
Emerging Roles for Ion Channels in Ovarian Cancer: Pathomechanisms and Pharmacological Treatment

 ,
,  , and
, and
Abstract
Simple Summary
Abstract
1. Introduction
2. Sodium Channels

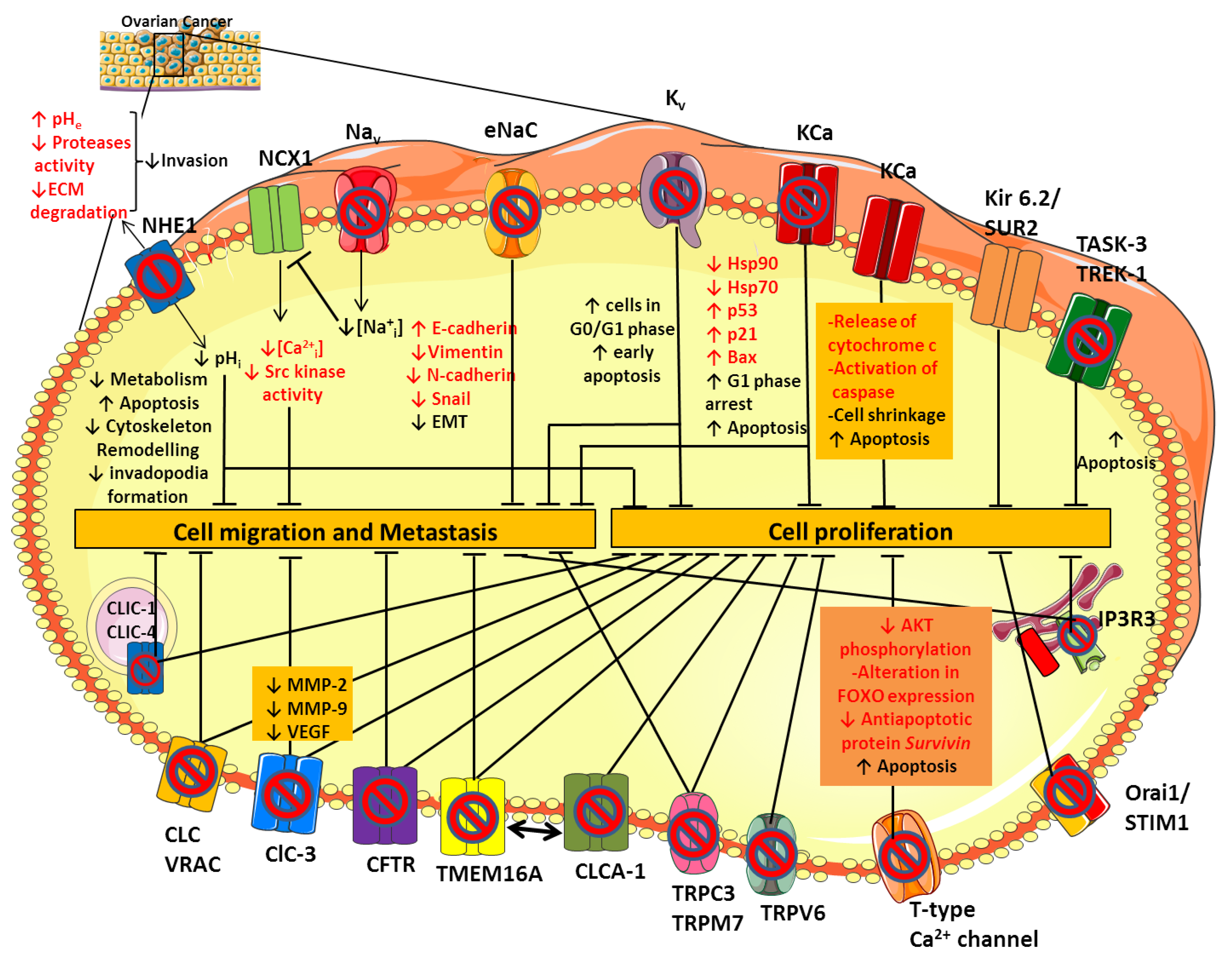{kind=link}
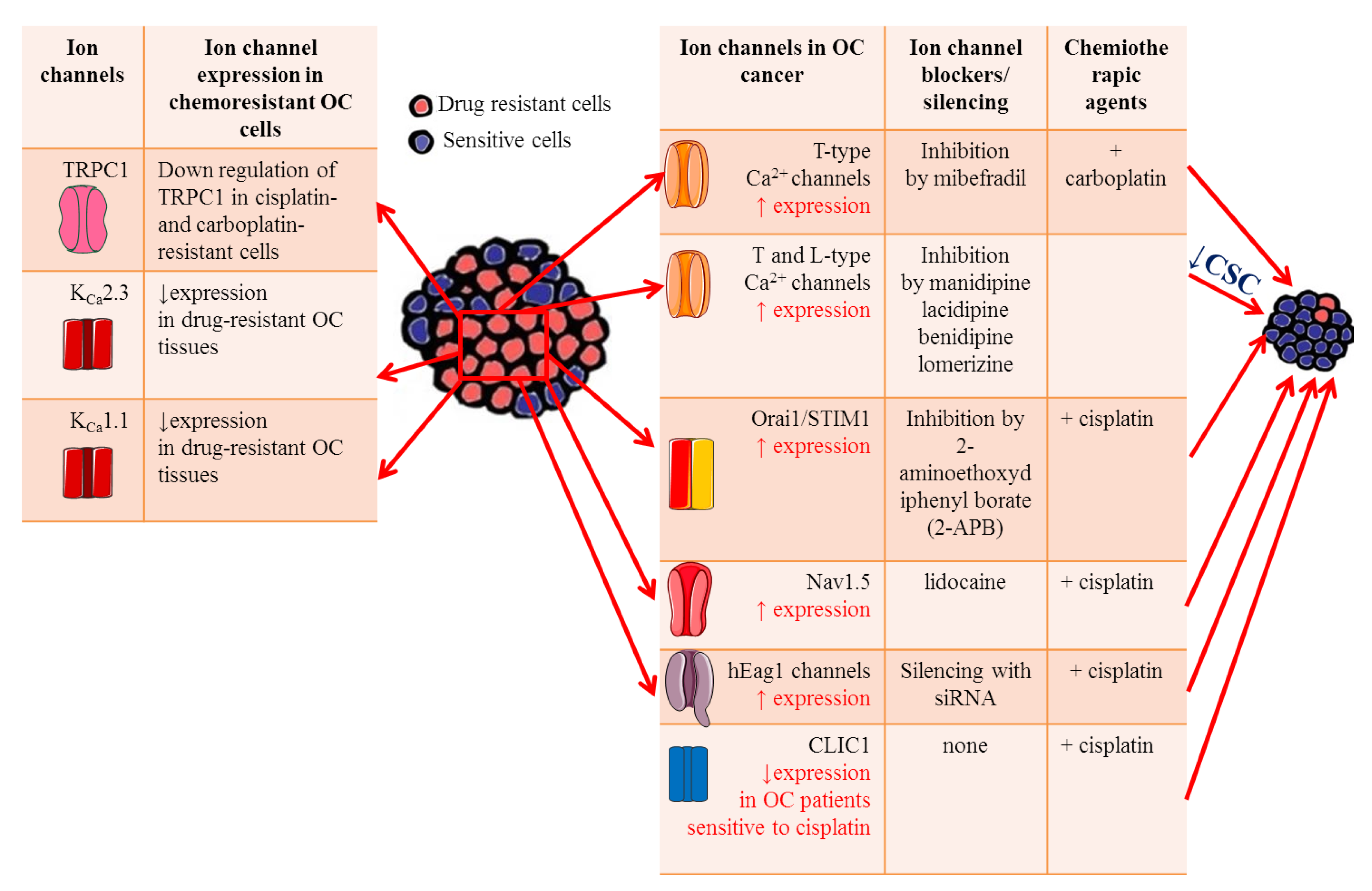{kind=link}
| Drug | Clinical Indications | Ion Channel Target | Biological Effects in OC | Other Known Effects/Uses | Ref. |
|---|---|---|---|---|---|
| TTX | Natural toxin | VGSC blocker | In vitro: reduction of OC cells migration and invasion | - | [28] |
| EPA | Omega-3 fatty acids | Nav1.5 blocker | In vitro: suppression of growth and reduction of invasivity | Cardioprotective effects, cognitive function improvement, fatigue recovery and endurance performance improvement, maintenance of immune function | [42,43,51] |
| Topiramate | Antiepileptic drug | VGSC blocker | In vitro: reduction of cell proliferation and migration | Off-label uses: neuropathic pain, psychotropic drug-induced weight gain, alcohol use disorders with tobacco dependence, binge eating disorder, bulimia nervosa, obesity with hypertension, adjunctive therapy in bipolar disorder, unipolar depression, borderline personality disorder, obsessive-compulsive disorder, posttraumatic stress disorder, Tourette syndrome, Prader-Willie syndrome, essential tremor. Carbonic anhydrase inhibitor | [45,52] |
| Bupivacaine | Local anesthetic drug | VGSC blocker | In vitro: reduction of OC cell proliferation and migration | - | [47] |
| Lidocaine | Local anesthetic drug | VGSC blocker | In vitro: reduction of OC cell proliferation, migration, and invasion; increase of cisplatin efficiency in OC cells In vivo: increase of cisplatin efficiency in a murine ovarian metastatic model | Intractable cough and asthma, and chronic (including neuropathic) pain. | [48,53,54] |
| E3Ab | Polyclonal antibody | Nav1.5 | In vitro: reduction of OC cell proliferation and invasive capacity | - | [49,50] |
| Imipramine | Tricyclic antidepressant drug | Eag blocker | In vitro: early apoptosis | Inhibitor of the reuptake of norepinephrine and serotonin; Antagonist of D2 dopamine, muscarinic, α1 and α2 adrenergic, and H1 histamine receptors. Off-label uses: chronic neuropathic pain and panic disorder. | [18,55,56,57] |
| Clofilium | Antiarrhythmic drug | Eag blocker | In vitro: early apoptosis | In mammalian cardiac myocytes, clofilium also affects Na+ and L-type Ca2+ currents. NMDA glutamate Rc antagonist. | [55,58,59] |
| Berberine | Natural alkaloid | hERG blocker | In vitro: inhibition of OC cells proliferation | Antiviral activity against Herpes simplex virus 1 and 2 by modulating host cell activation of the NF-kB and MAPK pathway. | [60,61] |
| mAb56 | Monoclonal antibody | Eag blocker | In vitro: inhibition of OC cell growth | - | [62] |
| Tetrandrine | Natural alkaloid | BK channels blocker | In vitro: reduction of OC cell proliferation by inducing G1 phase arrest and accelerating apoptosis | Anti-hypertensive effects through T-type Ca2+ channel blocking; Anti-inflammatory, immunosuppressant and antiallergic effects mediated by tetrandrine inhibition of ILs, TNF-a, prostaglandin, cycloxygenase-2 and T cells; Antioxidant activity by scavenging the superoxide anion radicals; Anticancer agent; Antimicrobial activity. | [63,64] |
| Iberiotoxin | Natural toxin | BK channels blocker | In vitro: reduction of OC cell proliferation by inducing G1 phase arrest | - | [63] |
| NS1619 | Experimental compound | BK channels opener | In vitro: inhibition of OC cell proliferation by cell shrinkage and release of cytochrome c and activation of caspase, inducing apoptosis | Protection of cardiac muscle against ischaemia and reperfusion injury; Cytoprotective effect through directly inhibition of SERCA activity. | [65,66] |
| Minoxidil | Antihypertensive drug | Kir6.2/SUR2 activator | In vitro: reduction of OC cell proliferation In vivo: Reduction of tumor growth in OC xenograft model | Treatment for androgenetic alopecia and off-label uses for other hair loss conditions | [67,68] |
| Methanandamide | Cannabinoid receptor agonist | TASK-3 blocker | In vitro: reduction of OC cell proliferation and increasing in apoptosis | Anti- and pronociceptive effects by activating both cannabinoid (CB1) and vanilloid (TRPV1) receptors of nociceptive primary afferents. | [69,70] |
| Zinc | Chemical element | TASK-3 blocker | In vitro: reduction of OC cells proliferation and increase of apoptosis | Anti-diarrheal effects by inhibiting cyclic adenosine monophosphate (cAMP), calcium, and nitric oxide; antioxidative and antimicrobial effects through the antioxidant activity of cysteine-rich metallothioneins | [69,71] |
| Curcumin | Natural phenolic antioxidant | TREK-1 blocker | In vitro: reduction of OC cells proliferation and increase of late apoptosis processes | Antioxidant, anti-inflammatory, antimicrobial, antidiabetic, anti-aging effects. Multi-ion channel blocker such as voltage-gated Na+, Ca2+, K+ channels Activation of various TRP channels. | [72,73,74,75] |
| Amphotericin | Antifungal | K+ ionophore | In vitro: sensitize cells to cisplatin and other platinum-based agents | Production of free radicals and subsequently oxidative damage; stimulation of phagocytic cells | [76,77] |
| Bumetanide | Diuretic | Na-K-Cl cotransporter inhibitor | In vitro: boost cisplatin-induced apoptosis | - | [78] |
| Niflumic acid | Nonsteroidal anti-inflammatory drug | CLC and VRAC blocker | In vitro: inhibition of OC cell proliferation, adhesion, and invasion | Blocking effect on TRPV1 channels, T-type calcium channels, CFTR, TRPA1, and several voltage-gated potassium channels. | [79,80,81,82,83,84] |
| CLCA1 blocker | In vitro: Reduced ability of OC cells to form multicellular aggregates | [85] | |||
| Tamoxifen | Non-steroidal antiestrogen | CLC and VRAC blocker | In vitro: Inhibition of OC cell proliferation and suppression of cell adhesion and invasion | Treatment of hormone-sensitive breast cancer | [79] |
| Nitro-2-(3-phenylpropylamino)-benzoate (NPPB) | Experimental compound | CLC and VRAC blocker | In vitro: Inhibition of OC cell proliferation and suppression of cell adhesion and invasion | - | [80,86] |
| Mibefradil | In a phase I clinical trial for the treatment of gliomas | T-type Ca2+ channel blocker | In vitro: Alteration of cell-cycle progression, slowing of proliferation, and increase of cell death processes Inhibition of growth and increase of apoptosis in platinum-resistant OC cells In vivo: mibefradil sensitizes platinum-resistant OC to carboplatin in a mouse model of peritoneal metastasis due to OC | Blocking effects on Orai channels (204 Li et al., 2019); Inhibition of CYP3A cytochrome (205 Foti et al., 2011); inhibition of Kv10.1 (206 Gómez-Lagunas et al., 2017) In a phase I clinical trial for the treatment of gliomas | [87,88,89,90,91,92] |
| Nifedipine | antihypertensive and antianginal | Voltage-gated Ca2+ channel blocker | In vitro: Inhibition of LPA-induced OC cell migration and adhesion; Increase of cytosolic calcium levels in A2780 cells | Off-label uses: Raynaud phenomenon; Tocolysis; Distal ureteric calculi | [93,94,95,96] |
| Sulforaphane (SFN) | Natural isothiocyanate compound | IP3R1 | In vitro: Inhibition of OC cell proliferation | Antioxidant, antidiabetic, antiinflammatory effects | [97,98,99,100] |
| Melatonin | Hormone | IP3R1 | In vitro: Inhibition of OC cell proliferation | Delayed sleep phase syndrome treatment | [101,102] |
| 3,4-dihydroquinazoline derivatives | Experimental compounds | T-type Ca2+ channel blocker | In vitro: Alteration of cell-cycle progression, slowing of proliferation, and increased cell death processes | - | [88] |
| lomerizine | Antimigrainous drug | L and T-type Ca2+ channel blocker | In vitro: reduction of stemness and induction of apoptosis in CSCs | Antihistamine and anti-serotonin effects | [103,104] |
| manidipine, lacidipine, benidipine | Antihypertensive drugs | L-type Ca2+ channel blockers | In vitro: reduction of stemness and induction of apoptosis in CSCs | - | [103] |
| Soricidin, SOR-C13, and SOR-C27 | Natural oligopeptide and analogs | TRPV6 | In vivo: reduction of growth of OC cell tumor xenografts in mice | - | [105] |
3. Potassium Channels
4. Chloride Channels
5. Calcium Channels
6. TRP Channels
7. Ion Channels and Drug Resistance
8. Concluding Remarks and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Lheureux, S.; Braunstein, M.; Oza, A.M. Epithelial Ovarian Cancer: Evolution of Management in the Era of Precision Medicine. CA Cancer J. Clin. 2019, 69, 280–304. [Google Scholar] [CrossRef] [PubMed]
- Trabert, B.; Poole, E.M.; White, E.; Visvanathan, K.; Adami, H.O.; Anderson, G.L.; Brasky, T.M.; Brinton, L.A.; Fortner, R.T.; Gaudet, M.; et al. Analgesic use and ovarian cancer risk: An analysis in the Ovarian Cancer Cohort Consortium. J. Natl. Cancer Inst. 2019, 111, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Pavelka, J.C.; Li, A.J.; Karlan, B.Y. Hereditary ovarian cancer—Assessing risk and prevention strategies. Obstet. Gynecol. Clin. North. Am. 2007, 34, 651–665. [Google Scholar] [CrossRef]
- Tschernichovsky, R.; Goodman, A. Risk-reducing strategies for ovarian cancer in BRCA mutation carriers: A balancing act. Oncologist 2017, 22, 450–459. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bast, R.C.J.; Hennessy, B.; Mills, G.B. The biology of ovarian cancer: New opportunities for translation. Nat. Rev. Cancer 2009, 9, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Dembo, A.J.; Davy, M.; Stenwig, A.E.; Berle, E.J.; Bush, R.S.; Kjorstad, K. Prognostic factors in patients with stage I epithelial ovarian cancer. Obstet. Gynecol. 1990, 75, 263–273. [Google Scholar] [PubMed]
- Goff, B.A.; Mandel, L.S.; Drescher, C.W.; Urban, N.; Gough, S.; Schurman, K.M.; Patras, J.; Mahony, B.S.; Andersen, M.R. Development of an ovarian cancer symptom index: Possibilities for earlier detection. Cancer 2007, 109, 221–227. [Google Scholar] [CrossRef]
- Stewart, C.; Ralyea, C.; Lockwood, S. Ovarian Cancer: An Integrated Review. Semin. Oncol. Nurs. 2019, 35, 151–156. [Google Scholar] [CrossRef]
- Banerjee, S.; Kaye, S.B. New strategies in the treatment of ovarian cancer: Current clinical perspectives and future potential. Clin. Cancer Res. 2013, 19, 961–968. [Google Scholar] [CrossRef]
- Du Bois, A.; Reuss, A.; Pujade-Lauraine, E.; Harter, P.; Ray-Coquard, I.; Pfisterer, J. Role of surgical outcome as prognostic factor in advanced epithelial ovarian cancer: A combined exploratory analysis of 3 prospectively randomized phase 3 multicenter trials: By the Arbeitsgemeinschaft Gynaekologische Onkologie Studiengruppe Ovarialkarzinom (AGO-OVAR) and the Groupe d’Investigateurs Nationaux Pour les Etudes des Cancers de l’Ovaire (GINECO). Cancer 2009, 115, 1234–1244. [Google Scholar]
- Lastraioli, E.; Iorio, J.; Arcangeli, A. Ion channel expression as promising cancer biomarker. Biochim. Biophys. Acta 2015, 1848, 2685–2702. [Google Scholar] [CrossRef]
- Litan, A.; Langhans, S. Cancer as a channelopathy: Ion channels and pumps in tumor development and progression. Front. Cell Neurosci. 2015, 9, 86. [Google Scholar] [CrossRef] [PubMed]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Ion Channels in Cancer: Are Cancer Hallmarks Oncochannelopathies? Physiol. Rev. 2018, 98, 559–621. [Google Scholar] [CrossRef] [PubMed]
- Frede, J.; Fraser, S.P.; Oskay-Özcelik, G.; Hong, Y.; Braicu, E.I.; Sehouli, J.; Gabra, H.; Djamgoz, M.B.A. Ovarian cancer: Ion channel and aquaporin expression as novel targets of clinical potential. Eur. J. Cancer 2013, 49, 2331–2344. [Google Scholar] [CrossRef]
- Schnipper, J.; Dhennin-Duthille, I.; Ahidouch, A.; Ouadid-Ahidouch, H. Ion Channel Signature in Healthy Pancreas and Pancreatic Ductal Adenocarcinoma. Front. Pharmacol. 2020, 11, 568993. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, M.; Li, W.; Edwards, M.J.; Ahmad, S.A.; Patel, S.; Szabo, I.; Gulbins, E. Voltage-Gated Potassium Channels as Regulators of Cell Death. Front. Cell Dev. Biol. 2020, 8, 611853. [Google Scholar] [CrossRef]
- Rosendo-Pineda, M.J.; Moreno, C.M.; Vaca, L. Role of ion channels during cell division. Cell Calcium 2020, 91, 102258. [Google Scholar] [CrossRef]
- Djamgoz, M.B.; Coombes, R.C.; Schwab, A. Ion transport and cancer: From initiation to metastasis. Philos. Trans. R Soc. Lond. B Biol. Sci. 2014, 369, 20130092. [Google Scholar] [CrossRef] [PubMed]
- Imbrici, P.; Liantonio, A.; Camerino, G.M.; De Bellis, M.; Camerino, C.; Mele, A.; Giustino, A.; Pierno, S.; De Luca, A.; Tricarico, D.; et al. Therapeutic Approaches to Genetic Ion Channelopathies and Perspectives in Drug Discovery. Front. Pharmacol. 2016, 7, 121. [Google Scholar] [CrossRef] [PubMed]
- Altamura, C.; Desaphy, J.F.; Conte, D.; De Luca, A.; Imbrici, P. Skeletal muscle ClC-1 chloride channels in health and diseases. Pflug. Arch. 2020, 472, 961–975. [Google Scholar] [CrossRef]
- Hernandez, C.M.; Richards, J.R. Physiology, Sodium Channels; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Djamgoz, M.B.A.; Fraser, S.P.; Brackenbury, W.J. In Vivo Evidence for Voltage-Gated Sodium Channel Expression in Carcinomas and Potentiation of Metastasis. Cancers 2019, 11, 1675. [Google Scholar] [CrossRef]
- Nelson, M.; Yang, M.; Millican-Slater, R.; Brackenbury, W. Nav1.5 regulates breast tumor growth and metastatic dissemination in vivo. Oncotarget 2015, 6, 32914–32929. [Google Scholar] [CrossRef]
- House, C.; Vaske, C.; Schwartz, A.; Obias, V.; Frank, B.; Luu, T.; Sarvazyan, N.; Irby, R.; Strausberg, R.L.; Hales, T.G.; et al. Voltage gated Na+ channel SCN5A is a key regulator of a gene transcriptional network that controls colon cancer invasion. Cancer Res. 2010, 70, 6957–6967. [Google Scholar] [CrossRef]
- Suy, S.; Hansen, T.; Auto, H.D.; Kallakury, B.V.S.; Dailey, V.; Danner, M.; Macarthur, L.; Zhang, Y.; Miessau, M.J.; Collins, S.P.; et al. Expression of Voltage-gated sodium channel Na1.8 in human prostate cancer is associated with high histological grade. J. Clin. Exp. Oncol. 2012, 1, 1–13. [Google Scholar] [CrossRef]
- He, M.; Liu, S.; Gallolu Kankanamalage, S.; Borromeo, M.D.; Girard, L.; Gazdar, A.F.; Minna, J.D.; Johnson, J.E.; Cobb, M.H. The Epithelial Sodium Channel (αENaC) Is a Downstream Therapeutic Target of ASCL1 in Pulmonary Neuroendocrine Tumors. Transl. Oncol. 2018, 11, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Shen, Y.; Cai, J.; Lei, M. Expression of voltage-gated sodium channel·subunit in human ovarian cancer. Oncol. Rep. 2010, 23, 1293–1299. [Google Scholar] [PubMed]
- Gillet, L.; Roger, S.; Besson, P.; Lecaille, F.; Gore, J.; Bougnoux, P.; Lalmanach, G.; Le Guennec, J.Y. Voltage-gated sodium channel activity promotes cysteine cathepsin-dependent invasiveness and colony growth of human cancer cells. J. Biol. Chem. 2009, 284, 8680–8691. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Kozminski, D.J.; Wold, L.A.; Modak, R.; Calhoun, J.D.; Isom, L.L.; Brackenbury, W.J. Therapeutic potential for phenytoin: Targeting Na(v)1.5 sodium channels to reduce migration and invasion in metastatic breast cancer. Breast Cancer Res. Treat. 2012, 134, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Liskova, V.; Hudecova, S.; Lencesova, L.; Iuliano, F.; Sirova, M.; Ondrias, K.; Pastorekova, S.; Krizanova, S. Type 1 Sodium Calcium Exchanger Forms a Complex with Carbonic Anhydrase IX and Via Reverse Mode Activity Contributes to pH Control in Hypoxic Tumors. Cancers 2019, 11, 1139. [Google Scholar] [CrossRef] [PubMed]
- Besson, P.; Driffort, V.; Bon, É.; Gradek, F.; Chevalier, S.; Roger, S. How do voltage-gated sodium channels enhance migration and invasiveness in cancer cells? Biochim. Biophys. Acta 2015, 1848, 2493–2501. [Google Scholar] [CrossRef] [PubMed]
- Greco, M.R.; Antelmi, E.; Busco, G.; Guerra, L.; Rubino, R.; Casavola, V.; Reshkin, S.J.; Cardone, R.A. Protease activity at invadopodial focal digestive areas is dependent on NHE1-driven acidic pHe. Oncol. Rep. 2014, 31, 940–946. [Google Scholar] [CrossRef] [PubMed]
- Matarrese, P.; Ascione, B.; Ciarlo, L.; Vona, R.; Leonetti, C.; Scarsella, M.; Mileo, A.M.; Catricalà, C.; Paggi, M.G.; Malorni, W. Cathepsin B inhibition interferes with metastatic potential of human melanoma: An in vitro and in vivo study. Mol. Cancer 2010, 9, 207. [Google Scholar] [CrossRef] [PubMed]
- Brisson, L.; Driffort, V.; Benoist, L.; Poet, M.; Counillon, L.; Antelmi, E.; Rubino, R.; Besson, P.; Labbal, F.; Chevalier, S.; et al. NaV1.5 Na⁺ channels allosterically regulate the NHE-1 exchanger and promote the activity of breast cancer cell invadopodia. J. Cell Sci. 2013, 126, 4835–4842. [Google Scholar] [CrossRef] [PubMed]
- Harguindey, S.; Arranz, J.L.; Polo Orozco, J.D.; Rauch, C.; Fais, S.; Cardone, R.A.; Reshkin, S.J. Cariporide and other new and powerful NHE1 inhibitors as potentially selective anticancer drugs—An integral molecular/biochemical/metabolic/clinical approach after one hundred years of cancer research. J. Transl. Med. 2013, 11, 282. [Google Scholar] [CrossRef]
- Sanhueza, C.; Araos, J.; Naranjo, L.; Toledo, F.; Beltrán, A.R.; Ramírez, M.A.; Gutiérrez, J.; Pardo, F.; Leiva, A.; Sobrevia, L. Sodium/proton exchanger isoform 1 regulates intracellular pH and cell proliferation in human ovarian cancer. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 81–91. [Google Scholar] [CrossRef]
- Yi, Y.H.; Ho, P.Y.; Chen, T.W.; Lin, W.J.; Gukassyan, V.; Tsai, T.H.; Wang, D.W.; Lew, T.S.; Tang, C.Y.; Lo, S.J.; et al. Membrane targeting and coupling of NHE1-integrinαIIbβ3-NCX1 by lipid rafts following integrin-ligand interactions trigger Ca2+ oscillations. J. Biol. Chem. 2009, 284, 3855–3864. [Google Scholar] [CrossRef]
- Xu, J.; Ji, B.; Wen, G.; Yang, Y.; Jin, H.; Liu, X.; Xie, R.; Song, W.; Song, P.; Dong, H.; et al. Na+/H+ exchanger 1, Na+/Ca2+ exchanger 1 and calmodulin complex regulates interleukin 6-mediated cellular behavior of human hepatocellular carcinoma. Carcinogenesis 2016, 37, 290–300. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bosetti, C.; Negri, E.; Franceschi, S.; Pelucchi, C.; Talamini, R.; Montella, M.; Conti, E.; La Vecchia, C. Diet and ovarian cancer risk: A case-control study in Italy. Int. J. Cancer 2001, 93, 911–915. [Google Scholar] [CrossRef]
- Merritt, M.A.; Cramer, D.W.; Missmer, S.A.; Vitonis, A.F.; Titus, L.J.; Terry, K.L. Dietary fat intake and risk of epithelial ovarian cancer by tumor histology. Br. J. Cancer 2014, 110, 1392–1401. [Google Scholar] [CrossRef]
- Sharma, A.; Belna, J.; Logan, J.; Espat, J.; Hurteau, J.A. The effects of ω-3 fatty acids on growth regulation of epithelial ovarian cancer cell lines. Gynecol. Oncol. 2005, 99, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, D.; Liu, J.; Zhao, C.; Yao, S.; Hong, L. Blocking the Nav1.5 channel using eicosapentaenoic acid reduces migration and proliferation of ovarian cancer cells. Int. J. Oncol. 2018, 53, 855–865. [Google Scholar] [CrossRef] [PubMed]
- Martin, F.; Ufodiama, C.; Watt, I.; Bland, M.; Brackenbury, W.J. Therapeutic value of voltage gated sodium channel inhibitors in breast, colorectal and prostate cancer: A systematic review. Front. Pharmacol. 2015, 6, 273. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Fang, Z.; Clark, L.; Sun, W.; Yin, Y.; Zhang, R.; Sullivan, S.; Tran, A.; Kong, W.; Wang, J.; et al. Topiramate exhibits anti-tumorigenic and metastatic effects in ovarian cancer cells. Am. J. Transl. Res. 2018, 10, 1663–1676. [Google Scholar] [PubMed]
- Exadaktylos, A.K.; Buggy, D.J.; Moriarty, D.C.; Mascha, E.; Sessler, D.I. Can anesthetic technique for primary breast cancer surgery affect recurrence ormetastasis? Anesthesiology 2006, 105, 660–664. [Google Scholar] [CrossRef] [PubMed]
- Xuan, W.; Zhao, H.; Hankin, J.; Chen, L.; Yao, S.; Ma, D. Local anesthetic bupivacaine induced ovarian and prostate cancer apoptotic cell death and underlying mechanisms in vitro. Sci. Rep. 2016, 6, 26277. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Yu, M.; Li, Y.; Wang, H.; Xu, C.; Zhang, X.; Li, M.; Guo, H.; Ma, D.; Guo, X. Lidocaine inhibits the metastatic potential of ovarian cancer by blocking Na V 1.5-mediated EMT and FAK/Paxillin signaling pathway. Cancer Med. 2020. [Google Scholar] [CrossRef]
- Xu, S.Z.; Zeng, F.; Lei, M.; Li, J.; Gao, B.; Xiong, C.; Sivaprasadarao, A.; Beech, D.J. Generation of functional ion-channel tools by E3 targeting. Nat. Biotechnol. 2005, 23, 1289–1293. [Google Scholar] [CrossRef]
- Gao, R.; Cao, T.; Chen, H.; Cai, J.; Lei, M.; Wang, Z. Nav1.5-E3 antibody inhibits cancer progression. Transl. Cancer Res. 2019, 8, 44–50. [Google Scholar] [CrossRef]
- Calder, P.C. Functional Roles of Fatty Acids and Their Effects on Human Health. JPEN J. Parenter. Enter. Nutr. 2015, 39, S18–S32. [Google Scholar] [CrossRef]
- Fariba, K.; Saadabadi, A. Topiramate; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Slaton, R.M.; Thomas, R.H.; Mbathi, J.W. Evidence for therapeutic uses of nebulized lidocaine in the treatment of intractable cough and asthma. Ann. Pharmacother. 2013, 47, 578–585. [Google Scholar] [CrossRef]
- Kandil, E.; Melikman, E.; Adinoff, B. Lidocaine Infusion: A Promising Therapeutic Approach for Chronic Pain. J. Anesth. Clin. Res. 2017, 8, 697. [Google Scholar] [CrossRef]
- Asher, V.; Warren, A.; Shaw, R.; Sowter, H.; Bali, A.; Khan, R. The role of Eag and HERG channels in cell proliferation and apoptotic cell death in SK-OV-3 ovarian cancer cell line. Cancer Cell Int. 2011, 11, 6. [Google Scholar] [CrossRef]
- Fayez, R.; Gupta, V. Imipramine; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Hearn, L.; Derry, S.; Phillips, T.; Moore, R.A.; Wiffen, P.J. Imipramine for neuropathic pain in adults. Cochrane Database Syst. Rev. 2014. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Himmel, H.M.; Ravens, U. Selectivity of class-III antiarrhythmic action of clofilium in guinea pig ventricular myocytes. J. Cardiovasc. Pharmacol. 1996, 27, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Wang, X.Q.; Sun, C.S.; Wei, L.; Yu, S.P. Inhibitory effects of clofilium on membrane currents associated with Ca channels, NMDA receptor channels and Na+, K+-ATPase in cortical neurons. Pharmacology 2005, 73, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Zhi, D.; Zhou, K.; Yu, D.; Fan, X.; Zhang, J.; Li, X.; Dong, M. hERG1 is involved in the pathophysiological process and inhibited by berberine in SKOV3 cells. Oncol. Lett. 2019, 17, 5653–5661. [Google Scholar] [CrossRef]
- Song, S.; Qiu, M.; Chu, Y.; Chen, D.; Wang, X.; Su, A.; Wu, Z. Downregulation of cellular c-Jun N-terminal protein kinase and NF-κB activation by berberine may result in inhibition of herpes simplex virus replication. Antimicrob. Agents Chemother. 2014, 58, 5068–5078. [Google Scholar] [CrossRef]
- Gómez-Varela, D.; Zwick-Wallasch, E.; Knötgen, H.; Sánchez, A.; Hettmann, T.; Ossipov, D.; Weseloh, R.; Contreras-Jurado, C.; Rothe, M.; Stühmer, W.; et al. Monoclonal antibody blockade of the human Eag1 potassium channel function exerts antitumor activity. Cancer Res. 2007, 67, 7343–7349. [Google Scholar] [CrossRef]
- Han, X.; Wang, F.; Yao, W.; Xing, H.; Weng, D.; Song, X.; Chen, G.; Xi, L.; Zhu, T.; Zhou, J.; et al. Heat shock proteins and p53 play a critical role in K+ channel-mediated tumor cell proliferation and apoptosis. Apoptosis 2007, 12, 1837–1846. [Google Scholar] [CrossRef]
- Bhagya, N.; Chandrashekar, K.R. Tetrandrine—A molecule of wide bioactivity. Phytochemistry 2016, 125, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Xi, L.; Wang, H.; Huang, X.; Ma, X.; Han, Z.; Wu, P.; Ma, X.; Lu, Y.; Wang, G.; et al. The potassium ion channel opener NS1619 inhibits proliferation and induces apoptosis in A2780 ovarian cancer cells. Biochem. Biophys. Res. Commun. 2008, 375, 205–209. [Google Scholar] [CrossRef]
- Wrzosek, A. The potassium channel opener NS1619 modulates calcium homeostasis in muscle cells by inhibiting SERCA. Cell Calcium 2014, 56, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Fukushiro-Lopes, D.; Hegel, A.D.; Russo, A.; Senyuk, V.; Liotta, M.; Beeson, G.C.; Beeson, C.C.; Burdette, J.; Potkul, R.K.; Gentile, S. Repurposing Kir6/SUR2 Channel Activator Minoxidil to Arrests Growth of Gynecologic Cancers. Front. Pharmacol. 2020, 11, 577. [Google Scholar] [CrossRef]
- Suchonwanit, P.; Thammarucha, S.; Leerunyakul, K. Minoxidil and its use in hair disorders: A review. Drug Des. Dev. Ther. 2019, 13, 2777–2786. [Google Scholar] [CrossRef]
- Innamaa, A.; Jackson, L.; Asher, V.; Van Shalkwyk, G.; Warren, A.; Hay, D.; Bali, A.; Sowter, H.; Khan, R. Expression and prognostic significance of the oncogenic K2P potassium channel KCNK9 (TASK-3) in ovarian carcinoma. Anticancer Res. 2013, 33, 1401–1408. [Google Scholar]
- Fischer, M.J.M.; Messlinger, K. Cannabinoid and vanilloid effects of R(+)-methanandamide in the hemisected meningeal preparation. Cephalalgia 2007, 27, 422–428. [Google Scholar] [CrossRef]
- Rabinovich, D.; Smadi, Y. Zinc; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Innamaa, A.; Jackson, L.; Asher, V.; Van Schalkwyk, G.; Warren, A.; Keightley, A.; Hay, D.; Bali, A.; Sowter, H.; Khan, R. Expression and effects of modulation of the K2P potassium channels TREK-1 (KCNK2) and TREK-2 (KCNK10) in the normal human ovary and epithelial ovarian cancer. Clin. Transl. Oncol. 2013, 15, 910–918. [Google Scholar] [CrossRef]
- Kotha, R.R.; Luthria, D.L. Curcumin: Biological, Pharmaceutical, Nutraceutical, and Analytical Aspects. Molecules 2019, 24, 2930. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Zhang, Z.F.; Hu, L.K.; Zhang, P.H.; Cao, Z.Z.; Liu, Z.P.; Zhang, P.P.; Ma, J.H. Curcumin, a Multi-Ion Channel Blocker That Preferentially Blocks Late Na + Current and Prevents I/R-Induced Arrhythmias. Front. Physiol. 2020, 11, 978. [Google Scholar] [CrossRef]
- Kütük, S.G.; Gökçe, G.; Kütük, M.; Gurses Cila, H.A.; Naziroğlu, M. Curcumin enhances cisplatin-induced human laryngeal squamous cancer cell death through activation of TRPM2 channel and mitochondrial oxidative stress. Sci. Rep. 2019, 9, 17784. [Google Scholar] [CrossRef]
- Sharp, S.Y.; Mistry, P.; Valenti, M.R.; Bryant, A.P.; Kelland, L.R. Selective potentiation of platinum drug cytotoxicity in cisplatin-sensitive and -resistant human ovarian carcinoma cell lines by amphotericin B. Cancer Chemother. Pharmacol. 1994, 35, 137–143. [Google Scholar] [CrossRef]
- Mesa-Arango, A.C.; Scorzoni, L.; Zaragoza, O. It only takes one to do many jobs: Amphotericin B as antifungal and immunomodulatory drug. Front. Microbiol. 2012, 3, 286. [Google Scholar] [CrossRef]
- Marklund, L.; Henriksson, R.; Grankvist, K. Cisplatin-induced apoptosis of mesothelioma cells is affected by potassium ion flux modulator amphotericin B and bumetanide. Int. J. Cancer 2001, 93, 577–583. [Google Scholar] [CrossRef]
- Li, M.; Wang, Q.; Lin, W.; Wang, B. Regulation of ovarian cancer cell adhesion and invasion by chloride channels. Int. J. Gynecol. Cancer 2009, 19, 526–530. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wu, D.B.; Wang, J.; Chen, L. CLC-3 Cl-channel-mediated invasion and migration of human ovarian cancer cells. Eur J. Gynaecol. Oncol. 2016, 37, 689–695. [Google Scholar] [PubMed]
- Marwaha, L.; Bansal, Y.; Singh, R.; Saroj, P.; Sodhi, R.K.; Kuhad, A. Niflumic acid, a TRPV1 channel modulator, ameliorates stavudine-induced neuropathic pain. Inflammopharmacology 2016, 24, 319–334. [Google Scholar] [CrossRef]
- Balderas, E.; Arteaga-Tlecuitl, R.; Rivera, M.; Gomora, J.C.; Darszon, A. Niflumic acid blocks native and recombinant T-type channels. J. Cell Physiol. 2012, 227, 2542–2555. [Google Scholar] [CrossRef] [PubMed]
- Scott-Ward, T.S.; Li, H.; Schmidt, A.; Cai, Z.; Sheppard, D.N. Direct block of the cystic fibrosis transmembrane conductance regulator Cl(−) channel by niflumic acid. Mol. Membr Biol. 2004, 21, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Tian, J.; Zhu, Y.; Wang, C.; Xiao, R.; Herz, J.M.; Wood, J.D.; Zhu, M.X. Activation of TRPA1 channels by fenamate nonsteroidal anti-inflammatory drugs. Pflug. Arch. Eur. J. Physiol. 2010, 459, 579–592. [Google Scholar] [CrossRef]
- Musrap, N.; Tuccitto, A.; Karagiannis, G.S.; Saraon, P.; Batruch, I.; Diamandis, E.P. Comparative proteomics of ovarian cancer aggregate formation reveals an increased expression of calcium activated chloride channel regulator 1 (ClCa1). J. Biol. Chem. 2015, 290, 17218–17227. [Google Scholar] [CrossRef]
- Tizzano, E.F.; Silver, M.M.; Chitayat, D.; Benichou, J.C.; Buchwald, M. Differential cellular expression of cystic fibrosis transmembrane regulator in human reproductive tissues. Clues for the infertility in patients with cystic fibrosis. Am. J. Pathol. 1994, 144, 906–914. [Google Scholar]
- Li, W.; Zhang, S.L.; Wang, N.; Zhang, B.B.; Li, M. Blockade of T-type Ca(2+) channels inhibits human ovarian cancer cell proliferation. Cancer Investig. 2011, 29, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.J.; Choi, H.W.; Choi, D.L.; Cho, S.; Rim, H.K.; Choi, H.E.; Kim, K.S.; Huang, M.; Rhim, H.; Lee, K.T.; et al. In vitro cytotoxicity on human ovarian cancer cells by T-type calcium channel blockers. Bioorg. Med. Chem. Lett. 2013, 23, 6656–6662. [Google Scholar] [CrossRef] [PubMed]
- Dziegielewska, B.; Casarez, E.V.; Yang, W.Z.; Gray, L.S.; Dziegielewski, J.; Slack-Davis, J.K. T-type Ca2+ channel inhibition sensitizes ovarian cancer to carboplatin. Mol. Cancer Ther. 2016, 15, 460–470. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Rubaiy, H.N.; Chen, G.L.; Hallett, T.; Zaibi, N.; Zeng, B.; Saurabh, R.; Xu, S.Z. Mibefradil, a T-type Ca2+ channel blocker also blocks Orai channels by action at the extracellular surface. Br. J. Pharmacol. 2019, 176, 3845–3856. [Google Scholar] [CrossRef] [PubMed]
- Foti, R.S.; Rock, D.A.; Pearson, J.T.; Wahlstrom, J.L.; Wienkers, L.C. Mechanism-based inactivation of cytochrome P450 3A4 by mibefradil through heme destruction. Drug Metab. Dispos. 2011, 39, 1188–1195. [Google Scholar] [CrossRef]
- Gómez-Lagunas, F.; Barriga-Montoya, C. Mibefradil inhibition of the Cole-Moore shift and K+-conductance of the tumor-related Kv10.1 channel. Channels 2017, 11, 373–376. [Google Scholar] [CrossRef]
- Kim, E.K.; Ha, J.M.; Kim, Y.W.; Jin, S.Y.; Ha, H.K.; Bae, S.S. Inhibitory role of polyunsaturated fatty acids on lysophosphatidic acid-induced cancer cell migration and adhesion. FEBS Lett. 2014, 588, 2971–2977. [Google Scholar] [CrossRef]
- Chovancova, B.; Liskova, V.; Miklikova, S.; Hudecova, S.; Babula, P.; Penesova, A.; Sevcikova, A.; Durinikiva, E.; Novakova, M.; Matuskova, M.; et al. Calcium signaling affects migration and proliferation differently in individual cancer cells due to nifedipine treatment. Biochem. Pharmacol. 2020, 171, 113695. [Google Scholar] [CrossRef]
- Xu, S.K.; Huang, Q.F.; Zeng, W.F.; Sheng, C.S.; Li, Y.; Wang, J.G. A randomized multicenter study on ambulatory blood pressure and arterial stiffness in patients treated with valsartan/amlodipine or nifedipine GITS. J. Clin. Hypertens. 2019, 21, 252–261. [Google Scholar] [CrossRef]
- Ye, Z.; Yang, H.; Li, H.; Zhang, X.; Deng, Y.; Zeng, G.; Chen, L.; Cheng, Y.; Yang, J.; Mi, Q.; et al. A multicentre, prospective, randomized trial: Comparative efficacy of tamsulosin and nifedipine in medical expulsive therapy for distal ureteric stones with renal colic. BJU Int. 2011, 8, 276–279. [Google Scholar] [CrossRef]
- Hudecova, S.; Markova, J.; Simko, V.; Csaderova, L.; Stracina, T.; Sirova, M.; Fojtu, M.; Svastova, E.; Gronesova, P.; Pastorek, M. Sulforaphane-induced apoptosis involves the type 1 IP3 receptor. Oncotarget 2016, 7, 61403–61418. [Google Scholar] [CrossRef][Green Version]
- Sun, B.; Zhang, X.; Yin, Y.; Sun, H.; Ge, H.; Li, W. Effects of sulforaphane and vitamin E on cognitive disorder and oxidative damage in lead-exposed mice hippocampus at lactation. J. Trace Elem. Med. Biol. 2017, 44, 88–92. [Google Scholar] [CrossRef]
- Axelsson, A.S.; Tubbs, E.; Mecham, B.; Chacko, S.; Nenonen, H.A.; Tang, Y.; Wahey, J.; Derry, J.M.J.; Wollheim, C.B.; Wierup, N.; et al. Sulforaphane reduces hepatic glucose production and improves glucose control in patients with type 2 diabetes. Sci. Transl. 2017, 9, eaah4477. [Google Scholar] [CrossRef]
- Yu, C.; He, Q.; Zheng, J.; Li, L.Y.; Hou, Y.H.; Song, F.Z. Sulforaphane improves outcomes and slows cerebral ischemic/reperfusion injury via inhibition of NLRP3 inflammasome activation in rats. Int Immunopharmacol. 2017, 45, 74–78. [Google Scholar] [CrossRef]
- Chovancova, B.; Hudecova, S.; Lencesova, L.; Babula, P.; Rezuchova, I.; Penesova, A.; Grman, M.; Moravcik, R.; Zeman, M.; Krizanova, O. Melatonin-induced changes in cytosolic calcium might be responsible for apoptosis induction in tumour cells. Cell Physiol. Biochem. 2017, 44, 763–777. [Google Scholar] [CrossRef] [PubMed]
- Mundey, K.; Benloucif, S.; Harsanyi, K.; Dubocovich, M.L.; Zee, P.C. Phase-Dependent Treatment of Delayed Sleep Phase Syndrome with Melatonin. Sleep 2005, 28, 1271–1278. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Kim, J.W.; Kim, D.K.; Choi, D.K.; Lee, S.; Yu, J.H.; Kwon, O.B.; Lee, J.; Lee, D.S.; Kim, J.H.; et al. Calcium Channels as Novel Therapeutic Targets for Ovarian Cancer Stem Cells. Int. J. Mol. Sci. 2020, 21, 2327. [Google Scholar] [CrossRef] [PubMed]
- Chu, M.K.; Wang, S.J. Nomenclature for flunarizine, cinnarizine, and lomerizine. Cephalalgia 2020, 40, 321–322. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.L.; Cao, Q.; Zhou, K.C.; Feng, Y.J.; Wang, Y.Z. Transient receptor potential channel C3 contributes to the progression of human ovarian cancer. Oncogene 2009, 28, 1320–1328. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Ling, Z.H.; Wang, H.; Wang, X.Y.; Gui, J. Upregulation of SCNN1A Promotes Cell Proliferation, Migration, and Predicts Poor Prognosis in Ovarian Cancer Through Regulating Epithelial–Mesenchymal Transformation. Cancer Biother. Radiopharm. 2019, 34, 642–649. [Google Scholar] [CrossRef] [PubMed]
- Putzke, A.P.; Ventura, A.P.; Bailey, A.M.; Akture, C.; Opoku-Ansah, J.; Celiktas, M.; Hwang, M.S.; Darling, D.S.; Coleman, I.L.; Nelson, P.S.; et al. Metastatic progression of prostate cancer and E-cadherin: Regulation by Zeb1 and Src family kinases. Am. J. Pathol. 2011, 179, 400. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Tokura, Y. Epithelial–mesenchymal transition in the skin. J. Dermatol. Sci. 2011, 61, 7. [Google Scholar] [CrossRef] [PubMed]
- Kuang, Q.; Purhonen, P.; Hebert, H. Structure of potassium channels. Cell Mol. Life Sci. 2015, 72, 3677–3693. [Google Scholar] [CrossRef]
- Zhanping, W.; Xiaoyu, P.; Na, C.; Shenglan, W.; Bo, W. Voltage-gated K+ channels are associated with cell proliferation and cell cycle of ovarian cancer cell. Gynecol. Oncol. 2007, 104, 455–460. [Google Scholar] [CrossRef]
- Asher, V.; Khan, R.; Warren, A.; Shaw, R.; Schalkwyk, G.V.; Bali, A.; Sowter, H.M. The Eag potassium channel as a new prognostic marker in ovarian cancer. Diagn. Pathol. 2010, 5, 78. [Google Scholar] [CrossRef]
- Wonderlin, W.M.; Strobl, J.S. Potassium channels, proliferation and G1 progression. J. Membr. Biol. 1996, 154, 91–107. [Google Scholar] [CrossRef]
- Pardo, L.A. Voltage-gated potassium channels in cell proliferation. Physiology 2004, 19, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Iamshanova, O.; Fiorio Pla, A.; Prevarskaya, N. Molecular mechanisms of tumour invasion: Regulation by calcium signals. J. Physiol. 2017, 595, 3063–3075. [Google Scholar] [CrossRef]
- He, S.; Moutaoufik, M.; Islam, S.; Persad, A.; Wu, A.; Aly, K.A.; Fonge, H.; Babu, M.; Cayabyab, F.S. HERG channel and cancer: A mechanistic review of carcinogenic processes and therapeutic potential. Biochim. Biophys. Acta Rev. Cancer 2020, 1873, 188355. [Google Scholar] [CrossRef]
- Arcangeli, A.; Crociani, O.; Lastraioli, E.; Masi, A.; Pillozzi, S.; Becchetti, A. Targeting ion channels in cancer: A novel frontier in antineoplastic therapy. Curr. Med. Chem. 2009, 16, 66–93. [Google Scholar] [CrossRef]
- Chelintseva, E.; Djamgoz, M.B.A. Mesenchymal stem cell differentiation: Control by calcium-activated potassium channels. J. Cell Physiol. 2018, 233, 3755–3768. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Chang, Y.; Reinhart, P.H.; Sontheimer, H.; Chang, Y. Cloning and characterization of glioma BK, a novel BK channel isoform highly expressed in human glioma cells. J. Neurosci. 2002, 22, 1840–1849. [Google Scholar] [CrossRef] [PubMed]
- Bloch, M.; Ousingsawat, J.; Simon, R.; Schraml, P.; Gasser, T.C.; Mihatsch, M.J.; Kunzelmann, K.; Bubendorf, L. KCNMA1 gene amplification promotes tumor cell proliferation in human prostate cancer. Oncogene 2007, 26, 2525–2534. [Google Scholar] [CrossRef]
- Oeggerli, M.; Tian, Y.; Ruiz, C.; Wijker, B.; Sauter, G.; Obermann, E.; Güth, U.; Zlobec, I.; Sausbier, M.; Kunzelmann, K.; et al. Role of KCNMA1 in breast cancer. PLoS ONE 2012, 7, e41664. [Google Scholar] [CrossRef] [PubMed]
- Zylicz, M.; King, F.W.; Wawrzynow, A. Hsp70 interactions with the p53 tumour suppressor protein. Embo J. 2001, 20, 4634–4638. [Google Scholar] [CrossRef]
- Liu, X.; Wei, L.; Zhao, B.; Cai, X.; Dong, C.; Yin, F. Low expression of KCNN3 may affect drug resistance in ovarian cancer. Mol. Med. Rep. 2018, 18, 1377–1386. [Google Scholar] [CrossRef] [PubMed]
- Robles-Martinez, L.; Garay, E.; Martel-Gallegos, M.G.; Cisneros-Mejorado, A.; Pérez-Montiel, D.; Lara, A.; Arellano, R. Kca3.1 Activation Via P2y2 Purinergic Receptors Promotes Human Ovarian Cancer Cell (Skov-3) Migration. Sci. Rep. 2017, 7, 4340. [Google Scholar] [CrossRef] [PubMed]
- Tinker, A.; Aziz, Q.; Li, Y.; Specterman, M. ATP-Sensitive Potassium Channels and Their Physiological and Pathophysiological Roles. Compr. Physiol. 2018, 8, 1463–1511. [Google Scholar]
- Mu, D.; Chen, L.; Zhang, X.; See, L.H.; Koch, C.M.; Yen, C.; Tong, J.J.; Spiegel, L.; Nguyen, K.C.Q.; Servoss, A.; et al. Genomic amplification and oncogenic properties of the KCNK9 potassium channel gene. Cancer Cell. 2003, 3, 297–302. [Google Scholar] [CrossRef]
- Pei, L.; Wiser, O.; Slavin, A.; Mu, D.; Powers, S.; Jan, L.Y.; Hoey, T. Oncogenic potential of TASK3 (KCNK9) depends on K+ channel function. Proc. Natl. Acad. Sci. USA 2003, 100, 7803–7807. [Google Scholar] [CrossRef] [PubMed]
- Giordano, A.; Tommonaro, G. Curcumin and Cancer. Nutrients 2019, 11, 2376. [Google Scholar] [CrossRef]
- Cuddapah, V.A.; Sontheimer, H. Ion channels and transporters (corrected) in cancer. 2. Ion channels and the control of cancer cell migration. Am. J. Physiol. Cell Physiol. 2011, 301, C541–C549. [Google Scholar] [CrossRef]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Ion channels and the hallmarks of cancer. Trends Mol. Med. 2010, 16, 107–121. [Google Scholar] [CrossRef]
- Verkman, A.; Galietta, L. Chloride channels as drug targets. Nat. Rev. Drug Discov. 2009, 8, 153–171. [Google Scholar] [CrossRef]
- Ashley, R.H. Challenging accepted ion channel biology: p64 and the CLIC family of putative intracellular anion channel proteins (Review). Mol. Membr. Biol. 2003, 20, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ye, D.; Luo, H.; Lai, Z.; Zou, L.; Zhu, L.; Mao, J.; Jacob, T.; Ye, W.; Wang, L.; Chenc, L. ClC-3 Chloride Channel Proteins Regulate the Cell Cycle by Up-regulating cyclin D1-CDK4/6 through Suppressing p21/p27 Expression in Nasopharyngeal Carcinoma Cells. Sci. Rep. 2016, 6, 30276. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wang, B.; Lin, W. Cl-channel blockers inhibit cell proliferation and arrest the cell cycle of human ovarian cancer cells. Eur. J. Gynaecol. Oncol. 2008, 29, 267–271. [Google Scholar]
- Xu, J.; Yong, M.; Li, J.; Dong, X.; Yu, T.; Fu, X.; Hu, L. High level of CFTR expression is associated with tumor aggression and knockdown of CFTR suppresses proliferation of ovarian cancer in vitro and in vivo. Oncol. Rep. 2015, 33, 2227–2234. [Google Scholar] [CrossRef]
- Xu, J.; Lin, L.; Yong, M.; Dong, X.; Yu, T.; Hu, L. Adenovirus-mediated overexpression of cystic fibrosis transmembrane conductance regulator enhances invasiveness and motility of serous ovarian cancer cells. Mol. Med. Rep. 2016, 13, 265–272. [Google Scholar] [CrossRef][Green Version]
- Lang, F.; Stournaras, C. Ion channels in cancer: Future perspectives and clinical potential. Philos. Trans. R Soc. Lond. B Biol. Sci. 2014, 369, 20130108. [Google Scholar] [CrossRef] [PubMed]
- Stock, C.; Schwab, A. Ion channels and transporters in metastasis. Biochim. Biophys. Acta 2015, 1848, 2638–2646. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Lu, M.; Liu, B.; Huang, Y.; Wang, K.W. Inhibition of Ca(2+)-activated Cl(−) channel ANO1/TMEM16A expression suppresses tumor growth and invasiveness in human prostate carcinoma. Cancer Lett. 2012, 326, 41–51. [Google Scholar] [CrossRef]
- Britschgi, A.; Bill, A.; Brinkhaus, H.; Rothwell, C.; Clay, I.; Duss, S.; Rebhan, M.; Raman, P.; Guy, C.T.; Wetzel, K. Calcium activated chloride channel ANO1 promotes breast cancer progression by activating EGFR and CAMK signaling. Proc. Natl. Acad. Sci. USA 2013, 110, 1026–1034. [Google Scholar] [CrossRef]
- Bill, A.; Gutierrez, A.; Kulkarni, S.; Kemp, C.; Bonenfant, D.; Voshol, H.; Duvvuri, U.; Gaither, A. ANO1/ TMEM16A interacts with EGFR and correlates with sensitivity to EGFR-targeting therapy in head and neck cancer. Oncotarget 2015, 6, 9173–9188. [Google Scholar] [CrossRef]
- Jia, L.; Liu, W.; Guan, L.; Lu, M.; Wang, K.W. Inhibition of calcium activated Chloride Channel ANO1/TMEM16A suppresses tumor growth and invasion in human lung cancer. PLoS ONE 2015, 10, e0136584. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Cao, Q.H.; Lu, D.J.; Luo, B.; Lu, X.F.; Luo, R.C.; Wang, X.C. TMEM16A overexpression contributes to tumor invasion and poor prognosis of human gastric cancer through TGF β signaling. Oncotarget 2015, 6, 11585–11599. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Yang, J.; Chen, H.; Ma, B.; Pan, K.; Su, C.; Xu, F.; Zhang, J. Knockdown of TMEM16A suppressed MAPK and inhibited cell proliferation and migration in hepatocellular carcinoma. OncoTargets Ther. 2016, 9, 325–333. [Google Scholar]
- Liu, Z.; Zhang, S.; Hou, F.; Zhang, C.; Gao, J.; Wang, K. Inhibition of Ca2+–activated chloride channel ANO1 suppresses ovarian cancer through inactivating PI3K/Akt signaling. Int. J. Cancer 2019, 144, 2215–2226. [Google Scholar] [CrossRef]
- Schreiber, R.; Ousingsawat, J.; Wanitchakool, P.; Sirianant, L.; Benedetto, R.; Reiss, K.; Kunzelmann, K. Regulation of TMEM16A/ANO1 and TMEM16F/ANO6 ion currents and phospholipid scrambling by Ca2+ and plasma membrane lipid. J. Physiol. 2018, 596, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Crottès, D.; Yeh Jan, L. The multifaceted role of TMEM16A in cancer. Cell Calcium 2019, 82, 102050. [Google Scholar] [CrossRef]
- Gueguinou, M.; Gambade, A.; Felix, R.; Chantome, A.; Fourbon, Y.; Bougnoux, P.; Weber, G.; Potier-Cartereau, M.; Vandier, C. Lipid rafts, KCa/ClCa/Ca2+ channel complexes and EGFR signaling: Novel targets to reduce tumor development by lipids? Biochim. Biophys. Acta 2015, 1848, 2603–2620. [Google Scholar] [CrossRef] [PubMed]
- Ye, W.; Han, T.W.; Nassar, L.M.; Zubia, M.; Jan, Y.N.; Jan, L.Y. Phosphatidylinositol-(4, 5)-bisphosphate regulates calcium gating of small-conductance cation channel TMEM16F. Proc. Natl. Acad. Sci. USA 2018, 115, E1667–E1674. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Ansari, D.; Bauden, M.; Zhou, Q.; Andersson, R. The Emerging Role of Calcium-activated Chloride Channel Regulator 1 in Cancer. Anticancer Res. 2019, 39, 1661–1666. [Google Scholar] [CrossRef] [PubMed]
- Peretti, M.; Angelini, M.; Savalli, N.; Florio, T.; Yuspa, S.H.; Mazzanti, M. Chloride channels in cancer: Focus on chloride intracellular channel 1 and 4 (CLIC1 AND CLIC4) proteins in tumor development and as novel therapeutic targets. Biochim. Biophys. Acta 2014, 1848, 2523–2531. [Google Scholar] [CrossRef]
- Valenzuela, S.M.; Martin, D.K.; Por, S.B.; Robbins, J.M.; Warton, K.; Bootcov, M.R.; Schofield, P.R.; Campbell, T.J.; Breit, S.N. Molecular cloning and expression of a chloride ion channel of cell nuclei. J. Biol. Chem. 1997, 272, 12575–12582. [Google Scholar] [CrossRef]
- Tang, H.Y.; Beer, L.A.; Chang-Wong, T.; Hammond, R.; Gimotty, P.; Coukos, G.; Speicher, D.W. A xenograft mouse model coupled with in-depth plasma proteome analysis facilitates identification of novel serum biomarkers for human ovarian cancer. J. Proteome Res. 2012, 11, 678–691. [Google Scholar] [CrossRef]
- Tang, H.Y.; Beer, L.A.; Tanyi, J.L.; Zhang, R.; Liu, Q.; Speicher, D.W. Protein isoform-specific validation defines multiple chloride intracellular channel and tropomyosin isoforms as serological biomarkers of ovarian cancer. J. Proteom. 2013, 89, 165–178. [Google Scholar] [CrossRef]
- Yu, W.; Cui, R.; Qu, H.; Liu, C.; Deng, H.; Zhang, Z. Expression and prognostic value of CLIC1 in epithelial ovarian cancer. Exp. Ther. Med. 2018, 15, 4943–4949. [Google Scholar] [CrossRef]
- Qu, H.; Chen, Y.; Cao, G.; Liu, C.; Xu, J.; Deng, H.; Zhang, Z. Identification and validation of differentially expressed proteins in epithelial ovarian cancers using quantitative proteomics. Oncotarget 2016, 7, 83187–83199. [Google Scholar] [CrossRef]
- Singha, B.; Harper, S.L.; Goldman, A.R.; Bitler, B.G.; Aird, K.M.; Borowsky, M.; Cadungog, M.G.; Liu, Q.; Zhang, R.; Jean, S.; et al. CLIC1 and CLIC4 complement CA125 as a diagnostic biomarker panel for all subtypes of epithelial ovarian cancer. Sci. Rep. 2018, 8, 14725. [Google Scholar] [CrossRef]
- Hernandez-Fernaud, J.R.; Ruengeler, E.; Casazza, A.; Neilson, L.J.; Pulleine, E.; Santi, A.; Ismail, S.; Lilla, S.; Dhayade, S.; MacPherson, I.R.; et al. Secreted CLIC3 drives cancer progression through its glutathione-dependent oxidoreductase activity. Nat. Commun. 2017, 8, 14206. [Google Scholar] [CrossRef] [PubMed]
- Argenzio, E.; Moolenaar, W.H. Emerging biological roles of Cl-intracellular channel proteins. J. Cell Sci. 2016, 129, 4165–4174. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium signalling: Dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 2003, 4, 517–529. [Google Scholar] [CrossRef] [PubMed]
- Clapham, D.E. Calcium signaling. Cell 1995, 80, 259. [Google Scholar] [CrossRef]
- Mertens-Walker, I.; Bolitho, C.; Baxter, R.C.; Marsh, D.J. Gonadotropin-induced ovarian cancer cell migration and proliferation require extracellular signal-regulated kinase 1/2 activation regulated by calcium and protein kinase Cδ. Endocr. Relat. Cancer 2010, 17, 335–349. [Google Scholar] [CrossRef]
- Kouba, S.; Ouldamer, L.; Garcia, C.; Fontaine, D.; Chantome, A.; Vandier, C.; Goupille, C.; Potier-Cartereau, M. Lipid metabolism and Calcium signaling in epithelial ovarian cancer. Cell Calcium 2019, 81, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Gaudette, D.C.; Boynton, J.D.; Frankel, A.; Fang, X.J.; Sharma, A.; Hurteau, J.; Casey, G.; Goodbody, A.; Mellors, A.; et al. Characterization of an ovarian cancer activating factor in ascites from ovarian cancer patients. Clin. Cancer Res. 1995, 1, 1223–1232. [Google Scholar] [PubMed]
- Yu, X.; Zhang, Y.; Chen, H. LPA receptor 1 mediates LPA-induced ovarian cancer metastasis: An in vitro and in vivo study. BMC Cancer 2016, 16, 846. [Google Scholar] [CrossRef]
- Zhang, H.; Zhou, L.; Shi, W.; Song, N.; Yu, K.; Gu, Y. A mechanism underlying the effects of polyunsaturated fatty acids on breast cancer. Int. J. Mol. Med. 2012, 30, 487–494. [Google Scholar] [CrossRef]
- Lencesova, L.; Krizanova, O. IP(3) receptors, stress and apoptosis. Gen. Physiol. Biophys. 2012, 31, 119–130. [Google Scholar] [CrossRef][Green Version]
- Shibao, K.; Fiedler, M.J.; Nagata, J.; Minagawa, N.; Hirata, K.; Nakayama, Y.; Iwakiri, Y.; Nathanson, M.H.; Yamaguchi, K. The type III inositol 1, 4, 5-trisphosphate receptor is associated with aggressiveness of colorectal carcinoma. Cell Calcium 2010, 48, 315–323. [Google Scholar] [CrossRef]
- Rezuchova, I.; Hudecova, S.; Soltysova, A.; Matuskova, M.; Durinikova, E.; Chovancova, B.; Zuzcak, M.; Cihova, M.; Burikova, M.; Penesova, A.; et al. Type 3 inositol 1,4,5-trisphosphate receptor has antiapoptoticand proliferative role in cancer cells. Cell Death Dis. 2019, 10, 186. [Google Scholar] [CrossRef] [PubMed]
- Mound, A.; Vautrin-Glabik, A.; Foulon, A.; Botia, B.; Hague, F.; Parys, J.B.; Ouadid-Ahidouch, H.; Rodat-Despoix, L. Downregulation of type 3 inositol (1, 4, 5)-trisphosphate receptor decreases breast cancer cell migration through an oscillatory Ca2+ signal. Oncotarget 2017, 8, 72324. [Google Scholar] [CrossRef]
- Kang, S.S.; Han, K.S.; Ku, B.M.; Lee, Y.K.; Hong, J.; Shin, H.Y.; Almonte, A.G.; Woo, D.H.; Brat, D.J.; Hwang, E.M.; et al. Caffeine-mediated inhibition of calcium release channel inositol 1, 4, 5-trisphosphatereceptor subtype 3 blocks glioblastoma invasion and extends survival. Cancer Res. 2010, 70, 1173–1183. [Google Scholar] [CrossRef]
- Lewis, R.S. Store-operated calcium channels: New perspectives on mechanism and function. Cold Spring Harb. Perspect. Biol. 2011, 3, a003970. [Google Scholar] [CrossRef]
- Yang, S.; Zhang, J.J.; Huang, X.Y. Orai1 and STIM1 are critical for breast tumor cell migration and metastasis. Cancer Cell 2009, 15, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.F.; Chiu, W.T.; Chen, Y.T.; Lin, P.Y.; Huang, H.J.; Chou, C.Y.; Chang, H.C.; Tang, M.J.; Shen, M.R. Calcium store sensor stromal-interaction molecule 1-dependent signaling plays an important role in cervicalcancer growth, migration, and angiogenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 15225–15230. [Google Scholar] [CrossRef] [PubMed]
- Abdelazeem, K.N.M.; Droppova, B.; Sukkar, B.; Al-Maghout, T.; Pelzl, L.; Zacharopoulou, N.; Hassan Ali Hassan, N.; Abdel-Fattah, K.I.; Stournaras, C.; Lang, F. Upregulation of Orai1 and STIM1 expression as well as store-operated Ca 2+ entry in ovary carcinoma cells by placental growth factor. Biochem. Biophys. Res. Commun. 2019, 512, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, S.F.; Owsianik, G.; Nilius, B. TRP channels: An overview. Cell Calcium 2005, 38, 233–252. [Google Scholar] [CrossRef]
- Zhuang, L.; Peng, J.B.; Tou, L.; Takanaga, H.; Adam, R.M.; Hediger, M.A.; Freeman, M.R. Calcium-selective ion channel, CaT1, is apically localized in gastrointestinal tract epithelia and is aberrantly expressed in human malignancies. Lab. Investig. 2002, 82, 1755–1764. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Chay, D.B.; Nam, S.; Cho, H.; Chung, J.Y.; Kim, J.H. Prognostic Significance of Transient Receptor Potential Vanilloid Type 1 (TRPV1) and Phosphatase and Tension Homolog (PTEN) in Epithelial Ovarian Cancer. Cancer Genom. Proteom. 2020, 17, 309–319. [Google Scholar] [CrossRef]
- Xue, H.; Wang, Y.; MacCormack, T.J.; Lutes, T.; Rice, C.; Davey, M.; Dugourd, D.; Ilenchuk, T.T.; Stewart, J.M. Inhibition of Transient Receptor Potential Vanilloid 6 channel, elevated in human ovarian cancers, reduces tumour growth in a xenograft model. J. Cancer 2018, 9, 3196–3207. [Google Scholar] [CrossRef] [PubMed]
- Lehen’kyi, V.; Flourakis, M.; Skryma, R.; Prevarskaya, N. TRPV6 channel controls prostate cancer cell proliferation via Ca(2+)/NFAT-dependent pathways. Oncogene 2007, 26, 7380–7385. [Google Scholar] [CrossRef]
- Quang, C.T.; Leboucher, S.; Passaro, D.; Fuhrmann, L.; Nourieh, M.; Vincent-Salomon, A.; Ghysdael, J. The calcineurin/NFAT pathway is activated in diagnostic breast cancer cases and is essential to survival and metastasis of mammary cancer cells. Cell Death Dis. 2015, 6, e1658. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Hirte, H.; Welch, S.; Ilenchuk, T.T.; Lutes, T.; Rice, C.; Fields, N.; Nemet, A.; Dugourd, D.; Piha-Paul, S.; et al. First-in-human phase I study of SOR-C13, a TRPV6 calcium channel inhibitor, in patients with advanced solid tumors. Investig. New Drugs 2017, 35, 324–333. [Google Scholar] [CrossRef]
- Faouzi, M.; Hague, F.; Geerts, D.; Ay, A.S.; Potier-Cartereau, M.; Ahidouch, A.; Ouadid-Ahidouch, H. Functional cooperation between KCa3.1 and TRPC1 channels in human breast cancer: Role in cell proliferation and patient prognosis. Oncotarget 2016, 7, 36419–36435. [Google Scholar] [CrossRef]
- Chen, J.; Luan, Y.; Yu, R.; Zhang, Z.; Zhang, J.; Wang, W. Transient receptor potential (TRP) channels, promising potential diagnostic and therapeutic tools for cancer. Biosci. Trends 2014, 8, 1–10. [Google Scholar] [CrossRef]
- Villalobos, C.; Hernandez-Morales, M.; Gutierrez, L.G.; Nunez, L. TRPC1 and ORAI1 channels in colon cancer. Cell Calcium 2019, 81, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Asghar, M.Y.; Magnusson, M.; Kemppainen, K.; Sukumaran, P.; Lof, C.; Pulli, I.; Kalhori, V.; Tornquist, K. Transient Receptor Potential Canonical 1 (TRPC1) Channels as Regulators of Sphingolipid and VEGF Receptor Expression: Implication for thyroid cancer cell migration and proliferation. J. Biol. Chem. 2015, 290, 16116–16131. [Google Scholar] [CrossRef]
- Dong, H.; Shim, K.N.; Li, J.M.; Estrema, C.; Ornelas, T.A.; Nguyen, F.; Liu, S.; Ramamoorthy, S.L.; Ho, S.; Carethers, J.M.; et al. Molecular mechanisms underlying Ca2+–mediated motility of human pancreatic duct cells. Am. J. Physiol. Cell Physiol. 2010, 299, C1493–C1503. [Google Scholar] [CrossRef]
- Zeng, B.; Yuan, C.; Yang, X.; Atkin, S.L.; Xu, S.Z. TRPC Channels and their splice variants are essential for promoting human ovarian cancer cell proliferation and tumorigenesis. Curr. Cancer Drug Targets 2013, 13, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Jiang, K.; Li, J.; Hao, X.; Chu, W.; Luo, C.; Zhu, Y.; Xie, R.; Chen, B. Estrogen enhances the proliferation and migration of ovarian cancer cells by activating transient receptor potential channel C3. J. Ovarian Res. 2020, 13, 20. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liao, Q.J.; Zhang, Y.; Zhou, H.; Luo, C.H.; Tang, J.; Wang, Y.; Tang, Y.; Zhao, M.; Zhao, X.H.; et al. TRPM7 is required for ovarian cancer cell growth, migration and invasion. Biochem. Biophys. Res. Commun. 2014, 454, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Wang, X.; Chen, M.; Ouyang, K.; Song, L.S.; Cheng, H. Calcium flickers steer cell migration. Nature 2009, 457, 901–905. [Google Scholar] [CrossRef]
- Yang, D.; Kim, J. Emerging role of transient receptor potential (TRP) channels in cancer progression. BMB Rep. 2020, 53, 125–132. [Google Scholar] [CrossRef]
- Ottevanger, P.B. Ovarian cancer stem cells more questions than answers. Semin. Cancer Biol. 2017, 44, 67–71. [Google Scholar] [CrossRef]
- French, B.; Gasch, C.; O’Leary, J.J.; Gallagher, M.F. Developing ovarian cancer stem cell models: Laying the pipeline from discovery to clinical intervention. Mol. Cancer 2014, 13, 262. [Google Scholar] [CrossRef]
- Yang, X.; Zheng, F.; Xing, H.; Gao, Q.; Wei, W.; Lu, Y.; Wang, S.; Zhou, J.; Hu, W.; Ma, D. Resistance to chemotherapy-induced apoptosis via decreased caspase-3 activity and overexpression of antiapoptotic proteins in ovarian cancer. J. Cancer Res. Clin. Oncol. 2004, 130, 423–428. [Google Scholar] [CrossRef]
- Schmidt, S.; Liu, G.; Liu, G.; Yang, W.; Honisch, S.; Pantelakos, S.; Stournaras, C.; Höning, A.; Lang, F. Enhanced Orai1 and STIM1 expression as well as store operated Ca2+ entry in therapy resistant ovary carcinoma cells. Oncotarget 2014, 5, 4799–4810. [Google Scholar] [CrossRef]
- Mir, R.; Stanzani, E.; Martinez-Soler, F.; Villanueva, A.; Vidal, A.; Condom, E.; Ponce, J.; Gil, J.; Tortosa, A.; Giménez-Bonafé, P. YM155 sensitizes ovarian cancer cells to cisplatin inducing apoptosis and tumor regression. Gynecol. Oncol. 2014, 132, 211–220. [Google Scholar] [CrossRef]
- Liu, X.; Zou, J.; Su, J.; Lu, Y.; Zhang, J.; Li, L.; Yin, F. Downregulation of transient receptor potential cation channel, subfamily C, member 1 contributes to drug resistance and high histological grade in ovarian cancer. Int. J. Oncol. 2016, 48, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Jaber, N.; Dou, Z.; Lin, R.Z.; Zhang, J.; Zong, W.X. Mammalian PIK3C3/VPS34: The key to autophagic processing in liver and heart. Autophagy 2012, 8, 707–708. [Google Scholar] [CrossRef] [PubMed]
- Yin, F.; Liu, X.; Li, D.; Wang, Q.; Zhang, W.; Li, L. Bioinformatic analysis of chemokine (C-C motif) ligand 21 and SPARC-like protein 1 revealing their associations with drug resistance in ovarian cancer. Int. J. Oncol. 2013, 42, 1305–1316. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.; Fojo, T.; Bates, S. Tumour stem cells and drug resistance. Nat. Rev. Cancer 2005, 5, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Marklund, L.; Andersson, B.; Behnam-Motlagh, P.; Sandstrom, P.E.; Henriksson, R.; Grankvist, K. Cellular potassium ion deprivation enhances apoptosis induced by cisplatin. Basic Clin. Pharmacol. Toxicol. 2004, 94, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Hui, C.; Lan, Z.; Yue-li, L.; Li-lin, H. Knockdown of Eag1 Expression by RNA Interference Increases Chemosensitivity to Cisplatin in Ovarian Cancer Cells. Reprod. Sci. 2015, 22, 1618–1626. [Google Scholar] [CrossRef]
- Lloyd, K.L.; Cree, I.A.; Savage, R.S. Prediction of resistance to chemotherapy in ovarian cancer: A systematic review. BMC Cancer 2015, 15, 117. [Google Scholar] [CrossRef]
- Gueguinou, M.; Harnois, T.; Crottes, D.; Uguen, A.; Deliot, N.; Gambade, A.; Chantôme, A.; Haelters, J.P.; Jaffrès, P.A.; Jourdan, M.L.; et al. SK3/TRPC1/Orai1 complex regulates SOCE-dependent colon cancer cell migration: A novel oppor-tunity to modulate anti-EGFR mAb action by the alkyl-lipid Ohmline. Oncotarget 2016, 7, 36168–36184. [Google Scholar] [CrossRef]
- Samuel, P.; Pink, R.C.; Caley, D.P.; Currie, J.M.; Brooks, S.A.; Carter, D.R. Over-expression of miR-31 or loss of KCNMA1 leads to increased cisplatin resistance in ovarian cancer cells. Tumour Biol. 2016, 37, 2565–2573. [Google Scholar] [CrossRef] [PubMed]
- Caravia, L.; Staicu, C.E.; Radu, B.M.; Condrat, C.E.; Cretoiu, D.; Bacalbasa, N.; Suciu, N.; Cretoiu, S.M.; Voinea, S.C. Altered Organelle Calcium Transport in Ovarian Physiology and Cancer. Cancers 2020, 12, 2232. [Google Scholar] [CrossRef]
- Pillozzi, S.; D’Amico, M.; Bartoli, G.; Gasparoli, L.; Petroni, G.; Crociani, O.; Marzo, T.; Guerriero, A.; Messori, L.; Severi, M.; et al. The combined activation of KCa 3.1 and inhibition of K v 11.1/hERG1 currents contribute to overcome Cisplatin resistance in colorectal cancer cells. Br. J. Cancer 2018, 118, 200–212. [Google Scholar] [CrossRef]
- Haustrate, A.; Hantute-Ghesquier, A.; Prevarskaya, N.; Lehen’kyi, V. Monoclonal Antibodies Targeting Ion Channels and Their Therapeutic Potential. Front. Pharmacol. 2019, 10, 606. [Google Scholar] [CrossRef]
- Hernandez-Resendiz, I.; Hartung, F.; Pardo, L.A. Antibodies Targeting KV Potassium Channels: A Promising Treatment for Cancer. Bioelectricity 2019, 1, 3. [Google Scholar] [CrossRef]
- Duranti, C.; Arcangeli, A. Ion Channel Targeting with Antibodies and Antibody Fragments for Cancer Diagnosis. Antibodies 2019, 8, 33. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Altamura, C.; Greco, M.R.; Carratù, M.R.; Cardone, R.A.; Desaphy, J.-F. Emerging Roles for Ion Channels in Ovarian Cancer: Pathomechanisms and Pharmacological Treatment. Cancers 2021, 13, 668. https://doi.org/10.3390/cancers13040668
Altamura C, Greco MR, Carratù MR, Cardone RA, Desaphy J-F. Emerging Roles for Ion Channels in Ovarian Cancer: Pathomechanisms and Pharmacological Treatment. Cancers. 2021; 13(4):668. https://doi.org/10.3390/cancers13040668
Chicago/Turabian StyleAltamura, Concetta, Maria Raffaella Greco, Maria Rosaria Carratù, Rosa Angela Cardone, and Jean-François Desaphy. 2021. "Emerging Roles for Ion Channels in Ovarian Cancer: Pathomechanisms and Pharmacological Treatment" Cancers 13, no. 4: 668. https://doi.org/10.3390/cancers13040668
APA StyleAltamura, C., Greco, M. R., Carratù, M. R., Cardone, R. A., & Desaphy, J.-F. (2021). Emerging Roles for Ion Channels in Ovarian Cancer: Pathomechanisms and Pharmacological Treatment. Cancers, 13(4), 668. https://doi.org/10.3390/cancers13040668