
Targeting Oncoimmune Drivers of Cancer Metastasis

 , and
, and {kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Initiation of Tumor Escape through EMT
3. EMT-Induced CSCs
4. Tumor Regeneration after EMT
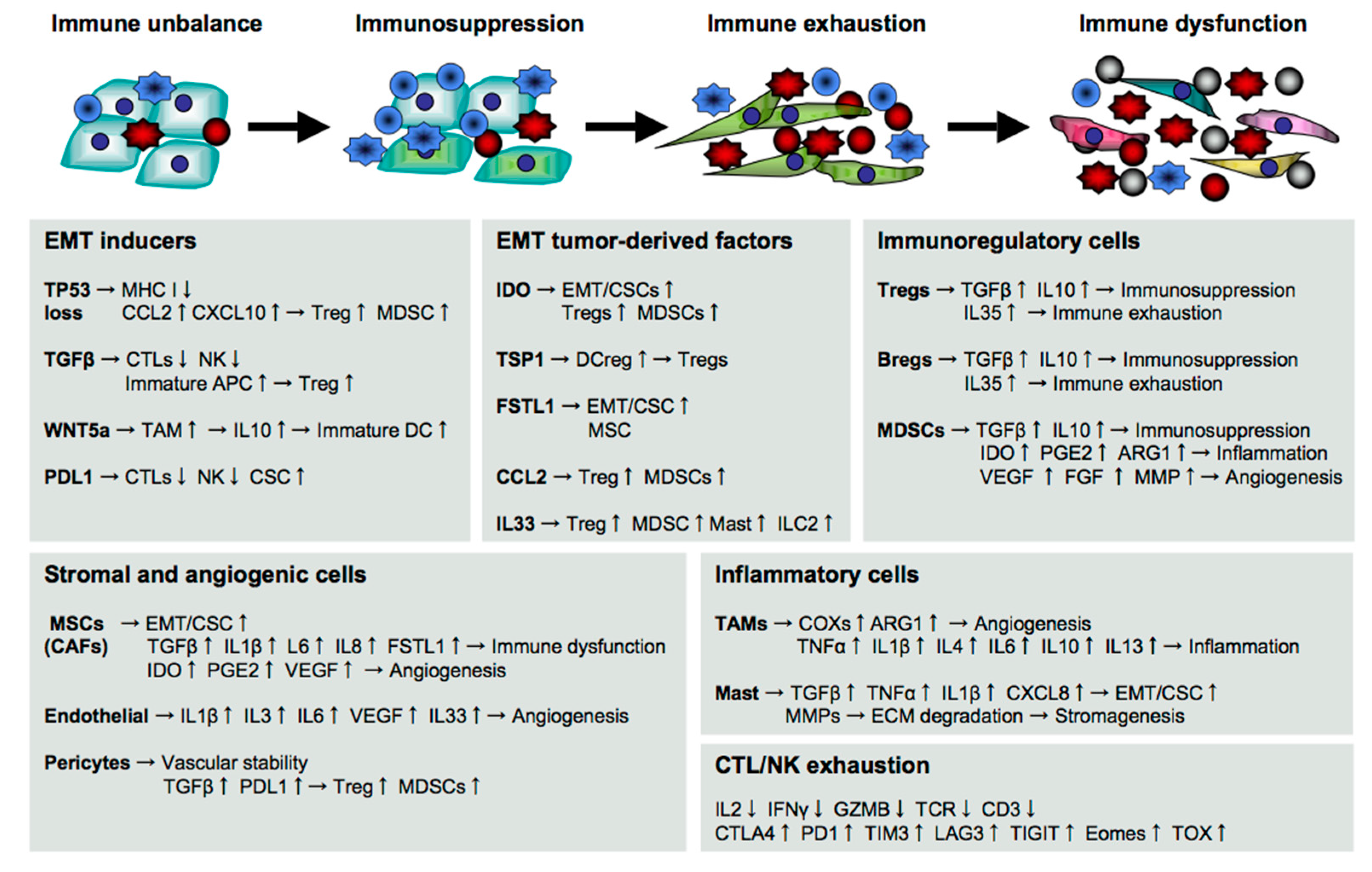
5. Immune Supporters for Cancer Metastasis
5.1. Immune Evasion at the Beginning of Cancer Metastasis
5.2. Immunosuppression during Cancer Metastasis
5.3. Stromagenesis and Angiogenesis for Cancer Metastasis
5.4. Immune Exhaustion and Dysfunction for Cancer Metastasis
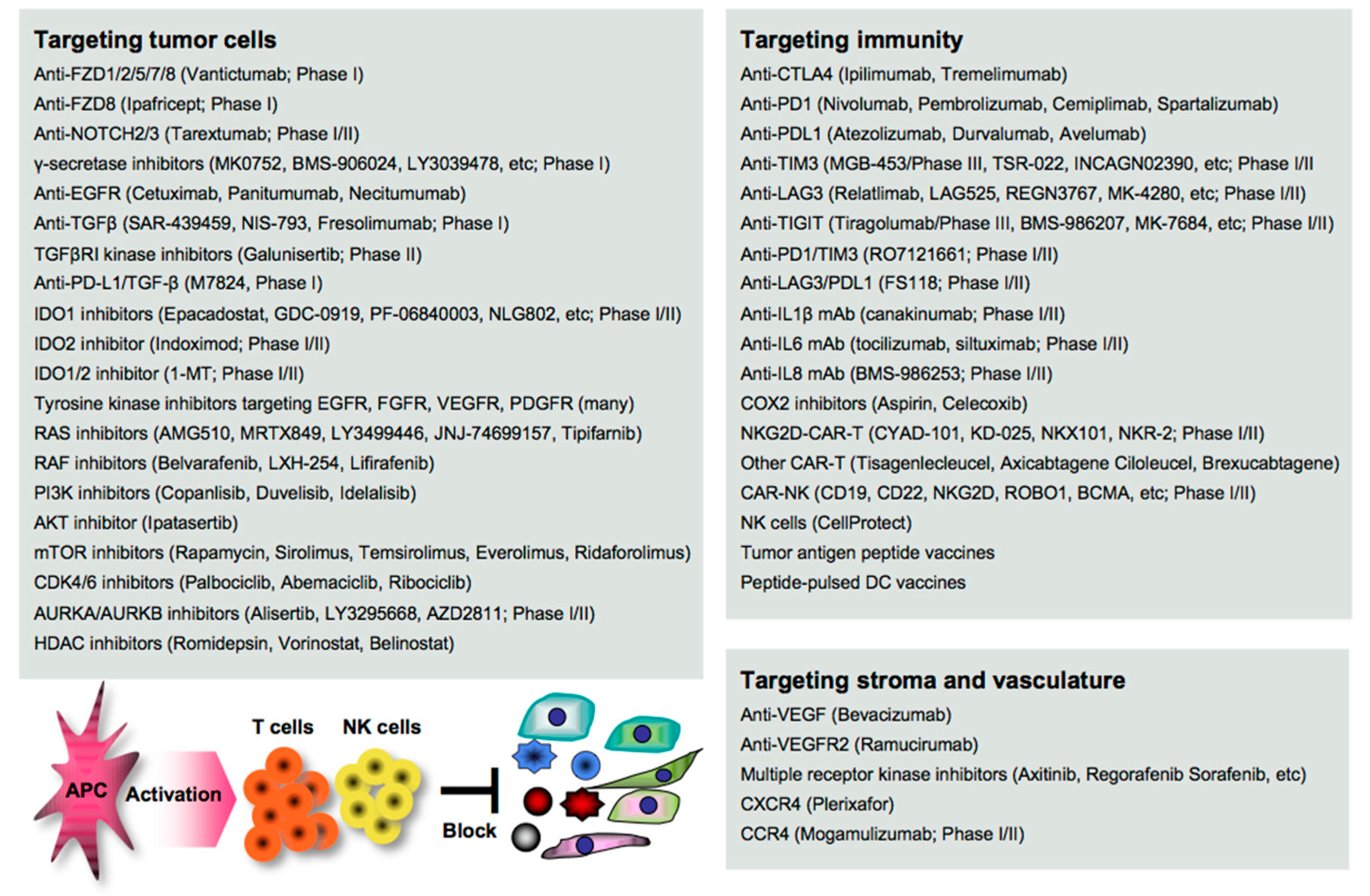
6. Treatments for Cancer Metastasis
6.1. Targeting EMT/CSC Inducers
6.2. Targeting Stromagenesis and Angiogenesis
6.3. Targeting Immune Determinants
7. Conclusions
Funding
Conflicts of Interest
References
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef] [PubMed]
- Bakir, B.; Chiarella, A.M.; Pitarresi, J.R.; Rustgi, A.K. EMT, MET, Plasticity, and Tumor Metastasis. Trends Cell Biol. 2020, 30, 764–776. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-Mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-Mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Makena, M.R.; Ranjan, A.; Thirumala, V.; Reddy, A.P. Cancer stem cells: Road to therapeutic resistance and strategies to overcome resistance. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165339. [Google Scholar] [CrossRef]
- Coward, J.; Harding, A. Size Does Matter: Why Polyploid Tumor Cells are Critical Drug Targets in the War on Cancer. Front. Oncol. 2014, 4, 123. [Google Scholar] [CrossRef]
- Zhang, S.; Mercado-Uribe, I.; Xing, Z.; Sun, B.; Kuang, J.; Liu, J. Generation of cancer stem-like cells through the formation of polyploid giant cancer cells. Oncogene 2014, 33, 116–128. [Google Scholar] [CrossRef]
- Amend, S.R.; Torga, G.; Lin, K.C.; Kostecka, L.G.; de Marzo, A.; Austin, R.H.; Pienta, K.J. Polyploid giant cancer cells: Unrecognized actuators of tumorigenesis, metastasis, and resistance. Prostate 2019, 79, 1489–1497. [Google Scholar] [CrossRef]
- Liu, Y.; Cao, X. Immunosuppressive cells in tumor immune escape and metastasis. J. Mol. Med. 2016, 94, 509–522. [Google Scholar] [CrossRef]
- Lorenzo-Sanz, L.; Munoz, P. Tumor-Infiltrating Immunosuppressive Cells in Cancer-Cell Plasticity, Tumor Progression and Therapy Response. Cancer Microenviron. 2019, 12, 119–132. [Google Scholar] [CrossRef]
- Aguirre-Ghiso, J.A. How dormant cancer persists and reawakens. Science 2018, 361, 1314–1315. [Google Scholar] [CrossRef] [PubMed]
- Popovic, A.; Jaffee, E.M.; Zaidi, N. Emerging strategies for combination checkpoint modulators in cancer immunotherapy. J. Clin. Invest. 2018, 128, 3209–3218. [Google Scholar] [CrossRef]
- Varricchi, G.; Galdiero, M.R.; Marone, G.; Criscuolo, G.; Triassi, M.; Bonaduce, D.; Marone, G.; Tocchetti, C.G. Cardiotoxicity of immune checkpoint inhibitors. ESMO Open 2017, 2, e000247. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, R.; Mezquita, L.; Texier, M.; Lahmar, J.; Audigier-Valette, C.; Tessonnier, L.; Mazieres, J.; Zalcman, G.; Brosseau, S.; Le Moulec, S.; et al. Hyperprogressive Disease in Patients with Advanced Non-Small Cell Lung Cancer Treated With PD-1/PD-L1 Inhibitors or With Single-Agent Chemotherapy. JAMA Oncol. 2018, 4, 1543–1552. [Google Scholar] [CrossRef]
- Frelaut, M.; Le Tourneau, C.; Borcoman, E. Hyperprogression under Immunotherapy. Int. J. Mol. Sci. 2019, 20, 2674. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Lei, Y.; Gao, X.; Liang, Q.; Li, L.; Feng, J.; Hou, P.; Han, L.; Zhang, Y.; Huang, B.; et al. p53 Attenuates the oncogenic Ras-induced epithelial-mesenchymal transition in human mammary epithelial cells. Biochem. Biophys. Res. Commun. 2013, 434, 606–613. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, D.M.; Medici, D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci. Signal. 2014, 7, re8. [Google Scholar] [CrossRef]
- Moore, A.R.; Rosenberg, S.C.; McCormick, F.; Malek, S. RAS-Targeted therapies: Is the undruggable drugged? Nat. Rev. Drug Discov. 2020, 19, 533–552. [Google Scholar] [CrossRef]
- Gujral, T.S.; Chan, M.; Peshkin, L.; Sorger, P.K.; Kirschner, M.W.; MacBeath, G. A noncanonical Frizzled2 pathway regulates epithelial-mesenchymal transition and metastasis. Cell 2014, 159, 844–856. [Google Scholar] [CrossRef]
- Lopez-Bergami, P.; Barbero, G. The emerging role of Wnt5a in the promotion of a pro-inflammatory and immunosuppressive tumor microenvironment. Cancer Metastasis Rev. 2020, 39, 933–952. [Google Scholar] [CrossRef]
- Liu, T.; Yu, J.; Deng, M.; Yin, Y.; Zhang, H.; Luo, K.; Qin, B.; Li, Y.; Wu, C.; Ren, T.; et al. CDK4/6-Dependent activation of DUB3 regulates cancer metastasis through SNAIL1. Nat. Commun. 2017, 8, 13923. [Google Scholar] [CrossRef] [PubMed]
- Siemens, H.; Jackstadt, R.; Hunten, S.; Kaller, M.; Menssen, A.; Gotz, U.; Hermeking, H. miR-34 and SNAIL form a double-negative feedback loop to regulate epithelial-mesenchymal transitions. Cell Cycle 2011, 10, 4256–4271. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.P.; Wang, W.L.; Chang, Y.L.; Wu, C.T.; Chao, Y.C.; Kao, S.H.; Yuan, A.; Lin, C.W.; Yang, S.C.; Chan, W.K.; et al. p53 controls cancer cell invasion by inducing the MDM2-mediated degradation of Slug. Nat. Cell Biol. 2009, 11, 694–704. [Google Scholar] [CrossRef] [PubMed]
- Noman, M.Z.; Janji, B.; Abdou, A.; Hasmim, M.; Terry, S.; Tan, T.Z.; Mami-Chouaib, F.; Thiery, J.P.; Chouaib, S. The immune checkpoint ligand PD-L1 is upregulated in EMT-activated human breast cancer cells by a mechanism involving ZEB-1 and miR-200. Oncoimmunology 2017, 6, e1263412. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.M.; Xia, W.; Hsu, Y.H.; Chan, L.C.; Yu, W.H.; Cha, J.H.; Chen, C.T.; Liao, H.W.; Kuo, C.W.; Khoo, K.H.; et al. STT3-dependent PD-L1 accumulation on cancer stem cells promotes immune evasion. Nat. Commun. 2018, 9, 1908. [Google Scholar] [CrossRef] [PubMed]
- Kudo-Saito, C.; Shirako, H.; Ohike, M.; Tsukamoto, N.; Kawakami, Y. CCL2 is critical for immunosuppression to promote cancer metastasis. Clin. Exp. Metastasis 2013, 30, 393–405. [Google Scholar] [CrossRef]
- Zhuang, H.; Cao, G.; Kou, C.; Liu, T. CCL2/CCR2 axis induces hepatocellular carcinoma invasion and epithelial-mesenchymal transition in vitro through activation of the Hedgehog pathway. Oncol. Rep. 2018, 39, 21–30. [Google Scholar] [CrossRef]
- Volonte, A.; Di Tomaso, T.; Spinelli, M.; Todaro, M.; Sanvito, F.; Albarello, L.; Bissolati, M.; Ghirardelli, L.; Orsenigo, E.; Ferrone, S.; et al. Cancer-Initiating cells from colorectal cancer patients escape from T cell-mediated immunosurveillance in vitro through membrane-bound IL-4. J. Immunol. 2014, 192, 523–532. [Google Scholar] [CrossRef]
- Chen, J.; Gong, C.; Mao, H.; Li, Z.; Fang, Z.; Chen, Q.; Lin, M.; Jiang, X.; Hu, Y.; Wang, W.; et al. E2F1/SP3/STAT6 axis is required for IL-4-induced epithelial-mesenchymal transition of colorectal cancer cells. Int. J. Oncol. 2018, 53, 567–578. [Google Scholar] [CrossRef]
- Huang, C.; Li, N.; Li, Z.; Chang, A.; Chen, Y.; Zhao, T.; Li, Y.; Wang, X.; Zhang, W.; Wang, Z.; et al. Tumour-Derived Interleukin 35 promotes pancreatic ductal adenocarcinoma cell extravasation and metastasis by inducing ICAM1 expression. Nat. Commun. 2017, 8, 14035. [Google Scholar] [CrossRef]
- Kudo-Saito, C.; Fuwa, T.; Murakami, K.; Kawakami, Y. Targeting FSTL1 prevents tumor bone metastasis and consequent immune dysfunction. Cancer Res. 2013, 73, 6185–6193. [Google Scholar] [CrossRef] [PubMed]
- Lau, M.C.; Ng, K.Y.; Wong, T.L.; Tong, M.; Lee, T.K.; Ming, X.Y.; Law, S.; Lee, N.P.; Cheung, A.L.; Qin, Y.R.; et al. FSTL1 Promotes Metastasis and Chemoresistance in Esophageal Squamous Cell Carcinoma through NFκB-BMP Signaling Cross-Talk. Cancer Res. 2017, 77, 5886–5899. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, G.M.; Ismail, H.M.; Bashir, M.; Muhuri, M.; Vaz, C.; Nama, S.; Ow, G.S.; Vladimirovna, I.A.; Ramalingam, R.; Burke, B.; et al. EGF hijacks miR-198/FSTL1 wound-healing switch and steers a two-pronged pathway toward metastasis. J. Exp. Med. 2017, 214, 2889–2900. [Google Scholar] [CrossRef] [PubMed]
- Gu, C.; Wang, X.; Long, T.; Wang, X.; Zhong, Y.; Ma, Y.; Hu, Z.; Li, Z. FSTL1 interacts with VIM and promotes colorectal cancer metastasis via activating the focal adhesion signalling pathway. Cell Death Dis. 2018, 9, 654. [Google Scholar] [CrossRef] [PubMed]
- Kudo-Saito, C.; Ishida, A.; Shouya, Y.; Teramoto, K.; Igarashi, T.; Kon, R.; Saito, K.; Awada, C.; Ogiwara, Y.; Toyoura, M. Blocking the FSTL1-DIP2A Axis Improves Anti-Tumor Immunity. Cell Rep. 2018, 24, 1790–1801. [Google Scholar] [CrossRef]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef]
- Powell, E.; Piwnica-Worms, D.; Piwnica-Worms, H. Contribution of p53 to metastasis. Cancer Discov. 2014, 4, 405–414. [Google Scholar] [CrossRef]
- Zhu, P.; Fan, Z. Cancer stem cells and tumorigenesis. Biophys. Rep. 2018, 4, 178–188. [Google Scholar] [CrossRef]
- Almozyan, S.; Colak, D.; Mansour, F.; Alaiya, A.; Al-Harazi, O.; Qattan, A.; Al-Mohanna, F.; Al-Alwan, M.; Ghebeh, H. PD-L1 promotes OCT4 and Nanog expression in breast cancer stem cells by sustaining PI3K/AKT pathway activation. Int. J. Cancer 2017, 141, 1402–1412. [Google Scholar] [CrossRef]
- Katoh, M. Canonical and non-canonical WNT signaling in cancer stem cells and their niches: Cellular heterogeneity, omics reprogramming, targeted therapy and tumor plasticity (Review). Int. J. Oncol. 2017, 51, 1357–1369. [Google Scholar] [CrossRef]
- Skandalis, S.S.; Karalis, T.T.; Chatzopoulos, A.; Karamanos, N.K. Hyaluronan-CD44 axis orchestrates cancer stem cell functions. Cell. Signal. 2019, 63, 109377. [Google Scholar] [CrossRef] [PubMed]
- Eyvazi, S.; Kazemi, B.; Dastmalchi, S.; Bandehpour, M. Involvement of CD24 in Multiple Cancer Related Pathways Makes It an Interesting New Target for Cancer Therapy. Curr. Cancer Drug Targets 2018, 18, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Barzegar Behrooz, A.; Syahir, A.; Ahmad, S. CD133: Beyond a cancer stem cell biomarker. J. Drug Target 2019, 27, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Pan, Q.; Sun, F.; Yu, Y.; Wang, J. Cluster of differentiation 166 (CD166) regulates cluster of differentiation (CD44) via NF-κB in liver cancer cell line Bel-7402. Biochem. Biophys. Res. Commun. 2014, 451, 334–338. [Google Scholar] [CrossRef] [PubMed]
- Darvishi, B.; Boroumandieh, S.; Majidzadeh, A.K.; Salehi, M.; Jafari, F.; Farahmand, L. The role of activated leukocyte cell adhesion molecule (ALCAM) in cancer progression, invasion, metastasis and recurrence: A novel cancer stem cell marker and tumor-specific prognostic marker. Exp. Mol. Pathol. 2020, 115, 104443. [Google Scholar] [CrossRef] [PubMed]
- Redmer, T.; Welte, Y.; Behrens, D.; Fichtner, I.; Przybilla, D.; Wruck, W.; Yaspo, M.L.; Lehrach, H.; Schafer, R.; Regenbrecht, C.R. The nerve growth factor receptor CD271 is crucial to maintain tumorigenicity and stem-like properties of melanoma cells. PLoS ONE 2014, 9, e92596. [Google Scholar] [CrossRef] [PubMed]
- Chung, M.K.; Jung, Y.H.; Lee, J.K.; Cho, S.Y.; Murillo-Sauca, O.; Uppaluri, R.; Shin, J.H.; Sunwoo, J.B. CD271 Confers an Invasive and Metastatic Phenotype of Head and Neck Squamous Cell Carcinoma through the Upregulation of Slug. Clin. Cancer Res. 2018, 24, 674–683. [Google Scholar] [CrossRef]
- Davis, J.E., Jr.; Kirk, J.; Ji, Y.; Tang, D.G. Tumor Dormancy and Slow-Cycling Cancer Cells. Adv. Exp. Med. Biol. 2019, 1164, 199–206. [Google Scholar]
- Rehman, S.K.; Haynes, J.; Collignon, E.; Brown, K.R.; Wang, Y.; Nixon, A.M.L.; Bruce, J.P.; Wintersinger, J.A.; Singh Mer, A.; Lo, E.B.L.; et al. Colorectal Cancer Cells Enter a Diapause-like DTP State to Survive Chemotherapy. Cell 2021, 184, 226–242.e21. [Google Scholar] [CrossRef]
- Meireson, A.; Devos, M.; Brochez, L. IDO Expression in Cancer: Different Compartment, Different Functionality? Front. Immunol. 2020, 11, 531491. [Google Scholar] [CrossRef]
- Litzenburger, U.M.; Opitz, C.A.; Sahm, F.; Rauschenbach, K.J.; Trump, S.; Winter, M.; Ott, M.; Ochs, K.; Lutz, C.; Liu, X.; et al. Constitutive IDO expression in human cancer is sustained by an autocrine signaling loop involving IL-6, STAT3 and the AHR. Oncotarget 2014, 5, 1038–1051. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Baker, D.; Ten Dijke, P. TGF-Β-Mediated Epithelial-Mesenchymal Transition and Cancer Metastasis. Int. J. Mol. Sci. 2019, 20, 2767. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Singhal, U.; Qiao, Y.; Kasputis, T.; Chung, J.S.; Zhao, H.; Chammaa, F.; Belardo, J.A.; Roth, T.M.; Zhang, H.; et al. Wnt Signaling Drives Prostate Cancer Bone Metastatic Tropism and Invasion. Transl. Oncol. 2020, 13, 100747. [Google Scholar] [CrossRef] [PubMed]
- Xiang, L.; Gilkes, D.M. The Contribution of the Immune System in Bone Metastasis Pathogenesis. Int. J. Mol. Sci. 2019, 20, 999. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.; Mishra, L.; Li, S. EMT, CTCs and CSCs in tumor relapse and drug-resistance. Oncotarget 2015, 6, 10697–10711. [Google Scholar] [CrossRef] [PubMed]
- Alix-Panabieres, C.; Mader, S.; Pantel, K. Epithelial-Mesenchymal plasticity in circulating tumor cells. J. Mol. Med. 2017, 95, 133–142. [Google Scholar] [CrossRef]
- Garg, M. Epithelial, mesenchymal and hybrid epithelial/mesenchymal phenotypes and their clinical relevance in cancer metastasis. Expert Rev. Mol. Med. 2017, 19, e3. [Google Scholar] [CrossRef]
- Blaylock, R.L. Cancer microenvironment, inflammation and cancer stem cells: A hypothesis for a paradigm change and new targets in cancer control. Surg. Neurol. Int. 2015, 6, 92. [Google Scholar] [CrossRef]
- Albrengues, J.; Shields, M.A.; Ng, D.; Park, C.G.; Ambrico, A.; Poindexter, M.E.; Upadhyay, P.; Uyeminami, D.L.; Pommier, A.; Kuttner, V.; et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science 2018, 361, eaao4227. [Google Scholar] [CrossRef]
- Lee, C.C.; Lin, J.C.; Hwang, W.L.; Kuo, Y.J.; Chen, H.K.; Tai, S.K.; Lin, C.C.; Yang, M.H. Macrophage-Secreted interleukin-35 regulates cancer cell plasticity to facilitate metastatic colonization. Nat. Commun. 2018, 9, 3763. [Google Scholar] [CrossRef]
- Cowan, R.W.; Singh, G. Giant cell tumor of bone: A basic science perspective. Bone 2013, 52, 238–246. [Google Scholar] [CrossRef] [PubMed]
- War, A.R.; Dang, K.; Jiang, S.; Xiao, Z.; Miao, Z.; Yang, T.; Li, Y.; Qian, A. Role of cancer stem cells in the development of giant cell tumor of bone. Cancer Cell Int. 2020, 20, 135. [Google Scholar] [CrossRef] [PubMed]
- Fei, F.; Zhang, D.; Yang, Z.; Wang, S.; Wang, X.; Wu, Z.; Wu, Q.; Zhang, S. The number of polyploid giant cancer cells and epithelial-mesenchymal transition-related proteins are associated with invasion and metastasis in human breast cancer. J. Exp. Clin. Cancer Res. 2015, 34, 158. [Google Scholar] [CrossRef] [PubMed]
- Xuan, B.; Ghosh, D.; Cheney, E.M.; Clifton, E.M.; Dawson, M.R. Dysregulation in Actin Cytoskeletal Organization Drives Increased Stiffness and Migratory Persistence in Polyploidal Giant Cancer Cells. Sci. Rep. 2018, 8, 11935. [Google Scholar] [CrossRef]
- Wu, C.C.; Yu, C.T.; Chang, G.C.; Lai, J.M.; Hsu, S.L. Aurora-A promotes gefitinib resistance via a NF-κB signaling pathway in p53 knockdown lung cancer cells. Biochem. Biophys. Res. Commun. 2011, 405, 168–172. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Ma, C.A.; Zhao, Y.; Jain, A. Aurora B interacts with NIR-p53, leading to p53 phosphorylation in its DNA-binding domain and subsequent functional suppression. J. Biol. Chem. 2011, 286, 2236–2244. [Google Scholar] [CrossRef]
- Saddic, L.A.; Wirt, S.; Vogel, H.; Felsher, D.W.; Sage, J. Functional interactions between retinoblastoma and c-MYC in a mouse model of hepatocellular carcinoma. PLoS ONE 2011, 6, e19758. [Google Scholar] [CrossRef]
- Vazquez-Martin, A.; Anatskaya, O.V.; Giuliani, A.; Erenpreisa, J.; Huang, S.; Salmina, K.; Inashkina, I.; Huna, A.; Nikolsky, N.N.; Vinogradov, A.E. Somatic polyploidy is associated with the upregulation of c-MYC interacting genes and EMT-like signature. Oncotarget 2016, 7, 75235–75260. [Google Scholar] [CrossRef]
- Kudo-Saito, C.; Miyamoto, T.; Imazeki, H.; Shoji, H.; Aoki, K.; Boku, N. IL33 Is a Key Driver of Treatment Resistance of Cancer. Cancer Res. 2020, 80, 1981–1990. [Google Scholar] [CrossRef]
- Fang, M.; Li, Y.; Huang, K.; Qi, S.; Zhang, J.; Zgodzinski, W.; Majewski, M.; Wallner, G.; Gozdz, S.; Macek, P.; et al. IL33 Promotes Colon Cancer Cell Stemness via JNK Activation and Macrophage Recruitment. Cancer Res. 2017, 77, 2735–2745. [Google Scholar] [CrossRef]
- Zhao, R.; Yu, Z.; Li, M.; Zhou, Y. Interleukin-33/ST2 Signaling Promotes Hepatocellular Carcinoma Cell Stemness Expansion Through Activating c-Jun N-terminal Kinase Pathway. Am. J. Med. Sci. 2019, 358, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Corse, E.; Gottschalk, R.A.; Allison, J.P. Strength of TCR-peptide/MHC interactions and in vivo T cell responses. J. Immunol. 2011, 186, 5039–5045. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Niu, D.; Lai, L.; Ren, E.C. p53 increases MHC class I expression by upregulating the endoplasmic reticulum aminopeptidase ERAP1. Nat. Commun. 2013, 4, 2359. [Google Scholar] [CrossRef] [PubMed]
- Ropero, S.; Esteller, M. The role of histone deacetylases (HDACs) in human cancer. Mol. Oncol. 2007, 1, 19–25. [Google Scholar] [CrossRef]
- Morrison, B.J.; Steel, J.C.; Morris, J.C. Reduction of MHC-I expression limits T-lymphocyte-mediated killing of Cancer-initiating cells. BMC Cancer 2018, 18, 469. [Google Scholar] [CrossRef]
- Blagih, J.; Zani, F.; Chakravarty, P.; Hennequart, M.; Pilley, S.; Hobor, S.; Hock, A.K.; Walton, J.B.; Morton, J.P.; Gronroos, E.; et al. Cancer-Specific Loss of p53 Leads to a Modulation of Myeloid and T Cell Responses. Cell Rep. 2020, 30, 481–496.e6. [Google Scholar] [CrossRef]
- Blagih, J.; Buck, M.D.; Vousden, K.H. p53, cancer and the immune response. J. Cell Sci. 2020, 133, jcs237453. [Google Scholar] [CrossRef]
- Josefowicz, S.Z.; Lu, L.F.; Rudensky, A.Y. Regulatory T cells: Mechanisms of differentiation and function. Annu. Rev. Immunol. 2012, 30, 531–564. [Google Scholar] [CrossRef]
- Schulke, S. Induction of Interleukin-10 Producing Dendritic Cells as a Tool to Suppress Allergen-Specific T Helper 2 Responses. Front. Immunol. 2018, 9, 455. [Google Scholar] [CrossRef]
- Yang, L.; Dong, Y.; Li, Y.; Wang, D.; Liu, S.; Wang, D.; Gao, Q.; Ji, S.; Chen, X.; Lei, Q.; et al. IL-10 derived from M2 macrophage promotes cancer stemness via JAK1/STAT1/NF-κB/Notch1 pathway in non-small cell lung cancer. Int. J. Cancer 2019, 145, 1099–1110. [Google Scholar] [CrossRef]
- Dong, P.; Xiong, Y.; Yue, J.; Hanley, S.J.B.; Watari, H. Tumor-Intrinsic PD-L1 Signaling in Cancer Initiation, Development and Treatment: Beyond Immune Evasion. Front. Oncol. 2018, 8, 386. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Zhan, H. Communication between EMT and PD-L1 signaling: New insights into tumor immune evasion. Cancer Lett. 2020, 468, 72–81. [Google Scholar] [CrossRef]
- Hao, N.; Whitelaw, M.L. The emerging roles of AhR in physiology and immunity. Biochem. Pharmacol. 2013, 86, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Jayachandran, A.; Anaka, M.; Prithviraj, P.; Hudson, C.; McKeown, S.J.; Lo, P.H.; Vella, L.J.; Goding, C.R.; Cebon, J.; Behren, A. Thrombospondin 1 promotes an aggressive phenotype through epithelial-to-mesenchymal transition in human melanoma. Oncotarget 2014, 5, 5782–5797. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Xu, D.; Liu, Z.; Li, Y.; Zhang, C.; Gong, Y.; Jiang, Y.; Xing, B. THBS1 facilitates colorectal liver metastasis through enhancing epithelial-mesenchymal transition. Clin. Transl. Oncol. 2020, 22, 1730–1740. [Google Scholar] [CrossRef] [PubMed]
- Kudo-Saito, C.; Shirako, H.; Takeuchi, T.; Kawakami, Y. Cancer metastasis is accelerated through immunosuppression during Snail-induced EMT of cancer cells. Cancer Cell 2009, 15, 195–206. [Google Scholar] [CrossRef]
- Wilson, D.C.; Marinov, A.D.; Blair, H.C.; Bushnell, D.S.; Thompson, S.D.; Chaly, Y.; Hirsch, R. Follistatin-Like protein 1 is a mesenchyme-derived inflammatory protein and may represent a biomarker for systemic-onset juvenile rheumatoid arthritis. Arthritis Rheum. 2010, 62, 2510–2516. [Google Scholar] [CrossRef]
- Li, D.; Wang, Y.; Xu, N.; Wei, Q.; Wu, M.; Li, X.; Zheng, P.; Sun, S.; Jin, Y.; Zhang, G.; et al. Follistatin-Like protein 1 is elevated in systemic autoimmune diseases and correlated with disease activity in patients with rheumatoid arthritis. Arthritis Res. Ther. 2011, 13, R17. [Google Scholar] [CrossRef]
- Fan, N.; Sun, H.; Wang, Y.; Wang, Y.; Zhang, L.; Xia, Z.; Peng, L.; Hou, Y.; Shen, W.; Liu, R.; et al. Follistatin-Like 1: A potential mediator of inflammation in obesity. Mediators Inflamm. 2013, 2013, 752519. [Google Scholar] [CrossRef]
- Mattiotti, A.; Prakash, S.; Barnett, P.; van den Hoff, M.J.B. Follistatin-Like 1 in development and human diseases. Cell Mol. Life Sci. 2018, 75, 2339–2354. [Google Scholar] [CrossRef]
- Lim, S.Y.; Yuzhalin, A.E.; Gordon-Weeks, A.N.; Muschel, R.J. Targeting the CCL2-CCR2 signaling axis in cancer metastasis. Oncotarget 2016, 7, 28697–28710. [Google Scholar] [CrossRef] [PubMed]
- Acharyya, S.; Oskarsson, T.; Vanharanta, S.; Malladi, S.; Kim, J.; Morris, P.G.; Manova-Todorova, K.; Leversha, M.; Hogg, N.; Seshan, V.E.; et al. A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell 2012, 150, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Taki, M.; Abiko, K.; Baba, T.; Hamanishi, J.; Yamaguchi, K.; Murakami, R.; Yamanoi, K.; Horikawa, N.; Hosoe, Y.; Nakamura, E.; et al. Snail promotes ovarian cancer progression by recruiting myeloid-derived suppressor cells via CXCR2 ligand upregulation. Nat. Commun. 2018, 9, 1685. [Google Scholar] [CrossRef] [PubMed]
- Paluskievicz, C.M.; Cao, X.; Abdi, R.; Zheng, P.; Liu, Y.; Bromberg, J.S. T Regulatory Cells and Priming the Suppressive Tumor Microenvironment. Front. Immunol. 2019, 10, 2453. [Google Scholar] [CrossRef] [PubMed]
- Walunas, T.L.; Lenschow, D.J.; Bakker, C.Y.; Linsley, P.S.; Freeman, G.J.; Green, J.M.; Thompson, C.B.; Bluestone, J.A. CTLA-4 can function as a negative regulator of T cell activation. Immunity 1994, 1, 405–413. [Google Scholar] [CrossRef]
- Krummel, M.F.; Allison, J.P. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J. Exp. Med. 1995, 182, 459–465. [Google Scholar] [CrossRef]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef]
- Chemnitz, J.M.; Parry, R.V.; Nichols, K.E.; June, C.H.; Riley, J.L. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J. Immunol. 2004, 173, 945–954. [Google Scholar] [CrossRef]
- Chen, X.; Du, Y.; Lin, X.; Qian, Y.; Zhou, T.; Huang, Z. CD4 + CD25 + regulatory T cells in tumor immunity. Int. Immunopharmacol. 2016, 34, 244–249. [Google Scholar] [CrossRef]
- Xue, W.; Yan, D.; Kan, Q. Interleukin-35 as an Emerging Player in Tumor Microenvironment. J. Cancer 2019, 10, 2074–2082. [Google Scholar] [CrossRef]
- Turnis, M.E.; Sawant, D.V.; Szymczak-Workman, A.L.; Andrews, L.P.; Delgoffe, G.M.; Yano, H.; Beres, A.J.; Vogel, P.; Workman, C.J.; Vignali, D.A. Interleukin-35 Limits Anti-Tumor Immunity. Immunity 2016, 44, 316–329. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I. Myeloid-Derived Suppressor Cells. Cancer Immunol. Res. 2017, 5, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wei, G.; Cheng, W.A.; Dong, Z.; Sun, H.; Lee, V.Y.; Cha, S.C.; Smith, D.L.; Kwak, L.W.; Qin, H. Targeting myeloid-derived suppressor cells for cancer immunotherapy. Cancer Immunol. Immunother. 2018, 67, 1181–1195. [Google Scholar] [CrossRef]
- Schwartz, M.; Zhang, Y.; Rosenblatt, J.D. B cell regulation of the anti-tumor response and role in carcinogenesis. J. Immunother. Cancer 2016, 4, 40. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Xia, L.; Jia, D.; Zou, H.; Jin, G.; Qian, W.; Xu, H.; Li, T. PD-L1+ regulatory B cells act as a T cell suppressor in a PD-L1-dependent manner in melanoma patients with bone metastasis. Mol. Immunol. 2020, 119, 83–91. [Google Scholar] [CrossRef]
- Choi, J.K.; Egwuagu, C.E. Interleukin 35 Regulatory B Cells. J. Mol. Biol. 2020, 433, 166607. [Google Scholar] [CrossRef]
- Regulski, M.J. Mesenchymal Stem Cells: “Guardians of Inflammation”. Wounds 2017, 29, 20–27. [Google Scholar]
- English, K. Mechanisms of mesenchymal stromal cell immunomodulation. Immunol. Cell Biol. 2013, 91, 19–26. [Google Scholar] [CrossRef]
- Kudo-Saito, C. Cancer-Associated mesenchymal stem cells aggravate tumor progression. Front. Cell Dev. Biol. 2015, 3, 23. [Google Scholar] [CrossRef]
- Ridge, S.M.; Sullivan, F.J.; Glynn, S.A. Mesenchymal stem cells: Key players in cancer progression. Mol. Cancer 2017, 16, 31. [Google Scholar] [CrossRef]
- Ercolano, G.; Falquet, M.; Vanoni, G.; Trabanelli, S.; Jandus, C. ILC2s: New Actors in Tumor Immunity. Front. Immunol. 2019, 10, 2801. [Google Scholar] [CrossRef] [PubMed]
- Schuijs, M.J.; Png, S.; Richard, A.C.; Tsyben, A.; Hamm, G.; Stockis, J.; Garcia, C.; Pinaud, S.; Nicholls, A.; Ros, X.R.; et al. ILC2-driven innate immune checkpoint mechanism antagonizes NK cell antimetastatic function in the lung. Nat. Immunol. 2020, 21, 998–1009. [Google Scholar] [CrossRef] [PubMed]
- Knochelmann, H.M.; Dwyer, C.J.; Bailey, S.R.; Amaya, S.M.; Elston, D.M.; Mazza-McCrann, J.M.; Paulos, C.M. When worlds collide: Th17 and Treg cells in cancer and autoimmunity. Cell Mol. Immunol. 2018, 15, 458–469. [Google Scholar] [CrossRef] [PubMed]
- Golebski, K.; Ros, X.R.; Nagasawa, M.; van Tol, S.; Heesters, B.A.; Aglmous, H.; Kradolfer, C.M.A.; Shikhagaie, M.M.; Seys, S.; Hellings, P.W.; et al. IL-1β, IL-23, and TGF-beta drive plasticity of human ILC2s towards IL-17-producing ILCs in nasal inflammation. Nat. Commun. 2019, 10, 2162. [Google Scholar] [CrossRef]
- Johansson, A.; Hamzah, J.; Ganss, R. More than a scaffold: Stromal modulation of tumor immunity. Biochim. Biophys. Acta 2016, 1865, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Du, L.; Lin, L.; Wang, Y. Tumour-Associated mesenchymal stem/stromal cells: Emerging therapeutic targets. Nat. Rev. Drug Discov. 2017, 16, 35–52. [Google Scholar] [CrossRef]
- Guo, S.; Deng, C.X. Effect of Stromal Cells in Tumor Microenvironment on Metastasis Initiation. Int. J. Biol. Sci. 2018, 14, 2083–2093. [Google Scholar] [CrossRef]
- Kwa, M.Q.; Herum, K.M.; Brakebusch, C. Cancer-Associated fibroblasts: How do they contribute to metastasis? Clin. Exp. Metastasis 2019, 36, 71–86. [Google Scholar] [CrossRef]
- Varricchi, G.; Galdiero, M.R.; Loffredo, S.; Marone, G.; Iannone, R.; Marone, G.; Granata, F. Are Mast Cells MASTers in Cancer? Front. Immunol. 2017, 8, 424. [Google Scholar] [CrossRef]
- Varricchi, G.; de Paulis, A.; Marone, G.; Galli, S.J. Future Needs in Mast Cell Biology. Int. J. Mol. Sci. 2019, 20, 4397. [Google Scholar] [CrossRef]
- Pirtskhalaishvili, G.; Nelson, J.B. Endothelium-Derived factors as paracrine mediators of prostate cancer progression. Prostate 2000, 44, 77–87. [Google Scholar] [CrossRef]
- Butler, J.M.; Kobayashi, H.; Rafii, S. Instructive role of the vascular niche in promoting tumour growth and tissue repair by angiocrine factors. Nat. Rev. Cancer 2010, 10, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Melincovici, C.S.; Bosca, A.B.; Susman, S.; Marginean, M.; Mihu, C.; Istrate, M.; Moldovan, I.M.; Roman, A.L.; Mihu, C.M. Vascular endothelial growth factor (VEGF)—Key factor in normal and pathological angiogenesis. Rom. J. Morphol. Embryol. 2018, 59, 455–467. [Google Scholar] [PubMed]
- Zhou, Z.; Yan, F.; Liu, O. Interleukin (IL)-33: An orchestrator of immunity from host defence to tissue homeostasis. Clin. Transl. Immunol. 2020, 9, e1146. [Google Scholar] [CrossRef] [PubMed]
- Paiva, A.E.; Lousado, L.; Guerra, D.A.P.; Azevedo, P.O.; Sena, I.F.G.; Andreotti, J.P.; Santos, G.S.P.; Goncalves, R.; Mintz, A.; Birbrair, A. Pericytes in the Premetastatic Niche. Cancer Res. 2018, 78, 2779–2786. [Google Scholar] [CrossRef]
- Pieterse, Z.; Sinha, D.; Kaur, P. Pericytes in Metastasis. Adv. Exp. Med. Biol. 2019, 1147, 125–135. [Google Scholar]
- Domev, H.; Milkov, I.; Itskovitz-Eldor, J.; Dar, A. Immunoevasive pericytes from human pluripotent stem cells preferentially modulate induction of allogeneic regulatory T cells. Stem Cells Transl. Med. 2014, 3, 1169–1181. [Google Scholar] [CrossRef]
- Shalapour, S.; Karin, M. Immunity, inflammation, and cancer: An eternal fight between good and evil. J. Clin. Investig. 2015, 125, 3347–3355. [Google Scholar] [CrossRef]
- Pauken, K.E.; Wherry, E.J. Overcoming T cell exhaustion in infection and cancer. Trends Immunol. 2015, 36, 265–276. [Google Scholar] [CrossRef]
- Arasanz, H.; Gato-Canas, M.; Zuazo, M.; Ibanez-Vea, M.; Breckpot, K.; Kochan, G.; Escors, D. PD1 signal transduction pathways in T cells. Oncotarget 2017, 8, 51936–51945. [Google Scholar] [CrossRef]
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, Tim-3, and TIGIT: Co-Inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity 2016, 44, 989–1004. [Google Scholar] [CrossRef] [PubMed]
- Stanietsky, N.; Simic, H.; Arapovic, J.; Toporik, A.; Levy, O.; Novik, A.; Levine, Z.; Beiman, M.; Dassa, L.; Achdout, H.; et al. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 17858–17863. [Google Scholar] [CrossRef] [PubMed]
- Judge, S.J.; Murphy, W.J.; Canter, R.J. Characterizing the Dysfunctional NK Cell: Assessing the Clinical Relevance of Exhaustion, Anergy, and Senescence. Front. Cell Infect. Microbiol. 2020, 10, 49. [Google Scholar] [CrossRef]
- Khan, O.; Giles, J.R.; McDonald, S.; Manne, S.; Ngiow, S.F.; Patel, K.P.; Werner, M.T.; Huang, A.C.; Alexander, K.A.; Wu, J.E.; et al. TOX transcriptionally and epigenetically programs CD8+ T cell exhaustion. Nature 2019, 571, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Park, S.; Park, S.Y.; Kim, G.; Park, S.M.; Cho, J.W.; Kim, D.H.; Park, Y.M.; Koh, Y.W.; Kim, H.R.; et al. Single-Cell transcriptome analysis reveals TOX as a promoting factor for T cell exhaustion and a predictor for anti-PD-1 responses in human cancer. Genome Med. 2020, 12, 22. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; He, Q.; Shen, H.; Xia, A.; Tian, W.; Yu, W.; Sun, B. TOX promotes the exhaustion of antitumor CD8+ T cells by preventing PD1 degradation in hepatocellular carcinoma. J. Hepatol. 2019, 71, 731–741. [Google Scholar] [CrossRef]
- Hudson, W.H.; Gensheimer, J.; Hashimoto, M.; Wieland, A.; Valanparambil, R.M.; Li, P.; Lin, J.X.; Konieczny, B.T.; Im, S.J.; Freeman, G.J.; et al. Proliferating Transitory T Cells with an Effector-like Transcriptional Signature Emerge from PD-1+ Stem-like CD8+ T Cells during Chronic Infection. Immunity 2019, 51, 1043–1058.e4. [Google Scholar] [CrossRef]
- Stoletov, K.; Beatty, P.H.; Lewis, J.D. Novel therapeutic targets for cancer metastasis. Expert Rev. Anticancer Ther. 2020, 20, 97–109. [Google Scholar] [CrossRef]
- Cho, E.S.; Kang, H.E.; Kim, N.H.; Yook, J.I. Therapeutic implications of cancer epithelial-mesenchymal transition (EMT). Arch. Pharm. Res. 2019, 42, 14–24. [Google Scholar] [CrossRef]
- Jimeno, A.; Gordon, M.; Chugh, R.; Messersmith, W.; Mendelson, D.; Dupont, J.; Stagg, R.; Kapoun, A.M.; Xu, L.; Uttamsingh, S.; et al. A First-in-Human Phase I Study of the Anticancer Stem Cell Agent Ipafricept (OMP-54F28), a Decoy Receptor for Wnt Ligands, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2017, 23, 7490–7497. [Google Scholar] [CrossRef]
- Smith, D.C.; Chugh, R.; Patnaik, A.; Papadopoulos, K.P.; Wang, M.; Kapoun, A.M.; Xu, L.; Dupont, J.; Stagg, R.J.; Tolcher, A. A phase 1 dose escalation and expansion study of Tarextumab (OMP-59R5) in patients with solid tumors. Investig. New Drugs 2019, 37, 722–730. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.I.; Bendell, J.C.; Bullock, A.; LoConte, N.K.; Hatoum, H.; Ritch, P.; Hool, H.; Leach, J.W.; Sanchez, J.; Sohal, D.P.S.; et al. A randomized phase II trial of nab-paclitaxel and gemcitabine with tarextumab or placebo in patients with untreated metastatic pancreatic cancer. Cancer Med. 2019, 8, 5148–5157. [Google Scholar] [CrossRef] [PubMed]
- Cook, N.; Basu, B.; Smith, D.M.; Gopinathan, A.; Evans, J.; Steward, W.P.; Palmer, D.; Propper, D.; Venugopal, B.; Hategan, M.; et al. A phase I trial of the gamma-secretase inhibitor MK-0752 in combination with gemcitabine in patients with pancreatic ductal adenocarcinoma. Br. J. Cancer 2018, 118, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Gavai, A.V.; Quesnelle, C.; Norris, D.; Han, W.C.; Gill, P.; Shan, W.; Balog, A.; Chen, K.; Tebben, A.; Rampulla, R.; et al. Discovery of Clinical Candidate BMS-906024: A Potent Pan-Notch Inhibitor for the Treatment of Leukemia and Solid Tumors. ACS Med. Chem. Lett. 2015, 6, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Massard, C.; Azaro, A.; Soria, J.C.; Lassen, U.; Le Tourneau, C.; Sarker, D.; Smith, C.; Ohnmacht, U.; Oakley, G.; Patel, B.K.R.; et al. First-in-Human study of LY3039478, an oral Notch signaling inhibitor in advanced or metastatic cancer. Ann. Oncol. 2018, 29, 1911–1917. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Shimizu, F.; Hovinga, K.; Beal, K.; Karimi, S.; Droms, L.; Peck, K.K.; Gutin, P.; Iorgulescu, J.B.; Kaley, T.; et al. Molecular and Clinical Effects of Notch Inhibition in Glioma Patients: A Phase 0/I Trial. Clin. Cancer Res. 2016, 22, 4786–4796. [Google Scholar] [CrossRef]
- Pant, S.; Jones, S.F.; Kurkjian, C.D.; Infante, J.R.; Moore, K.N.; Burris, H.A.; McMeekin, D.S.; Benhadji, K.A.; Patel, B.K.R.; Frenzel, M.J.; et al. A first-in-Human phase I study of the oral Notch inhibitor, LY900009, in patients with advanced cancer. Eur. J. Cancer 2016, 56, 1–9. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Zhang, S.; Gong, Z.; Li, X.; Cao, K.; Deng, H.; He, Y.; et al. The role of microenvironment in tumor angiogenesis. J. Exp. Clin. Cancer Res. 2020, 39, 204. [Google Scholar] [CrossRef]
- Lugano, R.; Ramachandran, M.; Dimberg, A. Tumor angiogenesis: Causes, consequences, challenges and opportunities. Cell Mol. Life Sci. 2020, 77, 1745–1770. [Google Scholar] [CrossRef]
- Jiao, Q.; Bi, L.; Ren, Y.; Song, S.; Wang, Q.; Wang, Y.S. Advances in studies of tyrosine kinase inhibitors and their acquired resistance. Mol. Cancer 2018, 17, 36. [Google Scholar] [CrossRef]
- Huang, L.; Jiang, S.; Shi, Y. Tyrosine kinase inhibitors for solid tumors in the past 20 years (2001–2020). J. Hematol. Oncol. 2020, 13, 143. [Google Scholar] [CrossRef] [PubMed]
- Facchinetti, F.; Hollebecque, A.; Bahleda, R.; Loriot, Y.; Olaussen, K.A.; Massard, C.; Friboulet, L. Facts and New Hopes on Selective FGFR Inhibitors in Solid Tumors. Clin. Cancer Res. 2020, 26, 764–774. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.G.; Zhu, L.C.; Wang, Y.J.; Li, Y.Y.; Wang, D. Current Advance of Therapeutic Agents in Clinical Trials Potentially Targeting Tumor Plasticity. Front. Oncol. 2019, 9, 887. [Google Scholar] [CrossRef]
- Brandes, A.A.; Carpentier, A.F.; Kesari, S.; Sepulveda-Sanchez, J.M.; Wheeler, H.R.; Chinot, O.; Cher, L.; Steinbach, J.P.; Capper, D.; Specenier, P.; et al. A Phase II randomized study of galunisertib monotherapy or galunisertib plus lomustine compared with lomustine monotherapy in patients with recurrent glioblastoma. Neuro Oncol. 2016, 18, 1146–1156. [Google Scholar] [CrossRef] [PubMed]
- Kelley, R.K.; Gane, E.; Assenat, E.; Siebler, J.; Galle, P.R.; Merle, P.; Hourmand, I.O.; Cleverly, A.; Zhao, Y.; Gueorguieva, I.; et al. A Phase 2 Study of Galunisertib (TGF-β1 Receptor Type I Inhibitor) and Sorafenib in Patients With Advanced Hepatocellular Carcinoma. Clin. Transl. Gastroenterol. 2019, 10, e00056. [Google Scholar] [CrossRef] [PubMed]
- Principe, D.R.; Park, A.; Dorman, M.J.; Kumar, S.; Viswakarma, N.; Rubin, J.; Torres, C.; McKinney, R.; Munshi, H.G.; Grippo, P.J.; et al. TGFβ Blockade Augments PD-1 Inhibition to Promote T-Cell-Mediated Regression of Pancreatic Cancer. Mol. Cancer Ther. 2019, 18, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Jochems, C.; Tritsch, S.R.; Pellom, S.T.; Su, Z.; Soon-Shiong, P.; Wong, H.C.; Gulley, J.L.; Schlom, J. Analyses of functions of an anti-PD-L1/TGFβR2 bispecific fusion protein (M7824). Oncotarget 2017, 8, 75217–75231. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.M.T.; Dancsok, A.R.; Nielsen, T.O. Indoleamine Dioxygenase Inhibitors: Clinical Rationale and Current Development. Curr. Oncol. Rep. 2019, 21, 2. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Dummer, R.; Hamid, O.; Gajewski, T.F.; Caglevic, C.; Dalle, S.; Arance, A.; Carlino, M.S.; Grob, J.J.; Kim, T.M.; et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): A phase 3, randomised, double-blind study. Lancet Oncol. 2019, 20, 1083–1097. [Google Scholar] [CrossRef]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in cancer: Mechanisms and advances in clinical trials. Mol. Cancer 2019, 18, 26. [Google Scholar] [CrossRef]
- Qin, G.; Xu, F.; Qin, T.; Zheng, Q.; Shi, D.; Xia, W.; Tian, Y.; Tang, Y.; Wang, J.; Xiao, X.; et al. Palbociclib inhibits epithelial-mesenchymal transition and metastasis in breast cancer via c-Jun/COX-2 signaling pathway. Oncotarget 2015, 6, 41794–41808. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Wang, Y.; Lin, Y.; Liu, Y.; Wang, Y.; Jia, J.; Singh, P.; Chi, Y.I.; Wang, C.; Dong, C.; et al. Dub3 inhibition suppresses breast cancer invasion and metastasis by promoting Snail1 degradation. Nat. Commun. 2017, 8, 14228. [Google Scholar] [CrossRef] [PubMed]
- Goel, S.; DeCristo, M.J.; McAllister, S.S.; Zhao, J.J. CDK4/6 Inhibition in Cancer: Beyond Cell Cycle Arrest. Trends Cell Biol. 2018, 28, 911–925. [Google Scholar] [CrossRef] [PubMed]
- Kozyreva, V.K.; Kiseleva, A.A.; Ice, R.J.; Jones, B.C.; Loskutov, Y.V.; Matalkah, F.; Smolkin, M.B.; Marinak, K.; Livengood, R.H.; Salkeni, M.A.; et al. Combination of Eribulin and Aurora a Inhibitor MLN8237 Prevents Metastatic Colonization and Induces Cytotoxic Autophagy in Breast Cancer. Mol. Cancer Ther. 2016, 15, 1809–1822. [Google Scholar] [CrossRef] [PubMed]
- Bavetsias, V.; Linardopoulos, S. Aurora Kinase Inhibitors: Current Status and Outlook. Front. Oncol. 2015, 5, 278. [Google Scholar] [CrossRef]
- Vela, M.; Aris, M.; Llorente, M.; Garcia-Sanz, J.A.; Kremer, L. Chemokine receptor-specific antibodies in cancer immunotherapy: Achievements and challenges. Front. Immunol. 2015, 6, 12. [Google Scholar] [CrossRef]
- Miao, M.; De Clercq, E.; Li, G. Clinical significance of chemokine receptor antagonists. Expert Opin. Drug Metab. Toxicol. 2020, 16, 11–30. [Google Scholar] [CrossRef]
- Havel, J.J.; Chowell, D.; Chan, T.A. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat. Rev. Cancer 2019, 19, 133–150. [Google Scholar] [CrossRef]
- Park, Y.; Koh, J.; Na, H.Y.; Kwak, Y.; Lee, K.W.; Ahn, S.H.; Park, D.J.; Kim, H.H.; Lee, H.S. PD-L1 Testing in Gastric Cancer by the Combined Positive Score of the 22C3 PharmDx and SP263 Assay with Clinically Relevant Cut-offs. Cancer Res. Treat. 2020, 52, 661–670. [Google Scholar] [CrossRef]
- Xin Yu, J.; Hubbard-Lucey, V.M.; Tang, J. Immuno-Oncology drug development goes global. Nat. Rev. Drug Discov. 2019, 18, 899–900. [Google Scholar] [CrossRef]
- Schenk, K.M.; Reuss, J.E.; Choquette, K.; Spira, A.I. A review of canakinumab and its therapeutic potential for non-small cell lung cancer. Anticancer Drugs 2019, 30, 879–885. [Google Scholar] [CrossRef] [PubMed]
- Rossi, J.F.; Lu, Z.Y.; Jourdan, M.; Klein, B. Interleukin-6 as a therapeutic target. Clin. Cancer Res. 2015, 21, 1248–1257. [Google Scholar] [CrossRef] [PubMed]
- Bilusic, M.; Heery, C.R.; Collins, J.M.; Donahue, R.N.; Palena, C.; Madan, R.A.; Karzai, F.; Marte, J.L.; Strauss, J.; Gatti-Mays, M.E.; et al. Phase I trial of HuMax-IL8 (BMS-986253), an anti-IL-8 monoclonal antibody, in patients with metastatic or unresectable solid tumors. J. Immunother. Cancer 2019, 7, 240. [Google Scholar] [CrossRef] [PubMed]
- Stasinopoulos, I.; Shah, T.; Penet, M.F.; Krishnamachary, B.; Bhujwalla, Z.M. COX-2 in cancer: Gordian knot or Achilles heel? Front. Pharmacol. 2013, 4, 34. [Google Scholar] [CrossRef] [PubMed]
- Lucotti, S.; Cerutti, C.; Soyer, M.; Gil-Bernabe, A.M.; Gomes, A.L.; Allen, P.D.; Smart, S.; Markelc, B.; Watson, K.; Armstrong, P.C.; et al. Aspirin blocks formation of metastatic intravascular niches by inhibiting platelet-derived COX-1/thromboxane A2. J. Clin. Invest. 2019, 129, 1845–1862. [Google Scholar] [CrossRef] [PubMed]
- Tougeron, D.; Sha, D.; Manthravadi, S.; Sinicrope, F.A. Aspirin and colorectal cancer: Back to the future. Clin. Cancer Res. 2014, 20, 1087–1094. [Google Scholar] [CrossRef] [PubMed]
- Hamada, T.; Cao, Y.; Qian, Z.R.; Masugi, Y.; Nowak, J.A.; Yang, J.; Song, M.; Mima, K.; Kosumi, K.; Liu, L.; et al. Aspirin Use and Colorectal Cancer Survival According to Tumor CD274 (Programmed Cell Death 1 Ligand 1) Expression Status. J. Clin. Oncol. 2017, 35, 1836–1844. [Google Scholar] [CrossRef]
- Jin, M.Z.; Jin, W.L. The updated landscape of tumor microenvironment and drug repurposing. Signal Transduct. Target Ther. 2020, 5, 166. [Google Scholar] [CrossRef]
- Houssaini, M.S.; Damou, M.; Ismaili, N. Advances in the management of non-small cell lung cancer (NSCLC): A new practice changing data from asco 2020 annual meeting. Cancer Treat. Res. Commun. 2020, 25, 100239. [Google Scholar] [CrossRef]
- Peng, M.; Mo, Y.; Wang, Y.; Wu, P.; Zhang, Y.; Xiong, F.; Guo, C.; Wu, X.; Li, Y.; Li, X.; et al. Neoantigen vaccine: An emerging tumor immunotherapy. Mol. Cancer 2019, 18, 128. [Google Scholar] [CrossRef]
- Aldous, A.R.; Dong, J.Z. Personalized neoantigen vaccines: A new approach to cancer immunotherapy. Bioorg. Med. Chem. 2018, 26, 2842–2849. [Google Scholar] [CrossRef] [PubMed]
- McClure, J.J.; Li, X.; Chou, C.J. Advances and Challenges of HDAC Inhibitors in Cancer Therapeutics. Adv. Cancer Res. 2018, 138, 183–211. [Google Scholar]
- Wawruszak, A.; Kalafut, J.; Okon, E.; Czapinski, J.; Halasa, M.; Przybyszewska, A.; Miziak, P.; Okla, K.; Rivero-Muller, A.; Stepulak, A. Histone Deacetylase Inhibitors and Phenotypical Transformation of Cancer Cells. Cancers 2019, 11, 148. [Google Scholar] [CrossRef] [PubMed]
- Woods, D.M.; Sodre, A.L.; Villagra, A.; Sarnaik, A.; Sotomayor, E.M.; Weber, J. HDAC Inhibition Upregulates PD-1 Ligands in Melanoma and Augments Immunotherapy with PD-1 Blockade. Cancer Immunol. Res. 2015, 3, 1375–1385. [Google Scholar] [CrossRef]
- Martinez, M.; Moon, E.K. CAR T Cells for Solid Tumors: New Strategies for Finding, Infiltrating, and Surviving in the Tumor Microenvironment. Front. Immunol. 2019, 10, 128. [Google Scholar] [CrossRef]
- Beyar-Katz, O.; Gill, S. Advances in chimeric antigen receptor T cells. Curr. Opin. Hematol. 2020, 27, 368–377. [Google Scholar] [CrossRef] [PubMed]
- Dhar, P.; Wu, J.D. NKG2D and its ligands in cancer. Curr. Opin. Immunol. 2018, 51, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Baumeister, S.H.; Murad, J.; Werner, L.; Daley, H.; Trebeden-Negre, H.; Gicobi, J.K.; Schmucker, A.; Reder, J.; Sentman, C.L.; Gilham, D.E.; et al. Phase I Trial of Autologous CAR T Cells Targeting NKG2D Ligands in Patients with AML/MDS and Multiple Myeloma. Cancer Immunol. Res. 2019, 7, 100–112. [Google Scholar] [CrossRef]
- Luna, J.I.; Grossenbacher, S.K.; Murphy, W.J.; Canter, R.J. Targeting Cancer Stem Cells with Natural Killer Cell Immunotherapy. Expert Opin. Biol. Ther. 2017, 17, 313–324. [Google Scholar] [CrossRef]
- Dianat-Moghadam, H.; Mahari, A.; Heidarifard, M.; Parnianfard, N.; Pourmousavi-Kh, L.; Rahbarghazi, R.; Amoozgar, Z. NK cells-directed therapies target circulating tumor cells and metastasis. Cancer Lett. 2021, 497, 41–53. [Google Scholar] [CrossRef]
- Wang, W.; Jiang, J.; Wu, C. CAR-NK for tumor immunotherapy: Clinical transformation and future prospects. Cancer Lett. 2020, 472, 175–180. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kudo-Saito, C.; Ozaki, Y.; Imazeki, H.; Hayashi, H.; Masuda, J.; Ozawa, H.; Ogiwara, Y. Targeting Oncoimmune Drivers of Cancer Metastasis. Cancers 2021, 13, 554. https://doi.org/10.3390/cancers13030554
Kudo-Saito C, Ozaki Y, Imazeki H, Hayashi H, Masuda J, Ozawa H, Ogiwara Y. Targeting Oncoimmune Drivers of Cancer Metastasis. Cancers. 2021; 13(3):554. https://doi.org/10.3390/cancers13030554
Chicago/Turabian StyleKudo-Saito, Chie, Yukinori Ozaki, Hiroshi Imazeki, Hideyuki Hayashi, Jun Masuda, Hiroki Ozawa, and Yamato Ogiwara. 2021. "Targeting Oncoimmune Drivers of Cancer Metastasis" Cancers 13, no. 3: 554. https://doi.org/10.3390/cancers13030554
APA StyleKudo-Saito, C., Ozaki, Y., Imazeki, H., Hayashi, H., Masuda, J., Ozawa, H., & Ogiwara, Y. (2021). Targeting Oncoimmune Drivers of Cancer Metastasis. Cancers, 13(3), 554. https://doi.org/10.3390/cancers13030554