
DPP4 Inhibitor Sitagliptin Enhances Lymphocyte Recruitment and Prolongs Survival in a Syngeneic Ovarian Cancer Mouse Model

 , , ,
, , ,  ,
, 
 and
and 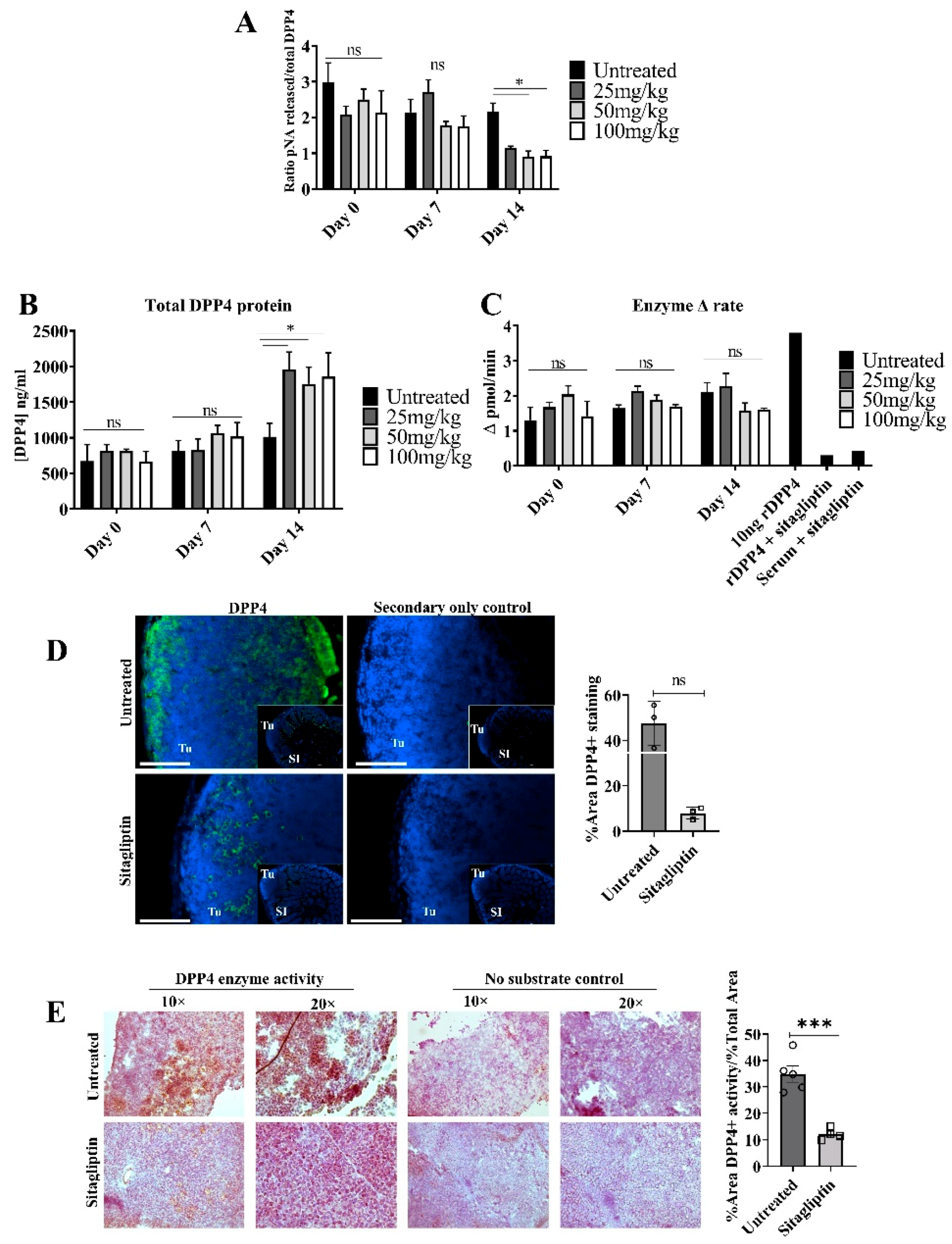{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Methods
2.1. Cell Culture
2.2. Animal Experiments
2.3. Intrabursal Implantation of ID8 pROSA-iRFP720 Tumours
2.4. In Vivo Fluorescence Imaging
2.5. Measurement of Plasma DPP4 Enzyme Activity
2.6. In Situ Staining of DPP4 Activity
2.7. Flow Cytometry
2.8. Tissue Immunofluorescent Staining
2.9. Chemokine Luminex Assay
2.10. Statistical Analysis
3. Results
3.1. Sitagliptin Alters DPP4 Activity and Localisation in Tumour Tissue
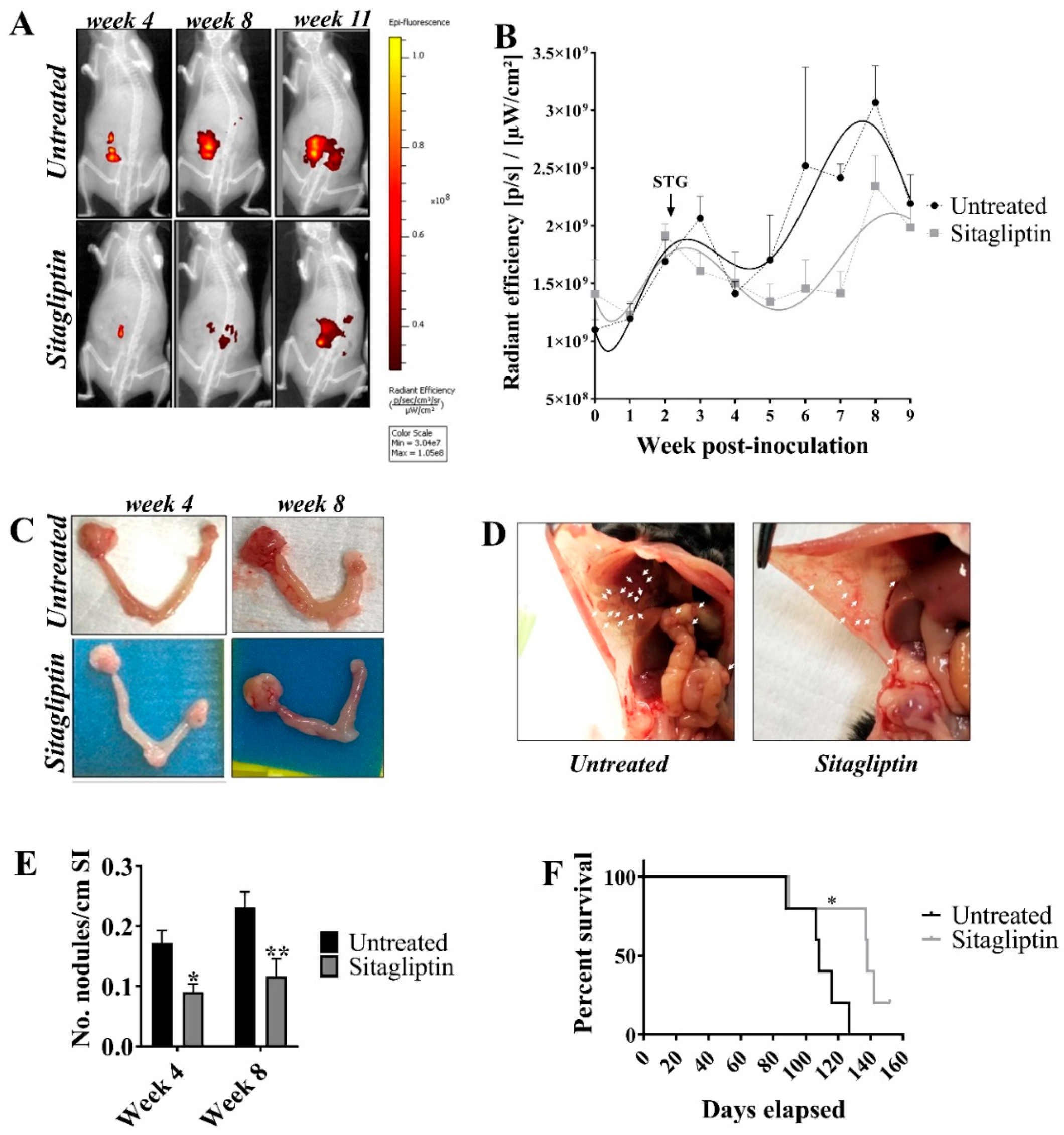
3.2. Sitagliptin Reduces Overall Tumour Burden and Prolongs Survival
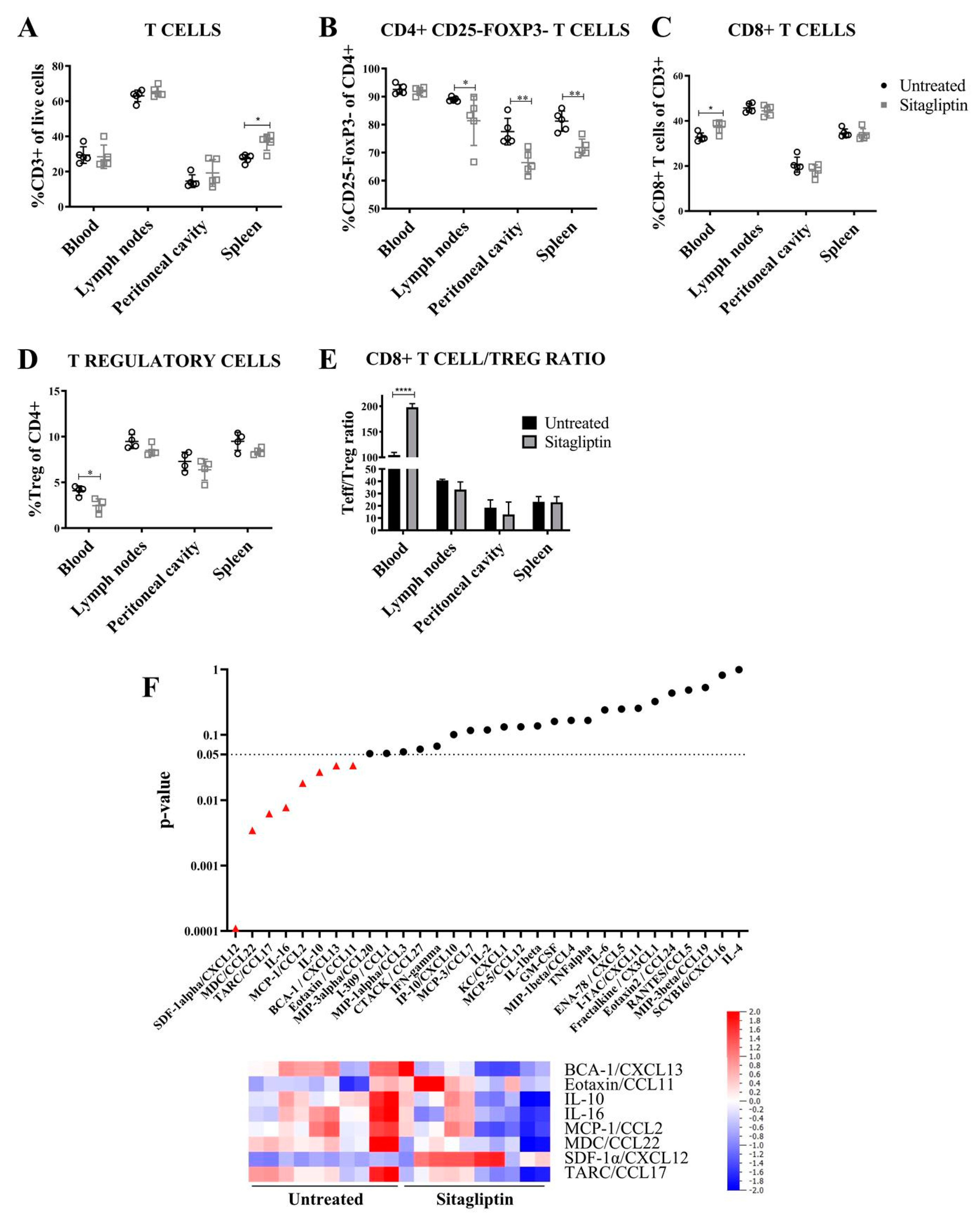
3.3. Sitagliptin Alters the Immune Landscape During Early-Stage Disease
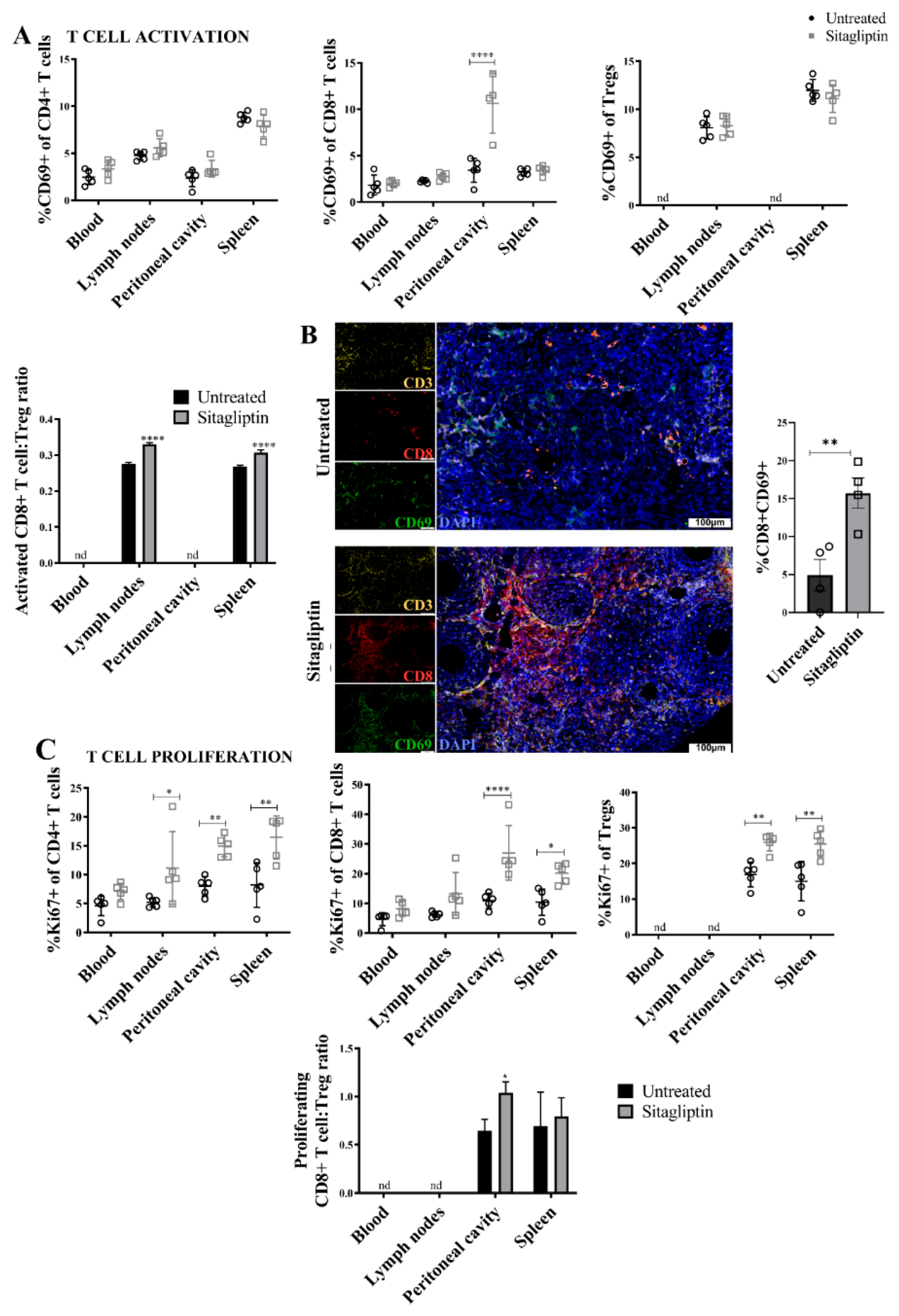
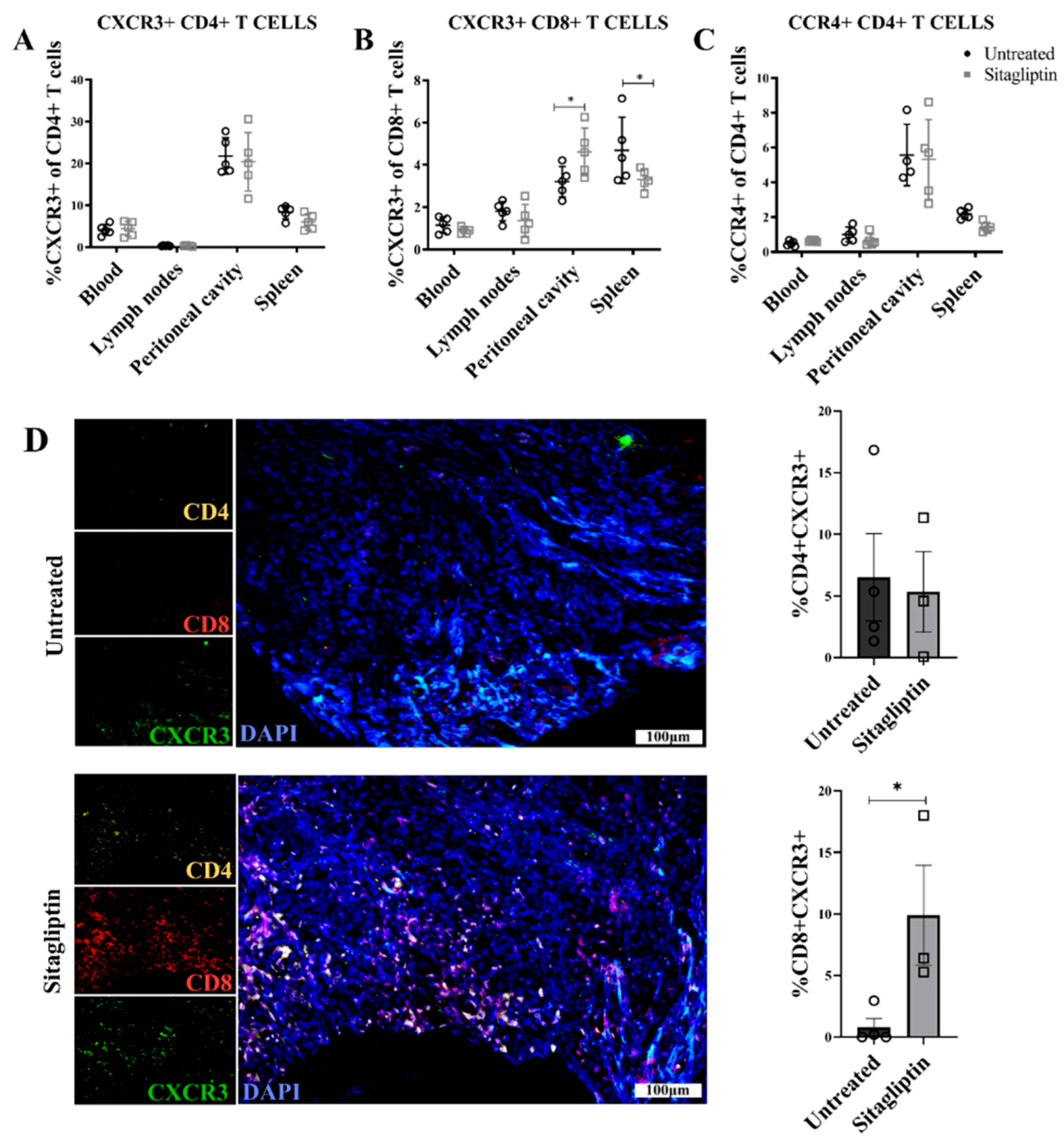
3.4. Sitagliptin Induces Activation and Proliferation of Peripheral and Intra-Tumoral CD8+ T Cells
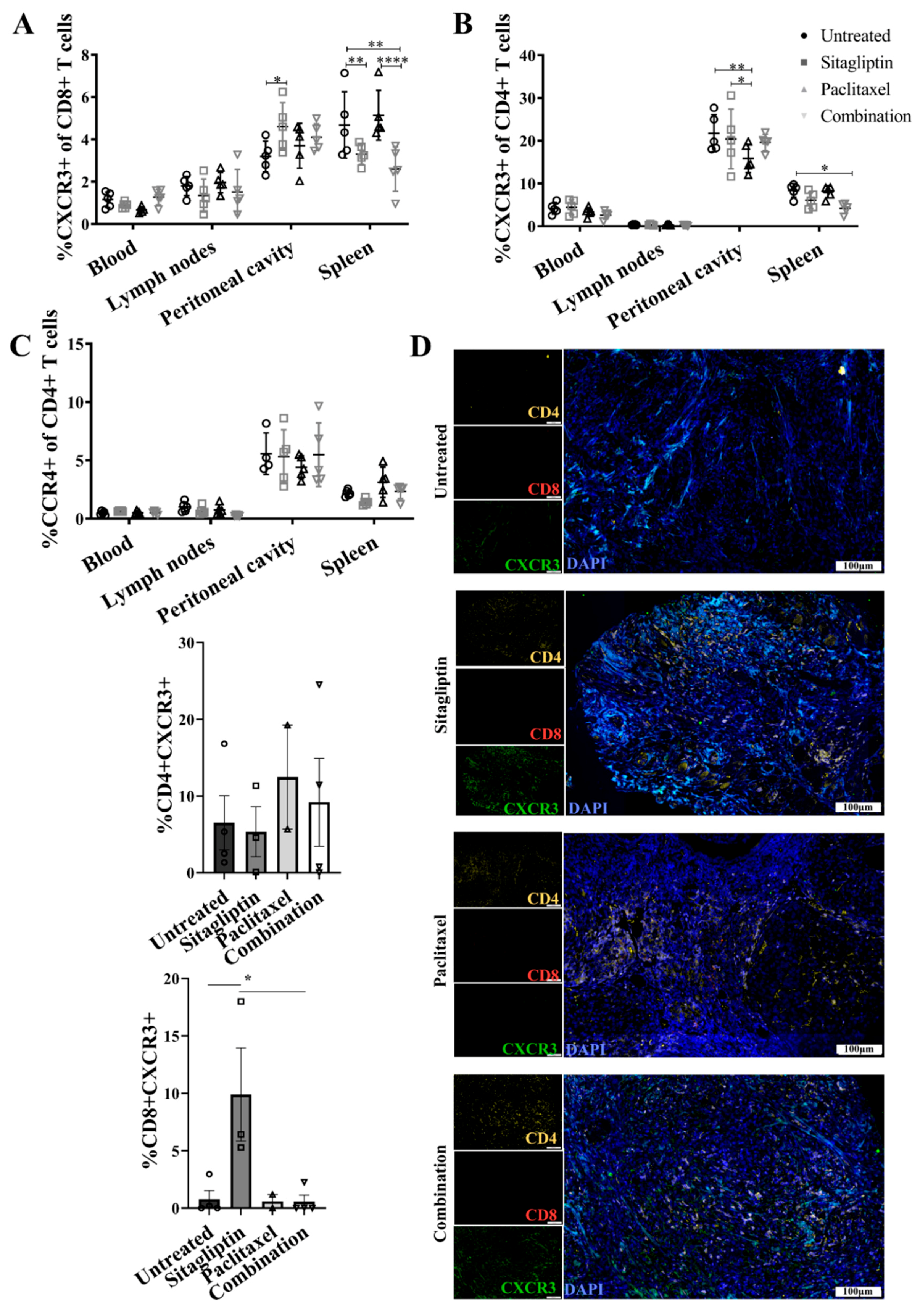
3.5. Enhanced CXCR3-Mediated Recruitment of CD8+ T Cells Is Abrogated by Paclitaxel
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wei, W.; Li, N.; Sun, Y.; Li, B.; Xu, L.; Wu, L. Clinical outcome and prognostic factors of patients with early-stage epithelial ovarian cancer. Oncotarget 2017, 8, 23862–23870. [Google Scholar] [CrossRef] [PubMed]
- Ozols, R.F.; Bundy, B.N.; Greer, B.E.; Fowler, J.M.; Clarke-Pearson, D.; Burger, R.A.; Mannel, R.S.; DeGeest, K.; Hartenbach, E.M.; Baergen, R.; et al. Phase III trial of carboplatin and paclitaxel compared with cisplatin and paclitaxel in patients with optimally resected stage III ovarian cancer: A Gynecologic Oncology Group study. J. Clin. Oncol. 2003, 21, 3194–3200. [Google Scholar] [CrossRef] [PubMed]
- Davidson, B.; Trope, C.G. Ovarian cancer: Diagnostic, biological and prognostic aspects. Womens Health 2014, 10, 519–533. [Google Scholar] [CrossRef] [PubMed]
- Paleari, L.; Gandini, S.; Provinciali, N.; Puntoni, M.; Colombo, N.; DeCensi, A. Clinical benefit and risk of death with endocrine therapy in ovarian cancer: A comprehensive review and meta-analysis. Gynecol. Oncol. 2017, 146, 504–513. [Google Scholar] [CrossRef]
- Wilson, A.L.; Plebanski, M.; Stephens, A.N. New Trends in Anti-Cancer Therapy: Combining Conventional Chemotherapeutics with Novel Immunomodulators. Curr. Med. Chem. 2018, 25, 4758–4784. [Google Scholar] [CrossRef]
- Curiel, T.J.; Coukos, G.; Zou, L.; Alvarez, X.; Cheng, P.; Mottram, P.; Evdemon-Hogan, M.; Conejo-Garcia, J.R.; Zhang, L.; Burow, M.; et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 2004, 10, 942–949. [Google Scholar] [CrossRef]
- Zhang, L.; Conejo-Garcia, J.R.; Katsaros, D.; Gimotty, P.A.; Massobrio, M.; Regnani, G.; Makrigiannakis, A.; Gray, H.; Schlienger, K.; Liebman, M.N.; et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N. Engl. J. Med. 2003, 348, 203–213. [Google Scholar] [CrossRef]
- Woo, E.Y.; Chu, C.S.; Goletz, T.J.; Schlienger, K.; Yeh, H.; Coukos, G.; Rubin, S.C.; Kaiser, L.R.; June, C.H. Regulatory CD4(+)CD25(+) T cells in tumors from patients with early-stage non-small cell lung cancer and late-stage ovarian cancer. Cancer Res. 2001, 61, 4766–4772. [Google Scholar]
- Giuntoli, R.L., 2nd; Webb, T.J.; Zoso, A.; Rogers, O.; Diaz-Montes, T.P.; Bristow, R.E.; Oelke, M. Ovarian cancer-associated ascites demonstrates altered immune environment: Implications for antitumor immunity. Anticancer Res. 2009, 29, 2875–2884. [Google Scholar]
- Liang, P.I.; Yeh, B.W.; Li, W.M.; Chan, T.C.; Chang, I.W.; Huang, C.N.; Li, C.C.; Ke, H.L.; Yeh, H.C.; Wu, W.J.; et al. DPP4/CD26 overexpression in urothelial carcinoma confers an independent prognostic impact and correlates with intrinsic biological aggressiveness. Oncotarget 2017, 8, 2995–3008. [Google Scholar] [CrossRef]
- Lee, J.J.; Wang, T.Y.; Liu, C.L.; Chien, M.N.; Chen, M.J.; Hsu, Y.C.; Leung, C.H.; Cheng, S.P. Dipeptidyl Peptidase IV as a Prognostic Marker and Therapeutic Target in Papillary Thyroid Carcinoma. J. Clin. Endocrinol. Metab. 2017, 102, 2930–2940. [Google Scholar] [CrossRef] [PubMed]
- Nazarian, A.; Lawlor, K.; Yi, S.S.; Philip, J.; Ghosh, M.; Yaneva, M.; Villanueva, J.; Saghatelian, A.; Assel, M.; Vickers, A.J.; et al. Inhibition of circulating dipeptidyl peptidase 4 activity in patients with metastatic prostate cancer. Mol. Cell. Proteomics 2014, 13, 3082–3096. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Xu, L.; Wang, X.; Sun, B.; Ding, J. Expression levels of seprase/FAPalpha and DPPIV/CD26 in epithelial ovarian carcinoma. Oncol. Lett. 2015, 10, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Ou, X.; O’Leary, H.A.; Broxmeyer, H.E. Implications of DPP4 modification of proteins that regulate stem/progenitor and more mature cell types. Blood 2013, 122, 161–169. [Google Scholar] [CrossRef]
- Sesti, G.; Avogaro, A.; Belcastro, S.; Bonora, B.M.; Croci, M.; Daniele, G.; Dauriz, M.; Dotta, F.; Formichi, C.; Frontoni, S.; et al. Ten years of experience with DPP-4 inhibitors for the treatment of type 2 diabetes mellitus. Acta Diabetol. 2019, 56, 605–617. [Google Scholar] [CrossRef]
- Klemann, C.; Wagner, L.; Stephan, M.; von Horsten, S. Cut to the chase: A review of CD26/dipeptidyl peptidase-4′s (DPP4) entanglement in the immune system. Clin. Exp. Immunol. 2016, 185, 1–21. [Google Scholar] [CrossRef]
- Rainczuk, A.; Rao, J.; Gathercole, J.; Stephens, A.N. The emerging role of CXC chemokines in epithelial ovarian cancer. Reproduction 2012, 144, 303–317. [Google Scholar] [CrossRef]
- Zhang, H.; Maqsudi, S.; Rainczuk, A.; Duffield, N.; Lawrence, J.; Keane, F.M.; Justa-Schuch, D.; Geiss-Friedlander, R.; Gorrell, M.D.; Stephens, A.N. Identification of novel dipeptidyl peptidase 9 substrates by two-dimensional differential in-gel electrophoresis. FEBS J. 2015, 282, 3737–3757. [Google Scholar] [CrossRef]
- Casrouge, A.; Decalf, J.; Ahloulay, M.; Lababidi, C.; Mansour, H.; Vallet-Pichard, A.; Mallet, V.; Mottez, E.; Mapes, J.; Fontanet, A.; et al. Evidence for an antagonist form of the chemokine CXCL10 in patients chronically infected with HCV. J. Clin. Investig. 2011, 121, 308–317. [Google Scholar] [CrossRef]
- Decalf, J.; Tarbell, K.V.; Casrouge, A.; Price, J.D.; Linder, G.; Mottez, E.; Sultanik, P.; Mallet, V.; Pol, S.; Duffy, D.; et al. Inhibition of DPP4 activity in humans establishes its in vivo role in CXCL10 post-translational modification: Prospective placebo-controlled clinical studies. EMBO Mol. Med. 2016, 8, 679–683. [Google Scholar] [CrossRef]
- Barreira da Silva, R.; Laird, M.E.; Yatim, N.; Fiette, L.; Ingersoll, M.A.; Albert, M.L. Dipeptidylpeptidase 4 inhibition enhances lymphocyte trafficking, improving both naturally occurring tumor immunity and immunotherapy. Nat. Immunol. 2015, 16, 850–858. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.L.; Wilson, K.L.; Bilandzic, M.; Moffitt, L.R.; Makanji, M.; Gorrell, M.D.; Oehler, M.K.; Rainczuk, A.; Stephens, A.N.; Plebanski, M. Non-Invasive Fluorescent Monitoring of Ovarian Cancer in an Immunocompetent Mouse Model. Cancers 2018, 11, 32. [Google Scholar] [CrossRef] [PubMed]
- Nunez-Cruz, S.; Connolly, D.C.; Scholler, N. An orthotopic model of serous ovarian cancer in immunocompetent mice for in vivo tumor imaging and monitoring of tumor immune responses. J. Vis. Exp. 2010. [Google Scholar] [CrossRef] [PubMed]
- Luippold, G.; Mark, M.; Klein, T.; Amann, K.; Daniel, C. Differences in kidney-specific DPP-4 inhibition by linagliptin and sitagliptin. Diabetes Res. Clin. Pract. 2018, 143, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Coquery, C.M.; Loo, W.; Buszko, M.; Lannigan, J.; Erickson, L.D. Optimized protocol for the isolation of spleen-resident murine neutrophils. Cytometry A 2012, 81, 806–814. [Google Scholar] [CrossRef]
- Manglani, M.; Rua, R.; Hendricksen, A.; Braunschweig, D.; Gao, Q.; Tan, W.; Houser, B.; McGavern, D.B.; Oh, K. Method to quantify cytokines and chemokines in mouse brain tissue using Bio-Plex multiplex immunoassays. Methods 2019, 158, 22–26. [Google Scholar] [CrossRef]
- Varin, E.M.; Mulvihill, E.E.; Beaudry, J.L.; Pujadas, G.; Fuchs, S.; Tanti, J.F.; Fazio, S.; Kaur, K.; Cao, X.; Baggio, L.L.; et al. Circulating Levels of Soluble Dipeptidyl Peptidase-4 Are Dissociated from Inflammation and Induced by Enzymatic DPP4 Inhibition. Cell Metab. 2019, 29, 320–334.e325. [Google Scholar] [CrossRef]
- Sato, E.; Olson, S.H.; Ahn, J.; Bundy, B.; Nishikawa, H.; Qian, F.; Jungbluth, A.A.; Frosina, D.; Gnjatic, S.; Ambrosone, C.; et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 18538–18543. [Google Scholar] [CrossRef]
- Chang, D.K.; Peterson, E.; Sun, J.; Goudie, C.; Drapkin, R.I.; Liu, J.F.; Matulonis, U.; Zhu, Q.; Marasco, W.A. Anti-CCR4 monoclonal antibody enhances antitumor immunity by modulating tumor-infiltrating Tregs in an ovarian cancer xenograft humanized mouse model. Oncoimmunology 2016, 5, e1090075. [Google Scholar] [CrossRef]
- Draghiciu, O.; Lubbers, J.; Nijman, H.W.; Daemen, T. Myeloid derived suppressor cells-An overview of combat strategies to increase immunotherapy efficacy. Oncoimmunology 2015, 4, e954829. [Google Scholar] [CrossRef]
- Kovacs, E. The serum levels of IL-12 and IL-16 in cancer patients. Relation to the tumour stage and previous therapy. Biomed. Pharmacother. 2001, 55, 111–116. [Google Scholar] [CrossRef]
- Focosi, D.; Kast, R.E.; Metelli, M.R.; Benedetti, E.; Galimberti, S.; Papineschi, F.; Petrini, M. Enhancement of hematopoietic stem cell engraftment by inhibition of CXCL12 proteolysis with sitagliptin, an oral dipeptidyl-peptidase IV inhibitor: A report in a case of delayed graft failure. Leuk. Res. 2009, 33, 178–181. [Google Scholar] [CrossRef] [PubMed]
- Papazafiropoulou, A.K.; Papanas, N.; Trikkalinou, A.; Fousteris, E.; Melidonis, A. The Oral Dipeptidyl-Peptidase-4 Inhibitor Sitagliptin Increases Circulating Levels Of Stromal-Derived Factor-1 Alpha. Exp. Clin. Endocrinol. Diabetes 2018, 126, 367–370. [Google Scholar] [CrossRef] [PubMed]
- Lambeir, A.M.; Durinx, C.; Scharpe, S.; De Meester, I. Dipeptidyl-peptidase IV from bench to bedside: An update on structural properties, functions, and clinical aspects of the enzyme DPP IV. Crit. Rev. Clin. Lab. Sci. 2003, 40, 209–294. [Google Scholar] [CrossRef]
- Rainczuk, A.; Rao, J.R.; Gathercole, J.L.; Fairweather, N.J.; Chu, S.; Masadah, R.; Jobling, T.W.; Deb-Choudhury, S.; Dyer, J.; Stephens, A.N. Evidence for the antagonistic form of CXC-motif chemokine CXCL10 in serous epithelial ovarian tumours. Int. J. Cancer 2014, 134, 530–541. [Google Scholar] [CrossRef]
- Halim, L.; Romano, M.; McGregor, R.; Correa, I.; Pavlidis, P.; Grageda, N.; Hoong, S.J.; Yuksel, M.; Jassem, W.; Hannen, R.F.; et al. An Atlas of Human Regulatory T Helper-like Cells Reveals Features of Th2-like Tregs that Support a Tumorigenic Environment. Cell Rep. 2017, 20, 757–770. [Google Scholar] [CrossRef]
- Zsiros, E.; Duttagupta, P.; Dangaj, D.; Li, H.; Frank, R.; Garrabrant, T.; Hagemann, I.S.; Levine, B.L.; June, C.H.; Zhang, L.; et al. The Ovarian Cancer Chemokine Landscape Is Conducive to Homing of Vaccine-Primed and CD3/CD28-Costimulated T Cells Prepared for Adoptive Therapy. Clin. Cancer Res. 2015, 21, 2840–2850. [Google Scholar] [CrossRef]
- Buda, A.; Floriani, I.; Rossi, R.; Colombo, N.; Torri, V.; Conte, P.F.; Fossati, R.; Ravaioli, A.; Mangioni, C. Randomised controlled trial comparing single agent paclitaxel vs epidoxorubicin plus paclitaxel in patients with advanced ovarian cancer in early progression after platinum-based chemotherapy: An Italian Collaborative Study from the Mario Negri Institute, Milan, G.O.N.O. (Gruppo Oncologico Nord Ovest) group and I.O.R. (Istituto Oncologico Romagnolo) group. Br. J. Cancer 2004, 90, 2112–2117. [Google Scholar] [CrossRef]
- Kandalaft, L.E.; Powell, D.J., Jr.; Singh, N.; Coukos, G. Immunotherapy for ovarian cancer: What’s next? J. Clin. Oncol. 2011, 29, 925–933. [Google Scholar] [CrossRef]
- Shao, S.; Wang, C.; Tian, J.; Zhang, H.; Wang, S.; Du, Y. Diagnostic and prognostic significance of serum CD26 level in Asian women with high-grade serous ovarian carcinoma. Future Oncol. 2019, 15, 1863–1871. [Google Scholar] [CrossRef]
- Qin, C.J.; Zhao, L.H.; Zhou, X.; Zhang, H.L.; Wen, W.; Tang, L.; Zeng, M.; Wang, M.D.; Fu, G.B.; Huang, S.; et al. Inhibition of dipeptidyl peptidase IV prevents high fat diet-induced liver cancer angiogenesis by downregulating chemokine ligand 2. Cancer Lett. 2018, 420, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.J.; Kim, J.Y.; Lim, S.C.; Kim, G.; Yun, H.J.; Choi, H.S. Dipeptidyl peptidase 4 promotes epithelial cell transformation and breast tumourigenesis via induction of PIN1 gene expression. Br. J. Pharmacol. 2015, 172, 5096–5109. [Google Scholar] [CrossRef]
- Piazza, G.A.; Callanan, H.M.; Mowery, J.; Hixson, D.C. Evidence for a role of dipeptidyl peptidase IV in fibronectin-mediated interactions of hepatocytes with extracellular matrix. Biochem. J. 1989, 262, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Röhrborn, D.; Eckel, J.; Sell, H. Shedding of dipeptidyl peptidase 4 is mediated by metalloproteases and up-regulated by hypoxia in human adipocytes and smooth muscle cells. FEBS Lett. 2014, 588, 3870–3877. [Google Scholar] [CrossRef]
- Fasolato, S.; Trevellin, E.; Ruvoletto, M.; Granzotto, M.; Zanus, G.; Boscaro, E.; Babetto, E.; Terrin, L.; Battocchio, M.A.; Ciscato, F.; et al. SerpinB3 induces dipeptidyl-peptidase IV/CD26 expression and its metabolic effects in hepatocellular carcinoma. Life Sci. 2018, 200, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Casrouge, A.; Sauer, A.V.; Barreira da Silva, R.; Tejera-Alhambra, M.; Sanchez-Ramon, S.; Icare, B.; Cancrini, C.; Ingersoll, M.A.; Aiuti, A.; Albert, M.L. Lymphocytes are a major source of circulating soluble dipeptidyl peptidase 4. Clin. Exp. Immunol. 2018, 194, 166–179. [Google Scholar] [CrossRef]
- Lettau, M.; Dietz, M.; Vollmers, S.; Armbrust, F.; Peters, C.; Dang, T.M.; Chitadze, G.; Kabelitz, D.; Janssen, O. Degranulation of human cytotoxic lymphocytes is a major source of proteolytically active soluble CD26/DPP4. Cell Mol Life Sci. 2020, 77, 751–764. [Google Scholar] [CrossRef]
- Nargis, T.; Kumar, K.; Ghosh, A.R.; Sharma, A.; Rudra, D.; Sen, D.; Chakrabarti, S.; Mukhopadhyay, S.; Ganguly, D.; Chakrabarti, P. KLK5 induces shedding of DPP4 from circulatory Th17 cells in type 2 diabetes. Mol. Metab. 2017, 6, 1529–1539. [Google Scholar] [CrossRef]
- Javidroozi, M.; Zucker, S.; Chen, W.T. Plasma seprase and DPP4 levels as markers of disease and prognosis in cancer. Dis. Markers 2012, 32, 309–320. [Google Scholar] [CrossRef]
- Etienne-Manneville, S. Microtubules in cell migration. Annu. Rev. Cell Dev. Biol. 2013, 29, 471–499. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wilson, A.L.; Moffitt, L.R.; Wilson, K.L.; Bilandzic, M.; Wright, M.D.; Gorrell, M.D.; Oehler, M.K.; Plebanski, M.; Stephens, A.N. DPP4 Inhibitor Sitagliptin Enhances Lymphocyte Recruitment and Prolongs Survival in a Syngeneic Ovarian Cancer Mouse Model. Cancers 2021, 13, 487. https://doi.org/10.3390/cancers13030487
Wilson AL, Moffitt LR, Wilson KL, Bilandzic M, Wright MD, Gorrell MD, Oehler MK, Plebanski M, Stephens AN. DPP4 Inhibitor Sitagliptin Enhances Lymphocyte Recruitment and Prolongs Survival in a Syngeneic Ovarian Cancer Mouse Model. Cancers. 2021; 13(3):487. https://doi.org/10.3390/cancers13030487
Chicago/Turabian StyleWilson, Amy L., Laura R. Moffitt, Kirsty L. Wilson, Maree Bilandzic, Mark D. Wright, Mark D. Gorrell, Martin K. Oehler, Magdalena Plebanski, and Andrew N. Stephens. 2021. "DPP4 Inhibitor Sitagliptin Enhances Lymphocyte Recruitment and Prolongs Survival in a Syngeneic Ovarian Cancer Mouse Model" Cancers 13, no. 3: 487. https://doi.org/10.3390/cancers13030487
APA StyleWilson, A. L., Moffitt, L. R., Wilson, K. L., Bilandzic, M., Wright, M. D., Gorrell, M. D., Oehler, M. K., Plebanski, M., & Stephens, A. N. (2021). DPP4 Inhibitor Sitagliptin Enhances Lymphocyte Recruitment and Prolongs Survival in a Syngeneic Ovarian Cancer Mouse Model. Cancers, 13(3), 487. https://doi.org/10.3390/cancers13030487