
Targeting and Reprograming Cancer-Associated Fibroblasts and the Tumor Microenvironment in Pancreatic Cancer




Abstract
Simple Summary
Abstract
1. Introduction
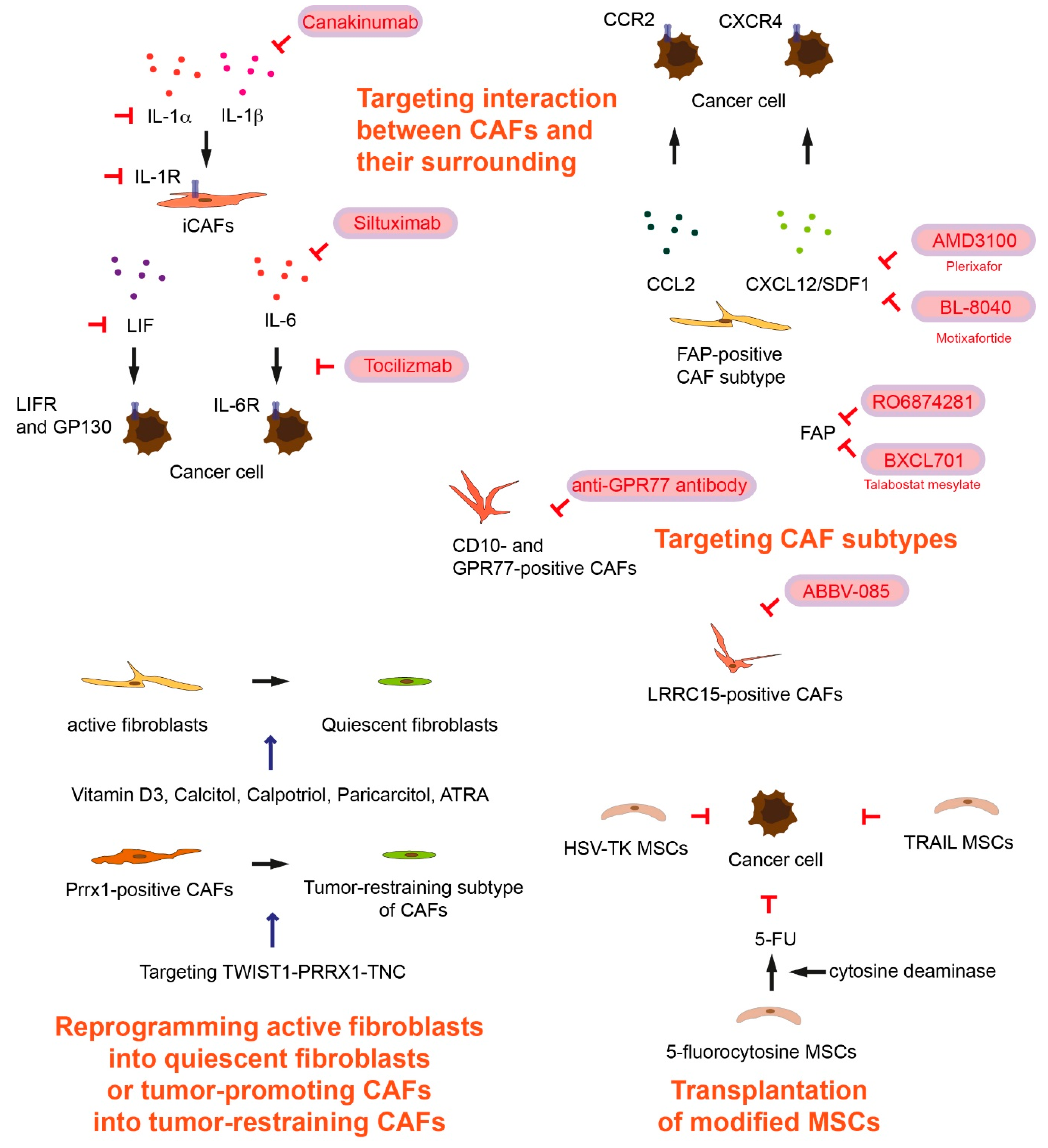
2. Targeting Cancer-Associated Fibroblast Subtypes
3. Targeting Interactions between Cancer-Associated Fibroblasts and Their Surrounding in Tumor Microenvironment
4. Transplantation of Modified Mesenchymal Stem Cells
5. Reprogramming Active Fibroblasts into Quiescent Fibroblasts or Tumor-Promoting Cancer-Associated Fibroblasts into Cancer-Restraining Cancer-Associated Fibroblasts
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef]
- Cunningham, D.; Chau, I.; Stocken, D.D.; Valle, J.W.; Smith, D.; Steward, W.; Harper, P.G.; Dunn, J.; Tudur-Smith, C.; West, J.; et al. Phase III Randomized Comparison of Gemcitabine Versus Gemcitabine Plus Capecitabine in Patients with Advanced Pancreatic Cancer. J. Clin. Oncol. 2009, 27, 5513–5518. [Google Scholar] [CrossRef]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.-L.; Gourgou-Bourgade, S.; De La Fouchardière, C.; et al. FOLFIRINOX versus Gemcitabine for Metastatic Pancreatic Cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased Survival in Pancreatic Cancer with nab-Paclitaxel plus Gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [PubMed]
- Neoptolemos, J.P.; Palmer, D.H.; Ghaneh, P.; Psarelli, E.E.; Valle, J.W.; Halloran, C.M.; Faluyi, O.; O’Reilly, D.A.; Cunningham, D.; Wadsley, J.; et al. Comparison of adjuvant gemcitabine and capecitabine with gemcitabine monotherapy in patients with resected pancreatic cancer (ESPAC-4): A multicentre, open-label, randomised, phase 3 trial. Lancet 2017, 389, 1011–1024. [Google Scholar] [CrossRef]
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.-L.; Choné, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med. 2018, 379, 2395–2406. [Google Scholar] [CrossRef] [PubMed]
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Prim. 2016, 2, 16022. [Google Scholar] [CrossRef] [PubMed]
- Sunami, Y.; Rebelo, A.; Kleeff, J. Lipid Metabolism and Lipid Droplets in Pancreatic Cancer and Stellate Cells. Cancers 2017, 10, 3. [Google Scholar] [CrossRef]
- Sunami, Y.; Häußler, J.; Kleeff, J. Cellular Heterogeneity of Pancreatic Stellate Cells, Mesenchymal Stem Cells, and Can-cer-Associated Fibroblasts in Pancreatic Cancer. Cancers 2020, 12, 3770. [Google Scholar] [CrossRef] [PubMed]
- Özdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.-C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of Carcinoma-Associated Fibroblasts and Fibrosis Induces Immunosuppression and Accelerates Pancreas Cancer with Reduced Survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef]
- Fujita, H.; Ohuchida, K.; Mizumoto, K.; Nakata, K.; Yu, J.; Kayashima, T.; Cui, L.; Manabe, T.; Ohtsuka, T.; Tanaka, M. α-Smooth Muscle Actin Expressing Stroma Promotes an Aggressive Tumor Biology in Pancreatic Ductal Adenocarcinoma. Pancreas 2010, 39, 1254–1262. [Google Scholar] [CrossRef] [PubMed]
- Underwood, T.J.; Hayden, A.L.; Derouet, M.; Garcia, E.; Noble, F.; White, M.J.; Thirdborough, S.; Mead, A.; Clemons, N.; Mellone, M.; et al. Cancer-associated fibroblasts predict poor outcome and promote periostin-dependent invasion in oesophageal adenocarcinoma. J. Pathol. 2015, 235, 466–477. [Google Scholar] [CrossRef] [PubMed]
- Hingorani, S.R.; Wang, L.; Multani, A.S.; Combs, C.; Deramaudt, T.B.; Hruban, R.H.; Rustgi, A.K.; Chang, S.; Tuveson, D.A. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005, 7, 469–483. [Google Scholar] [CrossRef]
- Sulzmaier, F.J.; Jean, C.; Schlaepfer, D.D. FAK in cancer: Mechanistic findings and clinical applications. Nat. Rev. Cancer 2014, 14, 598–610. [Google Scholar] [CrossRef] [PubMed]
- Demircioglu, F.; Wang, J.; Candido, J.; Costa, A.S.H.; Casado, P.; Delgado, B.D.L.; Reynolds, L.E.; Gomez-Escudero, J.; Newport, E.; Rajeeve, V.; et al. Cancer associated fibroblast FAK regulates malignant cell metabolism. Nat. Commun. 2020, 11, 1290. [Google Scholar] [CrossRef] [PubMed]
- Stoker, M.G.; Shearer, M.; O’Neill, C. Growth inhibition of polyoma-transformed cells by contact with static normal fibroblasts. J. Cell Sci. 1966, 1, 297–310. [Google Scholar]
- Klein, G. Evolutionary aspects of cancer resistance. Semin. Cancer Biol. 2014, 25, 10–14. [Google Scholar] [CrossRef]
- Miyai, Y.; Esaki, N.; Takahashi, M.; Enomoto, A. Cancer-associated fibroblasts that restrain cancer progression: Hypotheses and perspectives. Cancer Sci. 2020, 111, 1047–1057. [Google Scholar] [CrossRef]
- Fitzgerald, A.A.; Weiner, L.M. The role of fibroblast activation protein in health and malignancy. Cancer Metastasis Rev. 2020, 39, 783–803. [Google Scholar] [CrossRef]
- Cohen, S.J.; Alpaugh, R.K.; Palazzo, I.; Meropol, N.J.; Rogatko, A.; Xu, Z.; Hoffman, J.P.; Weiner, L.M.; Cheng, J.D. Fibroblast Activation protein and its relationship to clinical outcome in pancreatic adenocarcinoma. Pancreas 2008, 37, 154–158. [Google Scholar] [CrossRef] [PubMed]
- Lo, A.; Li, C.-P.; Buza, E.L.; Blomberg, R.; Govindaraju, P.; Avery, D.; Monslow, J.; Hsiao, M.; Puré, E. Fibroblast activation protein augments progression and metastasis of pancreatic ductal adenocarcinoma. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.B.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef] [PubMed]
- Ostermann, E.; Garin-Chesa, P.; Heider, K.H.; Kalat, M.; Lamche, H.; Puri, C.; Kerjaschki, D.; Rettig, W.J.; Adolf, G.R. Effective immunoconjugate therapy in cancer models targeting a serine protease of tumor fibroblasts. Clin. Cancer Res. 2008, 14, 4584–4592. [Google Scholar] [CrossRef]
- Kakarla, S.; Chow, K.K.H.; Mata, M.; Shaffer, D.R.; Song, X.-T.; Wu, M.-F.; Liu, H.; Wang, L.L.; Rowley, D.R.; Pfizenmaier, K.; et al. Antitumor effects of chimeric receptor engineered human T cells directed to tumor stroma. Mol. Ther. 2013, 21, 1611–1620. [Google Scholar] [CrossRef]
- Wang, L.-C.S.; Lo, A.; Scholler, J.; Sun, J.; Majumdar, R.S.; Kapoor, V.; Antzis, M.; Cotner, C.E.; Johnson, L.A.; Durham, A.C.; et al. Targeting Fibroblast Activation Protein in Tumor Stroma with Chimeric Antigen Receptor T Cells Can Inhibit Tumor Growth and Augment Host Immunity without Severe Toxicity. Cancer Immunol. Res. 2014, 2, 154–166. [Google Scholar] [CrossRef]
- Lo, A.; Wang, L.-C.S.; Scholler, J.; Monslow, J.; Avery, D.; Newick, K.; O’Brien, S.; Evans, R.A.; Bajor, D.J.; Clendenin, C.; et al. Tumor-Promoting Desmoplasia Is Disrupted by Depleting FAP-Expressing Stromal Cells. Cancer Res. 2015, 75, 2800–2810. [Google Scholar] [CrossRef]
- Tran, E.; Chinnasamy, D.; Yu, Z.; Morgan, R.A.; Lee, C.-C.R.; Restifo, N.P.; Rosenberg, S.A. Immune targeting of fibroblast activation protein triggers recognition of multipotent bone marrow stromal cells and cachexia. J. Exp. Med. 2013, 210, 1125–1135. [Google Scholar] [CrossRef]
- Kraman, M.; Bambrough, P.J.; Arnold, J.N.; Roberts, E.W.; Magiera, L.; Jones, J.O.; Gopinathan, A.; Tuveson, D.A.; Fearon, D.T. Suppression of Antitumor Immunity by Stromal Cells Expressing Fibroblast Activation Protein-alpha. Science 2010, 330, 827–830. [Google Scholar] [CrossRef]
- Roberts, E.W.; Deonarine, A.; Jones, J.O.; Denton, A.E.; Feig, C.; Lyons, S.K.; Espeli, M.; Kraman, M.; McKenna, B.; Wells, R.J.; et al. Depletion of stromal cells expressing fibroblast activation protein-α from skeletal muscle and bone marrow results in cachexia and anemia. J. Exp. Med. 2013, 210, 1137–1151. [Google Scholar] [CrossRef]
- Hofheinz, R.-D.; Al-Batran, S.-E.; Hartmann, F.; Hartung, G.; Jäger, D.; Renner, C.; Tanswell, P.; Kunz, U.; Amelsberg, A.; Kuthan, H.; et al. Stromal Antigen Targeting by a Humanised Monoclonal Antibody: An Early Phase II Trial of Sibrotuzumab in Patients with Metastatic Colorectal Cancer. Oncol. Res. Treat. 2003, 26, 44–48. [Google Scholar] [CrossRef]
- Ho, W.J.; Jaffee, E.M.; Zheng, L. The tumour microenvironment in pancreatic cancer—clinical challenges and opportunities. Nat. Rev. Clin. Oncol. 2020, 17, 527–540. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, C.C. Talabostat. Expert Opin. Investig. Drugs 2007, 16, 1459–1465. [Google Scholar] [CrossRef]
- Melero, I.; Alvarez, E.C.; Mau-Sorensen, M.; Lassen, U.; Lolkema, M.; Robbrecht, D.; Gomez-Roca, C.; Martin-Liberal, J.; Tabernero, J.; Ros, W.; et al. 412PD—Clinical activity, safety, and PK/PD from a phase I study of RO6874281, a fibroblast activation protein (FAP) targeted interleukin-2 variant (IL-2v). Ann. Oncol. 2018, 29, viii134–viii135. [Google Scholar] [CrossRef]
- Sunshine, J.C.; Taube, J.M. PD-1/PD-L1 inhibitors. Curr. Opin. Pharmacol. 2015, 23, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Gregory, R.K.; Smith, I.E. Vinorelbine—A clinical review. Br. J. Cancer 2000, 82, 1907–1913. [Google Scholar] [CrossRef]
- Yang, X.; Lin, Y.; Shi, Y.; Li, B.; Liu, W.; Ying-Hong, S.; Dang, Y.; Chu, Y.; Fan, J.; He, R. FAP Promotes Immunosuppression by Cancer-Associated Fibroblasts in the Tumor Microenvironment via STAT3–CCL2 Signaling. Cancer Res. 2016, 76, 4124–4135. [Google Scholar] [CrossRef]
- Tacke, F. Targeting hepatic macrophages to treat liver diseases. J. Hepatol. 2017, 66, 1300–1312. [Google Scholar] [CrossRef]
- Collins, M.A.; Brisset, J.-C.; Zhang, Y.; Bednar, F.; Pierre, J.; Heist, K.A.; Galbán, C.J.; Galbán, S.; Di Magliano, M.P. Metastatic Pancreatic Cancer Is Dependent on Oncogenic Kras in Mice. PLoS ONE 2012, 7, e49707. [Google Scholar] [CrossRef]
- Zhang, Y.; Velez-Delgado, A.; Mathew, E.; Li, D.; Mendez, F.M.; Flannagan, K.; Rhim, A.D.; Simeone, D.M.; Beatty, G.L.; Di Magliano, M.P. Myeloid cells are required for PD-1/PD-L1 checkpoint activation and the establishment of an immunosuppressive environment in pancreatic cancer. Gut 2017, 66, 124–136. [Google Scholar] [CrossRef]
- Sleightholm, R.L.; Neilsen, B.K.; Li, J.; Steele, M.M.; Singh, R.K.; Hollingsworth, M.A.; Oupický, D. Emerging roles of the CXCL12/CXCR4 axis in pancreatic cancer progression and therapy. Pharmacol. Ther. 2017, 179, 158–170. [Google Scholar] [CrossRef] [PubMed]
- Nagarsheth, N.; Wicha, M.S.; Zou, W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat. Rev. Immunol. 2017, 17, 559–572. [Google Scholar] [CrossRef] [PubMed]
- Biasci, D.; Smoragiewicz, M.; Connell, C.M.; Wang, Z.; Gao, Y.; Thaventhiran, J.E.D.; Basu, B.; Magiera, L.; Johnson, T.I.; Bax, L.; et al. CXCR4 inhibition in human pancreatic and colorectal cancers induces an integrated immune response. Proc. Natl. Acad. Sci. USA 2020, 117, 28960–28970. [Google Scholar] [CrossRef] [PubMed]
- Bockorny, B.; Semenisty, V.; Macarulla, T.; Borazanci, E.; Wolpin, B.M.; Stemmer, S.M.; Golan, T.; Geva, R.; Borad, M.J.; Pedersen, K.S.; et al. BL-8040, a CXCR4 antagonist, in combination with pembrolizumab and chemotherapy for pancreatic cancer: The COMBAT trial. Nat. Med. 2020, 26, 878–885. [Google Scholar] [CrossRef]
- Su, S.; Chen, J.; Yao, H.; Liu, J.; Yu, S.; Lao, L.; Wang, M.; Luo, M.; Xing, Y.; Chen, F.; et al. CD10+GPR77+ Cancer-Associated Fibroblasts Promote Cancer Formation and Chemoresistance by Sustaining Cancer Stemness. Cell 2018, 172, 841–856.e16. [Google Scholar] [CrossRef]
- Gerard, N.P.; Lu, B.; Liu, P.; Craig, S.; Fujiwara, Y.; Okinaga, S.; Gerard, C. An Anti-inflammatory Function for the Complement Anaphylatoxin C5a-binding Protein, C5L2. J. Biol. Chem. 2005, 280, 39677–39680. [Google Scholar] [CrossRef]
- Purcell, J.; Tanlimco, S.G.; Hickson, J.A.; Fox, M.; Sho, M.; Durkin, L.; Uziel, T.; Powers, R.; Foster, K.; McGonigal, T.; et al. LRRC15 Is a Novel Mesenchymal Protein and Stromal Target for Antibody–Drug Conjugates. Cancer Res. 2018, 78, 4059–4072. [Google Scholar] [CrossRef]
- Dominguez, C.X.; Müller, S.; Keerthivasan, S.; Koeppen, H.; Hung, J.; Gierke, S.; Breart, B.; Foreman, O.; Bainbridge, T.W.; Castiglioni, A.; et al. Single-Cell RNA Sequencing Reveals Stromal Evolution into LRRC15+ Myofibroblasts as a Determinant of Patient Response to Cancer Immunotherapy. Cancer Discov. 2019, 10, 232–253. [Google Scholar] [CrossRef]
- Demetri, G.D.; Luke, J.J.; Hollebecque, A.; Powderly, J.D.; Spira, A.I.; Subbiah, V.; Lai, D.W.; Yue, H.; Kasichayanula, S.; Gulbranson, S.; et al. First-in-human phase 1 study of ABBV-085, an antibody-drug conjugate (ADC) targeting LRRC15, in sarcomas and other advanced solid tumors. J. Clin. Oncol. 2019, 37, 3004. [Google Scholar] [CrossRef]
- Huang, H.; Zhang, Y.; Gallegos, V.; Sorrelle, N.; Zaid, M.M.; Toombs, J.; Du, W.; Wright, S.; Hagopian, M.; Wang, Z.; et al. Targeting TGF βR2-mutant tumors exposes vulnerabilities to stromal TGF β blockade in pancreatic cancer. EMBO Mol. Med. 2019, 11, e10515. [Google Scholar] [CrossRef]
- Mace, T.A.; Shakya, R.; Pitarresi, J.R.; Swanson, B.; McQuinn, C.W.; Loftus, S.; Nordquist, E.; Cruz-Monserrate, Z.; Yu, L.; Young, G.; et al. IL-6 and PD-L1 antibody blockade combination therapy reduces tumour progression in murine models of pancreatic cancer. Gut 2018, 67, 320–332. [Google Scholar] [CrossRef] [PubMed]
- Rossi, J.-F.; Lu, Z.-Y.; Jourdan, M.; Klein, B. Interleukin-6 as a Therapeutic Target. Clin. Cancer Res. 2015, 21, 1248–1257. [Google Scholar] [CrossRef]
- Sebba, A. Tocilizumab: The first interleukin-6-receptor inhibitor. Am. J. Health Pharm. 2008, 65, 1413–1418. [Google Scholar] [CrossRef] [PubMed]
- Biffi, G.; Oni, T.E.; Spielman, B.; Hao, Y.; Elyada, E.; Park, Y.; Preall, J.; Tuveson, D.A. IL1-Induced JAK/STAT Signaling Is Antagonized by TGFβ to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2019, 9, 282–301. [Google Scholar] [CrossRef] [PubMed]
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef] [PubMed]
- Schenk, K.M.; Reuss, J.E.; Choquette, K.; Spira, A.I. A review of canakinumab and its therapeutic potential for non-small cell lung cancer. Anti-Cancer Drugs 2019, 30, 879–885. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Gao, W.; Lytle, N.K.; Huang, P.; Yuan, X.; Dann, A.M.; Ridinger-Saison, M.; DelGiorno, K.E.; Antal, C.E.; Liang, G.; et al. Targeting LIF-mediated paracrine interaction for pancreatic cancer therapy and monitoring. Nat. Cell Biol. 2019, 569, 131–135. [Google Scholar] [CrossRef]
- Shi, Y.; Du, L.; Lin, L.; Wang, Y. Tumour-associated mesenchymal stem/stromal cells: Emerging therapeutic targets. Nat. Rev. Drug Discov. 2017, 16, 35–52. [Google Scholar] [CrossRef] [PubMed]
- Niess, H.; Von Einem, J.C.; Thomas, M.N.; Michl, M.; Angele, M.K.; Huss, R.; Günther, C.; Nelson, P.J.; Bruns, C.J.; Heinemann, V. Treatment of advanced gastrointestinal tumors with genetically modified autologous mesenchymal stromal cells (TREAT-ME1): Study protocol of a phase I/II clinical trial. BMC Cancer 2015, 15, 237. [Google Scholar] [CrossRef]
- Von Einem, J.C.; Peter, S.; Günther, C.; Volk, H.-D.; Grütz, G.; Salat, C.; Stoetzer, O.; Nelson, P.J.; Michl, M.; Modest, D.P.; et al. Treatment of advanced gastrointestinal cancer with genetically modified autologous mesenchymal stem cells—TREAT-ME-1—A phase I, first in human, first in class trial. Oncotarget 2017, 8, 80156–80166. [Google Scholar] [CrossRef]
- Einem, J.C.; Guenther, C.; Volk, H.; Grütz, G.; Hirsch, D.; Salat, C.; Stoetzer, O.; Nelson, P.J.; Michl, M.; Modest, D.P.; et al. Treatment of advanced gastrointestinal cancer with genetically modified autologous mesenchymal stem cells: Results from the phase 1/2 TREAT-ME-1 trial. Int. J. Cancer 2019, 145, 1538–1546. [Google Scholar] [CrossRef]
- Kucerova, L.; Altanerova, V.; Matuskova, M.; Tyciakova, S.; Altaner, C. Adipose Tissue–Derived Human Mesenchymal Stem Cells Mediated Prodrug Cancer Gene Therapy. Cancer Res. 2007, 67, 6304–6313. [Google Scholar] [CrossRef]
- Kucerova, L.; Matuskova, M.; Pastorakova, A.; Tyciakova, S.; Jakubikova, J.; Bohovic, R.; Altanerova, V.; Altaner, C. Cytosine deaminase expressing human mesenchymal stem cells mediated tumour regression in melanoma bearing mice. J. Gene Med. 2008, 10, 1071–1082. [Google Scholar] [CrossRef]
- Hutzen, B.; Pierson, C.R.; Russell, S.J.; Galanis, E.; Raffel, C.; Studebaker, A.W. Treatment of medulloblastoma using an oncolytic measles virus encoding the thyroidal sodium iodide symporter shows enhanced efficacy with radioiodine. BMC Cancer 2012, 12, 508. [Google Scholar] [CrossRef]
- Goyal, H.; Perisetti, A.; Rahman, M.R.; Levin, A.; Giuseppe, L. Vitamin D and Gastrointestinal Cancers: A Narrative Review. Dig. Dis. Sci. 2018, 64, 1098–1109. [Google Scholar] [CrossRef] [PubMed]
- Skinner, H.G.; Michaud, D.S.; Giovannucci, E.; Willett, W.C.; Colditz, G.A.; Fuchs, C.S. Vitamin D Intake and the Risk for Pancreatic Cancer in Two Cohort Studies. Cancer Epidemiol. Biomark. Prev. 2006, 15, 1688–1695. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.-D.; Ma, Y.; Flynn, G.; Muindi, J.R.; Kong, R.-X.; Trump, D.L.; Johnson, C.S. Calcitriol enhances gemcitabine antitumor activity in vitro and in vivo by promoting apoptosis in a human pancreatic carcinoma model system. Cell Cycle 2010, 9, 3094–3101. [Google Scholar] [CrossRef] [PubMed]
- Sherman, M.H.; Yu, R.T.; Engle, D.D.; Ding, N.; Atkins, A.R.; Tiriac, H.; Collisson, E.A.; Connor, F.; Van Dyke, T.; Kozlov, S.; et al. Vitamin D Receptor-Mediated Stromal Reprogramming Suppresses Pancreatitis and Enhances Pancreatic Cancer Therapy. Cell 2014, 159, 80–93. [Google Scholar] [CrossRef]
- Ferrer-Mayorga, G.; Gómez-López, G.; Barbáchano, A.; Fernández-Barral, A.; Peña, C.; Pisano, D.G.; Cantero, R.; Rojo, F.; Muñoz, A.; Larriba, M.J. Vitamin D receptor expression and associated gene signature in tumour stromal fibroblasts predict clinical outcome in colorectal cancer. Gut 2016, 66, 1449–1462. [Google Scholar] [CrossRef]
- Merrigan, S.; Kennedy, B. Vitamin D receptor agonists regulate ocular developmental angiogenesis and modulate expression of dre-miR-21 and VEGF. Br. J. Pharmacol. 2017, 174, 2636–2651. [Google Scholar] [CrossRef] [PubMed]
- Slavikova, V.; Chlapek, P.; Mazanek, P.; Sterba, J.; Veselska, R. Traffic lights for retinoids in oncology: Molecular markers of retinoid resistance and sensitivity and their use in the management of cancer differentiation therapy. BMC Cancer 2018, 18, 1–13. [Google Scholar] [CrossRef]
- Froeling, F.E.; Feig, C.; Chelala, C.; Dobson, R.; Mein, C.E.; Tuveson, D.; Clevers, H.; Hart, I.R.; Kocher, H.M. Retinoic Acid–Induced Pancreatic Stellate Cell Quiescence Reduces Paracrine Wnt–β-Catenin Signaling to Slow Tumor Progression. Gastroenterology 2011, 141, 1486–1497.e14. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Pramanik, D.; Mukherjee, R.; Campbell, N.R.; Elumalai, S.; De Wilde, R.F.; Hong, S.-M.; Goggins, M.G.; De Jesus-Acosta, A.; Laheru, D.; et al. Molecular Determinants of Retinoic Acid Sensitivity in Pancreatic Cancer. Clin. Cancer Res. 2012, 18, 280–289. [Google Scholar] [CrossRef] [PubMed]
- Noy, N. Non-classical Transcriptional Activity of Retinoic Acid. Macromol. Protein Complexes III Struct. Funct. 2016, 81, 179–199. [Google Scholar] [CrossRef]
- Han, X.; Li, Y.; Xu, Y.; Zhao, X.; Zhang, Y.; Yang, X.; Wang, Y.; Zhao, R.; Anderson, G.J.; Zhao, Y.; et al. Reversal of pancreatic desmoplasia by re-educating stellate cells with a tumour microenvironment-activated nanosystem. Nat. Commun. 2018, 9, 1–18. [Google Scholar] [CrossRef]
- Zhu, J.; Xiong, G.; Fu, H.; Evers, B.M.; Zhou, B.P.; Xu, R. Chaperone Hsp47 Drives Malignant Growth and Invasion by Modulating an ECM Gene Network. Cancer Res. 2015, 75, 1580–1591. [Google Scholar] [CrossRef]
- Ene–Obong, A.; Clear, A.J.; Watt, J.; Wang, J.; Fatah, R.; Riches, J.C.; Marshall, J.F.; Chin-Aleong, J.; Chelala, C.; Gribben, J.G.; et al. Activated Pancreatic Stellate Cells Sequester CD8+ T Cells to Reduce Their Infiltration of the Juxtatumoral Compartment of Pancreatic Ductal Adenocarcinoma. Gastroenterology 2013, 145, 1121–1132. [Google Scholar] [CrossRef]
- Kocher, H.M.; Basu, B.; Froeling, F.E.M.; Sarker, D.; Slater, S.; Carlin, D.; DeSouza, N.M.; De Paepe, K.N.; Goulart, M.R.; Hughes, C.; et al. Phase I clinical trial repurposing all-trans retinoic acid as a stromal targeting agent for pancreatic cancer. Nat. Commun. 2020, 11, 1–9. [Google Scholar] [CrossRef]
- Ocaña, O.H.; Córcoles, R.; Fabra, Á.; Moreno-Bueno, G.; Acloque, H.; Vega, S.; Barrallo-Gimeno, A.; Cano, A.; Nieto, M.A. Metastatic Colonization Requires the Repression of the Epithelial-Mesenchymal Transition Inducer Prrx1. Cancer Cell 2012, 22, 709–724. [Google Scholar] [CrossRef]
- Reichert, M.; Takano, S.; Von Burstin, J.; Kim, S.-B.; Lee, J.-S.; Ihida-Stansbury, K.; Hahn, C.; Heeg, S.; Schneider, G.; Rhim, A.D.; et al. The Prrx1 homeodomain transcription factor plays a central role in pancreatic regeneration and carcinogenesis. Genes Dev. 2013, 27, 288–300. [Google Scholar] [CrossRef]
- Takano, S.; Reichert, M.; Bakir, B.; Das, K.K.; Nishida, T.; Miyazaki, M.; Heeg, S.; Collins, M.A.; Marchand, B.; Hicks, P.D.; et al. Prrx1 isoform switching regulates pancreatic cancer invasion and metastatic colonization. Genes Dev. 2016, 30, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, K.; Maurer, C.; Peschke, K.; Teller, S.; Schuck, K.; Steiger, K.; Engleitner, T.; Öllinger, R.; Nomura, A.; Wirges, N.; et al. Mesenchymal Plasticity Regulated by Prrx1 Drives Aggressive Pancreatic Cancer Biology. Gastroenterology 2021, 160, 346–361. [Google Scholar] [CrossRef]
- Bailey, P.; Initiative, A.P.C.G.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.-M.; Gingras, M.-C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Lee, K.-W.; Yeo, S.-Y.; Sung, C.O.; Kim, S.-H. Twist1 Is a Key Regulator of Cancer-Associated Fibroblasts. Cancer Res. 2014, 75, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Yeo, S.-Y.; Lee, K.-W.; Shin, D.; An, S.; Cho, K.-H.; Kim, S.-H. A positive feedback loop bi-stably activates fibroblasts. Nat. Commun. 2018, 9, 1–16. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Intervention/Treatment | Condition or Disease | NCT Number | Stage of Clinical Trial | Recruitment Status (Recruiting, Completed, not yet Recruiting. Last Update) | Last Update |
|---|---|---|---|---|---|
| BIBH1 (Sibrotuzumab) | Metastatic colorectal cancer | NCT02198274 | Phase 2 | Completed | 23 July 2014 |
| Talabostat mesylate (BXCL701) Pembrolizumab | Prostate cancer | NCT03910660 | Phase 1/2 | Recruiting | 5 November 2020 |
| RO6874281 Atezolizumab (MPDL3280A), an engineered anti-PD-L1 antibody Gemcitabine Vinorelbine | Advanced/Metastatic head and neck, esophageal and cervical cancers | NCT03386721 | Phase 2 | Recruiting | 27 April 2020 |
| RO6874281 with Trastuzumab or Cetuximab | Solid tumor Breast cancer Cancer of head and neck | NCT02627274 | Phase 1 | Recruiting | 14 October 2020 |
| RO6874281 Pembrolizumab | Metastatic melanoma | NCT03875079 | Phase 1 | Recruiting | 9 October 2020 |
| Nab-Paclitaxel Gemcitabine Oxaliplatin Leucovorin Fluorouracil Atezolizumab Cobimetinib PEGGH20 BL-8040 Selicrelumab Bevacizumab RO6874281 AB928 Tiragolumab Tocilizumab | Metastatic pancreatic adenocarcinoma | NCT03193190 | Phase 1,2 | Recruiting | 27 October 2020 |
| Cemiplimab (REGN-2810, Libtayo) Plerixafor (AMD3100, Mozobil) | Metastatic pancreatic cancer | NCT04177810 | Phase 2 | Recruiting | 9 November 2020 |
| Plerixafor (Mozobil) | Metastatic pancreatic cancer Metastatic colorectal cancer Ovarian Serous Adenocarcinoma | NCT02179970 (CAM-PLEX) | Phase 1 | Completed | 23 July 2019 |
| BL-8040 Pembrolizumab Onivyde/5-FU/leucovorin | Metastatic pancreatic cancer | NCT02826486 (COMBAT) | Phase 2 | Active, not recruiting | 9 June 2020 |
| ABBV-085 | Advanced solid tumors | NCT02565758 | Phase 1 | Completed | 5 April 2019 |
| Intervention/Treatment | Condition or Disease | NCT Number | Stage of Clinical Trial | Recruitment Status (Recruiting, Completed, not yet Recruiting. Last Update) | Last Update |
|---|---|---|---|---|---|
| Siltuximab Spartalizumab | Metastatic pancreatic cancer | NCT04191421 | Phase 1,2 | Recruiting | 20 August 2020 |
| Tocilizumab Gemcitabine Nab-Paclitaxel | Unresectable pancreatic cancer | NCT02767557 (PACTO) | Phase 2 | Recruiting | 21 April 2020 |
| Ipilimumab Nivolumab Tocilizumab SBRT | Pancreatic cancer | NCT04258150 (TRIPPLE-R) | Phase 2 | Recruiting | 21 April 2020 |
| Canakinumab Spartalizumab Nab-paclitaxel Gemcitabine | Metastatic pancreatic cancer | NCT04581343 (PanCAN-SR1) | Phase 1 | Recruiting | 20 October 2020 |
| Intervention/Treatment | Condition or Disease | NCT Number | Stage of Clinical Trial | Recruitment Status (Recruiting, Completed, not yet Recruiting. Last Update) | Last Update |
|---|---|---|---|---|---|
| MSC_apceth_101 | Advanced gastrointestinal cancer | NCT02008539 | Phase 1, 2 | Terminated | 27 March 2017 |
| Oncolytic measles virus encoding thyroidal sodium iodide symporter infected MSCs | Recurrent ovarian, peritoneal or fallopian tube cancer | NCT02068794 | Phase 1, 2 | Recruiting | 3 December 2020 |
| MSCTRAIL | Lung cancer | NCT03298763 (TACTICAL) | Phase 1, 2 | Recruiting | 31 March 2020 |
| Intervention/Treatment | Condition or Disease | NCT Number | Stage of Clinical Trial | Recruitment Status (Recruiting, Completed, not yet Recruiting. Last Update) | Last Update |
|---|---|---|---|---|---|
| High-dose Vitamin D3 | Pancreatic cancer | NCT03472833 | Phase 3 | Recruiting | 14 September 2020 |
| Pembrolizumab paricalcitol | Pancreatic cancer, advanced pancreatic cancer, metastatic pancreatic cancer | NCT03331562 | Phase 2 | Completed | 8 October 2020 |
| Nivolumab Nab-Paclitaxel Gemcitabine Paricalcitol | Resectable pancreatic cancer | NCT03519308 | Early Phase 1 | Recruiting | July 30, 2020 |
| Paricalcitol Gemcitabine and Nab-paclitaxel | Advanced pancreatic cancer | NCT04617067 | Phase 2 | Recruiting | 9 November 2020 |
| 5-FU Leucovorin Liposomal irinotecan Paricalcitol | Advanced pancreatic cancer | NCT03883919 | Phase 1 | Recruiting | 27 August 2020 |
| Gemcitabine Hydroxychloroquine Nab-paclitaxel Paricalcitol | Advanced or metastatic pancreatic cancer | NCT04524702 | Phase 2 | Recruiting | 8 October 2020 |
| ATRA Gemcitabine Nab-paclitaxel | Pancreatic cancer | NCT03307148 (STAR_PAC) | Phase 1 | Completed | 22 January 2020 |
| ATRA Gemcitabine Nab-paclitaxel | Pancreatic cancer | NCT04241276 (STARPAC2) | Phase 2 | Not yet recruiting | 27 January 2020 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sunami, Y.; Böker, V.; Kleeff, J. Targeting and Reprograming Cancer-Associated Fibroblasts and the Tumor Microenvironment in Pancreatic Cancer. Cancers 2021, 13, 697. https://doi.org/10.3390/cancers13040697
Sunami Y, Böker V, Kleeff J. Targeting and Reprograming Cancer-Associated Fibroblasts and the Tumor Microenvironment in Pancreatic Cancer. Cancers. 2021; 13(4):697. https://doi.org/10.3390/cancers13040697
Chicago/Turabian StyleSunami, Yoshiaki, Viktoria Böker, and Jörg Kleeff. 2021. "Targeting and Reprograming Cancer-Associated Fibroblasts and the Tumor Microenvironment in Pancreatic Cancer" Cancers 13, no. 4: 697. https://doi.org/10.3390/cancers13040697
APA StyleSunami, Y., Böker, V., & Kleeff, J. (2021). Targeting and Reprograming Cancer-Associated Fibroblasts and the Tumor Microenvironment in Pancreatic Cancer. Cancers, 13(4), 697. https://doi.org/10.3390/cancers13040697