
Metformin and Androgen Receptor-Axis-Targeted (ARAT) Agents Induce Two PARP-1-Dependent Cell Death Pathways in Androgen-Sensitive Human Prostate Cancer Cells

 , ,
, ,  and
and 
Abstract
Simple Summary
Abstract
1. Introduction
2. Results
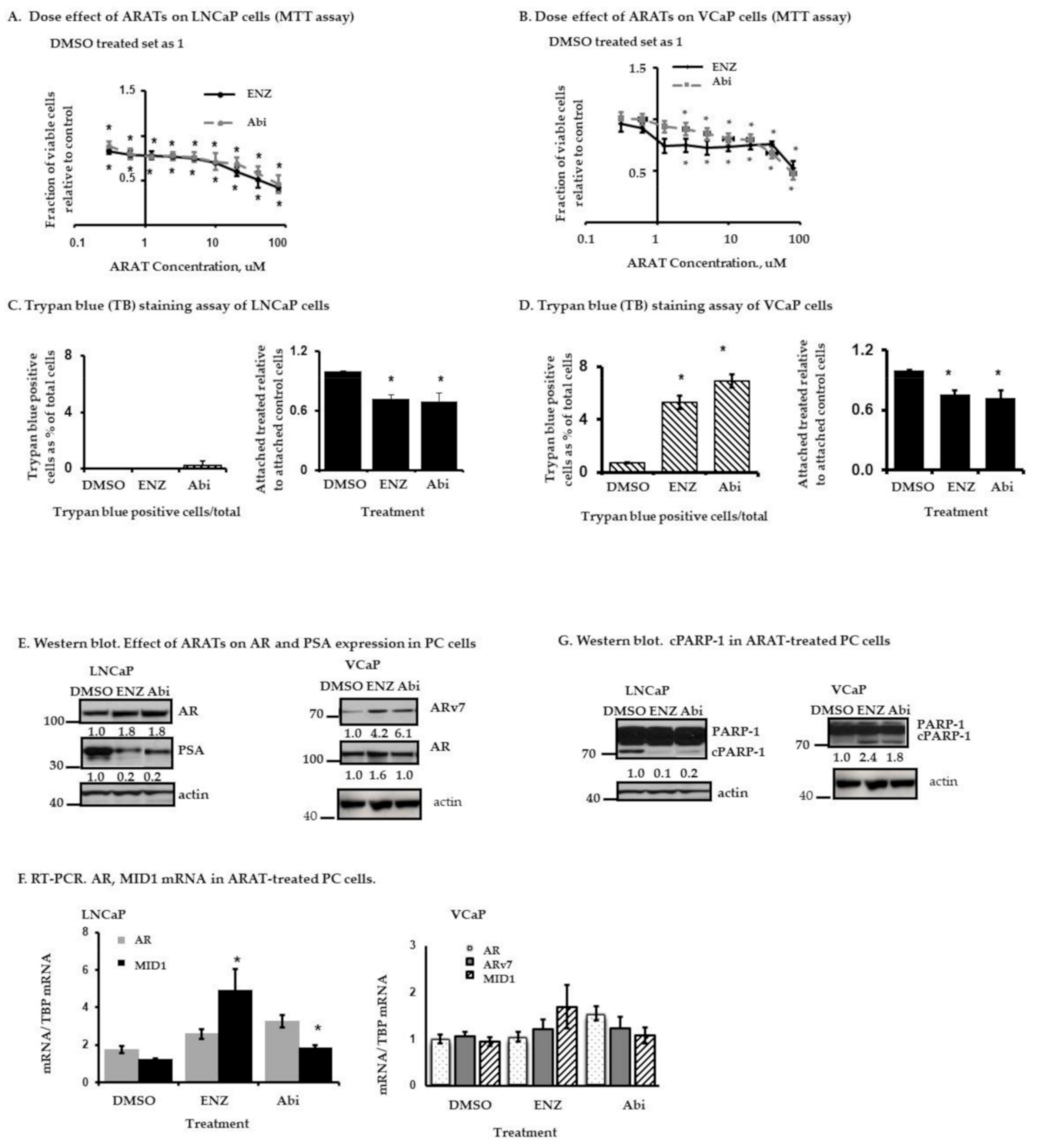
2.1. Effects of Enz and Abi on LNCaP and VCaP Cells
2.1.1. Response of LNCaP and VCaP Cells to Enz or Abi—Effects on Cell Proliferation and Cell Death
2.1.2. Effects of Enz and Abi on Prostate Serum Antigen (PSA), AR, AR-v7 Expression and PARP-1 Cleavage in LNCaP and VCaP Cells
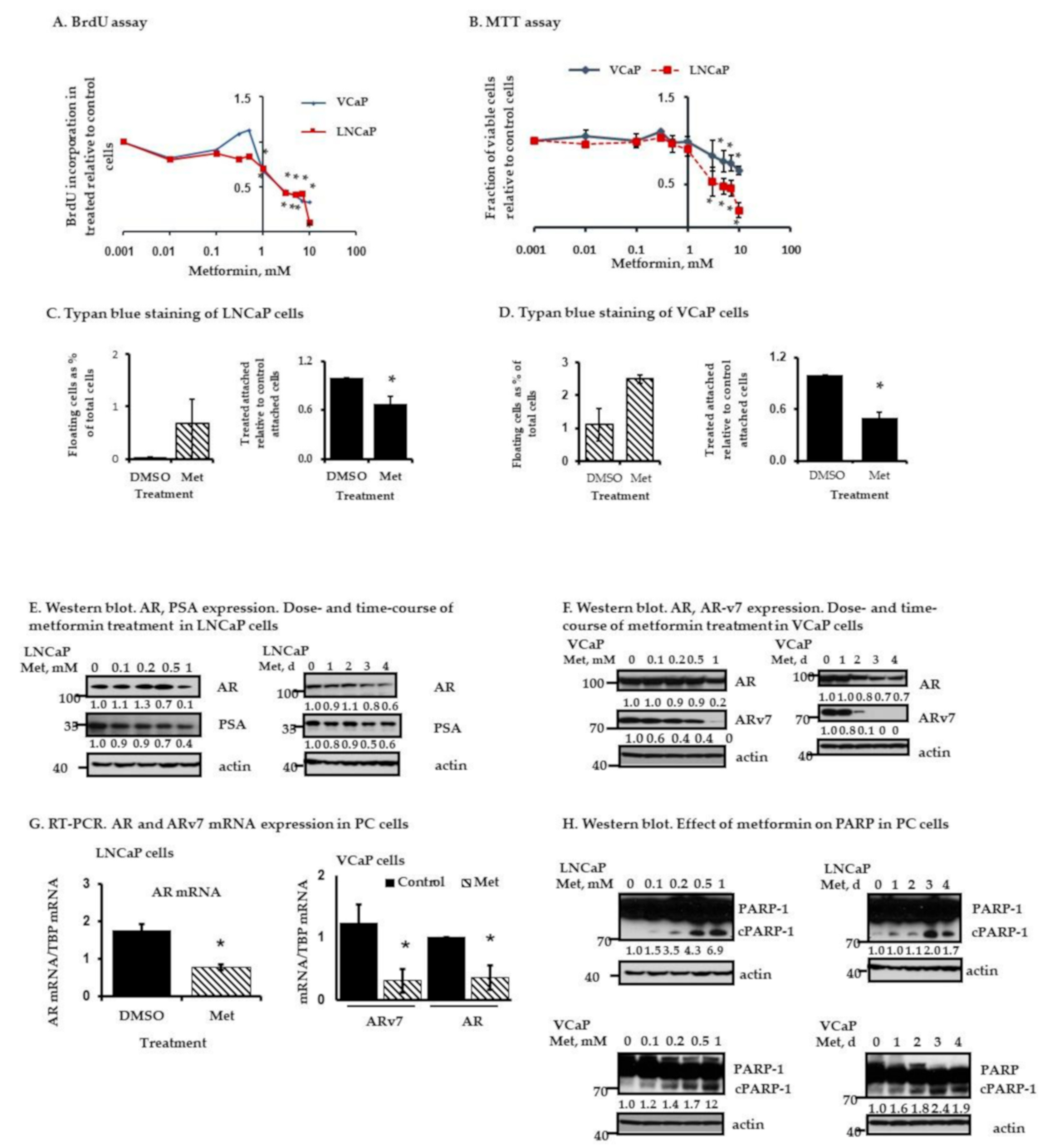
2.2. Metformin Inhibits AR and ARv7 Expression and Induces Cell Death in PC Cells
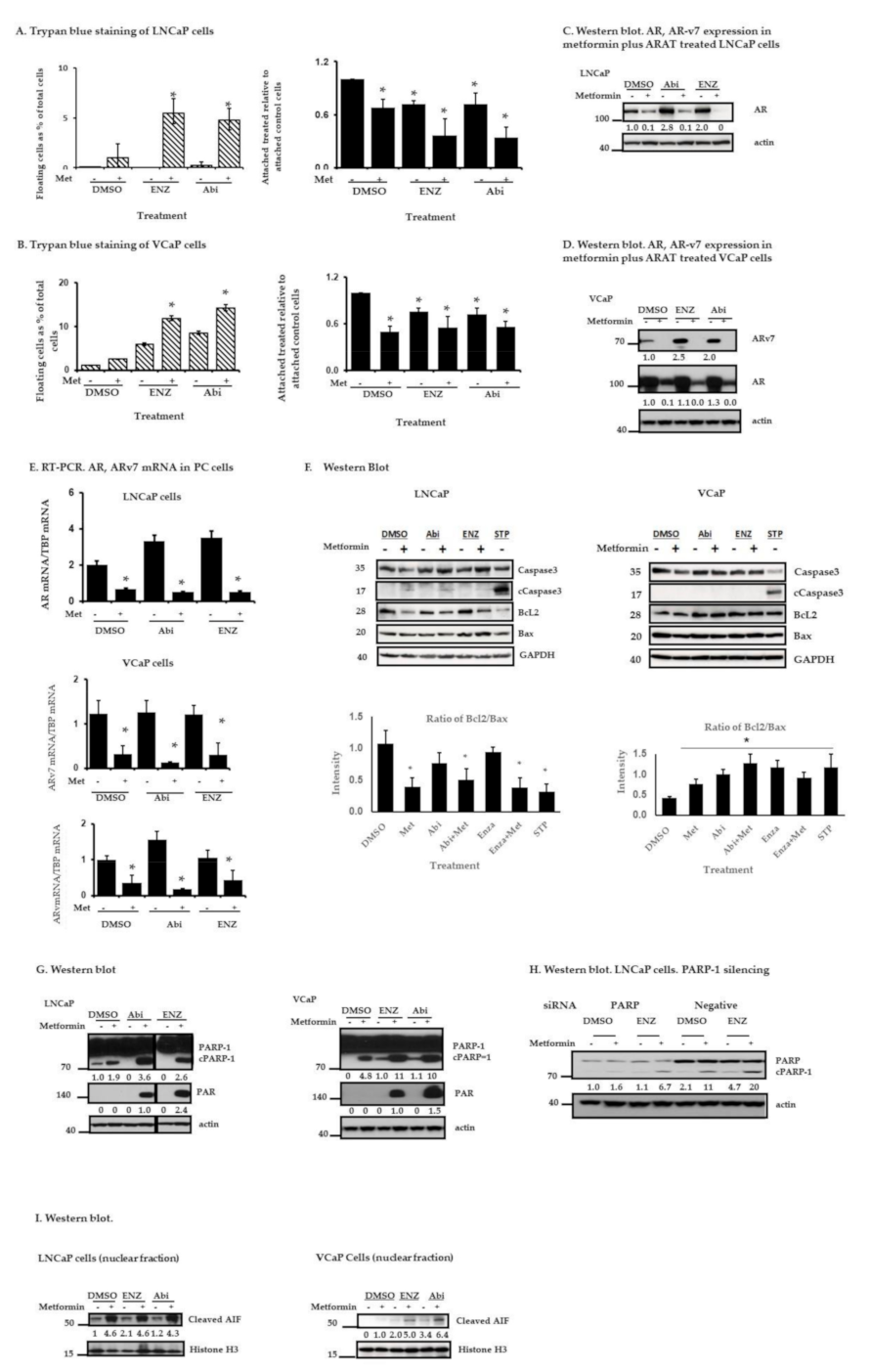
2.3. Metformin and ARAT Agents (Enz, Abi) Enhance Cell Death in PC Cells
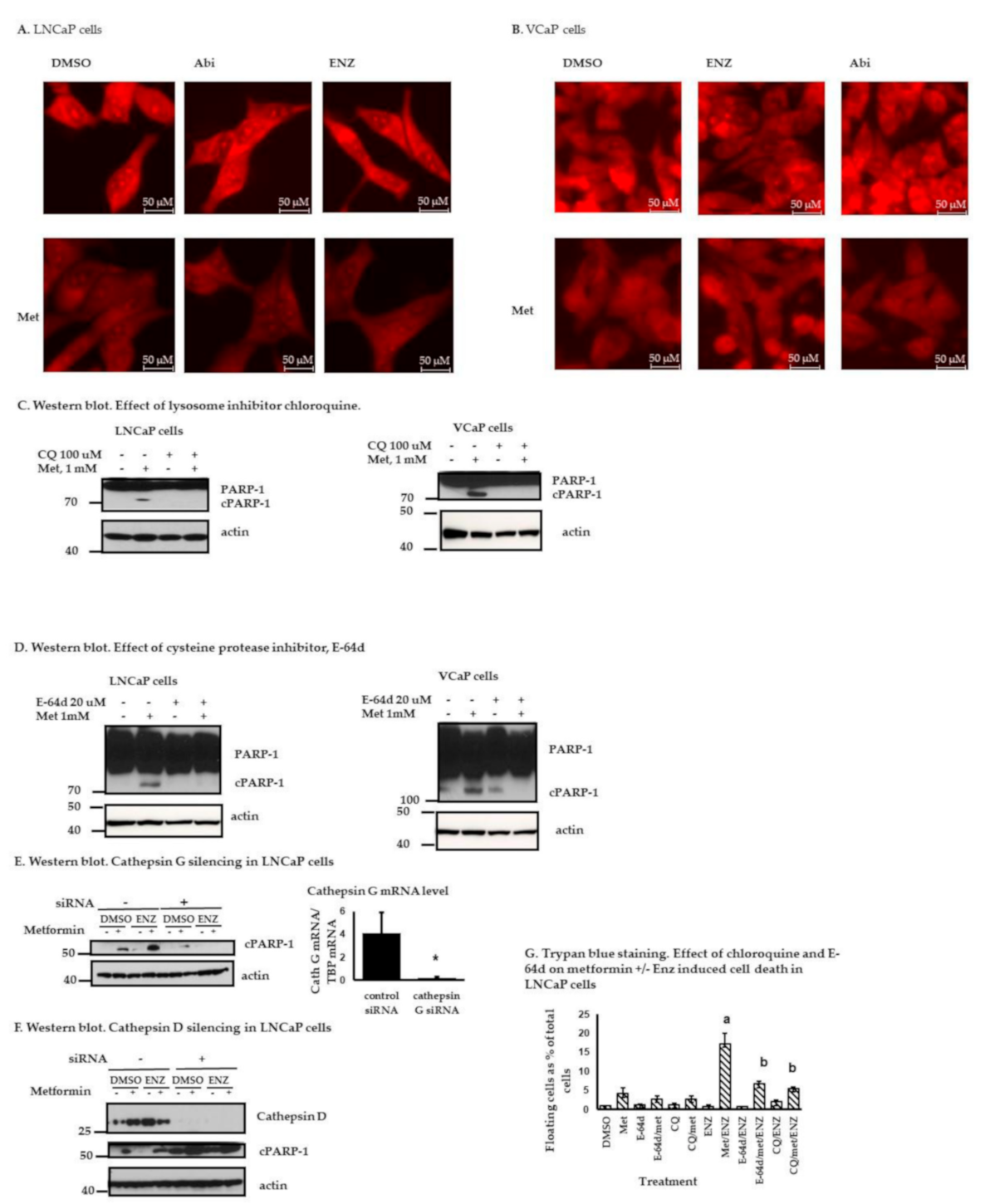
2.4. Metformin with or without ARATs Increases Lysosome Permeability and Cathepsin G-Mediated PARP-1 Cleavage in Androgen-Sensitive PC Cells
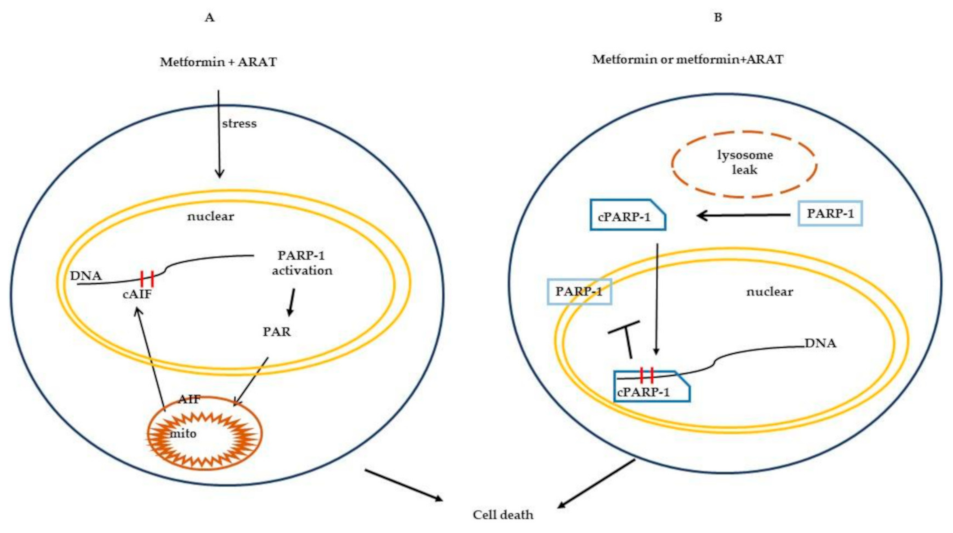
3. Discussion
4. Materials and Methods
4.1. Materials and Drugs
4.2. Cell Lines and Culture Conditions
4.3. RNA Preparation and Quantitative RT-PCR (qPCR)
4.4. Preparation of Protein Extracts and Western Blot Analysis
4.5. Isolation of Nuclear Fractions
4.6. MTT Assay
4.7. Trypan Blue Staining Assay
4.8. Annexin V Detection
4.9. Lysosome Staining by Acridine Orange
4.10. siRNA Interference
4.11. Drug Combination Index
4.12. 2-D Clonogenic Assays
4.13. Statistical Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Huang, Y.; Jiang, X.; Liang, X.; Jiang, G. Molecular and cellular mechanisms of castration resistant prostate cancer (Review). Oncol. Lett. 2018, 15, 6063–6076. [Google Scholar] [CrossRef]
- Rice, M.A.; Malhotra, S.V.; Stoyanova, T. Second-Generation Antiandrogens: From Discovery to Standard of Care in Castration Resistant Prostate Cancer. Front. Oncol. 2019, 9, 801. [Google Scholar] [CrossRef]
- Zheng, X.; Zhao, X.; Xu, H.; Han, X.; Xu, H.; Dong, X.; Peng, R.; Yang, L.; Wei, Q.; Ai, J. Efficacy and safety of abiraterone and enzalutamide for castration-resistant prostate cancer: A systematic review and meta-analysis of randomized controlled trials. Medicine 2019, 98, e17748. [Google Scholar] [CrossRef] [PubMed]
- Fang, M.; Nakazawa, M.; Antonarakis, E.S.; Li, C. Efficacy of Abiraterone and Enzalutamide in Pre- and Postdocetaxel Castration-Resistant Prostate Cancer: A Trial-Level Meta-Analysis. Prostate Cancer 2017, 2017, 8560827. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Zhu, J.; George, D.J.; Armstrong, A.J. Enzalutamide versus abiraterone acetate for the treatment of men with metastatic castration-resistant prostate cancer. Expert Opin. Pharmacother. 2015, 16, 473–485. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; He, B. Androgen Receptor Signaling in the Development of Castration-Resistant Prostate Cancer. Front. Oncol. 2019, 9, 858. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekar, T.; Yang, J.C.; Gao, A.C.; Evans, C.P. Targeting molecular resistance in castration-resistant prostate cancer. BMC Med. 2015, 13, 206. [Google Scholar] [CrossRef]
- Linder, S.; Van Der Poel, H.G.; Bergman, A.M.; Zwart, W.; Prekovic, S. Enzalutamide therapy for advanced prostate cancer: Efficacy, resistance and beyond. Endocr. Relat. Cancer 2019, 26, R31–R52. [Google Scholar] [CrossRef]
- Paschalis, A.; Sharp, A.; Welti, J.; Neeb, A.J.; Raj, G.V.; Luo, J.; Plymate, S.R.; De Bono, J.S. Alternative splicing in prostate cancer. Nat. Rev. Clin. Oncol. 2018, 15, 663–675. [Google Scholar] [CrossRef]
- Ugwueze, C.V.; Ogamba, O.J.; Young, E.E.; Onyenekwe, B.M.; Ezeokpo, B.C. Metformin: A Possible Option in Cancer Chemotherapy. Anal. Cell. Pathol. 2020, 2020, 7180923. [Google Scholar] [CrossRef]
- Margel, D.; Urbach, D.R.; Lipscombe, L.L.; Bell, C.M.; Kulkarni, G.; Austin, P.C.; Fleshner, N. Metformin Use and All-Cause and Prostate Cancer–Specific Mortality Among Men with Diabetes. J. Clin. Oncol. 2013, 31, 3069–3075. [Google Scholar] [CrossRef]
- Demir, Ü.; Köhler, A.; Schneider, R.; Schweiger, S.; Klocker, H. Metformin anti-tumor effect via disruption of the MID1 translational regulator complex and AR downregulation in prostate cancer cells. BMC Cancer 2014, 14, 52. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, G.; Tong, D.; Parmar, H.; Hasenmayer, D.; Yuan, W.; Zhang, D.; Jiang, J. Metformin represses androgen-dependent and androgen-independent prostate cancers by targeting androgen receptor. Prostate 2015, 75, 1187–1196. [Google Scholar] [CrossRef]
- Yang, J.; Wei, J.; Wu, Y.; Wang, Z.; Guo, Y.; Lee, P.; Li, X. Metformin induces ER stress-dependent apoptosis through miR-708-5p/NNAT pathway in prostate cancer. Oncogenesis 2015, 4, e158. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Ke, C.; Tang, Q.; Dong, H.; Zheng, X.; Lin, W.; Ke, J.; Huang, J.; Yeung, S.-C.J.; Zhang, H. Metformin promotes autophagy and apoptosis in esophageal squamous cell carcinoma by downregulating Stat3 signaling. Cell Death Dis. 2014, 5, e1088. [Google Scholar] [CrossRef] [PubMed]
- Salani, B.; Marini, C.; Del Rio, A.; Ravera, S.; Massollo, M.; Orengo, A.M.; Amaro, A.; Passalacqua, M.; Maffioli, S.; Pfeffer, U.; et al. Metformin Impairs Glucose Consumption and Survival in Calu-1 Cells by Direct Inhibition of Hexokinase-II. Sci. Rep. 2013, 3, srep02070. [Google Scholar] [CrossRef]
- Bröker, L.E.; Kruyt, F.A.; Giaccone, G. Cell Death Independent of Caspases: A Review. Clin. Cancer Res. 2005, 11, 3155–3162. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef] [PubMed]
- Yan, G.; Elbadawi, M.; Efferth, T. Multiple cell death modalities and their key features (Review). World Acad. Sci. J. 2020, 2, 39–48. [Google Scholar] [CrossRef]
- Weaver, A.N.; Yang, E.S. Beyond DNA Repair: Additional Functions of PARP-1 in Cancer. Front. Oncol. 2013, 3, 290. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, D.; Qiu, Y. Role of PARP-1 in prostate cancer. Am. J. Clin. Exp. Urol. 2015, 3, 1–12. [Google Scholar]
- Robinson, N.; Ganesan, R.; Hegedűs, C.; Kovács, K.; Kufer, T.A.; Virág, L. Programmed necrotic cell death of macrophages: Focus on pyroptosis, necroptosis, and parthanatos. Redox Biol. 2019, 26, 101239. [Google Scholar] [CrossRef] [PubMed]
- D’Amours, D.; Desnoyers, S.; D’Silva, I.; Poirier, G.G. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem. J. 1999, 342 Pt 2, 249–268. [Google Scholar] [CrossRef]
- Fatokun, A.A.; Dawson, V.L.; Dawson, T.M. Parthanatos: Mitochondrial-linked mechanisms and therapeutic opportunities. Br. J. Pharmacol. 2014, 171, 2000–2016. [Google Scholar] [CrossRef] [PubMed]
- David, K.K.; Andrabi, S.A.; Dawson, T.M.; Dawson, V.L. Parthanatos, a messenger of death. Front. Biosci. (Landmark. Ed.) 2015, 14, 1116–1128. [Google Scholar] [CrossRef]
- Park, H.; Kam, T.-I.; Dawson, T.M.; Dawson, V.L. Poly (ADP-ribose) (PAR)-dependent cell death in neurodegenerative diseases. Int. Rev. Cell Mol. Biol. 2020, 353, 1–29. [Google Scholar] [CrossRef]
- Wang, X.; Ge, P. Parthanatos in the pathogenesis of nervous system diseases. Neuroscience 2020, 449, 241–250. [Google Scholar] [CrossRef]
- Tran, C.; Ouk, S.; Clegg, N.J.; Chen, Y.; Watson, P.A.; Arora, V.; Wongvipat, J.; Smith-Jones, P.M.; Yoo, D.; Kwon, A.; et al. Development of a Second-Generation Antiandrogen for Treatment of Advanced Prostate Cancer. Science 2009, 324, 787–790. [Google Scholar] [CrossRef]
- Richards, J.; Lim, A.C.; Hay, C.W.; Taylor, A.E.; Wingate, A.; Nowakowska, K.; Pezaro, C.; Carreira, S.; Goodall, J.; Arlt, W.; et al. Interactions of Abiraterone, Eplerenone, and Prednisolone with Wild-type and Mutant Androgen Receptor: A Rationale for Increasing Abiraterone Exposure or Combining with MDV3100. Cancer Res. 2012, 72, 2176–2182. [Google Scholar] [CrossRef]
- Scher, H.I.; Beer, T.M.; Higano, C.S.; Anand, A.; Taplin, M.-E.; Efstathiou, E.; Rathkopf, D.; Shelkey, J.; Yu, E.Y.; Alumkal, J.; et al. Antitumour activity of MDV3100 in castration-resistant prostate cancer: A phase 1–2 study. Lancet 2010, 375, 1437–1446. [Google Scholar] [CrossRef]
- Köhler, A.; Demir, Ü.; Kickstein, E.; Krauss, S.; Aigner, J.; Aranda-Orgillés, B.; Karagiannidis, A.I.; Achmüller, C.; Bu, H.; Wunderlich, A.; et al. A hormone-dependent feedback-loop controls androgen receptor levels by limiting MID1, a novel translation enhancer and promoter of oncogenic signaling. Mol. Cancer 2014, 13, 146. [Google Scholar] [CrossRef] [PubMed]
- Colquhoun, A.J.; Venier, N.A.; VanderSluis, A.D.; Besla, R.; Sugar, L.M.; Kiss, A.; Fleshner, N.E.; Pollak, M.; Klotz, L.H.; Venkateswaran, V. Metformin enhances the antiproliferative and apoptotic effect of bicalutamide in prostate cancer. Prostate Cancer Prostatic Dis. 2012, 15, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Saranyutanon, S.; Deshmukh, S.K.; Dasgupta, S.; Pai, S.; Singh, A.P.; Singh, A.P. Cellular and Molecular Progression of Prostate Cancer: Models for Basic and Preclinical Research. Cancers 2020, 12, 2651. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.-C.; Talalay, P. Analysis of combined drug effects: A new look at a very old problem. Trends Pharmacol. Sci. 1983, 4, 450–454. [Google Scholar] [CrossRef]
- Groth-Pedersen, L.; Jäättelä, M. Combating apoptosis and multidrug resistant cancers by targeting lysosomes. Cancer Lett. 2013, 332, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Aits, S.; Jäättelä, M. Lysosomal cell death at a glance. J. Cell Sci. 2013, 126 Pt 9, 1905–1912. [Google Scholar] [CrossRef]
- Gobeil, S.; Boucher, C.C.; Nadeau, D.; Poirier, G.G. Characterization of the necrotic cleavage of poly(ADP-ribose) polymerase (PARP-1): Implication of lysosomal proteases. Cell Death Differ. 2001, 8, 588–594. [Google Scholar] [CrossRef]
- Wang, F.; Gómez-Sintes, R.; Boya, P. Lysosomal membrane permeabilization and cell death. Traffic 2018, 19, 918–931. [Google Scholar] [CrossRef]
- Saini, N.; Yang, X. Metformin as an anti-cancer agent: Actions and mechanisms targeting cancer stem cells. Acta Biochim. Biophys. Sin. 2018, 50, 133–143. [Google Scholar] [CrossRef]
- Yu, X.; Mao, W.; Zhai, Y.; Tong, C.; Liu, M.; Ma, L.; Yu, X.; Li, S. Anti-tumor activity of metformin: From metabolic and epigenetic perspectives. Oncotarget 2017, 8, 5619–5628. [Google Scholar] [CrossRef]
- Bahmad, H.F.; Elajami, M.K.; El Zarif, T.; Bou-Gharios, J.; Abou-Antoun, T.J.; Abou-Kheir, W. Drug repurposing towards targeting cancer stem cells in pediatric brain tumors. Cancer Metastasis Rev. 2020, 39, 127–148. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.-W.; Andrabi, S.A.; Wang, H.; Kim, N.S.; Poirier, G.G.; Dawson, T.M.; Dawson, V.L. Apoptosis-inducing factor mediates poly(ADP-ribose) (PAR) polymer-induced cell death. Proc. Natl. Acad. Sci. USA 2006, 103, 18314–18319. [Google Scholar] [CrossRef]
- Yung, T.M.C.; Satoh, M.S. Functional Competition between Poly(ADP-ribose) Polymerase and Its 24-kDa Apoptotic Fragment in DNA Repair and Transcription. J. Biol. Chem. 2001, 276, 11279–11286. [Google Scholar] [CrossRef]
- Veĭko, N.N.; Shubaeva, N.O.; Malashenko, A.M.; Beskova, T.B.; Agapova, R.K.; Liapunova, N.A. Ribosomal genes in inbred mouse strains: Interstrain and intrastrain variations of copy number and extent of methylation. Genetika 2007, 43, 1226–1238. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, S.; Yung, E.; Pintilie, M.; Muaddi, H.; Chaib, S.; Yeung, M.; Fusciello, M.; Sykes, J.; Pitcher, B.; Hagenkort, A.; et al. MATE2 Expression Is Associated with Cancer Cell Response to Metformin. PLoS ONE 2016, 11, e0165214. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Tong, D.; Liu, G.; Xu, J.; Do, K.; Geary, K.; Zhang, D.; Zhang, J.; Zhang, Y.; Li, Y.; et al. Metformin reverses prostate cancer resistance to enzalutamide by targeting TGF-beta1/STAT3 axis-regulated EMT. Cell Death Dis. 2017, 8, e3007. [Google Scholar] [CrossRef]
- Richards, K.A.; Liou, J.-I.; Cryns, V.L.; Downs, T.M.; Abel, E.J.; Jarrard, D.F. Metformin Use is Associated with Improved Survival for Patients with Advanced Prostate Cancer on Androgen Deprivation Therapy. J. Urol. 2018, 200, 1256–1263. [Google Scholar] [CrossRef]
- Guo, Z.; Yang, X.; Sun, F.; Jiang, R.; Linn, D.E.; Chen, H.; Chen, H.; Kong, X.; Melamed, J.; Tepper, C.G.; et al. A Novel Androgen Receptor Splice Variant Is Up-regulated during Prostate Cancer Progression and Promotes Androgen Depletion–Resistant Growth. Cancer Res. 2009, 69, 2305–2313. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Matthews, H.; Deakin, J.; Rajab, M.; Idris-Usman, M.; Nirmalan, N.J. Investigating antimalarial drug interactions of emetine dihydrochloride hydrate using CalcuSyn-based interactivity calculations. PLoS ONE 2017, 12, e0173303. [Google Scholar] [CrossRef] [PubMed]
- Nghia Ho. Available online: https://nghiaho.com/?page_id=1011 (accessed on 28 February 2020).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cells | DMSO | Metformin | ENZ | ENZ/met | Abi | Abi/met | STP |
|---|---|---|---|---|---|---|---|
| LNCaP cells | 9.6 ± 2.8 | 16.6 ± 3.3 * | 19.8 ± 9.8 | 27.2 ± 6.8 * | 16.3 ± 6.0 | 25.8 ± 4.85 * | 17.5 ± 2.7 * |
| VCaP cells | 3.7 ± 2.3 | 4.8 ± 3.2 | 4.5 ± 3.4 | 6.0 ± 4.2 | 4.7 ± 3.8 | 5.0 ± 3.4 | 5.7 ± 0.3 * |
| AR_Fw | gctctacttcgcccctgatc |
| AR_Rv | ttcggacacactggctgtac |
| MID1_Fw | ctgggtcagccattttgact |
| MID1_Rv | tatttcagggaggcagttgg |
| TBP_Fw | tgcccgaaacgccgaatata |
| TBP_Rv | cgtggttcgtggctctctta |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, Y.; Wang, L.; Khan, M.A.; Hamburger, A.W.; Guang, W.; Passaniti, A.; Munir, K.; Ross, D.D.; Dean, M.; Hussain, A. Metformin and Androgen Receptor-Axis-Targeted (ARAT) Agents Induce Two PARP-1-Dependent Cell Death Pathways in Androgen-Sensitive Human Prostate Cancer Cells. Cancers 2021, 13, 633. https://doi.org/10.3390/cancers13040633
Xie Y, Wang L, Khan MA, Hamburger AW, Guang W, Passaniti A, Munir K, Ross DD, Dean M, Hussain A. Metformin and Androgen Receptor-Axis-Targeted (ARAT) Agents Induce Two PARP-1-Dependent Cell Death Pathways in Androgen-Sensitive Human Prostate Cancer Cells. Cancers. 2021; 13(4):633. https://doi.org/10.3390/cancers13040633
Chicago/Turabian StyleXie, Yi, Linbo Wang, Mohammad A. Khan, Anne W. Hamburger, Wei Guang, Antonino Passaniti, Kashif Munir, Douglas D. Ross, Michael Dean, and Arif Hussain. 2021. "Metformin and Androgen Receptor-Axis-Targeted (ARAT) Agents Induce Two PARP-1-Dependent Cell Death Pathways in Androgen-Sensitive Human Prostate Cancer Cells" Cancers 13, no. 4: 633. https://doi.org/10.3390/cancers13040633
APA StyleXie, Y., Wang, L., Khan, M. A., Hamburger, A. W., Guang, W., Passaniti, A., Munir, K., Ross, D. D., Dean, M., & Hussain, A. (2021). Metformin and Androgen Receptor-Axis-Targeted (ARAT) Agents Induce Two PARP-1-Dependent Cell Death Pathways in Androgen-Sensitive Human Prostate Cancer Cells. Cancers, 13(4), 633. https://doi.org/10.3390/cancers13040633