweich, J.; Einsele, H.; Seggewiss-Bernhardt, R. Sorafenib paradoxically activates the RAS/RAF/ERK pathway in polyclonal human NK cells during expansion and thereby enhances effector functions in a dose- and time-dependent manner. Clin. Exp. Immunol. 2018, 193, 64–72. [Google Scholar] [CrossRef]
- Huang, Y.X.; Chen, X.T.; Guo, K.Y.; Li, Y.H.; Wu, B.Y.; Song, C.Y.; He, Y.J. Sunitinib induces NK-κB-dependent NKG2D ligand expression in nasopharyngeal carcinoma and hepatoma cells. J. Immunother. 2017, 40, 164–174. [Google Scholar] [CrossRef]
- Huang, Y.; Wang, Y.; Li, Y.; Guo, K.; He, Y. Role of sorafenib and sunitinib in the induction of expressions of NKG2D ligands in nasopharyngeal carcinoma with high expression of ABCG2. J. Cancer Res. Clin. Oncol. 2011, 137, 829–837. [Google Scholar] [CrossRef]
- Harrison, D.A. The JAK/STAT pathway. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef]
- Schönberg, K.; Rudolph, J.; Wolf, D. NK cell modulation by JAK inhibition. Oncoscience 2015, 2, 677. [Google Scholar] [CrossRef]
- Xu, L.J.; Chen, X.D.; Shen, M.J.; Yang, D.R.; Fang, L.; Weng, G.; Tsai, Y.; Keng, P.C.; Chen, Y.; Lee, S.O. Inhibition of IL-6-JAK/Stat3 signaling in castration-resistant prostate cancer cells enhances the NK cell-mediated cytotoxicity via alteration of PD-L1/NKG2D ligand levels. Mol. Oncol. 2018, 12, 269–286. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Wei, Y.; Wei, X. AXL receptor tyrosine kinase as a promising anti-cancer approach: Functions, molecular mechanisms and clinical applications. Mol. Cancer 2019, 18. [Google Scholar] [CrossRef] [PubMed]
- Lutz-Nicoladoni, C.; Wolf, D.; Sopper, S.; Sharabi, A.; Palmer, D. Modulation of immune cell functions by the E3 ligase Cbl-b. Front. Oncol. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Paolino, M.; Choidas, A.; Wallner, S.; Pranjic, B.; Uribesalgo, I.; Loeser, S.; Jamieson, A.M.; Langdon, W.Y.; Ikeda, F.; Fededa, J.P.; et al. The E3 ligase Cbl-b and TAM receptors regulate cancer metastasis via natural killer cells. Nature 2014, 507, 508–512. [Google Scholar] [CrossRef]
- Kaufman, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic viruses: A new class of immunotherapy drugs. Nat. Rev. Drug Discov. 2015, 14, 642–662. [Google Scholar] [CrossRef] [PubMed]
- Fukuhara, H.; Ino, Y.; Todo, T. Oncolytic virus therapy: A new era of cancer treatment at dawn. Cancer Sci. 2016, 107, 1373–1379. [Google Scholar] [CrossRef] [PubMed]
- Hamid, O.; Ismail, R.; Puzanov, I. Intratumoral Immunotherapy—Update 2019. Oncologist 2020, 25. [Google Scholar] [CrossRef]
- Samudio, I.; Hofs, E.; Cho, B.; Li, M.; Bolduc, K.; Bu, L.; Liu, G.; Lam, V.; Rennie, P.; Jia, W.; et al. UV Light-inactivated HSV-1 Stimulates Natural Killer Cell-induced Killing of Prostate Cancer Cells. J. Immunother. 2019, 42, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Bhat, R.; Rommelaere, J. NK-cell-dependent killing of colon carcinoma cells is mediated by natural cytotoxicity receptors (NCRs) and stimulated by parvovirus infection of target cells. BMC Cancer 2013, 13. [Google Scholar] [CrossRef] [PubMed]
- Ogbomo, H.; Michaelis, M.; Geiler, J.; van Rikxoort, M.; Muster, T.; Egorov, A.; Doerr, H.W.; Cinatl, J. Tumor cells infected with oncolytic influenza a virus prime natural killer cells for lysis of resistant tumor cells. Med. Microbiol. Immunol. 2010, 199, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Jarahian, M.; Watzl, C.; Fournier, P.; Arnold, A.; Djandji, D.; Zahedi, S.; Cerwenka, A.; Paschen, A.; Schirrmacher, V.; Momburg, F. Activation of Natural Killer Cells by Newcastle Disease Virus Hemagglutinin-Neuraminidase. J. Virol. 2009, 83, 8108–8121. [Google Scholar] [CrossRef] [PubMed]
- Ogbomo, H.; Zemp, F.J.; Lun, X.; Zhang, J.; Stack, D.; Rahman, M.M.; Mcfadden, G.; Mody, C.H.; Forsyth, P.A. Myxoma Virus Infection Promotes NK Lysis of Malignant Gliomas In Vitro and In Vivo. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Bhat, R.; Rommelaere, J. Emerging role of Natural killer cells in oncolytic virotherapy. Immuno Targets Ther. 2015, 4, 65–77. [Google Scholar] [CrossRef]
- Diaz, R.M.; Galivo, F.; Kottke, T.; Wongthida, P.; Qiao, J.; Thompson, J.; Valdes, M.; Barber, G.; Vile, R.G. Oncolytic immunovirotherapy for melanoma using vesicular stomatitis virus. Cancer Res. 2007, 67, 2840–2848. [Google Scholar] [CrossRef]
- Miller, C.G.; Fraser, N.W. Requirement of an integrated immune response for successful neuroattenuated HSV-1 therapy in an intracranial metastatic melanoma model. Mol. Ther. 2003, 7, 741–747. [Google Scholar] [CrossRef]
- Tai, L.H.; de Souza, C.T.; Bélanger, S.; Ly, L.; Alkayyal, A.A.; Zhang, J.; Rintoul, J.L.; Ananth, A.A.; Lam, T.; Breitbach, C.J.; et al. Preventing postoperative metastatic disease by inhibiting surgery-induced dysfunction in natural killer cells. Cancer Res. 2013, 73, 97–107. [Google Scholar] [CrossRef]
- Altomonte, J.; Wu, L.; Chen, L.; Meseck, M.; Ebert, O.; García-Sastre, A.; Fallon, J.; Woo, S.L. Exponential enhancement of oncolytic vesicular stomatitis virus potency by vector-mediated suppression of inflammatory responses in vivo. Mol. Ther. 2008, 16, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Ma, R.; Russell, L.; Yoo, J.Y.; Han, J.; Cui, H.; Yi, P.; Zhang, J.; Nakashima, H.; Dai, H.; et al. An oncolytic herpesvirus expressing E-cadherin improves survival in mouse models of glioblastoma. Nat. Biotechnol. 2019, 37, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Breckenridge, C.A.; Yu, J.; Price, R.; Wojton, J.; Pradarelli, J.; Mao, H.; Wei, M.; Wang, Y.; He, S.; Hardcastle, J.; et al. NK cells impede glioblastoma virotherapy through NKp30 and NKp46 natural cytotoxicity receptors. Nat. Med. 2012, 18, 1827–1834. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Breckenridge, C.A.; Yu, J.; Price, R.; Wei, M.; Wang, Y.; Nowicki, M.O.; Ha, Y.P.; Bergin, S.; Hwang, C.; Fernandez, S.A.; et al. The Histone Deacetylase Inhibitor Valproic Acid Lessens NK Cell Action against Oncolytic Virus-Infected Glioblastoma Cells by Inhibition of STAT5/T-BET Signaling and Generation of Gamma Interferon. J. Virol. 2012, 86, 4566–4577. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Yoo, J.Y.; Lee, T.J.; Liu, J.; Yu, J.; Caligiuri, M.A.; Kaur, B.; Friedman, A. Complex role of NK cells in regulation of oncolytic virus–bortezomib therapy. Proc. Natl. Acad. Sci. USA 2018, 115, 4927–4932. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toffoli, E.C.; Sheikhi, A.; Höppner, Y.D.; de Kok, P.; Yazdanpanah-Samani, M.; Spanholtz, J.; Verheul, H.M.W.; van der Vliet, H.J.; de Gruijl, T.D. Natural Killer Cells and Anti-Cancer Therapies: Reciprocal Effects on Immune Function and Therapeutic Response. Cancers 2021, 13, 711. https://doi.org/10.3390/cancers13040711
Toffoli EC, Sheikhi A, Höppner YD, de Kok P, Yazdanpanah-Samani M, Spanholtz J, Verheul HMW, van der Vliet HJ, de Gruijl TD. Natural Killer Cells and Anti-Cancer Therapies: Reciprocal Effects on Immune Function and Therapeutic Response. Cancers. 2021; 13(4):711. https://doi.org/10.3390/cancers13040711
Chicago/Turabian StyleToffoli, Elisa C., Abdolkarim Sheikhi, Yannick D. Höppner, Pita de Kok, Mahsa Yazdanpanah-Samani, Jan Spanholtz, Henk M. W. Verheul, Hans J. van der Vliet, and Tanja D. de Gruijl. 2021. "Natural Killer Cells and Anti-Cancer Therapies: Reciprocal Effects on Immune Function and Therapeutic Response" Cancers 13, no. 4: 711. https://doi.org/10.3390/cancers13040711
APA StyleToffoli, E. C., Sheikhi, A., Höppner, Y. D., de Kok, P., Yazdanpanah-Samani, M., Spanholtz, J., Verheul, H. M. W., van der Vliet, H. J., & de Gruijl, T. D. (2021). Natural Killer Cells and Anti-Cancer Therapies: Reciprocal Effects on Immune Function and Therapeutic Response. Cancers, 13(4), 711. https://doi.org/10.3390/cancers13040711