
Elucidating the Importance of DOT1L Recruitment in MLL-AF9 Leukemia and Hematopoiesis

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Results
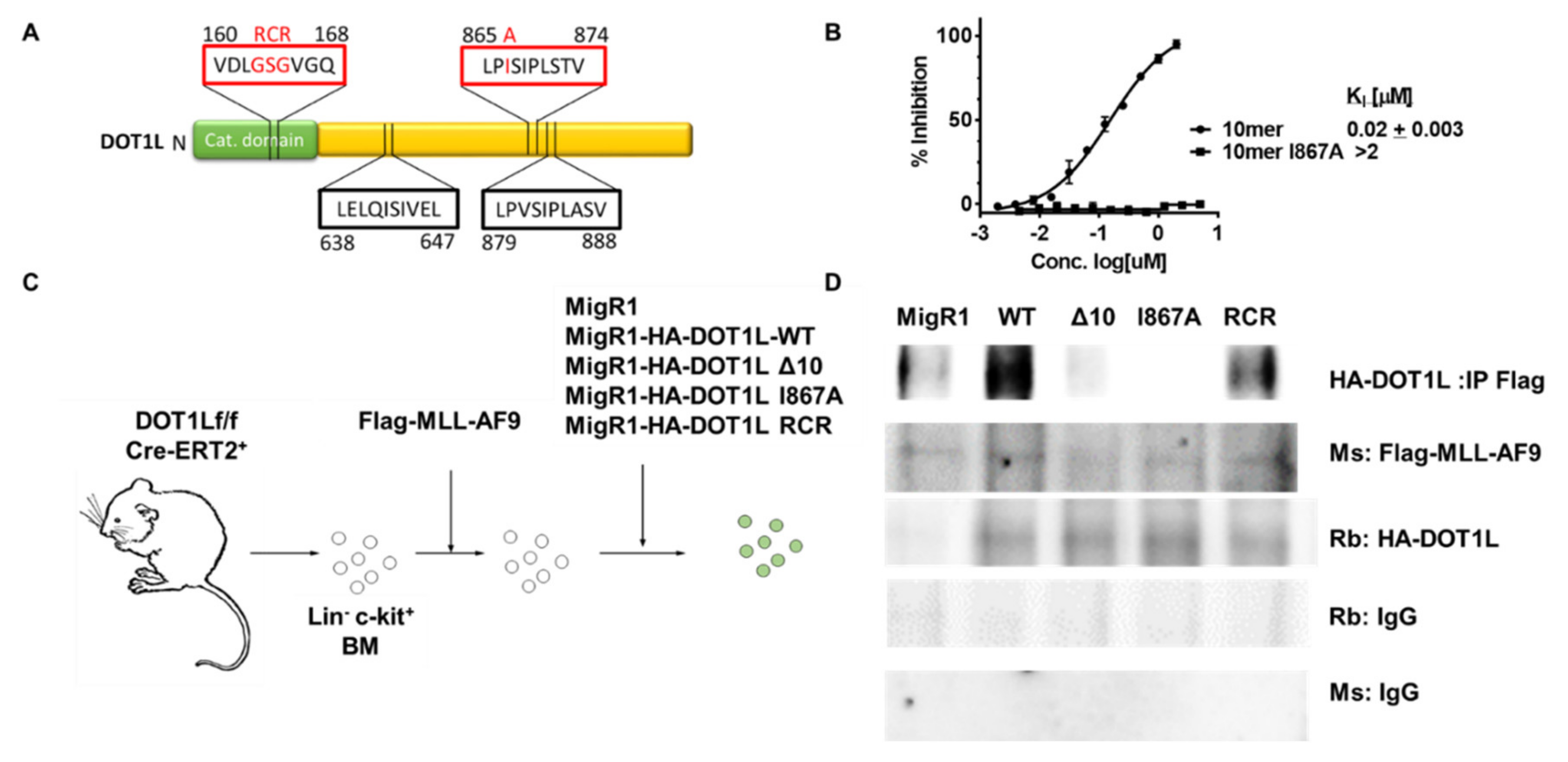
2.1. Characterization of MLL-AF9-Transformed Cells in the Presence of Different DOT1L Constructs
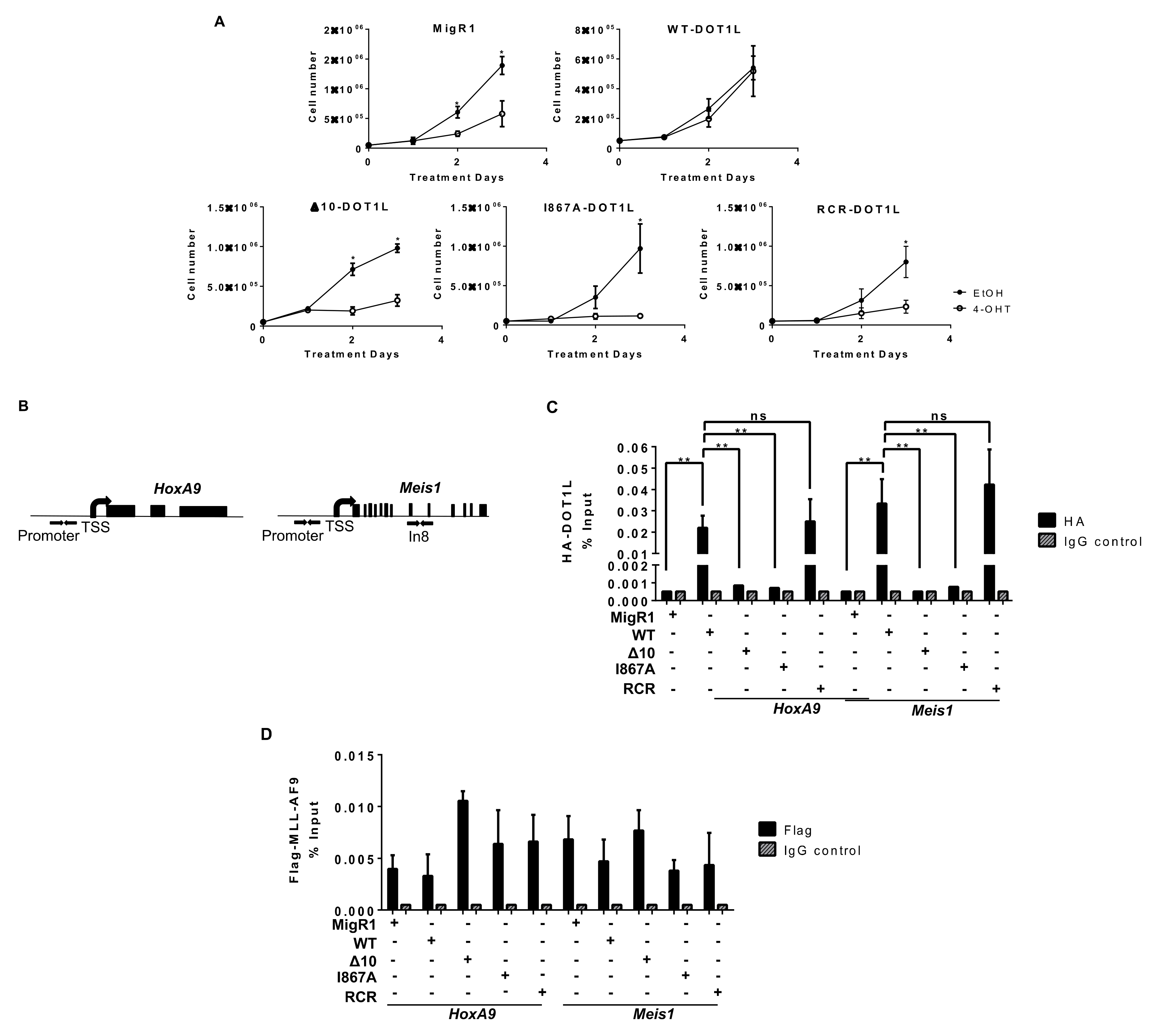
2.2. Targeted Disruption of Protein-Protein Interactions between MLL-AF9 and DOT1L Suppresses Leukemia Cell Growth and Promotes Their Differentiation
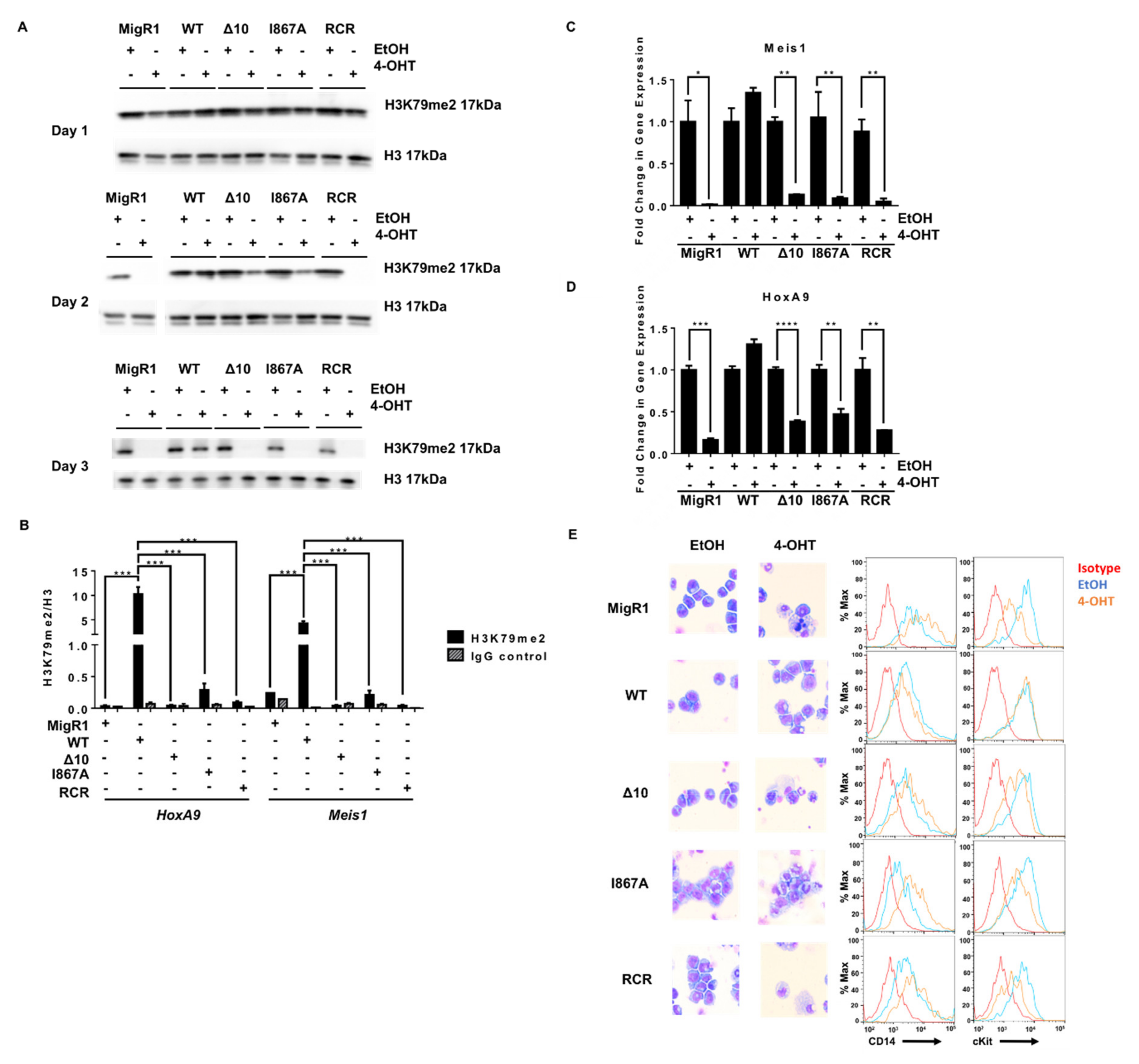
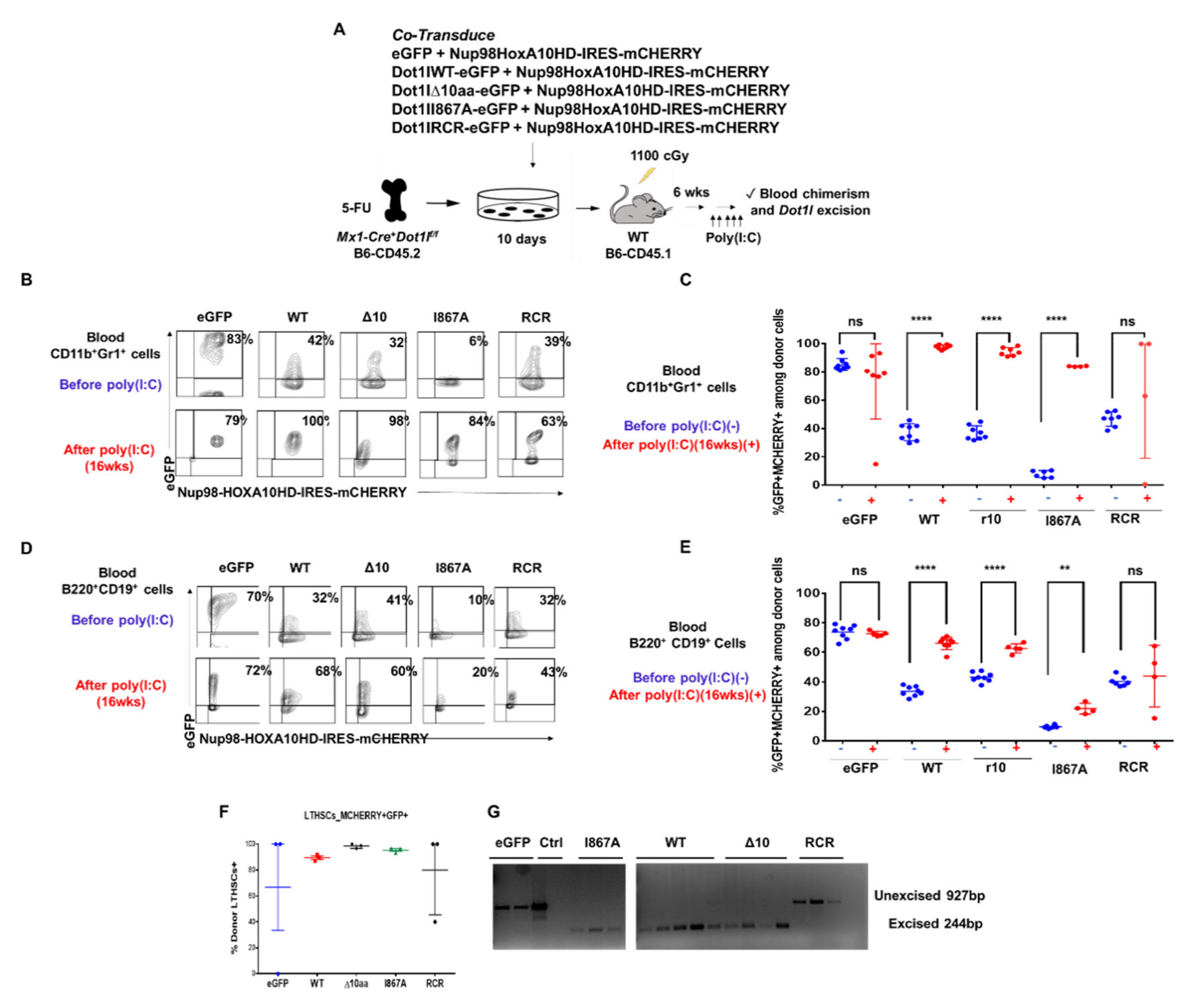
2.3. The Role of DOT1L, Its Enzymatic Activity and AF9-Binding Site in Non-Leukemic Hematopoiesis
3. Discussion
4. Materials and Methods
4.1. Fluorescence Polarization
4.2. Mice
4.3. Cell Line Generation
4.4. Cell Growth
4.5. Chromatin Immunoprecipitation
4.6. Co-Immunoprecipitation
4.7. Histone Methylation
4.8. Gene Expression
4.9. Differentiation
4.10. Flow Cytometry
4.11. Retroviral Transduction and Bone Marrow Transplantation
4.12. Western Blot
4.13. CFU-GM
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hsieh, J.J.; Cheng, E.H.; Korsmeyer, S.J. Taspase1: A threonine aspartase required for cleavage of MLL and proper HOX gene expression. Cell 2003, 115, 293–303. [Google Scholar] [CrossRef]
- Martin, C.; Zhang, Y. The diverse functions of histone lysine methylation. Nat. Rev. Mol. Cell Biol. 2005, 6, 838–849. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.; Kowarz, E.; Hofmann, J.; Renneville, A.; Zuna, J.; Trka, J.; Ben Abdelali, R.; Macintyre, E.; De Braekeleer, E.; De Braekeleer, M.; et al. New insights to the MLL recombinome of acute leukemias. Leukemia 2009, 23, 1490–1499. [Google Scholar] [CrossRef] [PubMed]
- Winters, A.C.; Bernt, K.M. MLL-Rearranged Leukemias-An Update on Science and Clinical Approaches. Front. Pediatr. 2017, 5, 4. [Google Scholar] [CrossRef] [PubMed]
- Andrea, M.M.; Peres, T.B.; Luchini, L.C.; Pettinelli, A., Jr. Impact of long-term pesticide applications on some soil biological parameters. J. Environ. Sci. Health B 2000, 35, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Pui, C.H.; Kane, J.R.; Crist, W.M. Biology and treatment of infant leukemias. Leukemia 1995, 9, 762–769. [Google Scholar] [PubMed]
- Meyer, C.; Burmeister, T.; Groger, D.; Tsaur, G.; Fechina, L.; Renneville, A.; Sutton, R.; Venn, N.C.; Emerenciano, M.; Pombo-de-Oliveira, M.S.; et al. The MLL recombinome of acute leukemias in 2017. Leukemia 2018, 32, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Poirel, H.; Rack, K.; Delabesse, E.; Radford-Weiss, I.; Troussard, X.; Debert, C.; Leboeuf, D.; Bastard, C.; Picard, F.; Veil-Buzyn, A.; et al. Incidence and characterization of MLL gene (11q23) rearrangements in acute myeloid leukemia M1 and M5. Blood 1996, 87, 2496–2505. [Google Scholar] [CrossRef]
- Yokoyama, A.; Lin, M.; Naresh, A.; Kitabayashi, I.; Cleary, M.L. A Higher-Order Complex Containing AF4 and ENL Family Proteins with P-TEFb Facilitates Oncogenic and Physiologic MLL-Dependent Transcription. Cancer Cell 2010, 17, 198–212. [Google Scholar] [CrossRef]
- Mueller, D.; Garcia-Cuellar, M.P.; Bach, C.; Buhl, S.; Maethner, E.; Slany, R.K. Misguided transcriptional elongation causes mixed lineage leukemia. PLoS Biol. 2009, 7, e1000249. [Google Scholar] [CrossRef]
- Mueller, D.; Bach, C.; Zeisig, D.; Garcia-Cuellar, M.P.; Monroe, S.; Sreekumar, A.; Zhou, R.; Nesvizhskii, A.; Chinnaiyan, A.; Hess, J.L.; et al. A role for the MLL fusion partner ENL in transcriptional elongation and chromatin modification. Blood 2007, 110, 4445–4454. [Google Scholar] [CrossRef]
- Mohan, M.; Herz, H.M.; Takahashi, Y.H.; Lin, C.; Lai, K.C.; Zhang, Y.; Washburn, M.P.; Florens, L.; Shilatifard, A. Linking H3K79 trimethylation to Wnt signaling through a novel Dot1-containing complex (DotCom). Genes Dev. 2010, 24, 574–589. [Google Scholar] [CrossRef]
- Biswas, D.; Milne, T.A.; Basrur, V.; Kim, J.; Elenitoba-Johnson, K.S.; Allis, C.D.; Roeder, R.G. Function of leukemogenic mixed lineage leukemia 1 (MLL) fusion proteins through distinct partner protein complexes. Proc. Natl. Acad. Sci. USA 2011, 108, 15751–15756. [Google Scholar] [CrossRef] [PubMed]
- Steger, D.J.; Lefterova, M.I.; Ying, L.; Stonestrom, A.J.; Schupp, M.; Zhuo, D.; Vakoc, A.L.; Kim, J.E.; Chen, J.; Lazar, M.A.; et al. DOT1L/KMT4 recruitment and H3K79 methylation are ubiquitously coupled with gene transcription in mammalian cells. Mol. Cell. Biol. 2008, 28, 2825–2839. [Google Scholar] [CrossRef]
- Bernt, K.M.; Zhu, N.; Sinha, A.U.; Vempati, S.; Faber, J.; Krivtsov, A.V.; Feng, Z.; Punt, N.; Daigle, A.; Bullinger, L.; et al. MLL-Rearranged Leukemia Is Dependent on Aberrant H3K79 Methylation by DOT1L. Cancer Cell 2011, 20, 66–78. [Google Scholar] [CrossRef]
- Nguyen, A.T.; Taranova, O.; He, J.; Zhang, Y. DOT1L, the H3K79 methyltransferase, is required for MLL-AF9–mediated leukemogenesis. Blood 2011, 117, 6912–6922. [Google Scholar] [CrossRef]
- Farooq, Z.; Banday, S.; Pandita, T.K.; Altaf, M. The many faces of histone H3K79 methylation. Mutat. Res. Rev. Mutat. Res. 2016, 768, 46–52. [Google Scholar] [CrossRef]
- Krivtsov, A.V.; Feng, Z.; Lemieux, M.E.; Faber, J.; Vempati, S.; Sinha, A.U.; Xia, X.; Jesneck, J.; Bracken, A.P.; Silverman, L.B.; et al. H3K79 methylation profiles define murine and human MLL-AF4 leukemias. Cancer Cell 2008, 14, 355–368. [Google Scholar] [CrossRef] [PubMed]
- Okada, Y.; Feng, Q.; Lin, Y.; Jiang, Q.; Li, Y.; Coffield, V.M.; Su, L.; Xu, G.; Zhang, Y. hDOT1L links histone methylation to leukemogenesis. Cell 2005, 121, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.-J.; Wu, H.; Achille, N.J.; Reisenauer, M.R.; Chou, C.-W.; Zeleznik-Le, N.J.; Hemenway, C.S.; Zhang, W. Histone H3 Lysine 79 Methyltransferase Dot1 Is Required for Immortalization by MLL Oncogenes. Cancer Res. 2010, 70, 10234–10242. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, C.W.; Armstrong, S.A. The role of DOT1L in the maintenance of leukemia gene expression. Curr Opin. Genet. Dev. 2016, 36, 68–72. [Google Scholar] [CrossRef]
- Annesley, C.E.; Brown, P. Novel agents for the treatment of childhood acute leukemia. Ther. Adv. Hematol. 2015, 6, 61–79. [Google Scholar] [CrossRef]
- Basavapathruni, A.; Olhava, E.J.; Daigle, S.R.; Therkelsen, C.A.; Jin, L.; Boriack-Sjodin, P.A.; Allain, C.J.; Klaus, C.R.; Raimondi, A.; Scott, M.P.; et al. Nonclinical pharmacokinetics and metabolism of EPZ-5676, a novel DOT1L histone methyltransferase inhibitor. Biopharm. Drug Dispos. 2014, 35, 237–252. [Google Scholar] [CrossRef] [PubMed]
- Daigle, S.R.; Olhava, E.J.; Therkelsen, C.A.; Basavapathruni, A.; Jin, L.; Boriack-Sjodin, P.A.; Allain, C.J.; Klaus, C.R.; Raimondi, A.; Scott, M.P.; et al. Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood 2013, 122, 1017–1025. [Google Scholar] [CrossRef] [PubMed]
- Stein, E.M.; Tallman, M.S. Mixed lineage rearranged leukaemia: Pathogenesis and targeting DOT1L. Curr. Opin. Hematol. 2015, 22, 92–96. [Google Scholar] [CrossRef]
- Stein, E.M.; Garcia-Manero, G.; Rizzieri, D.A.; Tibes, R.; Berdeja, J.G.; Jongen-Lavrencic, M.; Altman, J.K.; Dohner, H.; Thomson, B.; Blakemore, S.J.; et al. A Phase 1 Study of the DOT1L Inhibitor, Pinometostat (EPZ-5676), in Adults with Relapsed or Refractory Leukemia: Safety, Clinical Activity, Exposure and Target Inhibition. Blood 2015, 126, 2547. [Google Scholar] [CrossRef]
- Shukla, N.; Wetmore, C.; O’Brien, M.M.; Silverman, L.B.; Brown, P.; Cooper, T.M.; Thomson, B.; Blakemore, S.J.; Daigle, S.; Suttle, B.; et al. Final Report of Phase 1 Study of the DOT1L Inhibitor, Pinometostat (EPZ-5676), in Children with Relapsed or Refractory MLL-r Acute Leukemia. Blood 2016, 128, 2780. [Google Scholar] [CrossRef]
- Scholz, B.; Marschalek, R. Epigenetics and blood disorders. Br. J. Haematol. 2012, 158, 307–322. [Google Scholar] [CrossRef]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in cancer. Carcinogenesis 2010, 31, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Lund, A.H.; van Lohuizen, M. Epigenetics and cancer. Genes Dev. 2004, 18, 2315–2335. [Google Scholar] [CrossRef] [PubMed]
- Weinhold, B. Epigenetics: The science of change. Environ. Health Perspect. 2006, 114, A160–A167. [Google Scholar] [CrossRef] [PubMed]
- Beloqui, A.; Polaina, J.; Vieites, J.M.; Reyes-Duarte, D.; Torres, R.; Golyshina, O.V.; Chernikova, T.N.; Waliczek, A.; Aharoni, A.; Yakimov, M.M.; et al. Novel hybrid esterase-haloacid dehalogenase enzyme. Chembiochem 2010, 11, 1975–1978. [Google Scholar] [CrossRef] [PubMed]
- Carinci, F.; Pezzetti, F.; Spina, A.M.; Palmieri, A.; Carls, F.; Laino, G.; De Rosa, A.; Farina, E.; Illiano, F.; Stabellini, G.; et al. An in vitro model for dissecting distraction osteogenesis. J. Craniofac. Surg. 2005, 16, 71–78; discussion 78–79. [Google Scholar] [CrossRef] [PubMed]
- Jones, B.; Su, H.; Bhat, A.; Lei, H.; Bajko, J.; Hevi, S.; Baltus, G.A.; Kadam, S.; Zhai, H.; Valdez, R.; et al. The histone H3K79 methyltransferase Dot1L is essential for mammalian development and heterochromatin structure. PLoS Genet. 2008, 4, e1000190. [Google Scholar] [CrossRef]
- Jo, S.Y.; Granowicz, E.M.; Maillard, I.; Thomas, D.; Hess, J.L. Requirement for Dot1l in murine postnatal hematopoiesis and leukemogenesis by MLL translocation. Blood 2011, 117, 4759–4768. [Google Scholar] [CrossRef]
- Nguyen, A.T.; He, J.; Taranova, O.; Zhang, Y. Essential role of DOT1L in maintaining normal adult hematopoiesis. Cell Res. 2011, 21, 1370–1373. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Yang, Y.; Ortega, M.M.; Copeland, J.N.; Zhang, M.; Jacob, J.B.; Fields, T.A.; Vivian, J.L.; Fields, P.E. Early mammalian erythropoiesis requires the Dot1L methyltransferase. Blood 2010, 116, 4483–4491. [Google Scholar] [CrossRef]
- Shen, C.; Jo, S.Y.; Liao, C.; Hess, J.L.; Nikolovska-Coleska, Z. Targeting recruitment of disruptor of telomeric silencing 1-like (DOT1L): Characterizing the interactions between DOT1L and mixed lineage leukemia (MLL) fusion proteins. J. Biol. Chem. 2013, 288, 30585–30596. [Google Scholar] [CrossRef]
- Kuntimaddi, A.; Achille, N.J.; Thorpe, J.; Lokken, A.A.; Singh, R.; Hemenway, C.S.; Adli, M.; Zeleznik-Le, N.J.; Bushweller, J.H. Degree of recruitment of DOT1L to MLL-AF9 defines level of H3K79 Di- and tri-methylation on target genes and transformation potential. Cell Rep. 2015, 11, 808–820. [Google Scholar] [CrossRef]
- Du, L.; Grigsby, S.M.; Yao, A.; Chang, Y.; Johnson, G.; Sun, H.; Nikolovska-Coleska, Z. Peptidomimetics for Targeting Protein-Protein Interactions between DOT1L and MLL Oncofusion Proteins AF9 and ENL. ACS Med. Chem. Lett. 2018, 9, 895–900. [Google Scholar] [CrossRef]
- Maillard, I.; Weng, A.P.; Carpenter, A.C.; Rodriguez, C.G.; Sai, H.; Xu, L.; Allman, D.; Aster, J.C.; Pear, W.S. Mastermind critically regulates Notch-mediated lymphoid cell fate decisions. Blood 2004, 104, 1696–1702. [Google Scholar] [CrossRef]
- Feng, Q.; Wang, H.; Ng, H.H.; Erdjument-Bromage, H.; Tempst, P.; Struhl, K.; Zhang, Y. Methylation of H3-Lysine 79 Is Mediated by a New Family of HMTases without a SET Domain. Curr. Biol. 2002, 12, 1052–1058. [Google Scholar] [CrossRef]
- Min, J.; Feng, Q.; Li, Z.; Zhang, Y.; Xu, R.M. Structure of the catalytic domain of human DOT1L, a non-SET domain nucleosomal histone methyltransferase. Cell 2003, 112, 711–723. [Google Scholar] [CrossRef]
- Daigle, S.R.; Olhava, E.J.; Therkelsen, C.A.; Majer, C.R.; Sneeringer, C.J.; Song, J.; Johnston, L.D.; Scott, M.P.; Smith, J.J.; Xiao, Y.; et al. Selective Killing of Mixed Lineage Leukemia Cells by a Potent Small-Molecule DOT1L Inhibitor. Cancer Cell 2011, 20, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Milne, T.A.; Briggs, S.D.; Brock, H.W.; Martin, M.E.; Gibbs, D.; Allis, C.D.; Hess, J.L. MLL targets SET domain methyltransferase activity to Hox gene promoters. Mol. Cell 2002, 10, 1107–1117. [Google Scholar] [CrossRef]
- Monroe, S.C.; Jo, S.Y.; Sanders, D.S.; Basrur, V.; Elenitoba-Johnson, K.S.; Slany, R.K.; Hess, J.L. MLL-AF9 and MLL-ENL alter the dynamic association of transcriptional regulators with genes critical for leukemia. Exp. Hematol. 2011, 39, 77–86.e5. [Google Scholar] [CrossRef] [PubMed]
- Muntean, A.G.; Hess, J.L. The pathogenesis of mixed-lineage leukemia. Annu. Rev. Pathol. 2012, 7, 283–301. [Google Scholar] [CrossRef]
- Chen, C.W.; Armstrong, S.A. Targeting DOT1L and HOX gene expression in MLL-rearranged leukemia and beyond. Exp. Hematol. 2015, 43, 673–684. [Google Scholar] [CrossRef] [PubMed]
- McLean, C.M.; Karemaker, I.D.; van Leeuwen, F. The emerging roles of DOT1L in leukemia and normal development. Leukemia 2014, 28, 2131–2138. [Google Scholar] [CrossRef]
- Braun, B.S.; Tuveson, D.A.; Kong, N.; Le, D.T.; Kogan, S.C.; Rozmus, J.; Le Beau, M.M.; Jacks, T.E.; Shannon, K.M. Somatic activation of oncogenic Kras in hematopoietic cells initiates a rapidly fatal myeloproliferative disorder. Proc. Natl. Acad. Sci. USA 2004, 101, 597–602. [Google Scholar] [CrossRef]
- Kuhn, R.; Schwenk, F.; Aguet, M.; Rajewsky, K. Inducible gene targeting in mice. Science 1995, 269, 1427–1429. [Google Scholar] [CrossRef]
- Kiel, M.J.; Yilmaz, O.H.; Iwashita, T.; Yilmaz, O.H.; Terhorst, C.; Morrison, S.J. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell 2005, 121, 1109–1121. [Google Scholar] [CrossRef] [PubMed]
- Pronk, C.J.; Rossi, D.J.; Mansson, R.; Attema, J.L.; Norddahl, G.L.; Chan, C.K.; Sigvardsson, M.; Weissman, I.L.; Bryder, D. Elucidation of the phenotypic, functional, and molecular topography of a myeloerythroid progenitor cell hierarchy. Cell Stem Cell 2007, 1, 428–442. [Google Scholar] [CrossRef]
- Ohta, H.; Sekulovic, S.; Bakovic, S.; Eaves, C.J.; Pineault, N.; Gasparetto, M.; Smith, C.; Sauvageau, G.; Humphries, R.K. Near-maximal expansions of hematopoietic stem cells in culture using NUP98-HOX fusions. Exp. Hematol. 2007, 35, 817–830. [Google Scholar] [CrossRef]
- Abraham, A.; Kim, Y.S.; Zhao, H.; Humphries, K.; Persons, D.A. Increased Engraftment of Human Short Term Repopulating Hematopoietic Cells in NOD/SCID/IL2rgammanull Mice by Lentiviral Expression of NUP98-HOXA10HD. PLoS ONE 2016, 11, e0147059. [Google Scholar] [CrossRef]
- Deshpande, A.J.; Chen, L.; Fazio, M.; Sinha, A.U.; Bernt, K.M.; Banka, D.; Dias, S.; Chang, J.; Olhava, E.J.; Daigle, S.R.; et al. Leukemic transformation by the MLL-AF6 fusion oncogene requires the H3K79 methyltransferase Dot1l. Blood 2013, 121, 2533–2541. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Chory, E.J.; Wernimont, A.K.; Tempel, W.; Scopton, A.; Federation, A.; Marineau, J.J.; Qi, J.; Barsyte-Lovejoy, D.; Yi, J.; et al. Catalytic site remodelling of the DOT1L methyltransferase by selective inhibitors. Nat. Commun. 2012, 3, 1288. [Google Scholar] [CrossRef]
- Klaus, C.R.; Iwanowicz, D.; Johnston, D.; Campbell, C.A.; Smith, J.J.; Moyer, M.P.; Copeland, R.A.; Olhava, E.J.; Scott, M.P.; Pollock, R.M.; et al. DOT1L inhibitor EPZ-5676 displays synergistic antiproliferative activity in combination with standard of care drugs and hypomethylating agents in MLL-rearranged leukemia cells. J. Pharmacol. Exp. Ther. 2014, 350, 646–656. [Google Scholar] [CrossRef]
- Anglin, J.L.; Deng, L.; Yao, Y.; Cai, G.; Liu, Z.; Jiang, H.; Cheng, G.; Chen, P.; Dong, S.; Song, Y. Synthesis and structure-activity relationship investigation of adenosine-containing inhibitors of histone methyltransferase DOT1L. J. Med. Chem. 2012, 55, 8066–8074. [Google Scholar] [CrossRef]
- Deng, L.; Zhang, L.; Yao, Y.; Wang, C.; Redell, M.S.; Dong, S.; Song, Y. Synthesis, Activity and Metabolic Stability of Non-Ribose Containing Inhibitors of Histone Methyltransferase DOT1L. Medchemcomm 2013, 4, 822–826. [Google Scholar] [CrossRef]
- Mobitz, H.; Machauer, R.; Holzer, P.; Vaupel, A.; Stauffer, F.; Ragot, C.; Caravatti, G.; Scheufler, C.; Fernandez, C.; Hommel, U.; et al. Discovery of Potent, Selective, and Structurally Novel Dot1L Inhibitors by a Fragment Linking Approach. ACS Med. Chem. Lett. 2017, 8, 338–343. [Google Scholar] [CrossRef]
- Zeisig, D.T.; Bittner, C.B.; Zeisig, B.B.; Garcia-Cuellar, M.P.; Hess, J.L.; Slany, R.K. The eleven-nineteen-leukemia protein ENL connects nuclear MLL fusion partners with chromatin. Oncogene 2005, 24, 5525–5532. [Google Scholar] [CrossRef]
- Bitoun, E.; Oliver, P.L.; Davies, K.E. The mixed-lineage leukemia fusion partner AF4 stimulates RNA polymerase II transcriptional elongation and mediates coordinated chromatin remodeling. Hum. Mol. Genet. 2007, 16, 92–106. [Google Scholar] [CrossRef] [PubMed]
- Ayton, P.M.; Cleary, M.L. Molecular mechanisms of leukemogenesis mediated by MLL fusion proteins. Oncogene 2001, 20, 5695–5707. [Google Scholar] [CrossRef]
- Armstrong, S.A.; Staunton, J.E.; Silverman, L.B.; Pieters, R.; den Boer, M.L.; Minden, M.D.; Sallan, S.E.; Lander, E.S.; Golub, T.R.; Korsmeyer, S.J. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat. Genet. 2002, 30, 41–47. [Google Scholar] [CrossRef]
- Ross, M.E.; Mahfouz, R.; Onciu, M.; Liu, H.C.; Zhou, X.; Song, G.; Shurtleff, S.A.; Pounds, S.; Cheng, C.; Ma, J.; et al. Gene expression profiling of pediatric acute myelogenous leukemia. Blood 2004, 104, 3679–3687. [Google Scholar] [CrossRef] [PubMed]
- Paul, E.; Cronan, R.; Weston, P.J.; Boekelheide, K.; Sedivy, J.M.; Lee, S.Y.; Wiest, D.L.; Resnick, M.B.; Klysik, J.E. Disruption of Supv3L1 damages the skin and causes sarcopenia, loss of fat, and death. Mamm. Genome 2009, 20, 92–108. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gibbons, G.S.; Owens, S.R.; Fearon, E.R.; Nikolovska-Coleska, Z. Regulation of Wnt signaling target gene expression by the histone methyltransferase DOT1L. ACS Chem. Biol. 2015, 10, 109–114. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grigsby, S.M.; Friedman, A.; Chase, J.; Waas, B.; Ropa, J.; Serio, J.; Shen, C.; Muntean, A.G.; Maillard, I.; Nikolovska-Coleska, Z. Elucidating the Importance of DOT1L Recruitment in MLL-AF9 Leukemia and Hematopoiesis. Cancers 2021, 13, 642. https://doi.org/10.3390/cancers13040642
Grigsby SM, Friedman A, Chase J, Waas B, Ropa J, Serio J, Shen C, Muntean AG, Maillard I, Nikolovska-Coleska Z. Elucidating the Importance of DOT1L Recruitment in MLL-AF9 Leukemia and Hematopoiesis. Cancers. 2021; 13(4):642. https://doi.org/10.3390/cancers13040642
Chicago/Turabian StyleGrigsby, Sierrah M., Ann Friedman, Jennifer Chase, Bridget Waas, James Ropa, Justin Serio, Chenxi Shen, Andrew G. Muntean, Ivan Maillard, and Zaneta Nikolovska-Coleska. 2021. "Elucidating the Importance of DOT1L Recruitment in MLL-AF9 Leukemia and Hematopoiesis" Cancers 13, no. 4: 642. https://doi.org/10.3390/cancers13040642
APA StyleGrigsby, S. M., Friedman, A., Chase, J., Waas, B., Ropa, J., Serio, J., Shen, C., Muntean, A. G., Maillard, I., & Nikolovska-Coleska, Z. (2021). Elucidating the Importance of DOT1L Recruitment in MLL-AF9 Leukemia and Hematopoiesis. Cancers, 13(4), 642. https://doi.org/10.3390/cancers13040642