Diabetes and Pancreatic Cancer—A Dangerous Liaison Relying on Carbonyl Stress

 ,
, 

Simple Summary
Abstract
1. Introduction
2. Relationship between Diabetes and Pancreatic Cancer
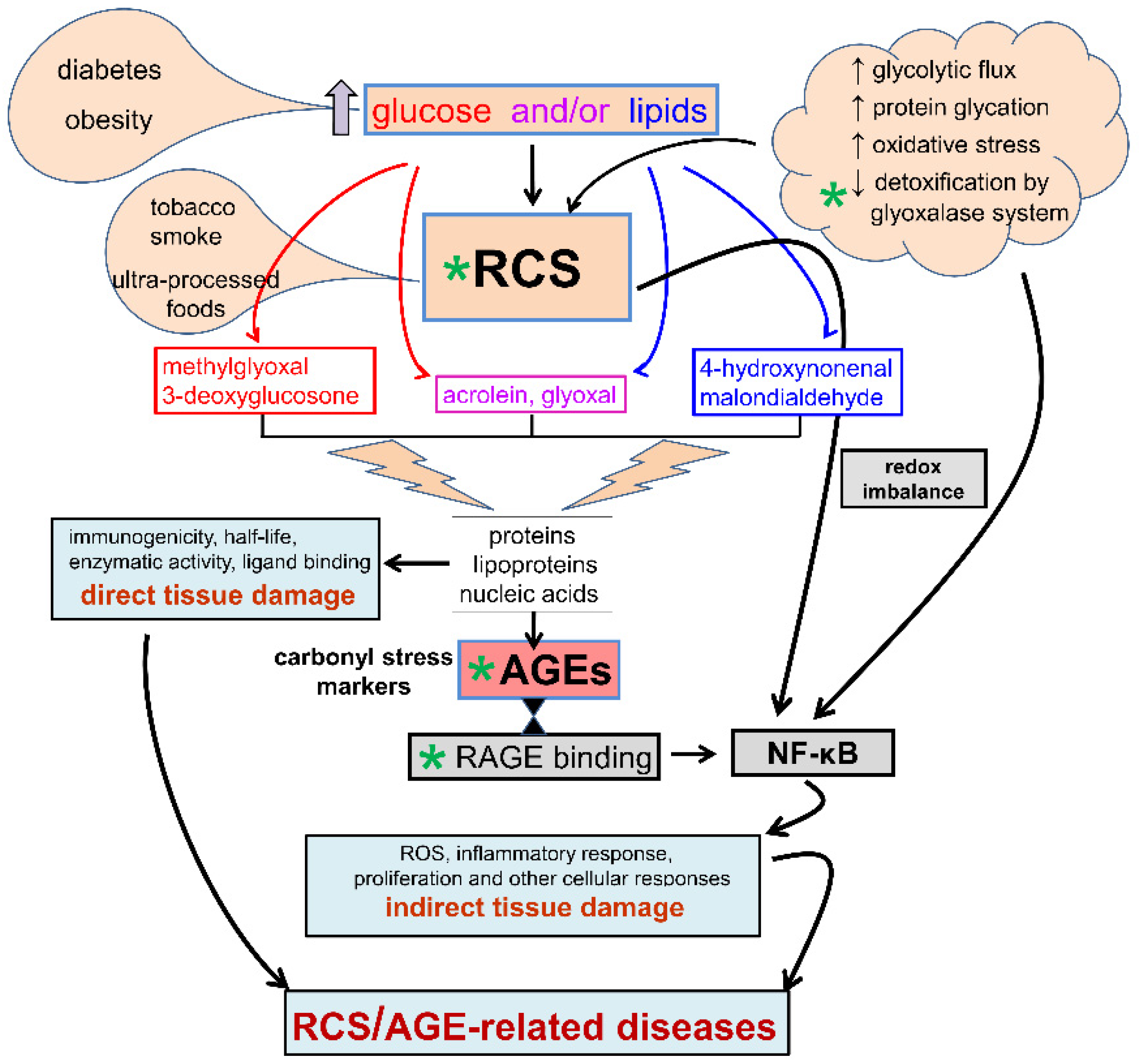
3. Diabetes, Carbonyl Stress, Advanced Glycation End-Products (AGEs) Formation, and Related Therapeutic Strategies
4. Carbonyl Stress in Cancer: A Possible Link between Metabolism and Malignances
5. RAGE, AGEs, and Their Carbonyl Precursors as Potential Targets in Pancreatic Cancer Associated with Diabetes and Other Carbonyl Stress-Related Conditions
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Primers 2016, 2, 16022. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Mizrahi, J.D.; Surana, R.; Valle, J.W.; Shroff, R.T. Pancreatic cancer. Lancet 2020, 395, 2008–2020. [Google Scholar] [CrossRef]
- Oberstein, P.E.; Olive, K.P. Pancreatic cancer: Why is it so hard to treat? Ther. Adv. Gastroenterol. 2013, 6, 321–337. [Google Scholar] [CrossRef]
- Andersen, D.K.; Korc, M.; Petersen, G.M.; Eibl, G.; Li, D.; Rickels, M.R.; Chari, S.T.; Abbruzzese, J.L. Diabetes, Pancreatogenic Diabetes, and Pancreatic Cancer. Diabetes 2017, 66, 1103–1110. [Google Scholar] [CrossRef]
- International Diabetes Federation IDF Diabetes Atlas, 9th ed.; International Diabetes Federation: Brussels, Belgium, 2019; Available online: https://diabetesatlas.org/en/ (accessed on 15 July 2020).
- Review Cells World Health Organization. Media Centre. Diabetes. Available online: https://www.who.int/news-room/fact-sheets/detail/diabetes (accessed on 30 October 2018).
- Ogurtsova, K.; da Rocha Fernandes, J.D.; Huang, Y.; Linnenkamp, U.; Guariguata, L.; Cho, N.H.; Cavan, D.; Shaw, J.E.; Makaroff, L.E. IDF Diabetes Atlas: Global estimates for the prevalence of diabetes for 2015 and 2040. Diabetes Res. Clin. Pract. 2017, 128, 40–50. [Google Scholar] [CrossRef]
- American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care 2014, 37 (Suppl. 1), S81–S90. [Google Scholar] [CrossRef]
- Menke, A.; Casagrande, S.; Geiss, L.; Cowie, C.C. Prevalence of and Trends in Diabetes among Adults in the United States, 1988–2012. JAMA 2015, 314, 1021–1029. [Google Scholar] [CrossRef]
- Harding, J.L.; Pavkov, M.E.; Magliano, D.J.; Shaw, J.E.; Gregg, E.W. Global trends in diabetes complications: A review of current evidence. Diabetologia 2019, 62, 3–16. [Google Scholar] [CrossRef]
- Dankner, R.; Boker, L.K.; Boffetta, P.; Balicer, R.D.; Murad, H.; Berlin, A.; Olmer, L.; Agai, N.; Freedman, L.S. A historical cohort study on glycemic-control and cancer-risk among patients with diabetes. Cancer Epidemiol. 2018, 57, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Carstensen, B.; Read, S.H.; Friis, S.; Sund, R.; Keskimäki, I.; Svensson, A.M.; Ljung, R.; Wild, S.H.; Kerssens, J.J.; Harding, J.L.; et al. Diabetes and Cancer Research Consortium. Cancer incidence in persons with type 1 diabetes: A five-country study of 9000 cancers in type 1 diabetic individuals. Diabetologia 2016, 59, 980–988. [Google Scholar] [CrossRef] [PubMed]
- Magruder, J.T.; Elahi, D.; Andersen, D.K. Diabetes and pancreatic cancer: Chicken or egg? Pancreas 2011, 40, 339–351. [Google Scholar] [CrossRef] [PubMed]
- Stevens, R.J.; Roddam, A.W.; Beral, V. Pancreatic cancer in type 1 and young-onset diabetes: Systematic review and meta-analysis. Br. J. Cancer 2007, 96, 507–509. [Google Scholar] [CrossRef]
- Morales-Oyarvide, V.; Mino-Kenudson, M.; Ferrone, C.R.; Sahani, D.V.; Pergolini, I.; Negreros-Osuna, A.A.; Warshaw, A.L.; Lillemoe, K.D.; Fernández-Del Castillo, C. Diabetes mellitus in intraductal papillary mucinous neoplasm of the pancreas is associated with high-grade dysplasia and invasive carcinoma. Pancreatology 2017, 17, 920–926. [Google Scholar] [CrossRef]
- Gallo, M.; Ruggeri, R.M.; Muscogiuri, G.; Pizza, G.; Faggiano, A.; Colao, A.; NIKE Group. Diabetes and pancreatic neuroendocrine tumours: Which interplays, if any? Cancer Treat. Rev. 2018, 67, 1–9. [Google Scholar] [CrossRef]
- Bright, R. Cases and Observations connected with Disease of the Pancreas and Duodenum. Med. Chir. Trans. 1833, 18, 1–56. [Google Scholar] [CrossRef]
- Batabyal, P.; Vander Hoorn, S.; Christophi, C.; Nikfarjam, M. Association of diabetes mellitus and pancreatic adenocarcinoma: A meta-analysis of 88 studies. Ann. Surg. Oncol. 2014, 21, 2453–2462. [Google Scholar] [CrossRef]
- Everhart, J.; Wright, D. Diabetes mellitus as a risk factor for pancreatic cancer. A meta-analysis. JAMA 1995, 273, 1605–1609. [Google Scholar] [CrossRef]
- Song, S.; Wang, B.; Zhang, X.; Hao, L.; Hu, X.; Li, Z.; Sun, S. Long-Term Diabetes Mellitus Is Associated with an Increased Risk of Pancreatic Cancer: A Meta-Analysis. PLoS ONE 2015, 10, e0134321. [Google Scholar] [CrossRef]
- Lu, Y.; García Rodríguez, L.A.; Malgerud, L.; González-Pérez, A.; Martín-Pérez, M.; Lagergren, J.; Bexelius, T.S. New-onset type 2 diabetes, elevated HbA1c, anti-diabetic medications, and risk of pancreatic cancer. Br. J. Cancer 2015, 113, 1607–1614. [Google Scholar] [CrossRef] [PubMed]
- Pannala, R.; Leirness, J.B.; Bamlet, W.R.; Basu, A.; Petersen, G.M.; Chari, S.T. Prevalence and clinical profile of pancreatic cancer-associated diabetes mellitus. Gastroenterology 2008, 134, 981–987. [Google Scholar] [CrossRef] [PubMed]
- Ben, Q.; Xu, M.; Ning, X.; Liu, J.; Hong, S.; Huang, W.; Zhang, H.; Li, Z. Diabetes mellitus and risk of pancreatic cancer: A meta-analysis of cohort studies. Eur. J. Cancer 2011, 47, 1928–1937. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Tang, H.; Hassan, M.M.; Holly, E.A.; Bracci, P.M.; Silverman, D.T. Diabetes and risk of pancreatic cancer: A pooled analysis of three large case-control studies. Cancer Causes Control 2011, 22, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Maisonneuve, P.; Lowenfels, A.B.; Bueno-de-Mesquita, H.B.; Ghadirian, P.; Baghurst, P.A.; Zatonski, W.A.; Miller, A.B.; Duell, E.J.; Boffetta, P.; Boyle, P. Past medical history and pancreatic cancer risk: Results from a multicenter case-control study. Ann. Epidemiol. 2010, 20, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Bian, X.; Wei, S.; He, M.; Yang, Y. The relationship between pancreatic cancer and type 2 diabetes: Cause and consequence. Cancer Manag. Res. 2019, 11, 8257–8268. [Google Scholar] [CrossRef]
- Wolpin, B.M.; Bao, Y.; Qian, Z.R.; Wu, C.; Kraft, P.; Ogino, S.; Stampfer, M.J.; Sato, K.; Ma, J.; Buring, J.E.; et al. Hyperglycemia, insulin resistance, impaired pancreatic β-cell function, and risk of pancreatic cancer. J. Natl. Cancer Inst. 2013, 105, 1027–1035. [Google Scholar] [CrossRef]
- Setiawan, V.W.; Stram, D.O.; Porcel, J.; Chari, S.T.; Maskarinec, G.; Le Marchand, L.; Wilkens, L.R.; Haiman, C.A.; Pandol, S.J.; Monroe, K.R. Pancreatic Cancer Following Incident Diabetes in African Americans and Latinos: The Multiethnic Cohort. J. Natl. Cancer Inst. 2019, 111, 27–33. [Google Scholar] [CrossRef]
- Dugnani, E.; Gandolfi, A.; Balzano, G.; Scavini, M.; Pasquale, V.; Aleotti, F.; Liberati, D.; Di Terlizzi, G.; Petrella, G.; Reni, M.; et al. Diabetes associated with pancreatic ductal adenocarcinoma is just diabetes: Results of a prospective observational study in surgical patients. Pancreatology 2016, 16, 844–852. [Google Scholar] [CrossRef]
- Sah, R.P.; Nagpal, S.J.; Mukhopadhyay, D.; Chari, S.T. New insights into pancreatic cancer-induced paraneoplastic diabetes. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 423–433. [Google Scholar] [CrossRef]
- Permert, J.; Ihse, I.; Jorfeldt, L.; von Schenck, H.; Arnquist, H.J.; Larsson, J. Improved glucose metabolism after subtotal pancreatectomy for pancreatic cancer. Br. J. Surg. 1993, 80, 1047–1050. [Google Scholar] [CrossRef] [PubMed]
- Balzano, G.; Dugnani, E.; Pasquale, V.; Capretti, G.; Radaelli, M.G.; Garito, T.; Stratta, G.; Nini, A.; Di Fenza, R.; Castoldi, R.; et al. Clinical signature and pathogenetic factors of diabetes associated with pancreas disease (T3cDM): A prospective observational study in surgical patients. Acta Diabetol. 2014, 51, 801–811. [Google Scholar] [CrossRef] [PubMed]
- Sala, P.C.; Torrinhas, R.S.; Heymsfield, S.B.; Waitzberg, D.L. Type 2 diabetes mellitus: A possible surgically reversible intestinal dysfunction. Obes. Surg. 2012, 22, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Koo, D.H.; Han, K.D.; Park, C.Y. The Incremental Risk of Pancreatic Cancer According to Fasting Glucose Levels: Nationwide Population-Based Cohort Study. J. Clin. Endocrinol. Metab. 2019, 104, 4594–4599. [Google Scholar] [CrossRef] [PubMed]
- Pang, Y.; Kartsonaki, C.; Guo, Y.; Bragg, F.; Yang, L.; Bian, Z.; Chen, Y.; Iona, A.; Millwood, I.Y.; Lv, J.; et al. Diabetes, plasma glucose and incidence of pancreatic cancer: A prospective study of 0.5 million Chinese adults and a meta-analysis of 22 cohort studies. Int. J. Cancer 2017, 140, 1781–1788. [Google Scholar] [CrossRef]
- Nagai, M.; Murakami, Y.; Tamakoshi, A.; Kiyohara, Y.; Yamada, M.; Ukawa, S.; Hirata, T.; Tanaka, S.; Miura, K.; Ueshima, H.; et al. Fasting but not casual blood glucose is associated with pancreatic cancer mortality in Japanese: EPOCH-JAPAN. Cancer Causes Control 2017, 28, 625–633. [Google Scholar] [CrossRef]
- Iarrobino, N.A.; Gill, B.S.; Klement, R.J.; Bernard, M.E.; Champ, C.E. The Impact of Serum Glucose in the Treatment of Locoregionally Advanced Pancreatic Cancer. Am. J. Clin. Oncol. 2019, 42, 692–697. [Google Scholar] [CrossRef]
- Hank, T.; Sandini, M.; Qadan, M.; Weniger, M.; Ciprani, D.; Li, A.; Ferrone, C.R.; Warshaw, A.L.; Lillemoe, K.D.; Fernández-Del Castillo, C. Diabetes mellitus is associated with unfavorable pathologic features, increased postoperative mortality, and worse long-term survival in resected pancreatic cancer. Pancreatology 2020, 20, 125–131. [Google Scholar] [CrossRef]
- Sharma, A.; Smyrk, T.C.; Levy, M.J.; Topazian, M.A.; Chari, S.T. Fasting Blood Glucose Levels Provide Estimate of Duration and Progression of Pancreatic Cancer Before Diagnosis. Gastroenterology 2018, 155, 490–500. [Google Scholar] [CrossRef]
- Huxley, R.; Ansary-Moghaddam, A.; Berrington de González, A.; Barzi, F.; Woodward, M. Type-II diabetes and pancreatic cancer: A meta-analysis of 36 studies. Br. J. Cancer 2005, 92, 2076–2083. [Google Scholar] [CrossRef]
- Shu, X.; Ji, J.; Li, X.; Sundquist, J.; Sundquist, K.; Hemminki, K. Cancer risk among patients hospitalized for Type 1 diabetes mellitus: A population-based cohort study in Sweden. Diabet. Med. 2010, 27, 791–797. [Google Scholar] [CrossRef] [PubMed]
- Renehan, A.G.; Tyson, M.; Egger, M.; Heller, R.F.; Zwahlen, M. Body-mass index and incidence of cancer: A systematic review and meta-analysis of prospective observational studies. Lancet 2008, 371, 569–578. [Google Scholar] [CrossRef]
- Aune, D.; Greenwood, D.C.; Chan, D.S.; Vieira, R.; Vieira, A.R.; Navarro Rosenblatt, D.A.; Cade, J.E.; Burley, V.J.; Norat, T. Body mass index, abdominal fatness and pancreatic cancer risk: A systematic review and non-linear dose-response meta-analysis of prospective studies. Ann. Oncol. 2012, 23, 843–852. [Google Scholar] [CrossRef] [PubMed]
- Urayama, K.Y.; Holcatova, I.; Janout, V.; Foretova, L.; Fabianova, E.; Adamcakova, Z.; Ryska, M.; Martinek, A.; Shonova, O.; Brennan, P.; et al. Body mass index and body size in early adulthood and risk of pancreatic cancer in a central European multicenter case-control study. Int. J. Cancer 2011, 129, 2875–2884. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Gentiluomo, M.; Lorenzo-Bermejo, J.; Morelli, L.; Obazee, O.; Campa, D.; Canzian, F. Mendelian randomisation study of the effects of known and putative risk factors on pancreatic cancer. J. Med. Genet. 2020, 57, 820–828. [Google Scholar] [CrossRef]
- Silverman, D.T. Risk factors for pancreatic cancer: A case-control study based on direct interviews. Teratog. Carcinog. Mutagen. 2001, 21, 7–25. [Google Scholar] [CrossRef]
- Colditz, G.A.; Willett, W.C.; Rotnitzky, A.; Manson, J.E. Weight gain as a risk factor for clinical diabetes mellitus in women. Ann. Intern. Med. 1995, 122, 481–486. [Google Scholar] [CrossRef]
- Jiao, L.; Flood, A.; Subar, A.F.; Hollenbeck, A.R.; Schatzkin, A.; Stolzenberg-Solomon, R. Glycemic index, carbohydrates, glycemic load, and the risk of pancreatic cancer in a prospective cohort study. Cancer Epidemiol. Biomark. Prev. 2009, 18, 1144–1151. [Google Scholar] [CrossRef]
- Molina-Montes, E.; Coscia, C.; Gómez-Rubio, P.; Fernández, A.; Boenink, R.; Rava, M.; Márquez, M.; Molero, X.; Löhr, M.; Sharp, L.; et al. Deciphering the complex interplay between pancreatic cancer, diabetes mellitus subtypes and obesity/BMI through causal inference and mediation analyses. Gut 2020. online ahead of print. [Google Scholar] [CrossRef]
- Paternoster, S.; Falasca, M. The intricate relationship between diabetes, obesity and pancreatic cancer. Biochim. Biophys. Acta Rev. Cancer 2020, 1873, 188326. [Google Scholar] [CrossRef]
- Quoc Lam, B.; Shrivastava, S.K.; Shrivastava, A.; Shankar, S.; Srivastava, R.K. The Impact of obesity and diabetes mellitus on pancreatic cancer: Molecular mechanisms and clinical perspectives. J. Cell. Mol. Med. 2020, 24, 7706–7716. [Google Scholar] [CrossRef] [PubMed]
- Carreras-Torres, R.; Johansson, M.; Gaborieau, V.; Haycock, P.C.; Wade, K.H.; Relton, C.L.; Martin, R.M.; Davey Smith, G.; Brennan, P. The role of obesity, Type 2 Diabetes, and metabolic factors in pancreatic cancer: A Mendelian Randomization Study. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.S.; Lee, J.S.; Song, E.; Kim, J.A.; Roh, E.; Yu, J.H.; Kim, N.H.; Yoo, H.J.; Seo, J.A.; Kim, S.G.; et al. Effect of metabolic health and obesity phenotype on the risk of pancreatic cancer: A nationwide population-based cohort study. Cancer Epidemiol. Biomark. Prev. 2020. [Google Scholar] [CrossRef] [PubMed]
- Philip, B.; Roland, C.L.; Daniluk, J.; Liu, Y.; Chatterjee, D.; Gomez, S.B.; Ji, B.; Huang, H.; Wang, H.; Fleming, J.B.; et al. A high-fat diet activates oncogenic Kras and COX2 to induce development of pancreatic ductal adenocarcinoma in mice. Gastroenterology 2013, 145, 1449–1458. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Yang, Y.; Liu, M.; Wang, D.; Wang, F.; Bi, Y.; Ji, J.; Li, S.; Liu, Y.; Chen, R.; et al. Oncogenic KRAS Reduces Expression of FGF21 in Acinar Cells to Promote Pancreatic Tumorigenesis in Mice on a High-Fat Diet. Gastroenterology 2019, 157, 1413–1428. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Bi, Y.; Hu, L.; Luo, Y.; Ji, J.; Mao, A.Z.; Logsdon, C.D.; Li, E.; Abbruzzese, J.L.; Li, Z.; et al. Obesogenic high-fat diet heightens aerobic glycolysis through hyperactivation of oncogenic KRAS. Cell Commun. Signal. 2019, 17, 19. [Google Scholar] [CrossRef]
- Chang, H.H.; Moro, A.; Chou, C.E.N.; Dawson, D.W.; French, S.; Schmidt, A.I.; Sinnett-Smith, J.; Hao, F.; Hines, O.J.; Eibl, G.; et al. Metformin Decreases the Incidence of Pancreatic Ductal Adenocarcinoma Promoted by Diet-induced Obesity in the Conditional KrasG12D Mouse Model. Sci. Rep. 2018, 8, 5899. [Google Scholar] [CrossRef]
- Incio, J.; Liu, H.; Suboj, P.; Chin, S.M.; Chen, I.X.; Pinter, M.; Ng, M.R.; Nia, H.T.; Grahovac, J.; Kao, S.; et al. Obesity-Induced Inflammation and Desmoplasia Promote Pancreatic Cancer Progression and Resistance to Chemotherapy. Cancer Discov. 2016, 6, 852–869. [Google Scholar] [CrossRef]
- Hertzer, K.M.; Xu, M.; Moro, A.; Dawson, D.W.; Du, L.; Li, G.; Chang, H.H.; Stark, A.P.; Jung, X.; Hines, O.J.; et al. Robust Early Inflammation of the Peripancreatic Visceral Adipose Tissue During Diet-Induced Obesity in the KrasG12D Model of Pancreatic Cancer. Pancreas 2016, 45, 458–465. [Google Scholar] [CrossRef]
- Chang, H.H.; Moro, A.; Takakura, K.; Su, H.Y.; Mo, A.; Nakanishi, M.; Waldron, R.T.; French, S.W.; Dawson, D.W.; Hines, O.J.; et al. Incidence of pancreatic cancer is dramatically increased by a high fat, high calorie diet in KrasG12D mice. PLoS ONE 2017, 12, e0184455. [Google Scholar] [CrossRef]
- Chung, K.M.; Singh, J.; Lawres, L.; Dorans, K.J.; Garcia, C.; Burkhardt, D.B.; Robbins, R.; Bhutkar, A.; Cardone, R.; Zhao, X.; et al. Endocrine-Exocrine Signaling Drives Obesity-Associated Pancreatic Ductal Adenocarcinoma. Cell 2020, 181, 832–847. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Chari, S.T. Pancreatic Cancer and Diabetes Mellitus. Curr. Treat. Options Gastroenterol. 2018, 16, 466–478. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.Y.; Yu, J.H.; Park, S.; Han, K.; Kim, N.H.; Yoo, H.J.; Choi, K.M.; Baik, S.H.; Seo, J.A. The influence of diabetes and antidiabetic medications on the risk of pancreatic cancer: A nationwide population-based study in Korea. Sci. Rep. 2018, 8, 9719. [Google Scholar] [CrossRef] [PubMed]
- Elena, J.W.; Steplowski, E.; Yu, K.; Hartge, P.; Tobias, G.S.; Brotzman, M.J.; Chanock, S.J.; Stolzenberg-Solomon, R.Z.; Arslan, A.A.; Bueno-de-Mesquita, H.B.; et al. Diabetes and risk of pancreatic cancer: A pooled analysis from the pancreatic cancer cohort consortium. Cancer Causes Control 2013, 24, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Bosetti, C.; Rosato, V.; Li, D.; Silverman, D.; Petersen, G.M.; Bracci, P.M.; Neale, R.E.; Muscat, J.; Anderson, K.; Gallinger, S.; et al. Diabetes, antidiabetic medications, and pancreatic cancer risk: An analysis from the International Pancreatic Cancer Case-Control Consortium. Ann. Oncol. 2014, 25, 2065–2072. [Google Scholar] [CrossRef]
- Yoo, D.; Kim, N.; Hwang, D.W.; Song, K.B.; Lee, J.H.; Lee, W.; Kwon, J.; Park, Y.; Hong, S.; Lee, J.W.; et al. Association between Metformin Use and Clinical Outcomes Following Pancreaticoduodenectomy in Patients with Type 2 Diabetes and Pancreatic Ductal Adenocarcinoma. J. Clin. Med. 2020, 9, 1953. [Google Scholar] [CrossRef]
- Broadhurst, P.J.; Hart, A.R. An observational study to justify and plan a future phase III randomized controlled trial of metformin in improving overall survival in patients with inoperable pancreatic cancer without liver metastases. J. Cancer Res. Clin. Oncol. 2020, 146, 1369–1375. [Google Scholar] [CrossRef] [PubMed]
- Toriola, A.T.; Luo, S.; Thomas, T.S.; Drake, B.F.; Chang, S.H.; Sanfilippo, K.M.; Carson, K.R. Metformin Use and Pancreatic Cancer Survival among Non-Hispanic White and African American U.S. Veterans with Diabetes Mellitus. Cancer Epidemiol. Biomark. Prev. 2020, 29, 169–175. [Google Scholar] [CrossRef]
- Wynn, A.; Vacheron, A.; Zuber, J.; Solomon, S.S. Metformin Associated With Increased Survival in Type 2 Diabetes Patients with Pancreatic Cancer and Lymphoma. Am. J. Med. Sci. 2019, 358, 200–203. [Google Scholar] [CrossRef]
- Cho, J.; Scragg, R.; Pandol, S.J.; Goodarzi, M.O.; Petrov, M.S. Antidiabetic Medications and Mortality Risk in Individuals with Pancreatic Cancer-Related Diabetes and Postpancreatitis Diabetes: A Nationwide Cohort Study. Diabetes Care 2019, 42, 1675–1683. [Google Scholar] [CrossRef]
- Wei, M.; Liu, Y.; Bi, Y.; Zhang, Z.J. Metformin and pancreatic cancer survival: Real effect or immortal time bias? Int. J. Cancer 2019, 145, 1822–1828. [Google Scholar] [CrossRef] [PubMed]
- Bonelli, L.; Aste, H.; Bovo, P.; Cavallini, G.; Felder, M.; Gusmaroli, R.; Morandini, E.; Ravelli, P.; Briglia, R.; Lombardo, L.; et al. Exocrine pancreatic cancer, cigarette smoking, and diabetes mellitus: A case-control study in northern Italy. Pancreas 2003, 27, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Galli, A.; Ceni, E.; Crabb, D.W.; Mello, T.; Salzano, R.; Grappone, C.; Milani, S.; Surrenti, E.; Surrenti, C.; Casini, A. Antidiabetic thiazolidinediones inhibit invasiveness of pancreatic cancer cells via PPARgamma independent mechanisms. Gut 2004, 53, 1688–1697. [Google Scholar] [CrossRef] [PubMed]
- Wen, W.; Wu, P.; Gong, J.; Zhao, M.; Zhang, Z.; Chen, R.; Chen, H.; Sun, J. Association of pioglitazone with increased risk of prostate cancer and pancreatic cancer: A functional network study. Diabetes Ther. 2018, 9, 2229–2243. [Google Scholar] [CrossRef]
- Lewis, J.D.; Habel, L.A.; Quesenberry, C.P.; Strom, B.L.; Peng, T.; Hedderson, M.M.; Ehrlich, S.F.; Mamtani, R.; Bilker, W.; Vaughn, D.J.; et al. Pioglitazone use and risk of bladder cancer and other common cancers in persons with diabetes. JAMA 2015, 314, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Rao Kondapally Seshasai, S.; Kaptoge, S.; Thompson, A.; Di Angelantonio, E.; Gao, P.; Sarwar, N.; Whincup, P.H.; Mukamal, K.J.; Gillum, R.F.; Holme, I.; et al. Diabetes mellitus, fasting glucose, and risk of cause-specific death. N. Engl. J. Med. 2011, 364, 829–841. [Google Scholar] [CrossRef]
- Stolzenberg-Solomon, R.Z.; Graubard, B.I.; Chari, S.; Limburg, P.; Taylor, P.R.; Virtamo, J.; Albanes, D. Insulin, glucose, insulin resistance, and pancreatic cancer in male smokers. JAMA 2005, 294, 2872–2878. [Google Scholar] [CrossRef]
- Wang, F.; Herrington, M.; Larsson, J.; Permert, J. The relationship between diabetes and pancreatic cancer. Mol. Cancer 2003, 2, 4. [Google Scholar] [CrossRef]
- Baynes, J.W.; Thorpe, S.R. Role of oxidative stress in diabetic complications: A new perspective on an old paradigm. Diabetes 1999, 48, 1–9. [Google Scholar] [CrossRef]
- Niwa, T.; Takeda, N.; Yoshizumi, H.; Tatematsu, A.; Ohara, M.; Tomiyama, S.; Niimura, K. Presence of 3-deoxyglucosone, a potent protein crosslinking intermediate of Maillard reaction, in diabetic serum. Biochem. Biophys. Res. Commun. 1993, 196, 837–843. [Google Scholar] [CrossRef]
- Brouwers, O.; Niessen, P.M.; Ferreira, I.; Miyata, T.; Scheffer, P.G.; Teerlink, T.; Schrauwen, P.; Brownlee, M.; Stehouwer, C.D.; Schalkwijk, C.G. Overexpression of glyoxalase-I reduces hyperglycemia-induced levels of advanced glycation end products and oxidative stress in diabetic rats. J. Biol. Chem. 2011, 286, 1374–1380. [Google Scholar] [CrossRef] [PubMed]
- Engelen, L.; Lund, S.S.; Ferreira, I.; Tarnow, L.; Parving, H.H.; Gram, J.; Winther, K.; Pedersen, O.; Teerlink, T.; Barto, R.; et al. Improved glycemic control induced by both metformin and repaglinide is associated with a reduction in blood levels of 3-deoxyglucosone in nonobese patients with type 2 diabetes. Eur. J. Endocrinol. 2011, 164, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.X.; Requena, J.R.; Jenkins, A.J.; Lyons, T.J.; Baynes, J.W.; Thorpe, S.R. The advanced glycation end product, Nepsilon-(carboxymethyl)lysine, is a product of both lipid peroxidation and glycoxidation reactions. J. Biol. Chem. 1996, 271, 9982–9986. [Google Scholar] [CrossRef] [PubMed]
- Bucala, R.; Makita, Z.; Koschinsky, T.; Cerami, A.; Vlassara, H. Lipid advanced glycosylation: Pathway for lipid oxidation in vivo. Proc. Natl. Acad. Sci. USA 1993, 90, 6434–6438. [Google Scholar] [CrossRef]
- Requena, J.R.; Ahmed, M.U.; Fountain, C.W.; Degenhardt, T.P.; Reddy, S.; Perez, C.; Lyons, T.J.; Jenkins, A.J.; Baynes, J.W.; Thorpe, S.R. Carboxymethylethanolamine, a biomarker of phospholipid modification during the maillard reaction in vivo. J. Biol. Chem. 1997, 272, 17473–17479. [Google Scholar] [CrossRef]
- Mistry, N.; Podmore, I.; Cooke, M.; Butler, P.; Griffiths, H.; Herbert, K.; Lunec, J. Novel monoclonal antibody recognition of oxidative DNA damage adduct, deoxycytidine-glyoxal. Lab. Investig. 2003, 83, 241–250. [Google Scholar] [CrossRef]
- Negre-Salvayre, A.; Coatrieux, C.; Ingueneau, C.; Salvayre, R. Advanced lipid peroxidation end products in oxidative damage to proteins. Potential role in diseases and therapeutic prospects for the inhibitors. Br. J. Pharmacol. 2008, 153, 6–20. [Google Scholar] [CrossRef]
- Mol, M.; Degani, G.; Coppa, C.; Baron, G.; Popolo, L.; Carini, M.; Aldini, G.; Vistoli, G.; Altomare, A. Advanced lipoxidation end products (ALEs) as RAGE binders: Mass spectrometric and computational studies to explain the reasons why. Redox Biol. 2019, 23, 101083. [Google Scholar] [CrossRef]
- Thornalley, P.J. Endogenous alpha-oxoaldehydes and formation of protein and nucleotide advanced glycation endproducts in tissue damage. Novartis Found. Symp. 2007, 285, 229–243. [Google Scholar] [CrossRef]
- Xue, M.; Weickert, M.O.; Qureshi, S.; Kandala, N.B.; Anwar, A.; Waldron, M.; Shafie, A.; Messenger, D.; Fowler, M.; Jenkins, G.; et al. Improved Glycemic Control and Vascular Function in Overweight and Obese Subjects by Glyoxalase 1 Inducer Formulation. Diabetes 2016, 65, 2282–2294. [Google Scholar] [CrossRef]
- Phillips, S.A.; Thornalley, P.J. The formation of methylglyoxal from triose phosphates. Investigation using a specific assay for methylglyoxal. Eur. J. Biochem. 1993, 212, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Reddy, S.; Bichler, J.; Wells-Knecht, K.J.; Thorpe, S.R.; Baynes, J.W. N epsilon-(carboxymethyl)lysine is a dominant advanced glycation end product (AGE) antigen in tissue proteins. Biochemistry 1995, 34, 10872–10878. [Google Scholar] [CrossRef] [PubMed]
- Vlassara, H. Advanced glycation in health and disease: Role of the modern environment. Ann. N. Y. Acad. Sci. 2005, 1043, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Cerami, C.; Founds, H.; Nicholl, I.; Mitsuhashi, T.; Giordano, D.; Vanpatten, S.; Lee, A.; Al-Abed, Y.; Vlassara, H.; Bucala, R.; et al. Tobacco smoke is a source of toxic reactive glycation products. Proc. Natl. Acad. Sci. USA 1997, 94, 13915–13920. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, S.; Matsui, T.; Nakamura, K. Possible involvement of tobacco-derived advanced glycation end products (AGEs) in an increased risk for developing cancers and cardiovascular disease in former smokers. Med. Hypotheses 2008, 71, 259–261. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N. Advanced glycation endproducts--role in pathology of diabetic complications. Diabetes Res. Clin. Pract. 2005, 67, 3–21. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Akhter, F.; Shahab, U.; Rafi, Z.; Khan, M.S.; Nabi, R.; Khan, M.S.; Ahmad, K.; Ashraf, J.M.; Moinuddin. Do all roads lead to the Rome? The glycation perspective! Semin. Cancer Biol. 2018, 49, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Khan, H.; Siddiqui, Z.; Khan, M.Y.; Rehman, S.; Shahab, U.; Godovikova, T.; Silnikov, V.; Moinuddin. AGEs, RAGEs and s-RAGE; friend or foe for cancer. Semin. Cancer Biol. 2018, 49, 44–55. [Google Scholar] [CrossRef]
- Jiao, L.; Stolzenberg-Solomon, R.; Zimmerman, T.P.; Duan, Z.; Chen, L.; Kahle, L.; Risch, A.; Subar, A.F.; Cross, A.J.; Hollenbeck, A.; et al. Dietary consumption of advanced glycation end products and pancreatic cancer in the prospective NIH-AARP Diet and Health Study. Am. J. Clin. Nutr. 2015, 101, 126–134. [Google Scholar] [CrossRef]
- Chavakis, T.; Bierhaus, A.; Al-Fakhri, N.; Schneider, D.; Witte, S.; Linn, T.; Nagashima, M.; Morser, J.; Arnold, B.; Preissner, K.T.; et al. The pattern recognition receptor (RAGE) is a counterreceptor for leukocyte integrins: A novel pathway for inflammatory cell recruitment. J. Exp. Med. 2003, 198, 1507–1515. [Google Scholar] [CrossRef]
- Teissier, T.; Boulanger, É. The receptor for advanced glycation end-products (RAGE) is an important pattern recognition receptor (PRR) for inflammaging. Biogerontology 2019, 20, 279–301. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Toyomura, T.; Wake, H.; Liu, K.; Teshigawara, K.; Takahashi, H.; Nishibori, M.; Mori, S. Differential contribution of possible pattern-recognition receptors to advanced glycation end product-induced cellular responses in macrophage-like RAW264.7 cells. Biotechnol. Appl. Biochem. 2020, 67, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Manigrasso, M.B.; Juranek, J.; Ramasamy, R.; Schmidt, A.M. Unlocking the biology of RAGE in diabetic microvascular complications. Trends Endocrinol. Metab. 2014, 25, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Soman, S.; Raju, R.; Sandhya, V.K.; Advani, J.; Khan, A.A.; Harsha, H.C.; Prasad, T.S.; Sudhakaran, P.R.; Pandey, A.; Adishesha, P.K. A multicellular signal transduction network of AGE/RAGE signaling. J. Cell Commun. Signal. 2013, 7, 19–23. [Google Scholar] [CrossRef]
- Aldini, G.; Facino, R.M.; Beretta, G.; Carini, M. Carnosine and related dipeptides as quenchers of reactive carbonyl species: From structural studies to therapeutic perspectives. Biofactors 2005, 24, 77–87. [Google Scholar] [CrossRef]
- Regazzoni, L.; de Courten, B.; Garzon, D.; Altomare, A.; Marinello, C.; Jakubova, M.; Vallova, S.; Krumpolec, P.; Carini, M.; Ukropec, J.; et al. A carnosine intervention study in overweight human volunteers: Bioavailability and reactive carbonyl species sequestering effect. Sci. Rep. 2016, 6, 27224. [Google Scholar] [CrossRef]
- Baye, E.; Ukropec, J.; de Courten, M.P.J.; Mousa, A.; Kurdiova, T.; Johnson, J.; Wilson, K.; Plebanski, M.; Aldini, G.; Ukropcova, B.; et al. Carnosine Supplementation Improves Serum Resistin Concentrations in Overweight or Obese Otherwise Healthy Adults: A Pilot Randomized Trial. Nutrients 2018, 10, 1258. [Google Scholar] [CrossRef]
- Anderson, E.J.; Vistoli, G.; Katunga, L.A.; Funai, K.; Regazzoni, L.; Monroe, T.B.; Gilardoni, E.; Cannizzaro, L.; Colzani, M.; De Maddis, D.; et al. A carnosine analog mitigates metabolic disorders of obesity by reducing carbonyl stress. J. Clin. Investig. 2018, 128, 5280–5293. [Google Scholar] [CrossRef]
- Haus, J.M.; Thyfault, J.P. Therapeutic potential of carbonyl-scavenging carnosine derivative in metabolic disorders. J. Clin. Investig. 2018, 128, 5198–5200. [Google Scholar] [CrossRef]
- Menini, S.; Iacobini, C.; Ricci, C.; Fantauzzi, C.B.; Pugliese, G. Protection from diabetes-induced atherosclerosis and renal disease by D-carnosine-octylester: Effects of early vs. late inhibition of advanced glycation end-products in Apoe-null mice. Diabetologia 2015, 58, 845–853. [Google Scholar] [CrossRef]
- Menini, S.; Iacobini, C.; Ricci, C.; Scipioni, A.; Blasetti Fantauzzi, C.; Giaccari, A.; Salomone, E.; Canevotti, R.; Lapolla, A.; Orioli, M.; et al. D-Carnosine octylester attenuates atherosclerosis and renal disease in ApoE null mice fed a Western diet through reduction of carbonyl stress and inflammation. Br. J. Pharmacol. 2012, 166, 1344–1356. [Google Scholar] [CrossRef] [PubMed]
- Iacobini, C.; Menini, S.; Blasetti Fantauzzi, C.; Pesce, C.M.; Giaccari, A.; Salomone, E.; Lapolla, A.; Orioli, M.; Aldini, G.; Pugliese, G. FL-926-16, a novel bioavailable carnosinase-resistant carnosine derivative, prevents onset and stops progression of diabetic nephropathy in db/db mice. Br. J. Pharmacol. 2018, 175, 53–66. [Google Scholar] [CrossRef] [PubMed]
- Menini, S.; Iacobini, C.; Fantauzzi, C.B.; Pugliese, G. L-carnosine and its Derivatives as New Therapeutic Agents for the Prevention and Treatment of Vascular Complications of Diabetes. Curr. Med. Chem. 2020, 27, 1744–1763. [Google Scholar] [CrossRef] [PubMed]
- Bakris, G.L.; Bank, A.J.; Kass, D.A.; Neutel, J.M.; Preston, R.A.; Oparil, S. Advanced glycation end-product cross-link breakers. A novel approach to cardiovascular pathologies related to the aging process. Am. J. Hypertens. 2004, 17, 23S–30S. [Google Scholar] [CrossRef] [PubMed]
- Reddy, V.P.; Beyaz, A. Inhibitors of the Maillard reaction and AGE breakers as therapeutics for multiple diseases. Drug Discov. Today 2006, 11, 646–654. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.F.; Ramasamy, R.; Naka, Y.; Schmidt, A.M. Glycation, inflammation, and RAGE: A scaffold for the macrovascular complications of diabetes and beyond. Circ. Res. 2003, 93, 1159–1169. [Google Scholar] [CrossRef]
- Ellis, E.M. Reactive carbonyls and oxidative stress: Potential for therapeutic intervention. Pharmacol. Ther. 2007, 115, 13–24. [Google Scholar] [CrossRef]
- Aldini, G.; Dalle-Donne, I.; Facino, R.M.; Milzani, A.; Carini, M. Intervention strategies to inhibit protein carbonylation by lipoxidation-derived reactive carbonyls. Med. Res. Rev. 2007, 27, 817–868. [Google Scholar] [CrossRef]
- Bolton, W.K.; Cattran, D.C.; Williams, M.E.; Adler, S.G.; Appel, G.B.; Cartwright, K.; Foiles, P.G.; Freedman, B.I.; Raskin, P.; Ratner, R.E.; et al. Randomized trial of an inhibitor of formation of advanced glycation end products in diabetic nephropathy. Am. J. Nephrol. 2004, 24, 32–40. [Google Scholar] [CrossRef]
- Raval, A.D.; Thakker, D.; Rangoonwala, A.N.; Gor, D.; Walia, R. Vitamin B and its derivatives for diabetic kidney disease. Cochrane Database Syst. Rev. 2015, 1, CD009403. [Google Scholar] [CrossRef]
- Williams, M.E.; Bolton, W.K.; Khalifah, R.G.; Degenhardt, T.P.; Schotzinger, R.J.; McGill, J.B. Effects of pyridoxamine in combined phase 2 studies of patients with type 1 and type 2 diabetes and overt nephropathy. Am. J. Nephrol. 2007, 27, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Lewis, E.J.; Greene, T.; Spitalewiz, S.; Blumenthal, S.; Berl, T.; Hunsicker, L.G.; Pohl, M.A.; Rohde, R.D.; Raz, I.; Yerushalmy, Y.; et al. Pyridorin in type 2 diabetic nephropathy. J. Am. Soc. Nephrol. 2012, 23, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, T.; Sadzuka, Y. In vitro anticancer activities of B6 vitamers: A mini-review. Anticancer Res. 2019, 39, 3429–3432. [Google Scholar] [CrossRef] [PubMed]
- Drozak, J.; Veiga-da-Cunha, M.; Vertommen, D.; Stroobant, V.; Van Schaftingen, E. Molecular identification of carnosine synthase as ATP-grasp domain-containing protein 1 (ATPGD1). J. Biol. Chem. 2010, 285, 9346–9356. [Google Scholar] [CrossRef]
- Peters, V.; Klessens, C.Q.; Baelde, H.J.; Singler, B.; Veraar, K.A.; Zutinic, A.; Drozak, J.; Zschocke, J.; Schmitt, C.P.; de Heer, E. Intrinsic carnosine metabolism in the human kidney. Amino Acids 2015, 47, 2541–2550. [Google Scholar] [CrossRef]
- Guiotto, A.; Ruzza, P.; Babizhayev, M.A.; Calderan, A. Malondialdehyde scavenging and aldose-derived Schiff bases’ transglycation properties of synthetic histidyl-hydrazide carnosine analogs. Bioorg. Med. Chem. 2007, 1, 6158–6163. [Google Scholar] [CrossRef]
- Nokin, M.J.; Durieux, F.; Peixoto, P.; Chiavarina, B.; Peulen, O.; Blomme, A.; Turtoi, A.; Costanza, B.; Smargiasso, N.; Baiwir, D.; et al. Methylglyoxal, a glycolysis side-product, induces Hsp90 glycation and YAP-mediated tumor growth and metastasis. Elife 2016, 5, e19375. [Google Scholar] [CrossRef]
- Weigand, T.; Singler, B.; Fleming, T.; Nawroth, P.; Klika, K.D.; Thiel, C.; Baelde, H.; Garbade, S.F.; Wagner, A.H.; Hecker, M.; et al. Carnosine Catalyzes the Formation of the Oligo/Polymeric Products of Methylglyoxal. Cell. Physiol. Biochem. 2018, 46, 713–726. [Google Scholar] [CrossRef]
- Sousa Silva, M.; Gomes, R.A.; Ferreira, A.E.; Ponces Freire, A.; Cordeiro, C. The glyoxalase pathway: The first hundred years… and beyond. Biochem. J. 2013, 453, 1–15. [Google Scholar] [CrossRef]
- Janssen, B.; Hohenadel, D.; Brinkkoetter, P.; Peters, V.; Rind, N.; Fischer, C.; Rychlik, I.; Cerna, M.; Romzova, M.; de Heer, E.; et al. Carnosine as a protective factor in diabetic nephropathy: Association with a leucine repeat of the carnosinase gene CNDP1. Diabetes 2005, 54, 2320–2327. [Google Scholar] [CrossRef]
- Albrecht, T.; Schilperoort, M.; Zhang, S.; Braun, J.D.; Qiu, J.; Rodriguez, A.; Pastene, D.O.; Krämer, B.K.; Köppel, H.; Baelde, H.; et al. Carnosine Attenuates the Development of both Type 2 Diabetes and Diabetic Nephropathy in BTBR ob/ob Mice. Sci. Rep. 2017, 7, 44492. [Google Scholar] [CrossRef] [PubMed]
- Menini, S.; Iacobini, C.; Pugliese, G.; Pesce, C. Dietary interventions to contrast the onset and progression of diabetic nephropathy: A critical survey of new data. Crit. Rev. Food Sci. Nutr. 2018, 58, 1671–1680. [Google Scholar] [CrossRef] [PubMed]
- Aldini, G.; Orioli, M.; Rossoni, G.; Savi, F.; Braidotti, P.; Vistoli, G.; Yeum, K.J.; Negrisoli, G.; Carini, M. The carbonyl scavenger carnosine ameliorates dyslipidaemia and renal function in Zucker obese rats. J. Cell. Mol. Med. 2011, 15, 1339–1354. [Google Scholar] [CrossRef] [PubMed]
- Sauerhöfer, S.; Yuan, G.; Braun, G.; Deinzer, M.; Neumaier, M.; Gretz, N.; Floege, J.; Kriz, W.; van der Woude, F.; Moeller, M. L-carnosine, a substrate of carnosinase-1, influences glucose metabolism. Diabetes 2007, 56, 2425–2432. [Google Scholar] [CrossRef]
- Alkhalaf, A.; Bakker, S.J.; Bilo, H.J.; Gans, R.O.; Navis, G.J.; Postmus, D.; Forsblom, C.; Groop, P.H.; Vionnet, N.; Hadjadj, S.; et al. A polymorphism in the gene encoding carnosinase (CNDP1) as a predictor of mortality and progression from nephropathy to end-stage renal disease in type 1 diabetes mellitus. Diabetologia 2010, 53, 2562–2568. [Google Scholar] [CrossRef]
- Vistoli, G.; Carini, M.; Aldini, G. Transforming dietary peptides in promising lead compounds: The case of bioavailable carnosine analogs. Amino Acids 2012, 43, 111–126. [Google Scholar] [CrossRef]
- Apple, M.A.; Greenberg, D.M. Inhibition of cancer growth in mice by a normal metabolite. Life Sci. 1967, 6, 2157–2160. [Google Scholar] [CrossRef]
- Kang, Y.; Edwards, L.G.; Thornalley, P.J. Effect of methylglyoxal on human leukaemia 60 cell growth: Modification of DNA G1 growth arrest and induction of apoptosis. Leuk. Res. 1996, 20, 397–405. [Google Scholar] [CrossRef]
- Calabrese, E.J.; Blain, R.B. The hormesis database: The occurrence of hormetic dose responses in the toxicological literature. Regul. Toxicol. Pharmacol. 2011, 61, 73–81. [Google Scholar] [CrossRef]
- Oliveira, M.F.; Geihs, M.A.; França, T.F.A.; Moreira, D.C.; Hermes-Lima, M. Is “Preparation for Oxidative Stress” a Case of Physiological Conditioning Hormesis? Front. Physiol. 2018, 9, 945. [Google Scholar] [CrossRef]
- Calabrese, E.J. Hormesis and medicine. Br. J. Clin. Pharmacol. 2008, 66, 594–617. [Google Scholar] [CrossRef] [PubMed]
- Nokin, M.J.; Durieux, F.; Bellier, J.; Peulen, O.; Uchida, K.; Spiegel, D.A.; Cochrane, J.R.; Hutton, C.A.; Castronovo, V.; Bellahcène, A. Hormetic potential of methylglyoxal, a side-product of glycolysis, in switching tumours from growth to death. Sci. Rep. 2017, 7, 11722. [Google Scholar] [CrossRef] [PubMed]
- Gentile, F.; Arcaro, A.; Pizzimenti, S.; Daga, M.; Cetrangolo, G.P.; Dianzani, C.; Lepore, A.; Graf, M.; Ames, P.R.J.; Barrera, G. DNA damage by lipid peroxidation products: Implications in cancer, inflammation and autoimmunity. AIMS Genet. 2017, 4, 103–137. [Google Scholar] [CrossRef] [PubMed]
- Zarkovic, N.; Ilic, Z.; Jurin, M.; Schaur, R.J.; Puhl, H.; Esterbauer, H. Stimulation of HeLa cell growth by physiological concentrations of 4-hydroxynonenal. Cell Biochem. Funct. 1993, 11, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef]
- Yan, L.; Raj, P.; Yao, W.; Ying, H. Glucose metabolism in pancreatic cancer. Cancers 2019, 11, 1460. [Google Scholar] [CrossRef]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.; et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012, 149, 656–670. [Google Scholar] [CrossRef]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef]
- Fantin, V.R.; St-Pierre, J.; Leder, P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell 2006, 9, 425–434. [Google Scholar] [CrossRef]
- Patra, K.C.; Wang, Q.; Bhaskar, P.T.; Miller, L.; Wang, Z.; Wheaton, W.; Chandel, N.; Laakso, M.; Muller, W.J.; Allen, E.L.; et al. Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer. Cancer Cell 2013, 24, 213–228. [Google Scholar] [CrossRef]
- Newsholme, E.A.; Crabtree, B.; Ardawi, M.S. The role of high rates of glycolysis and glutamine utilization in rapidly dividing cells. Biosci. Rep. 1985, 5, 393–400. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Chandel, N.S. We need to talk about the Warburg effect. Nat. Metab. 2020, 2, 127–129. [Google Scholar] [CrossRef] [PubMed]
- Ramteke, P.; Deb, A.; Shepal, V.; Bhat, M.K. Hyperglycemia Associated Metabolic and Molecular Alterations in Cancer Risk, Progression, Treatment, and Mortality. Cancers 2019, 11, 1402. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, N.; Thornalley, P.J. The critical role of methylglyoxal and glyoxalase 1 in diabetic nephropathy. Diabetes 2014, 63, 50–52. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Rask-Madsen, C.; King, G.L. Vascular complications of diabetes: Mechanisms of injury and protective factors. Cell Metab. 2013, 17, 20–33. [Google Scholar] [CrossRef]
- Chiavarina, B.; Nokin, M.J.; Durieux, F.; Bianchi, E.; Turtoi, A.; Peulen, O.; Peixoto, P.; Irigaray, P.; Uchida, K.; Belpomme, D.; et al. Triple negative tumors accumulate significantly less methylglyoxal specific adducts than other human breast cancer subtypes. Oncotarget 2014, 5, 5472–5482. [Google Scholar] [CrossRef]
- Nokin, M.J.; Bellier, J.; Durieux, F.; Peulen, O.; Rademaker, G.; Gabriel, M.; Monseur, C.; Charloteaux, B.; Verbeke, L.; van Laere, S.; et al. Methylglyoxal, a glycolysis metabolite, triggers metastasis through MEK/ERK/SMAD1 pathway activation in breast cancer. Breast Cancer Res. 2019, 21, 11. [Google Scholar] [CrossRef]
- Bellier, J.; Nokin, M.J.; Caprasse, M.; Tiamiou, A.; Blomme, A.; Scheijen, J.L.; Koopmansch, B.; MacKay, G.M.; Chiavarina, B.; Costanza, B.; et al. Methylglyoxal Scavengers Resensitize KRAS-Mutated Colorectal Tumors to Cetuximab. Cell Rep. 2020, 30, 1400–1416.e6. [Google Scholar] [CrossRef]
- Wondrak, G.T.; Jacobson, M.K.; Jacobson, E.L. Antimelanoma activity of apoptogenic carbonyl scavengers. J. Pharmacol. Exp. Ther. 2006, 316, 805–814. [Google Scholar] [CrossRef]
- Zhang, W.; Nandakumar, N.; Shi, Y.; Manzano, M.; Smith, A.; Graham, G.; Gupta, S.; Vietsch, E.E.; Laughlin, S.Z.; Wadhwa, M.; et al. Downstream of mutant KRAS, the transcription regulator YAP is essential for neoplastic progression to pancreatic ductal adenocarcinoma. Sci. Signal. 2014, 7, ra42. [Google Scholar] [CrossRef] [PubMed]
- Pan, D. The hippo signaling pathway in development and cancer. Dev. Cell 2010, 19, 491–505. [Google Scholar] [CrossRef] [PubMed]
- van Heijst, J.W.; Niessen, H.W.; Musters, R.J.; van Hinsbergh, V.W.; Hoekman, K.; Schalkwijk, C.G. Argpyrimidine-modified Heat shock protein 27 in human non-small cell lung cancer: A possible mechanism for evasion of apoptosis. Cancer Lett. 2006, 241, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Oya-Ito, T.; Naito, Y.; Takagi, T.; Handa, O.; Matsui, H.; Yamada, M.; Shima, K.; Yoshikawa, T. Heat-shock protein 27 (Hsp27) as a target of methylglyoxal in gastrointestinal cancer. Biochim. Biophys. Acta 2011, 1812, 769–781. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, H.; Mashima, T.; Yamamoto, K.; Tsuruo, T. Modulation of heat-shock protein 27 (Hsp27) anti-apoptotic activity by methylglyoxal modification. J. Biol. Chem. 2002, 277, 45770–45775. [Google Scholar] [CrossRef] [PubMed]
- Schröter, D.; Höhn, A. Role of Advanced Glycation End Products in Carcinogenesis and their Therapeutic Implications. Curr. Pharm. Des. 2018, 24, 5245–5251. [Google Scholar] [CrossRef]
- Nguyen, A.H.; Detty, S.Q.; Agrawal, D.K. Clinical Implications of High-mobility Group Box-1 (HMGB1) and the Receptor for Advanced Glycation End-products (RAGE) in Cutaneous Malignancy: A Systematic Review. Anticancer Res. 2017, 37, 1–7. [Google Scholar] [CrossRef]
- Sims, G.P.; Rowe, D.C.; Rietdijk, S.T.; Herbst, R.; Coyle, A.J. HMGB1 and RAGE in inflammation and cancer. Annu. Rev. Immunol. 2010, 28, 367–388. [Google Scholar] [CrossRef]
- Menini, S.; Iacobini, C.; de Latouliere, L.; Manni, I.; Ionta, V.; Fantauzzi, C.B.; Pesce, C.; Cappello, P.; Novelli, F.; Piaggio, G.; et al. The advanced glycation end-product N-epsilon-carboxymethyllysine promotes progression of pancreatic cancer: Implications for diabetes-associated risk and its prevention. J. Pathol. 2018, 245, 197–208. [Google Scholar] [CrossRef]
- Avgerinos, K.I.; Spyrou, N.; Mantzoros, C.S.; Dalamaga, M. Obesity and cancer risk: Emerging biological mechanisms and perspectives. Metabolism 2019, 92, 121–135. [Google Scholar] [CrossRef]
- Fiolet, T.; Srour, B.; Sellem, L.; Kesse-Guyot, E.; Allès, B.; Méjean, C.; Deschasaux, M.; Fassier, P.; Latino-Martel, P.; Beslay, M.; et al. Consumption of ultra-processed foods and cancer risk: Results from NutriNet-Santé prospective cohort. BMJ 2018, 360, k322. [Google Scholar] [CrossRef] [PubMed]
- Matou-Nasri, S.; Sharaf, H.; Wang, Q.; Almobadel, N.; Rabhan, Z.; Al-Eidi, H.; Yahya, W.B.; Trivilegio, T.; Ali, R.; Al-Shanti, N.; et al. Biological impact of advanced glycation endproducts on estrogen receptor-positive MCF-7 breast cancer cells. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2808–2820. [Google Scholar] [CrossRef] [PubMed]
- Takino, J.; Nagamine, K.; Hori, T.; Sakasai-Sakai, A.; Takeuchi, M. Contribution of the toxic advanced glycation end-products-receptor axis in nonalcoholic steatohepatitis-related hepatocellular carcinoma. World J. Hepatol. 2015, 7, 2459–2469. [Google Scholar] [CrossRef] [PubMed]
- Hollenbach, M. The Role of Glyoxalase-I (Glo-I), Advanced Glycation Endproducts (AGEs), and Their Receptor (RAGE) in Chronic Liver Disease and Hepatocellular Carcinoma (HCC). Int. J. Mol. Sci. 2017, 18, 2466. [Google Scholar] [CrossRef]
- Yamagishi, S.; Nakamura, K.; Inoue, H.; Kikuchi, S.; Takeuchi, M. Possible participation of advanced glycation end products in the pathogenesis of colorectal cancer in diabetic patients. Med. Hypotheses 2005, 64, 1208–1210. [Google Scholar] [CrossRef]
- Miki, S.; Kasayama, S.; Miki, S.; Nakamura, Y.; Yamamoto, M.; Sato, B.; Kishimoto, T. Expression of receptors for advanced glycosylation end products on renal cell carcinoma cells in vitro. Biochem. Biophys. Res. Commun. 1993, 196, 984–989. [Google Scholar] [CrossRef]
- Lin, L.; Zhong, K.; Sun, Z.; Wu, G.; Ding, G. Receptor for advanced glycation end products (RAGE) partially mediates HMGB1-ERKs activation in clear cell renal cell carcinoma. J. Cancer Res. Clin. Oncol. 2012, 138, 11–22. [Google Scholar] [CrossRef]
- Yan, S.F.; Barile, G.R.; D’Agati, V.; Du Yan, S.; Ramasamy, R.; Schmidt, A.M. The biology of RAGE and its ligands: Uncovering mechanisms at the heart of diabetes and its complications. Curr. Diabetes Rep. 2007, 7, 146–153. [Google Scholar] [CrossRef]
- Riehl, A.; Németh, J.; Angel, P.; Hess, J. The receptor RAGE: Bridging inflammation and cancer. Cell Commun. Signal. 2009, 7, 12. [Google Scholar] [CrossRef]
- Kang, R.; Hou, W.; Zhang, Q.; Chen, R.; Lee, Y.J.; Bartlett, D.L.; Lotze, M.T.; Tang, D.; Zeh, H.J. RAGE is essential for oncogenic KRAS-mediated hypoxic signaling in pancreatic cancer. Cell Death Dis. 2014, 5, e1480. [Google Scholar] [CrossRef]
- Gebhardt, C.; Riehl, A.; Durchdewald, M.; Németh, J.; Fürstenberger, G.; Müller-Decker, K.; Enk, A.; Arnold, B.; Bierhaus, A.; Nawroth, P.P.; et al. RAGE signaling sustains inflammation and promotes tumor development. J. Exp. Med. 2008, 205, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, A.; Blood, D.C.; del Toro, G.; Canet, A.; Lee, D.C.; Qu, W.; Tanji, N.; Lu, Y.; Lalla, E.; Fu, C.; et al. Blockade of RAGE-amphoterin signalling suppresses tumour growth and metastases. Nature 2000, 405, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Tang, D.; Schapiro, N.E.; Livesey, K.M.; Farkas, A.; Loughran, P.; Bierhaus, A.; Lotze, M.T.; Zeh, H.J. The receptor for advanced glycation end products (RAGE) sustains autophagy and limits apoptosis, promoting pancreatic tumor cell survival. Cell Death Differ. 2010, 17, 666–676. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.D.; Schmidt, A.M.; Anderson, G.M.; Zhang, J.; Brett, J.; Zou, Y.S.; Pinsky, D.; Stern, D. Enhanced cellular oxidant stress by the interaction of advanced glycation endproducts with their receptors/binding proteins. J. Biol. Chem. 1994, 269, 9889–9897. [Google Scholar] [CrossRef]
- Ott, C.; Jacobs, K.; Haucke, E.; Navarrete Santos, A.; Grune, T.; Simm, A. Role of advanced glycation end products in cellular signaling. Redox Biol. 2014, 2, 411–429. [Google Scholar] [CrossRef] [PubMed]
- Pusterla, T.; Nèmeth, J.; Stein, I.; Wiechert, L.; Knigin, D.; Marhenke, S.; Longerich, T.; Kumar, V.; Arnold, B.; Vogel, A.; et al. Receptor for advanced glycation endproducts (RAGE) is a key regulator of oval cell activation and inflammation-associated liver carcinogenesis in mice. Hepatology 2013, 58, 363–373. [Google Scholar] [CrossRef]
- Kajikawa, M.; Nakashima, A.; Fujimura, N.; Maruhashi, T.; Iwamoto, Y.; Iwamoto, A.; Matsumoto, T.; Oda, N.; Hidaka, T.; Kihara, Y.; et al. Ratio of serum levels of AGEs to soluble form of RAGE is a predictor of endothelial function. Diabetes Care 2015, 38, 119–125. [Google Scholar] [CrossRef]
- Jiao, L.; Weinstein, S.J.; Albanes, D.; Taylor, P.R.; Graubard, B.I.; Virtamo, J.; Stolzenberg-Solomon, R.Z. Evidence that serum levels of the soluble receptor for advanced glycation end products are inversely associated with pancreatic cancer risk: A prospective study. Cancer Res. 2011, 71, 3582–3589. [Google Scholar] [CrossRef]
- White, D.L.; Hoogeveen, R.C.; Chen, L.; Richardson, P.; Ravishankar, M.; Shah, P.; Tinker, L.; Rohan, T.; Whitsel, E.A.; El-Serag, H.B.; et al. A prospective study of soluble receptor for advanced glycation end products and adipokines in association with pancreatic cancer in postmenopausal women. Cancer Med. 2018, 7, 2180–2191. [Google Scholar] [CrossRef]
- Azizan, N.; Suter, M.A.; Liu, Y.; Logsdon, C.D. RAGE maintains high levels of NFκB and oncogenic Kras activity in pancreatic cancer. Biochem. Biophys. Res. Commun. 2017, 493, 592–597. [Google Scholar] [CrossRef]
- Kang, R.; Loux, T.; Tang, D.; Schapiro, N.E.; Vernon, P.; Livesey, K.M.; Krasinskas, A.; Lotze, M.T.; Zeh, H.J. The expression of the receptor for advanced glycation endproducts (RAGE) is permissive for early pancreatic neoplasia. Proc. Natl. Acad. Sci. USA 2012, 109, 7031–7036. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, T.; Ramachandran, V.; Gomez, S.B.; Schmidt, A.M.; Logsdon, C.D. S100P-derived RAGE antagonistic peptide reduces tumor growth and metastasis. Clin. Cancer Res. 2012, 18, 4356–4364. [Google Scholar] [CrossRef] [PubMed]
- Vernon, P.J.; Loux, T.J.; Schapiro, N.E.; Kang, R.; Muthuswamy, R.; Kalinski, P.; Tang, D.; Lotze, M.T.; Zeh, H.J. The receptor for advanced glycation end products promotes pancreatic carcinogenesis and accumulation of myeloid-derived suppressor cells. J. Immunol. 2013, 190, 1372–1379. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Tang, D.; Livesey, K.M.; Schapiro, N.E.; Lotze, M.T.; Zeh, H.J. The Receptor for Advanced Glycation End-products (RAGE) protects pancreatic tumor cells against oxidative injury. Antioxid. Redox Signal. 2011, 15, 2175–2184. [Google Scholar] [CrossRef]
- Kang, R.; Tang, D.; Lotze, M.T.; Zeh, H.J. AGER/RAGE-mediated autophagy promotes pancreatic tumorigenesis and bioenergetics through the IL6-pSTAT3 pathway. Autophagy 2012, 8, 989–991. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Tang, D.; Schapiro, N.E.; Loux, T.; Livesey, K.M.; Billiar, T.R.; Wang, H.; Van Houten, B.; Lotze, M.T.; Zeh, H.J. The HMGB1/RAGE inflammatory pathway promotes pancreatic tumor growth by regulating mitochondrial bioenergetics. Oncogene 2014, 33, 567–577. [Google Scholar] [CrossRef]
- Li, J.; Schmidt, A.M. Characterization and functional analysis of the promoter of RAGE, the receptor for advanced glycation endproducts. J. Biol. Chem. 1997, 272, 16498–16506. [Google Scholar] [CrossRef]
- von Bauer, R.; Oikonomou, D.; Sulaj, A.; Mohammed, S.; Hotz-Wagenblatt, A.; Gröne, H.J.; Arnold, B.; Falk, C.; Luethje, D.; Erhardt, A.; et al. CD166/ALCAM mediates proinflammatory effects of S100B in delayed type hypersensitivity. J. Immunol. 2013, 191, 369–377. [Google Scholar] [CrossRef]
- Münsterberg, J.; Loreth, D.; Brylka, L.; Werner, S.; Karbanova, J.; Gandrass, M.; Schneegans, S.; Besler, K.; Hamester, F.; Robador, J.R.; et al. ALCAM contributes to brain metastasis formation in non-small-cell lung cancer through interaction with the vascular endothelium. Neuro Oncol. 2020, 22, 955–966. [Google Scholar] [CrossRef]
- Kahlert, C.; Weber, H.; Mogler, C.; Bergmann, F.; Schirmacher, P.; Kenngott, H.G.; Matterne, U.; Mollberg, N.; Rahbari, N.N.; Hinz, U.; et al. Increased expression of ALCAM/CD166 in pancreatic cancer is an independent prognostic marker for poor survival and early tumour relapse. Br. J. Cancer 2009, 101, 457–464. [Google Scholar] [CrossRef]
- Menini, S.; Iacobini, C.; de Latouliere, L.; Manni, I.; Vitale, M.; Pilozzi, E.; Pesce, C.; Cappello, P.; Novelli, F.; Piaggio, G.; et al. Diabetes promotes invasive pancreatic cancer by increasing systemic and tumour carbonyl stress in Kras G12D/+ mice. J. Exp. Clin. Cancer Res. 2020, 39, 152. [Google Scholar] [CrossRef] [PubMed]
- Gruber, R.; Panayiotou, R.; Nye, E.; Spencer-Dene, B.; Stamp, G.; Behrens, A. YAP1 and TAZ Control Pancreatic Cancer Initiation in Mice by Direct Up-regulation of JAK-STAT3 Signaling. Gastroenterology 2016, 151, 526–539. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Chun, A.; Cheung, K.; Rashidi, B.; Yang, X. Tumor suppressor LATS1 is a negative regulator of oncogene YAP. J. Biol. Chem. 2008, 283, 5496–5509. [Google Scholar] [CrossRef] [PubMed]
- Navas, C.; Hernández-Porras, I.; Schuhmacher, A.J.; Sibilia, M.; Guerra, C.; Barbacid, M. EGF receptor signaling is essential for k-ras oncogene-driven pancreatic ductal adenocarcinoma. Cancer Cell 2012, 22, 318–330. [Google Scholar] [CrossRef]
- Blasco, M.T.; Navas, C.; Martín-Serrano, G.; Graña-Castro, O.; Lechuga, C.G.; Martín-Díaz, L.; Djurec, M.; Li, J.; Morales-Cacho, L.; Esteban-Burgos, L.; et al. Complete Regression of Advanced Pancreatic Ductal Adenocarcinomas upon Combined Inhibition of EGFR and C-RAF. Cancer Cell 2019, 35, 573–587.e6. [Google Scholar] [CrossRef]
- Harding, J.L.; Shaw, J.E.; Peeters, A.; Cartensen, B.; Magliano, D.J. Cancer risk among people with type 1 and type 2 diabetes: Disentangling true associations, detection bias, and reverse causation. Diabetes Care 2015, 38, 734–735. [Google Scholar] [CrossRef]
- Kozuka, S.; Sassa, R.; Taki, T.; Masamoto, K.; Nagasawa, S.; Saga, S.; Hasegawa, K.; Takeuchi, M. Relation of pancreatic duct hyperplasia to carcinoma. Cancer 1979, 43, 1418–1428. [Google Scholar] [CrossRef]
- Kanda, M.; Matthaei, H.; Wu, J.; Hong, S.M.; Yu, J.; Borges, M.; Hruban, R.H.; Maitra, A.; Kinzler, K.; Vogelstein, B.; et al. Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia. Gastroenterology 2012, 142, 730–733.e9. [Google Scholar] [CrossRef]
- Hu, C.M.; Tien, S.C.; Hsieh, P.K.; Jeng, Y.M.; Chang, M.C.; Chang, Y.T.; Chen, Y.J.; Lee, E.Y.P.; Lee, W.H. High Glucose Triggers Nucleotide Imbalance through O-GlcNAcylation of Key Enzymes and Induces KRAS Mutation in Pancreatic Cells. Cell Metab. 2019, 29, 1334–1349.e10. [Google Scholar] [CrossRef]
- Yachida, S.; Jones, S.; Bozic, I.; Antal, T.; Leary, R.; Fu, B.; Kamiyama, M.; Hruban, R.H.; Eshleman, J.R.; Nowak, M.A.; et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 2010, 467, 1114–1117. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study | Design/Intervention | Population/Animal or Cell Culture Model | Drug Target | Main Result |
|---|---|---|---|---|
| Clinical | ||||
| Jiao et al. [190] | prospective case-cohort study | Finnish male smokers | AGE/RAGE axis | pre-diagnostic sRAGE is inversely associate while CML AGE:sRAGE ratio is positively associated with PDAC risk |
| White et al. [191] | prospective nested case-control study | postmenopausal women | AGE/RAGE axis | pre-diagnostic sRAGE is inversely associated with pancreatic cancer risk |
| Kahlert et al. [202] | retrospective study | PDAC patients undergoing potentially curative resection | ALCAM/CD166 (RAGE homolog) | ALCAM/CD166 is an independent prognostic marker for survival and tumour relapse in PDAC |
| Jiao et al. [101] | prospective study | NIH-AARP Diet and Health Study participants | AGEs | dietary AGE consumption is associated with increased risk of PDAC |
| Experimental | ||||
| Kang et al. [193] | RAGE knock-down | murine model of Kras-driven PDAC and human PDAC tissue | RAGE-IL6-pSTAT3 pathway | RAGE ablation delays PDAC development by decreasing STAT3 signaling and autophagy |
| Arumugam J et al. [194] | RAGE blockade | pancreatic orthotopic model | RAGE-NF-κB axis | RAGE inhibition reduces PDAC growth and metastasis |
| Azizan et al. [192] | RAGE blockade | pancreatic orthotopic model | RAGE-NF-κB-KRAS axis | RAGE inhibition lowers oncogenic KRAS activity by preventing NF-κB activation |
| Vernon et al. [195] | RAGE knock-down | murine model of Kras-driven PDAC | RAGE-IL6 pathway | RAGE ablation delays PDAC development by reducing the accumulation of myeloid-derived suppressor cells |
| Kang et al. [196] | RAGE knock-down or inhibition by RNA interference | PDAC cells | ROS-NF-κB-RAGE axis | suppression and knockdown of RAGE increases the sensitivity of PDAC cells to oxidative injury |
| Kang et al. [197] | RAGE knock-down | murine model of Kras-driven PDAC | RAGE-IL6-pSTAT3 pathway | RAGE ablation increases apoptosis and decreases autophagy/proliferation in the emerging PDAC microenvironment |
| Kang et al. [198] | RAGE knock-down or inhibition of HMGB1 release | ectopic tumor xenograft model and human PDAC tissue | HMGB1–RAGE axis | lack of RAGE or inhibition of HMGB1 release slows PDAC growth in vitro and in vivo by diminishing ATP production |
| Kang et al. [182] | RAGE knock-down | murine model of Kras-driven PDAC | RAGE-NF-κB-KRAS pathway | binding of RAGE to oncogenic KRAS facilitates HIF-1α activation and promotes PDAC growth |
| Menini et al. [171] | exogenous AGE administration/RAGE blockade | murine model of Kras-driven PDAC | AGE-RAGE-ALCAM/CD166 axis | AGEs accelerate the progression of PDAC through receptor-mediated mechanisms |
| Menini et al. [203] | inhibition of AGE formation by RCS scavenging | diabetic murine model of Kras-driven PDAC and human PDAC tissue | RCS (AGE precursors) | circulating and tumor-derived RCS/AGEs generated by hyperglycemia promote invasive PDAC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Menini, S.; Iacobini, C.; Vitale, M.; Pesce, C.; Pugliese, G. Diabetes and Pancreatic Cancer—A Dangerous Liaison Relying on Carbonyl Stress. Cancers 2021, 13, 313. https://doi.org/10.3390/cancers13020313
Menini S, Iacobini C, Vitale M, Pesce C, Pugliese G. Diabetes and Pancreatic Cancer—A Dangerous Liaison Relying on Carbonyl Stress. Cancers. 2021; 13(2):313. https://doi.org/10.3390/cancers13020313
Chicago/Turabian StyleMenini, Stefano, Carla Iacobini, Martina Vitale, Carlo Pesce, and Giuseppe Pugliese. 2021. "Diabetes and Pancreatic Cancer—A Dangerous Liaison Relying on Carbonyl Stress" Cancers 13, no. 2: 313. https://doi.org/10.3390/cancers13020313
APA StyleMenini, S., Iacobini, C., Vitale, M., Pesce, C., & Pugliese, G. (2021). Diabetes and Pancreatic Cancer—A Dangerous Liaison Relying on Carbonyl Stress. Cancers, 13(2), 313. https://doi.org/10.3390/cancers13020313