
Loss of cIAP1 in Endothelial Cells Limits Metastatic Extravasation through Tumor-Derived Lymphotoxin Alpha

 , , ,
, , , 
Abstract
Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
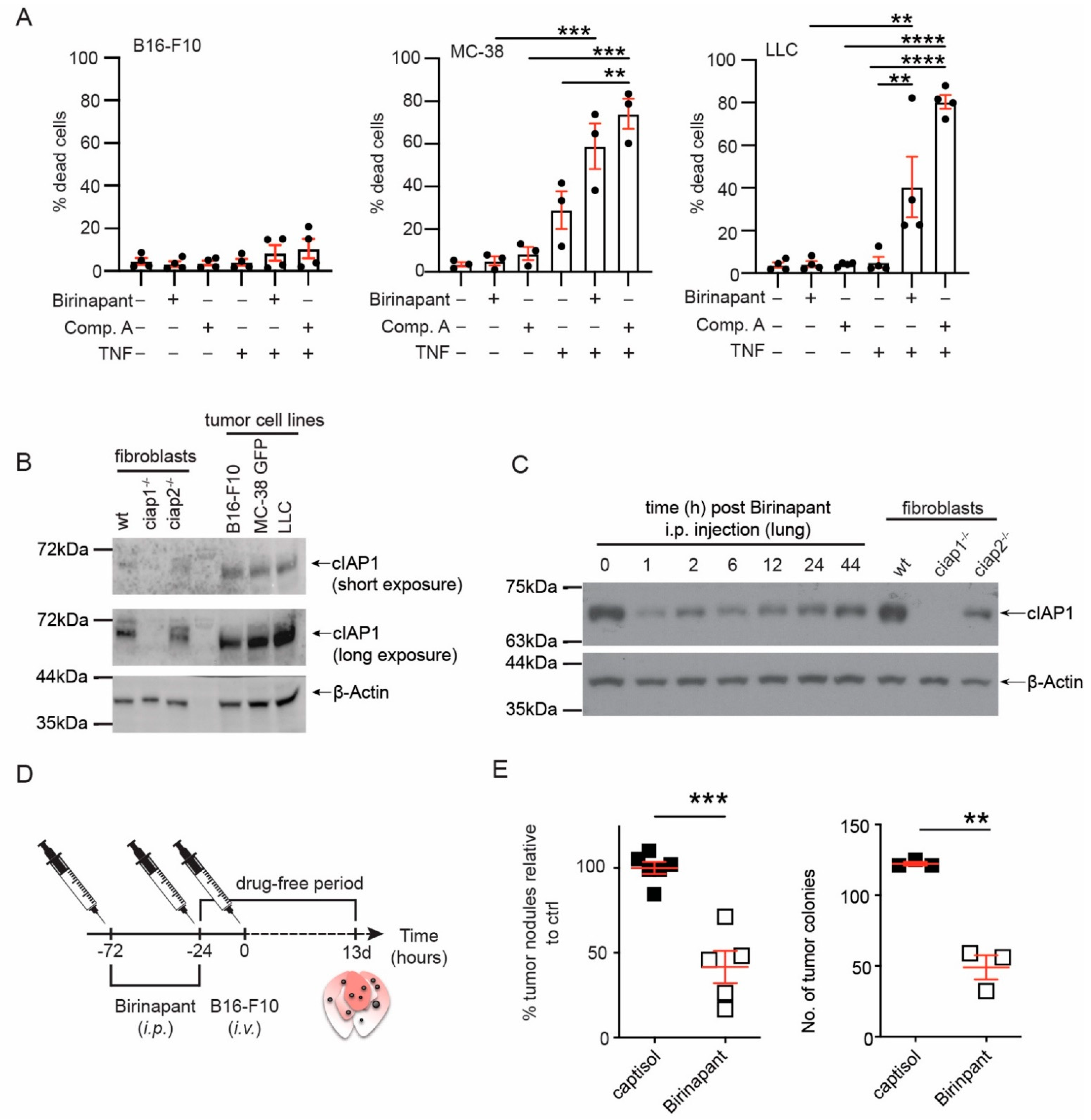
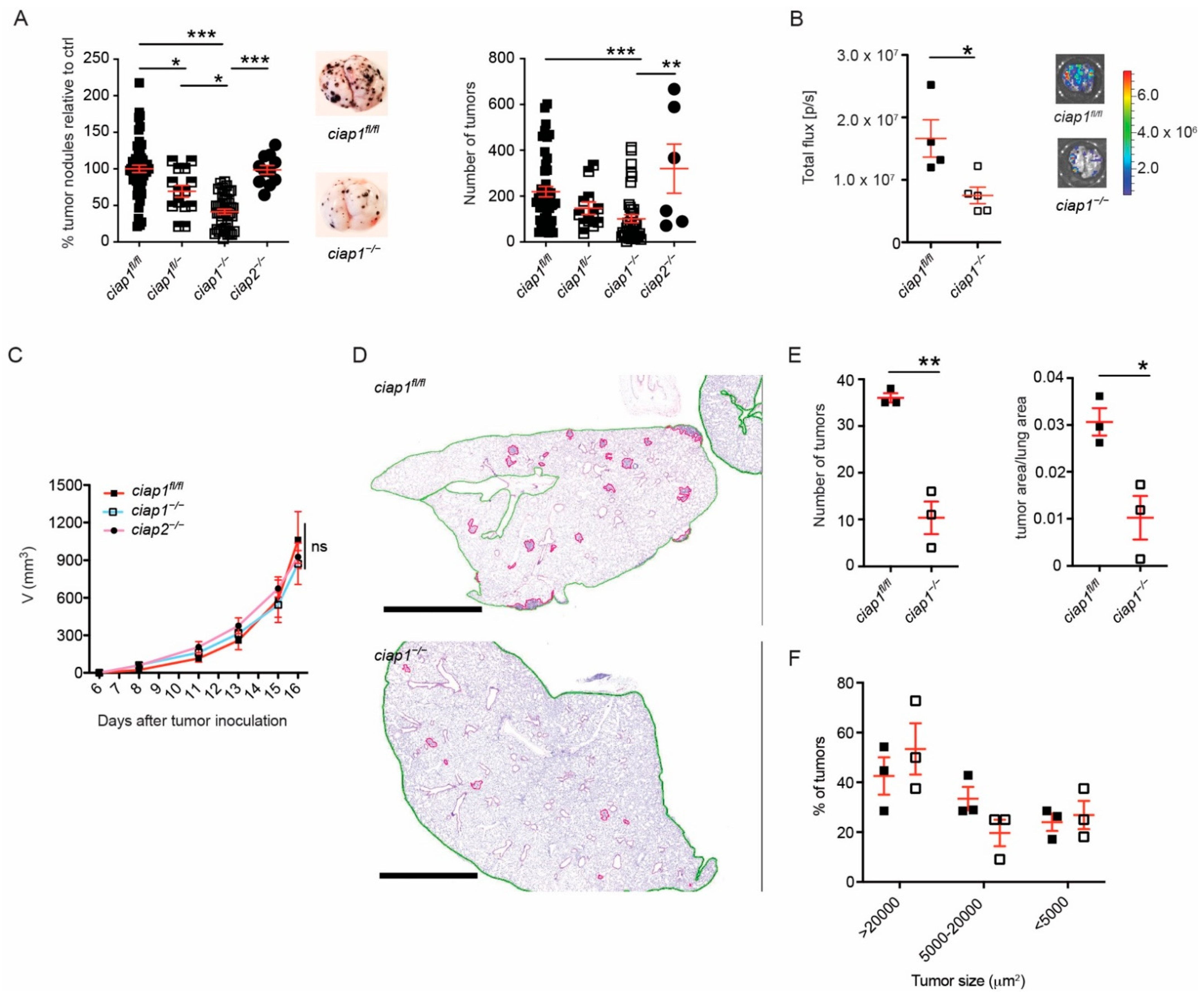
2.1. Smac Mimetic Use Reduces Tumor Nodule Counts in the Lung Due to cIAP1 Loss and Not cIAP2
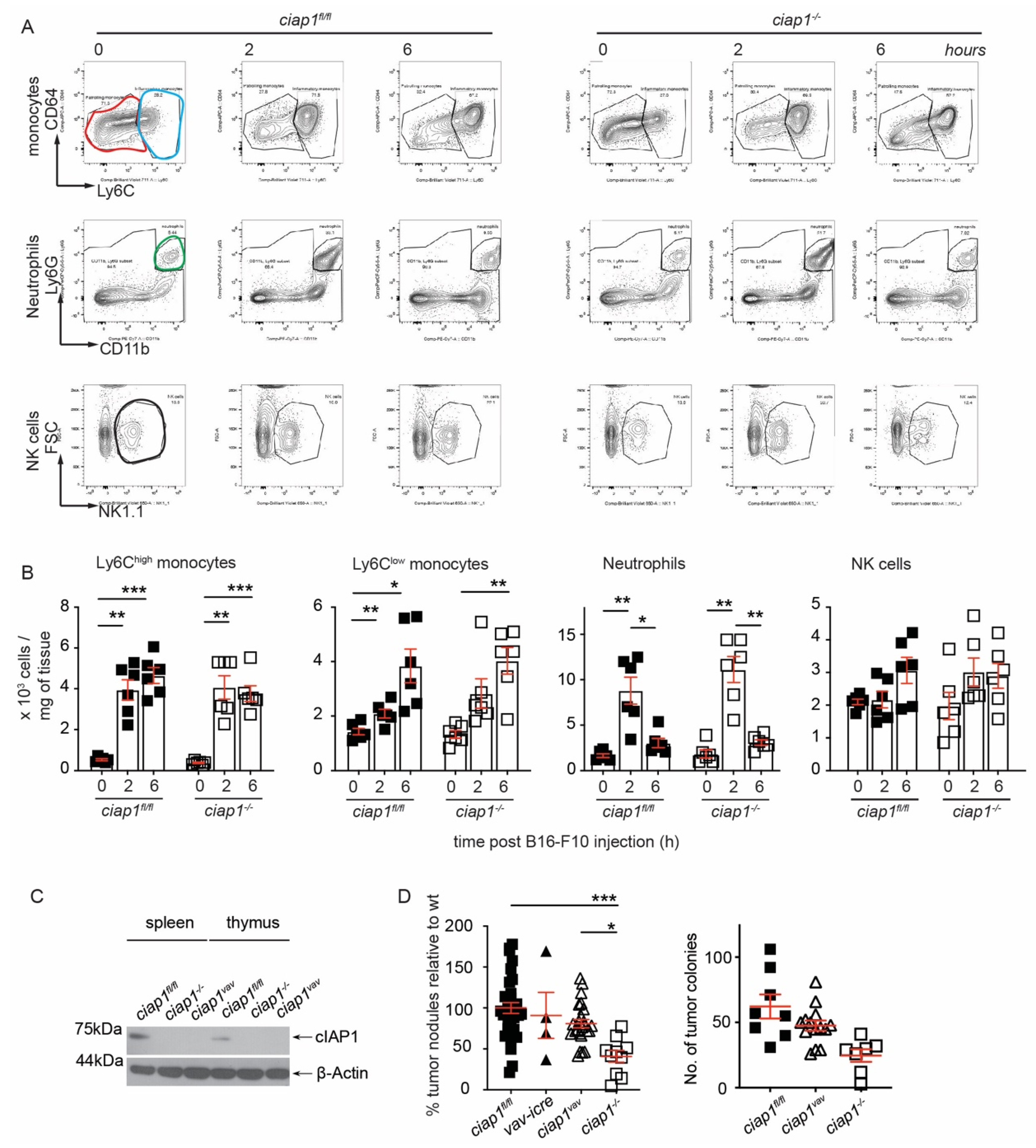
2.2. Loss of cIAP1 in the Hematopoietic Compartment Does Not Reduce Tumor Nodule Counts in the Lung
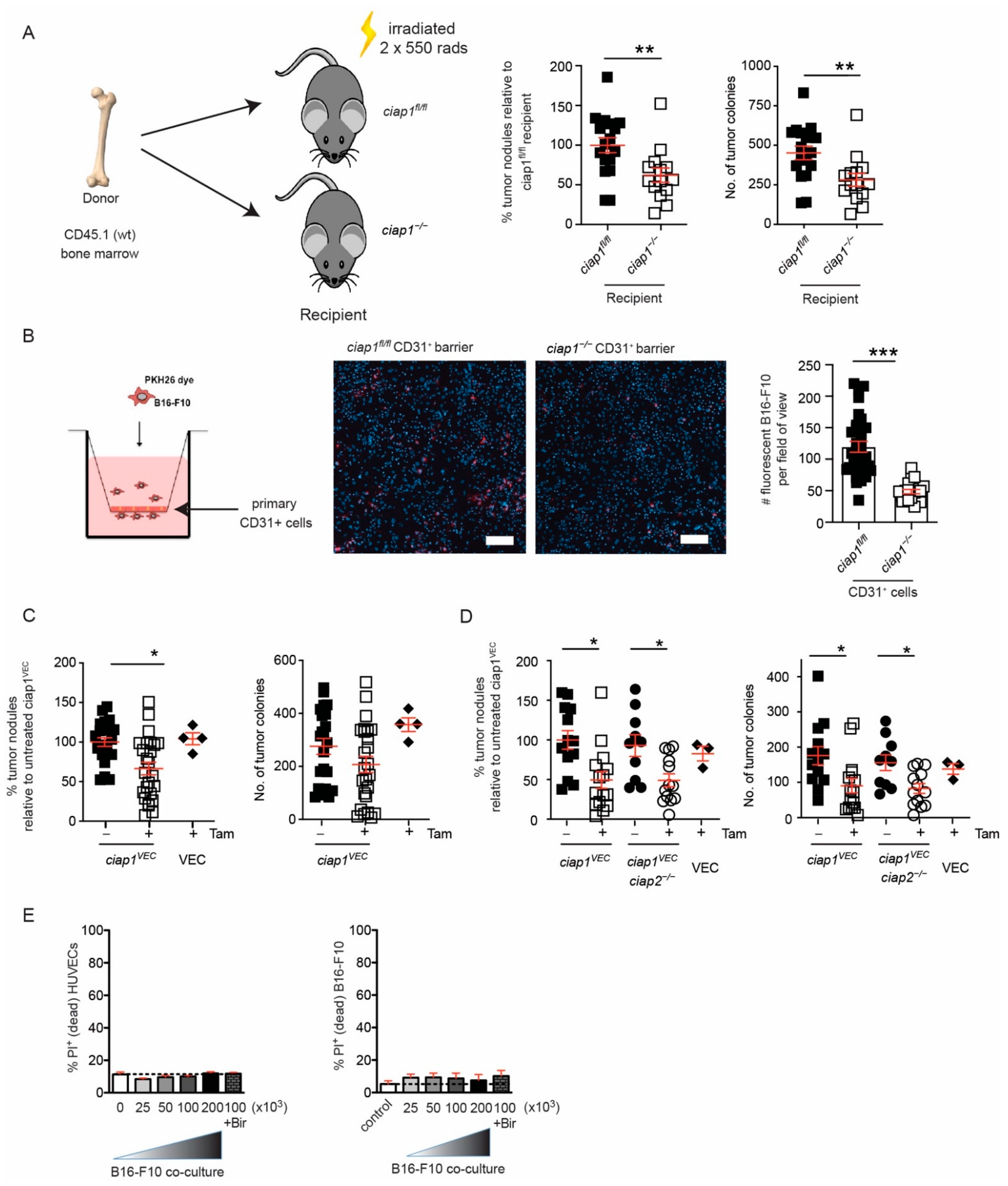
2.3. Loss of cIAP1 in the Endothelial Compartment Reduces Tumor Cell Transmigration Independent of Cell Death
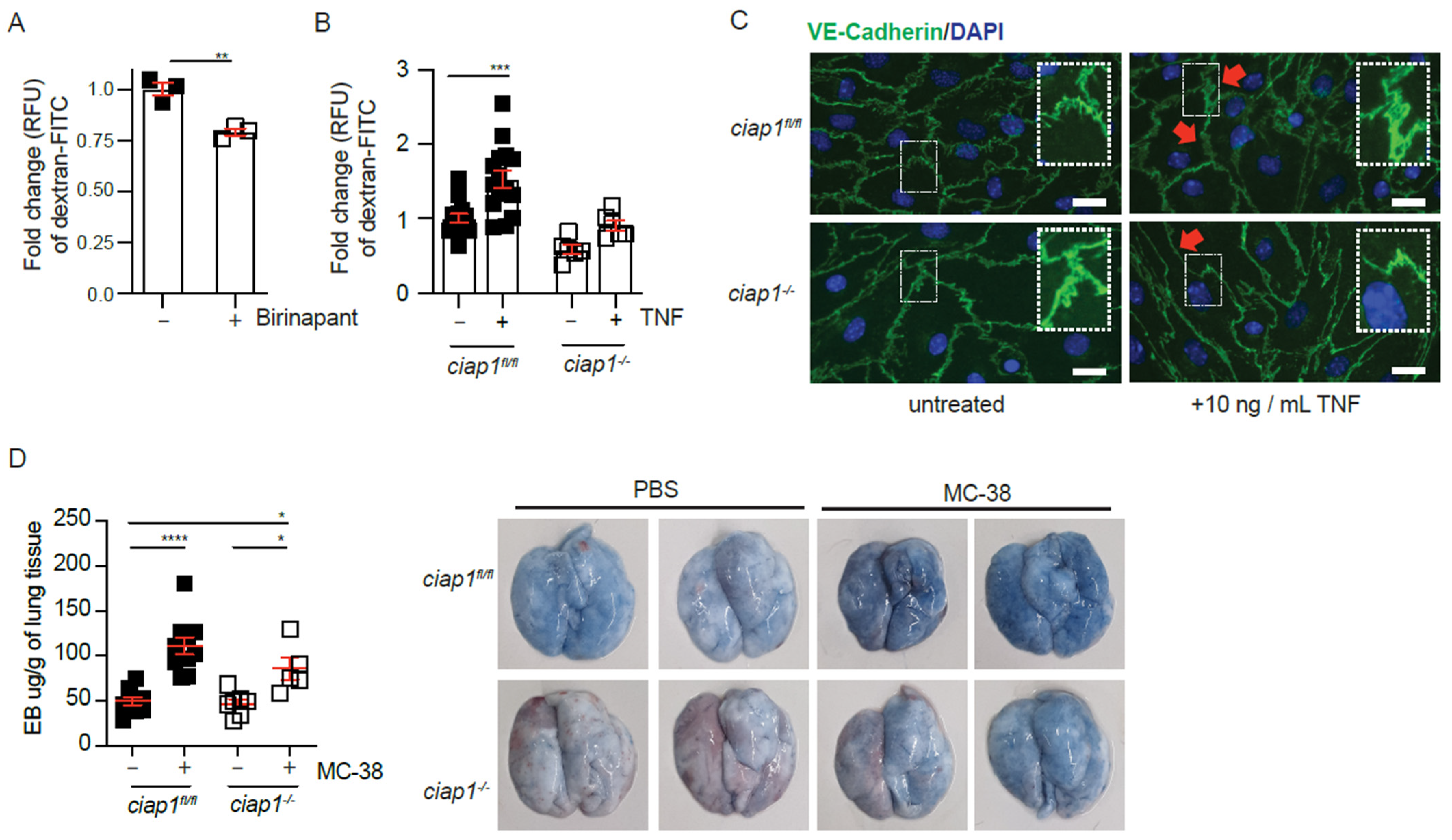
2.4. cIAP1 Aids in Tumor Extravasation by Promoting Permeability
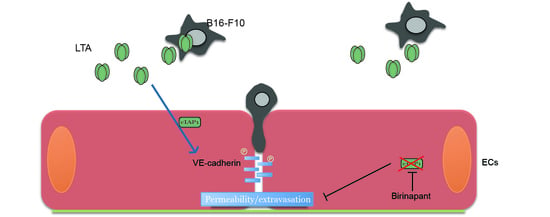
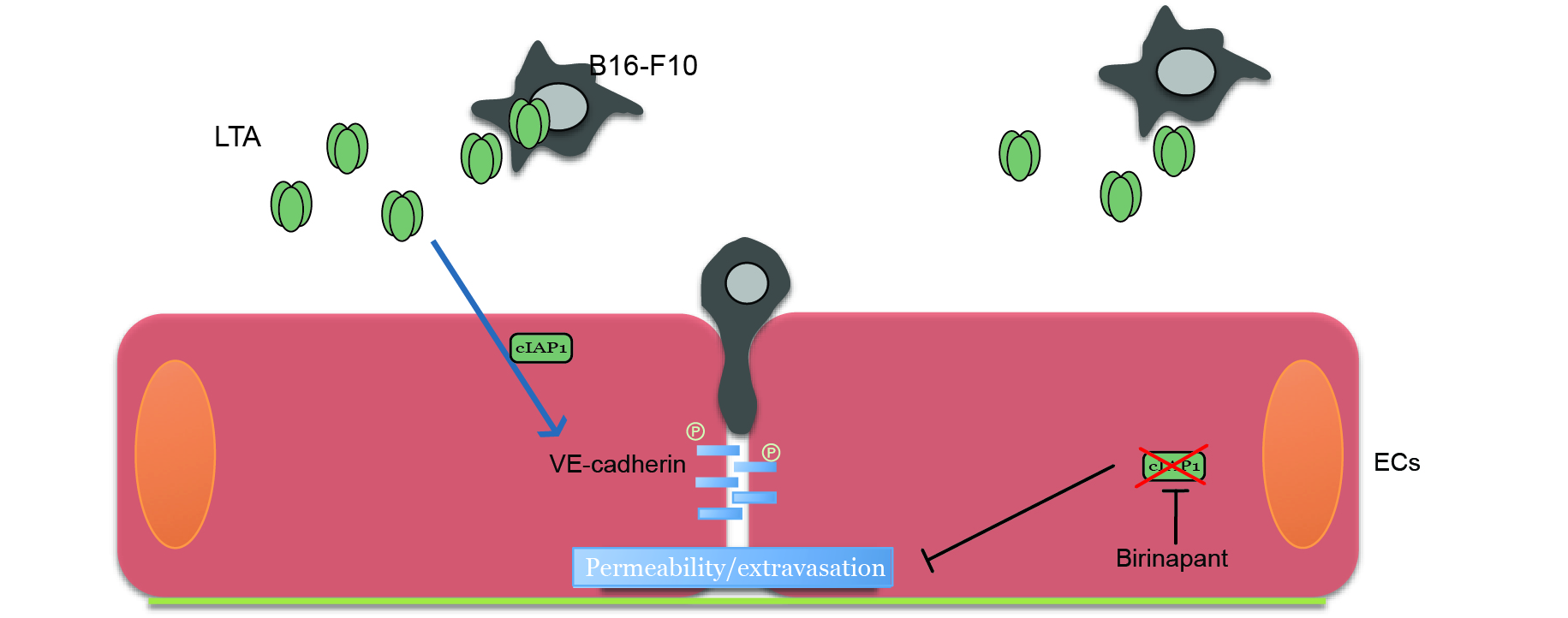
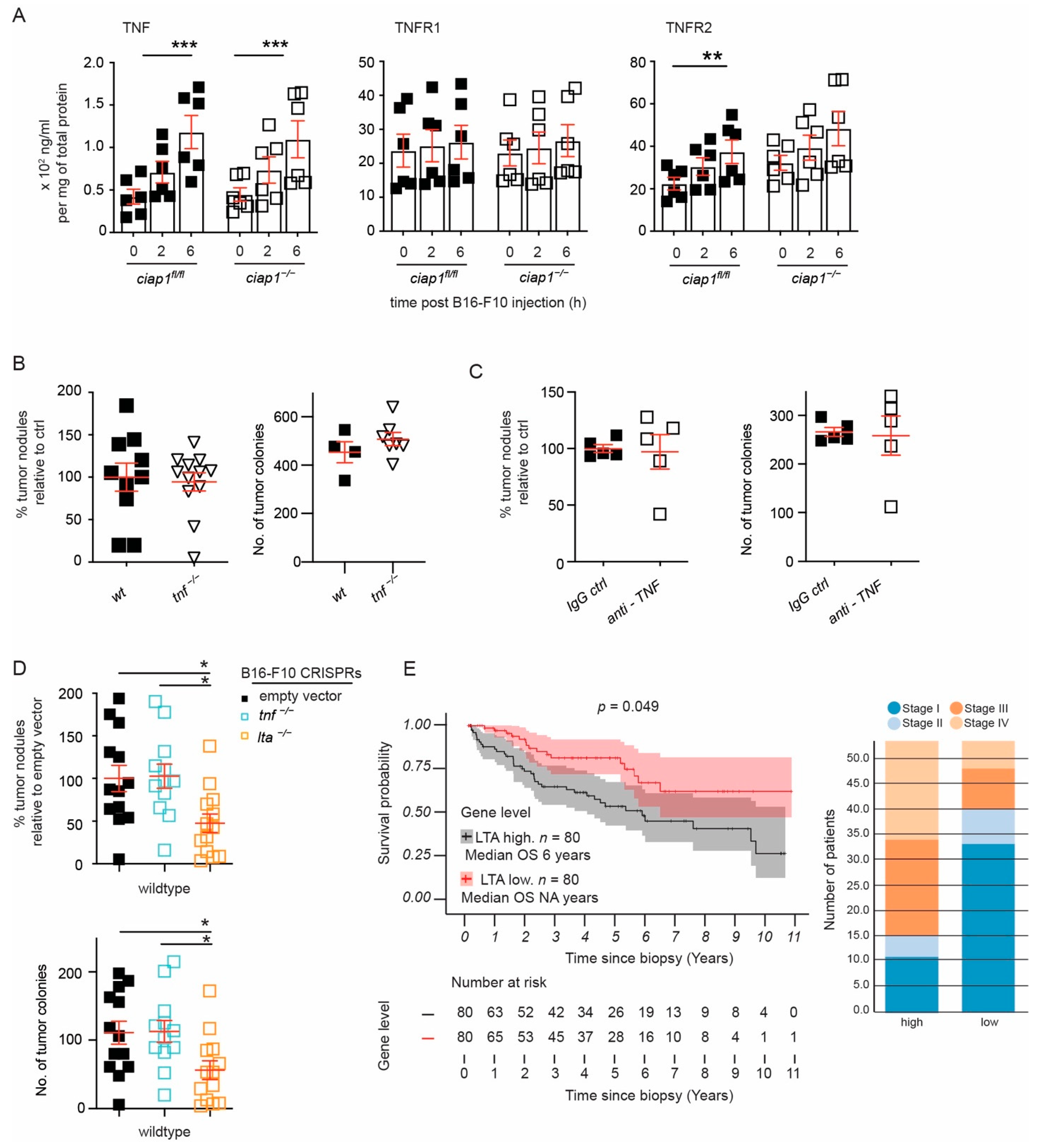
2.5. Tumor Derived Lymphotoxin Alpha Is Responsible for Tumor Cell Extravasation
3. Discussion
4. Materials and Methods
4.1. Animal Work
4.2. Cell Culture and Viability Assays
4.3. Primary Lung EC Isolation and VE-Cadherin Staining
4.4. Transendothelial Migration Assay
4.5. Vascular Permeability Assay
4.6. Multiplex Assay
4.7. Antibodies and Viability Dyes
4.8. Ligands and Inhibitors
4.9. Compounds Used In Vivo
4.10. Western Blotting
4.11. qPCR
4.12. Lentiviral CRISPR/Cas9 Constructs
4.13. mRNA Expression Level Analysis and Survival Curves
4.14. Statistical Analysis
4.15. Histology and Tumor Mass Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zender, L.; Spector, M.S.; Xue, W.; Flemming, P.; Cordon-Cardo, C.; Silke, J.; Fan, S.-T.; Luk, J.M.; Wigler, M.; Hannon, G.J.; et al. Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell 2006, 125, 1253–1267. [Google Scholar] [CrossRef]
- Dai, Z.; Zhu, W.-G.; Morrison, C.D.; Brena, R.M.; Smiraglia, D.J.; Raval, A.; Wu, Y.-Z.; Rush, L.J.; Ross, P.; Molina, J.R.; et al. A comprehensive search for DNA amplification in lung cancer identifies inhibitors of apoptosis cIAP1 and cIAP2 as candidate oncogenes. Hum. Mol. Genet. 2003, 12, 791–801. [Google Scholar] [CrossRef]
- Imoto, I.; Yang, Z.Q.; Pimkhaokham, A.; Tsuda, H.; Shimada, Y.; Imamura, M.; Ohki, M.; Inazawa, J. Identification of cIAP1 as a candidate target gene within an amplicon at 11q22 in esophageal squamous cell carcinomas. Cancer Res. 2001, 61, 6629–6634. [Google Scholar]
- Imoto, I.; Tsuda, H.; Hirasawa, A.; Miura, M.; Sakamoto, M.; Hirohashi, S.; Inazawa, J. Expression of cIAP1, a target for 11q22 amplification, correlates with resistance of cervical cancers to radiotherapy. Cancer Res. 2002, 62, 4860–4866. [Google Scholar]
- Du, C.; Fang, M.; Li, Y.; Li, L.; Wang, X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 2000, 102, 33–42. [Google Scholar] [CrossRef]
- Verhagen, A.M.; Ekert, P.G.; Pakusch, M.; Silke, J.; Connolly, L.M.; Reid, G.E.; Moritz, R.L.; Simpson, R.J.; Vaux, D.L. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell 2000, 102, 43–53. [Google Scholar] [CrossRef]
- Wu, G.; Chai, J.J.; Suber, T.L.; Wu, J.W.; Du, C.Y.; Wang, X.D.; Shi, Y.G. Structural basis of IAP recognition by Smac/DIABLO. Nature 2000, 408, 1008–1012. [Google Scholar] [CrossRef]
- Vince, J.E.; Wong, W.W.-L.; Khan, N.; Feltham, R.; Chau, D.; Ahmed, A.U.; Benetatos, C.A.; Chunduru, S.K.; Condon, S.M.; McKinlay, M.; et al. IAP antagonists target cIAP1 to induce TNFalpha-dependent apoptosis. Cell 2007, 131, 682–693. [Google Scholar] [CrossRef]
- Varfolomeev, E.; Blankenship, J.W.; Wayson, S.M.; Fedorova, A.V.; Kayagaki, N.; Garg, P.; Zobel, K.; Dynek, J.N.; Elliott, L.O.; Wallweber, H.J.A.; et al. IAP antagonists induce autoubiquitination of c-IAPs, NF-kappaB activation, and TNFalpha-dependent apoptosis. Cell 2007, 131, 669–681. [Google Scholar] [CrossRef]
- Gaither, A.; Porter, D.; Yao, Y.; Borawski, J.; Yang, G.; Donovan, J.; Sage, D.; Slisz, J.; Tran, M.; Straub, C.; et al. A Smac mimetic rescue screen reveals roles for inhibitor of apoptosis proteins in tumor necrosis factor-alpha signaling. Cancer Res. 2007, 67, 11493–11498. [Google Scholar] [CrossRef]
- Petersen, S.L.; Wang, L.; Yalcin-Chin, A.; Li, L.; Peyton, M.; Minna, J.; Harran, P.; Wang, X. Autocrine TNFalpha signaling renders human cancer cells susceptible to Smac-mimetic-induced apoptosis. Cancer Cell 2007, 12, 445–456. [Google Scholar] [CrossRef]
- Benetatos, C.A.; Mitsuuchi, Y.; Burns, J.M.; Neiman, E.M.; Condon, S.M.; Yu, G.; Seipel, M.E.; Kapoor, G.S.; Laporte, M.G.; Rippin, S.R.; et al. Birinapant (TL32711), a bivalent SMAC mimetic, targets TRAF2-associated cIAPs, abrogates TNF-induced NF-κB activation, and is active in patient-derived xenograft models. Mol. Cancer Ther. 2014, 13, 867–879. [Google Scholar] [CrossRef]
- Wong, W.W.-L.; Vince, J.E.; Lalaoui, N.; Lawlor, K.E.; Chau, D.; Bankovacki, A.; Anderton, H.; Metcalf, D.; O’Reilly, L.; Jost, P.J.; et al. cIAPs and XIAP regulate myelopoiesis through cytokine production in an RIPK1- and RIPK3-dependent manner. Blood 2014, 123, 2562–2572. [Google Scholar] [CrossRef]
- Lecis, D.; De Cesare, M.; Perego, P.; Conti, A.; Corna, E.; Drago, C.; Seneci, P.; Walczak, H.; Colombo, M.P.; Delia, D.; et al. Smac mimetics induce inflammation and necrotic tumour cell death by modulating macrophage activity. Cell Death Dis. 2013, 4, e920. [Google Scholar] [CrossRef]
- Chesi, M.; Mirza, N.N.; Garbitt, V.M.; Sharik, M.E.; Dueck, A.C.; Asmann, Y.W.; Akhmetzyanova, I.; Kosiorek, H.E.; Calcinotto, A.; Riggs, D.L.; et al. IAP antagonists induce anti-tumor immunity in multiple myeloma. Nat. Med. 2016, 22, 1411–1420. [Google Scholar] [CrossRef]
- Beug, S.T.; Tang, V.A.; LaCasse, E.C.; Cheung, H.H.; Beauregard, C.E.; Brun, J.; Nuyens, J.P.; Earl, N.; St-Jean, M.; Holbrook, J.; et al. Smac mimetics and innate immune stimuli synergize to promote tumor death. Nat. Biotechnol. 2014, 32, 182–190. [Google Scholar] [CrossRef]
- Beug, S.T.; Beauregard, C.E.; Healy, C.; Sanda, T.; St-Jean, M.; Chabot, J.; Walker, D.E.; Mohan, A.; Earl, N.; Lun, X.; et al. Smac mimetics synergize with immune checkpoint inhibitors to promote tumour immunity against glioblastoma. Nat. Commun. 2017, 8, 1–15. [Google Scholar] [CrossRef]
- Kearney, C.J.; Lalaoui, N.; Freeman, A.J.; Ramsbottom, K.M.; Silke, J.; Oliaro, J. PD-L1 and IAPs co-operate to protect tumors from cytotoxic lymphocyte-derived TNF. Cell Death Differ. 2017, 24, 1705–1716. [Google Scholar] [CrossRef]
- Witt, A.; Seeger, J.M.; Coutelle, O.; Zigrino, P.; Broxtermann, P.; Andree, M.; Brinkmann, K.; Jüngst, C.; Schauss, A.C.; Schüll, S.; et al. IAP antagonization promotes inflammatory destruction of vascular endothelium. EMBO Rep. 2015, 16, 719–727. [Google Scholar] [CrossRef][Green Version]
- Strilic, B.; Offermanns, S. Intravascular Survival and Extravasation of Tumor Cells. Cancer Cell 2017, 32, 282–293. [Google Scholar] [CrossRef]
- Santoro, M.M.; Samuel, T.; Mitchell, T.; Reed, J.C.; Stainier, D.Y.R. Birc2 (cIap1) regulates endothelial cell integrity and blood vessel homeostasis. Nat. Genet. 2007, 39, 1397–1402. [Google Scholar] [CrossRef]
- Moulin, M.; Anderton, H.; Voss, A.K.; Thomas, T.; Wong, W.W.-L.; Bankovacki, A.; Feltham, R.; Chau, D.; Cook, W.D.; Silke, J.; et al. IAPs limit activation of RIP kinases by TNF receptor 1 during development. EMBO J. 2012, 31, 1679–1691. [Google Scholar] [CrossRef]
- Strilic, B.; Yang, L.; Albarrán-Juárez, J.; Wachsmuth, L.; Han, K.; Müller, U.C.; Pasparakis, M.; Offermanns, S. Tumour-cell-induced endothelial cell necroptosis via death receptor 6 promotes metastasis. Nature 2016, 536, 215–218. [Google Scholar] [CrossRef]
- Tanimoto, T.; Tsuda, H.; Imazeki, N.; Ohno, Y.; Imoto, I.; Inazawa, J.; Matsubara, O. Nuclear expression of cIAP-1, an apoptosis inhibiting protein, predicts lymph node metastasis and poor patient prognosis in head and neck squamous cell carcinomas. Cancer Lett. 2005, 224, 141–151. [Google Scholar] [CrossRef]
- Shekhar, T.M.; Burvenich, I.J.G.; Harris, M.A.; Rigopoulos, A.; Zanker, D.; Spurling, A.; Parker, B.S.; Walkley, C.R.; Scott, A.M.; Hawkins, C.J. Smac mimetics LCL161 and GDC-0152 inhibit osteosarcoma growth and metastasis in mice. BMC Cancer 2019, 19, 924. [Google Scholar] [CrossRef]
- Lei, W.; Duan, R.; Li, J.; Liu, X.; Huston, A.; Boyce, B.F.; Yao, Z. The IAP Antagonist SM-164 Eliminates Triple-Negative Breast Cancer Metastasis to Bone and Lung in Mice. Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef]
- Gómez-Cuadrado, L.; Tracey, N.; Ma, R.; Qian, B.; Brunton, V.G. Mouse models of metastasis: Progress and prospects. Dis. Model. Mech. 2017, 10, 1061–1074. [Google Scholar] [CrossRef]
- Joyce, J.A.; Pollard, J.W. Microenvironmental regulation of metastasis. Nat. Rev. Cancer 2009, 9, 239–252. [Google Scholar] [CrossRef]
- Reymond, N.; d’Água, B.B.; Ridley, A.J. Crossing the endothelial barrier during metastasis. Nat. Rev. Cancer 2013, 13, 858–870. [Google Scholar] [CrossRef]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the immune system in cancer: From tumor initiation to metastatic progression. Genes Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef]
- Qian, B.-Z.; Li, J.; Zhang, H.; Kitamura, T.; Zhang, J.; Campion, L.R.; Kaiser, E.A.; Snyder, L.A.; Pollard, J.W. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011, 475, 222–225. [Google Scholar] [CrossRef]
- Sorensen, I.; Adams, R.H.; Gossler, A. DLL1-mediated Notch activation regulates endothelial identity in mouse fetal arteries. Blood 2009, 113, 5680–5688. [Google Scholar] [CrossRef]
- Kebers, F.; Lewalle, J.M.; Desreux, J.; Munaut, C.; Devy, L.; Foidart, J.M.; Noël, A. Induction of endothelial cell apoptosis by solid tumor cells. Exp. Cell Res. 1998, 240, 197–205. [Google Scholar] [CrossRef]
- Hornburger, M.C.; Mayer, B.A.; Leonhardt, S.; Willer, E.A.; Zahler, S.; Beyerle, A.; Rajalingam, K.; Vollmar, A.M.; Fürst, R. A novel role for inhibitor of apoptosis (IAP) proteins as regulators of endothelial barrier function by mediating RhoA activation. FASEB J. 2014, 28, 1938–1946. [Google Scholar] [CrossRef]
- Cheng, P.F.; Dummer, R.; Levesque, M.P. Data mining The Cancer Genome Atlas in the era of precision cancer medicine. Swiss Med. Wkly 2015, 145, w14183. [Google Scholar] [CrossRef]
- Fingas, C.D.; Blechacz, B.R.A.; Smoot, R.L.; Guicciardi, M.E.; Mott, J.; Bronk, S.F.; Werneburg, N.W.; Sirica, A.E.; Gores, G.J. A smac mimetic reduces TNF related apoptosis inducing ligand (TRAIL)-induced invasion and metastasis of cholangiocarcinoma cells. Hepatology 2010, 52, 550–561. [Google Scholar] [CrossRef]
- Angelini, D.J.; Hyun, S.-W.; Grigoryev, D.N.; Garg, P.; Gong, P.; Singh, I.S.; Passaniti, A.; Hasday, J.D.; Goldblum, S.E. TNF-α increases tyrosine phosphorylation of vascular endothelial cadherin and opens the paracellular pathway through fyn activation in human lung endothelia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 291, L1232–L1245. [Google Scholar] [CrossRef]
- Medvedev, A.E.; Espevik, T.; Ranges, G.; Sundan, A. Distinct roles of the two tumor necrosis factor (TNF) receptors in modulating TNF and lymphotoxin alpha effects. J. Biol. Chem. 1996, 271, 9778–9784. [Google Scholar] [CrossRef]
- Ferrero, E.; Zocchi, M.R.; Magni, E.; Panzeri, M.C.; Curnis, F.; Rugarli, C.; Ferrero, M.E.; Corti, A. Roles of tumor necrosis factor p55 and p75 receptors in TNF-alpha-induced vascular permeability. Am. J. Physiol. Cell Physiol. 2001, 281, C1173–C1179. [Google Scholar] [CrossRef]
- Morrish, E.; Brumatti, G.; Silke, J. Future Therapeutic Directions for Smac-Mimetics. Cells 2020, 9, 406. [Google Scholar] [CrossRef]
- Sun, X.-S.; Tao, Y.; Le Tourneau, C.; Pointreau, Y.; Sire, C.; Kaminsky, M.-C.; Coutte, A.; Alfonsi, M.; Boisselier, P.; Martin, L.; et al. Debio 1143 and high-dose cisplatin chemoradiotherapy in high-risk locoregionally advanced squamous cell carcinoma of the head and neck: A double-blind, multicentre, randomised, phase 2 study. Lancet Oncol. 2020, 21, 1173–1187. [Google Scholar] [CrossRef]
- Bardia, A.; Parton, M.; Kümmel, S.; Estévez, L.G.; Huang, C.-S.; Cortés, J.; Ruiz-Borrego, M.; Telli, M.L.; Martin-Martorell, P.; López, R.; et al. Paclitaxel With Inhibitor of Apoptosis Antagonist, LCL161, for Localized Triple-Negative Breast Cancer, Prospectively Stratified by Gene Signature in a Biomarker-Driven Neoadjuvant Trial. J. Clin. Oncol. 2018, 36, 3126–3133. [Google Scholar] [CrossRef]
- Wolf, M.J.; Hoos, A.; Bauer, J.; Boettcher, S.; Knust, M.; Weber, A.; Simonavicius, N.; Schneider, C.; Lang, M.; Stürzl, M.; et al. Endothelial CCR2 signaling induced by colon carcinoma cells enables extravasation via the JAK2-Stat5 and p38MAPK pathway. Cancer Cell 2012, 22, 91–105. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vasilikos, L.; Hänggi, K.; Spilgies, L.M.; Kisele, S.; Rufli, S.; Wong, W.W.-L. Loss of cIAP1 in Endothelial Cells Limits Metastatic Extravasation through Tumor-Derived Lymphotoxin Alpha. Cancers 2021, 13, 599. https://doi.org/10.3390/cancers13040599
Vasilikos L, Hänggi K, Spilgies LM, Kisele S, Rufli S, Wong WW-L. Loss of cIAP1 in Endothelial Cells Limits Metastatic Extravasation through Tumor-Derived Lymphotoxin Alpha. Cancers. 2021; 13(4):599. https://doi.org/10.3390/cancers13040599
Chicago/Turabian StyleVasilikos, Lazaros, Kay Hänggi, Lisanne M. Spilgies, Samanta Kisele, Stefanie Rufli, and W. Wei-Lynn Wong. 2021. "Loss of cIAP1 in Endothelial Cells Limits Metastatic Extravasation through Tumor-Derived Lymphotoxin Alpha" Cancers 13, no. 4: 599. https://doi.org/10.3390/cancers13040599
APA StyleVasilikos, L., Hänggi, K., Spilgies, L. M., Kisele, S., Rufli, S., & Wong, W. W.-L. (2021). Loss of cIAP1 in Endothelial Cells Limits Metastatic Extravasation through Tumor-Derived Lymphotoxin Alpha. Cancers, 13(4), 599. https://doi.org/10.3390/cancers13040599