
RUNX3 Transcript Variants Have Distinct Roles in Ovarian Carcinoma and Differently Influence Platinum Sensitivity and Angiogenesis

 ,
,  , , , , ,
, , , , ,
Abstract
Simple Summary
Abstract
1. Introduction
2. Methods
2.1. Vector Cloning
2.2. Cell Culture
2.3. Transfection, Single Cell Clone Generation and Transduction
2.4. Functional Analyses In Vitro
2.5. Expression Analyses
2.6. Fluorescence Microscopy-Based Analysis
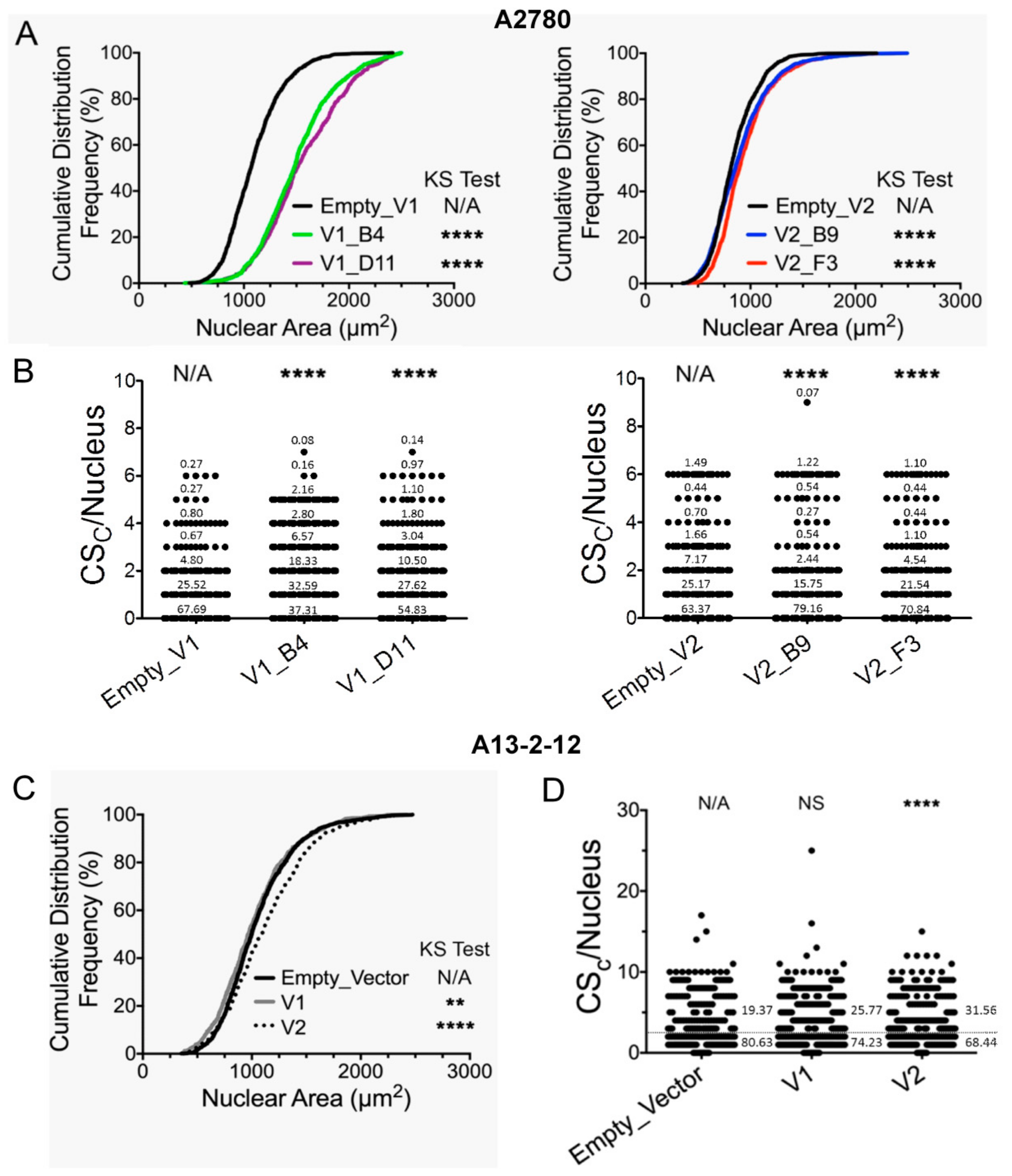
2.7. Chromosome Instability Assessments
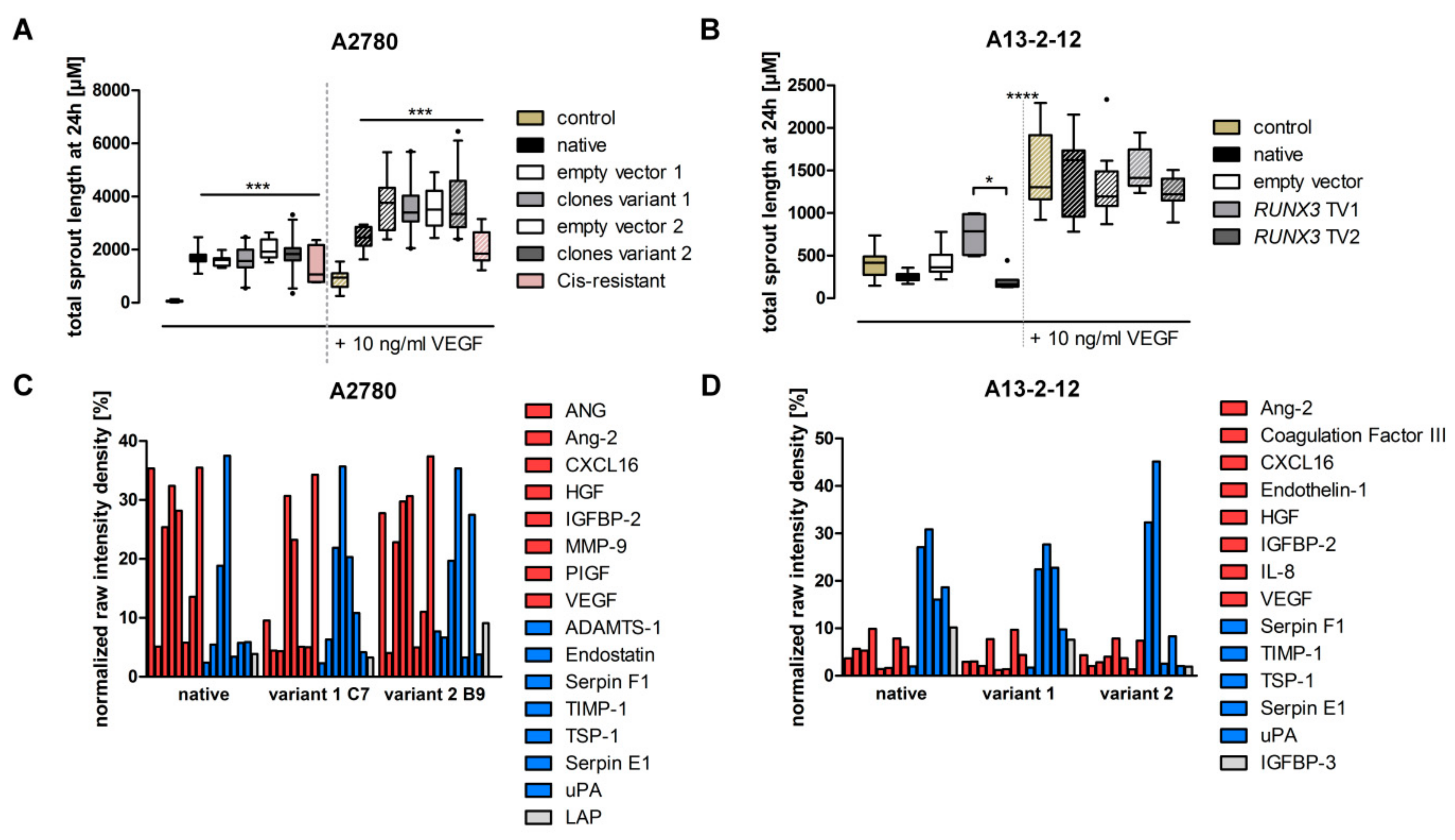
2.8. Assessment Angiogenic Potential
2.9. RNA Sequencing and Computational Analysis
2.10. Statistical Analysis
3. Results
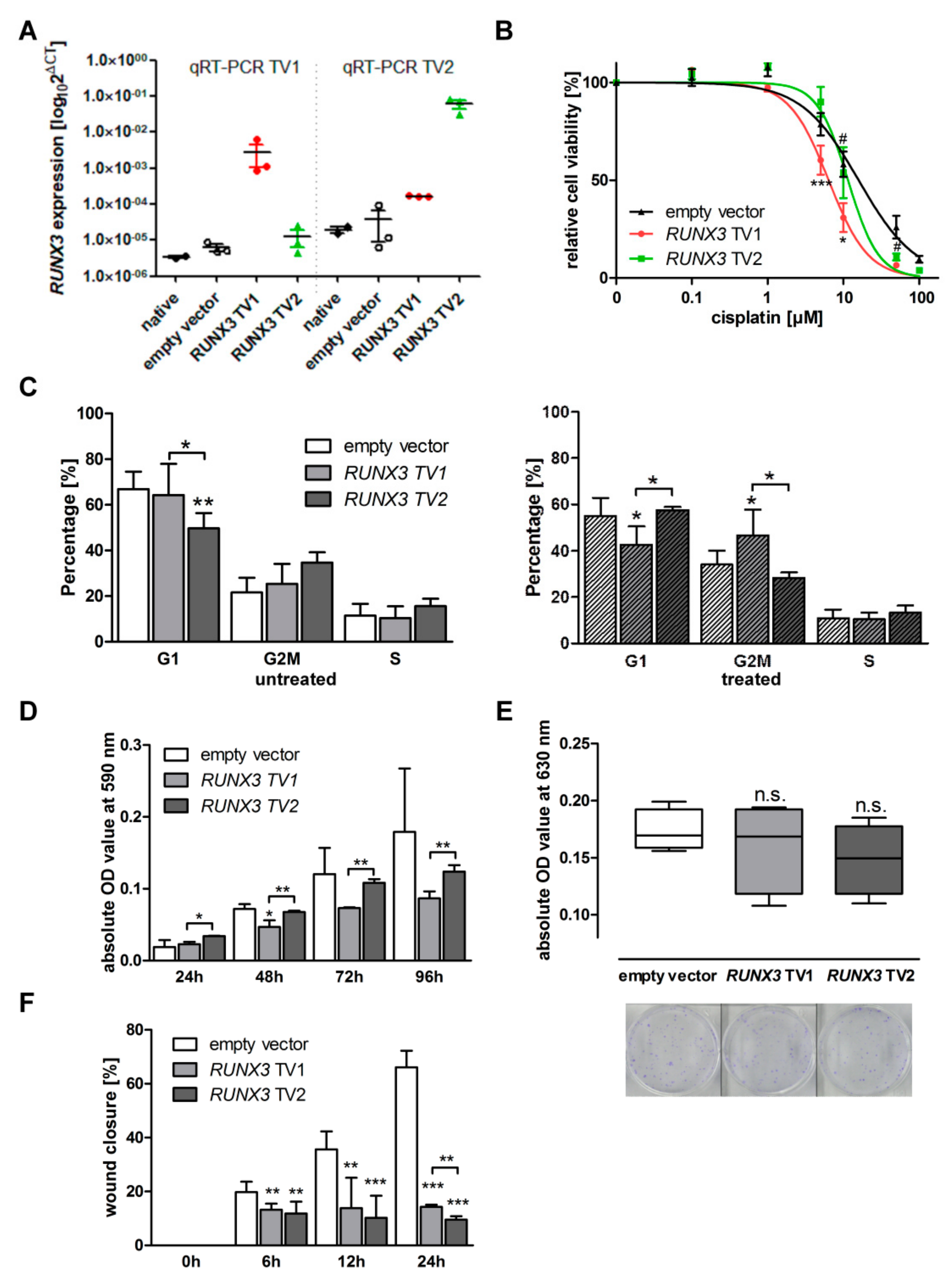
3.1. Cellular Phenotypes Are Differently Affected by RUNX3 Variants in Different EOC Models
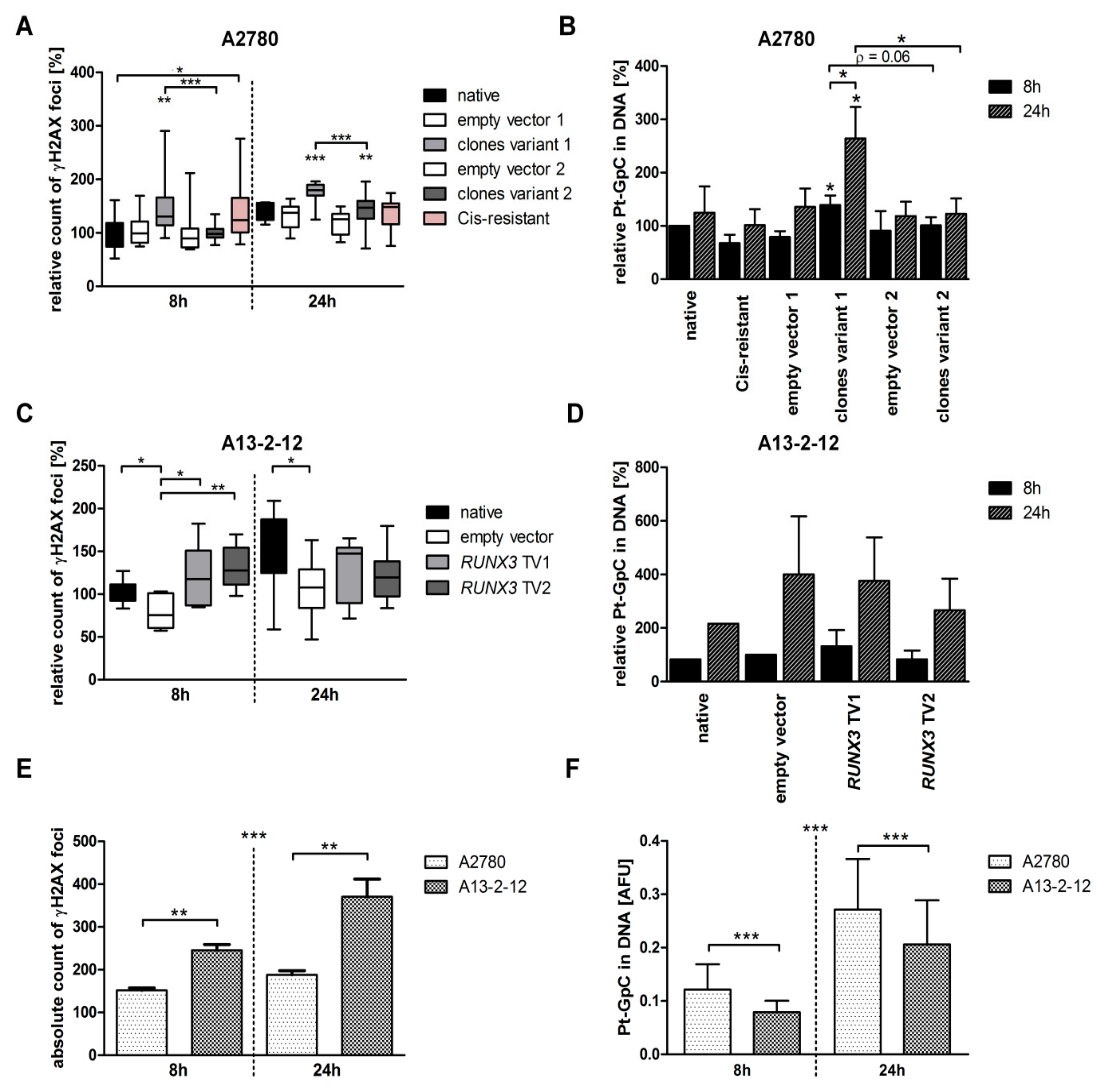
3.2. RUNX3 TV1 Increases Cisplatin-Induced DNA Damage in EOC Cells with BRCAwt
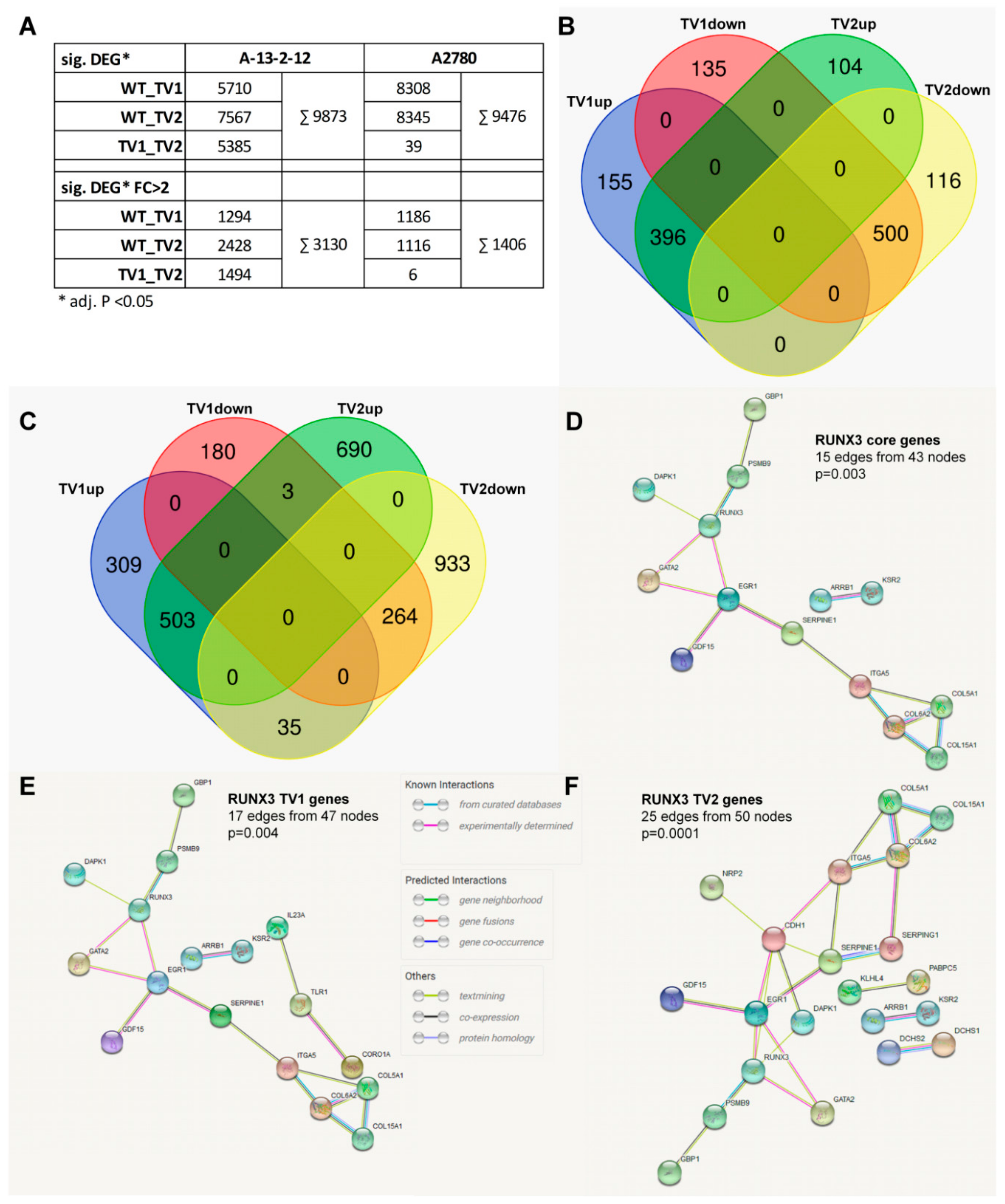
3.3. RUNX3 Variants Differentially Regulate the Transcriptome of EOC Cell Lines
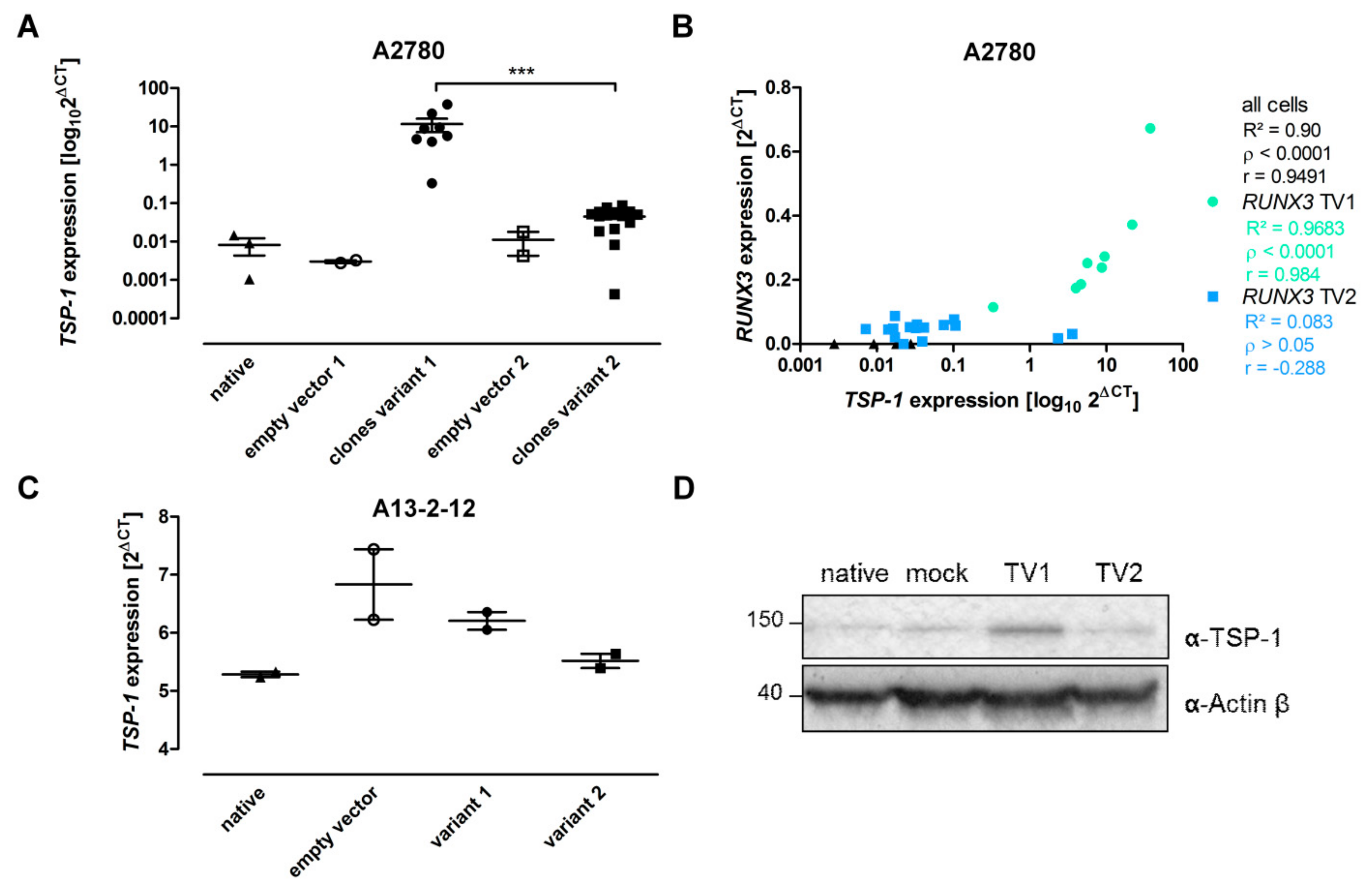
3.4. RUNX3 Variants Differentially Influence Thrombospondin-1 Expression and Angiogenic Effectors
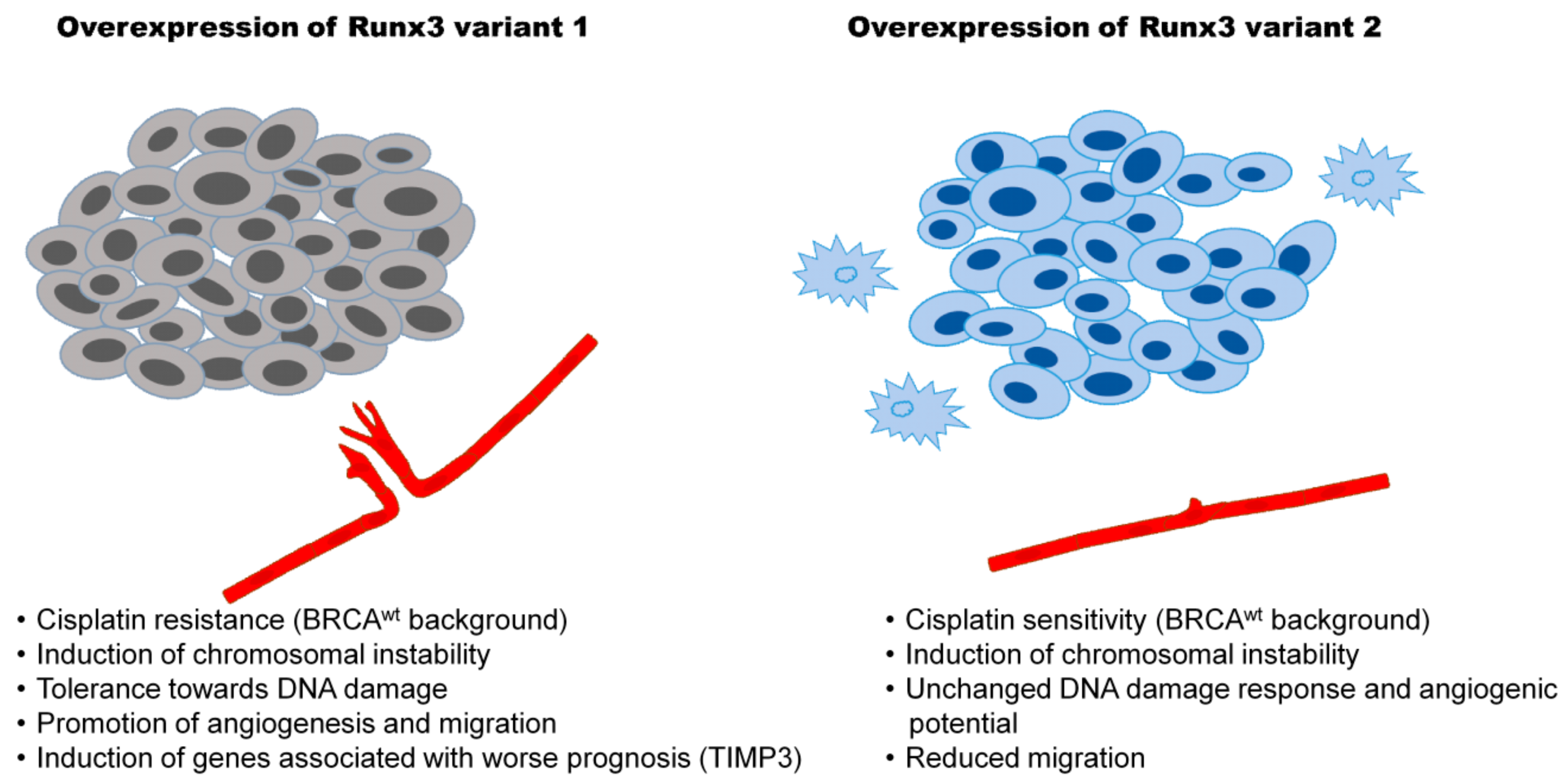
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vaughan, S.; Coward, J.I.; Bast, R.C., Jr.; Berchuck, A.; Berek, J.S.; Brenton, J.D.; Coukos, G.; Crum, C.C.; Drapkin, R.; Etemadmoghadam, D.; et al. Rethinking ovarian cancer: Recommendations for improving outcomes. Nat. Rev. Cancer 2011, 11, 719–725. [Google Scholar] [CrossRef] [PubMed]
- McCluggage, W.G. Morphological subtypes of ovarian carcinoma: A review with emphasis on new developments and pathogenesis. Pathology 2011, 43, 420–432. [Google Scholar] [CrossRef] [PubMed]
- Rescigno, P.; Cerillo, I.; Ruocco, R.; Condello, C.; De Placido, S.; Pensabene, M. New hypothesis on pathogenesis of ovarian cancer lead to future tailored approaches. Biomed. Res. Int. 2013, 2013, 852839. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Häfner, N.; Steinbach, D.; Jansen, L.; Diebolder, H.; Dürst, M.; Runnebaum, I.B. RUNX3 and CAMK2N1 hypermethylation as prognostic marker for epithelial ovarian cancer. Int. J. Cancer 2016, 138, 217–228. [Google Scholar] [CrossRef]
- Heinze, K.; Rengsberger, M.; Gajda, M.; Jansen, L.; Osmers, L.; Oliveira-Ferrer, L.; Schmalfeldt, B.; Dürst, M.; Häfner, N.; Runnebaum, I.B. CAMK2N1/RUNX3 methylation is an independent prognostic biomarker for progression-free and overall survival of platinum-sensitive epithelial ovarian cancer patients. Clin. Epigenetics 2021, 13, 15. [Google Scholar] [CrossRef]
- Heinze, K.; Kritsch, D.; Mosig, A.S.; Durst, M.; Hafner, N.; Runnebaum, I.B. Functional Analyses of RUNX3 and CaMKIINalpha in Ovarian Cancer Cell Lines Reveal Tumor-Suppressive Functions for CaMKIINalpha and Dichotomous Roles for RUNX3 Transcript Variants. Int. J. Mol. Sci. 2018, 19, 253. [Google Scholar] [CrossRef]
- Ito, Y. RUNX genes in development and cancer: Regulation of viral gene expression and the discovery of RUNX family genes. Adv. Cancer Res. 2008, 99, 33–76. [Google Scholar]
- Chuang, L.S.; Ito, K.; Ito, Y. RUNX family: Regulation and diversification of roles through interacting proteins. Int. J. Cancer 2013, 132, 1260–1271. [Google Scholar] [CrossRef]
- Taniuchi, I.; Osato, M.; Egawa, T.; Sunshine, M.J.; Bae, S.C.; Komori, T.; Ito, Y.; Littman, D.R. Differential requirements for Runx proteins in CD4 repression and epigenetic silencing during T lymphocyte development. Cell 2002, 111, 621–633. [Google Scholar] [CrossRef]
- Li, Q.L.; Ito, K.; Sakakura, C.; Fukamachi, H.; Inoue, K.; Chi, X.Z.; Lee, K.Y.; Nomura, S.; Lee, C.W.; Han, S.B.; et al. Causal relationship between the loss of RUNX3 expression and gastric cancer. Cell 2002, 109, 113–124. [Google Scholar] [CrossRef]
- Bagchi, A.; Mills, A.A. The quest for the 1p36 tumor suppressor. Cancer Res. 2008, 68, 2551–2556. [Google Scholar] [CrossRef] [PubMed]
- Levanon, D.; Groner, Y. Structure and regulated expression of mammalian RUNX genes. Oncogene 2004, 23, 4211–4219. [Google Scholar] [CrossRef] [PubMed]
- Ju, X.; Ishikawa, T.O.; Naka, K.; Ito, K.; Ito, Y.; Oshima, M. Context-dependent activation of Wnt signaling by tumor suppressor RUNX3 in gastric cancer cells. Cancer Sci. 2014, 105, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Chen, Y.; Wu, K.C.; Liu, J.; Zhao, Y.Q.; Pan, Y.L.; Du, R.; Zheng, G.R.; Xiong, Y.M.; Xu, H.L.; et al. RUNX3 directly interacts with intracellular domain of Notch1 and suppresses Notch signaling in hepatocellular carcinoma cells. Exp. Cell Res. 2010, 316, 149–157. [Google Scholar] [CrossRef]
- Li, H.; Li, D.; Meng, N. Effects of RUNX3 mediated Notch signaling pathway on biological characteristics of colorectal cancer cells. Int. J. Oncol. 2017, 50, 2059–2068. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tanaka, Y.; Imamura, J.; Kanai, F.; Ichimura, T.; Isobe, T.; Koike, M.; Kudo, Y.; Tateishi, K.; Ikenoue, T.; Ijichi, H.; et al. Runx3 interacts with DNA repair protein Ku70. Exp. Cell Res. 2007, 313, 3251–3260. [Google Scholar] [CrossRef] [PubMed]
- Tay, L.S.; Krishnan, V.; Sankar, H.; Chong, Y.L.; Chuang, L.S.H.; Tan, T.Z.; Kolinjivadi, A.M.; Kappei, D.; Ito, Y. RUNX Poly(ADP-Ribosyl)ation and BLM Interaction Facilitate the Fanconi Anemia Pathway of DNA Repair. Cell Rep. 2018, 24, 1747–1755. [Google Scholar] [CrossRef]
- Peng, Z.; Wei, D.; Wang, L.; Tang, H.; Zhang, J.; Le, X.; Jia, Z.; Li, Q.; Xie, K. RUNX3 inhibits the expression of vascular endothelial growth factor and reduces the angiogenesis, growth, and metastasis of human gastric cancer. Clin. Cancer Res. 2006, 12, 6386–6394. [Google Scholar] [CrossRef]
- Chen, F.; Wang, M.; Bai, J.; Liu, Q.; Xi, Y.; Li, W.; Zheng, J. Role of RUNX3 in suppressing metastasis and angiogenesis of human prostate cancer. PLoS ONE 2014, 9, e86917. [Google Scholar] [CrossRef]
- Lee, J.M.; Lee, D.J.; Bae, S.C.; Jung, H.S. Abnormal liver differentiation and excessive angiogenesis in mice lacking Runx3. Histochem. Cell Biol. 2013, 139, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Deepak, V.; Wang, L.; Ba, X.; Komori, T.; Zeng, X.; Liu, W. Thrombospondin-1 is a putative target gene of Runx2 and Runx3. Int. J. Mol. Sci. 2013, 14, 14321–14332. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.Y.; Lee, H.J.; Hwang, K.S.; Lee, M.; Kim, J.W.; Bang, Y.J.; Kang, G.H. Methylation of RUNX3 in various types of human cancers and premalignant stages of gastric carcinoma. Lab. Investig. 2004, 84, 479–484. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Subramaniam, M.M.; Chan, J.Y.; Yeoh, K.G.; Quek, T.; Ito, K.; Salto-Tellez, M. Molecular pathology of RUNX3 in human carcinogenesis. Biochim. Biophys. Acta 2009, 1796, 315–331. [Google Scholar] [CrossRef] [PubMed]
- Nevadunsky, N.S.; Barbieri, J.S.; Kwong, J.; Merritt, M.A.; Welch, W.R.; Berkowitz, R.S.; Mok, S.C. RUNX3 protein is overexpressed in human epithelial ovarian cancer. Gynecol. Oncol. 2009, 112, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Qin, X.; Wu, J.; Qi, B.; Tao, Y.; Wang, W.; Liu, F.; Li, H.; Zhao, B. Association of promoter methylation of RUNX3 gene with the development of esophageal cancer: A meta analysis. PLoS ONE 2014, 9, e107598. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Yan, Z.; Yin, Q.; Cao, K.; Wei, Y.; Rodriguez-Canales, J.; Ma, D.; Wu, Y. RUNX3 Epigenetic Inactivation Is Associated with Estrogen Receptor Positive Breast Cancer. J. Histochem. Cytochem. 2018, 66, 709–721. [Google Scholar] [CrossRef]
- Salto-Tellez, M.; Peh, B.K.; Ito, K.; Tan, S.H.; Chong, P.Y.; Han, H.C.; Tada, K.; Ong, W.Y.; Soong, R.; Voon, D.C.; et al. RUNX3 protein is overexpressed in human basal cell carcinomas. Oncogene 2006, 25, 7646–7649. [Google Scholar] [CrossRef]
- Tsunematsu, T.; Kudo, Y.; Iizuka, S.; Ogawa, I.; Fujita, T.; Kurihara, H.; Abiko, Y.; Takata, T. RUNX3 has an oncogenic role in head and neck cancer. PLoS ONE 2009, 4, e5892. [Google Scholar] [CrossRef]
- Whittle, M.C.; Izeradjene, K.; Rani, P.G.; Feng, L.; Carlson, M.A.; DelGiorno, K.E.; Wood, L.D.; Goggins, M.; Hruban, R.H.; Chang, A.E.; et al. RUNX3 Controls a Metastatic Switch in Pancreatic Ductal Adenocarcinoma. Cell 2015, 161, 1345–1360. [Google Scholar] [CrossRef]
- Zhang, S.; Wei, L.; Zhang, A.; Zhang, L.; Yu, H. RUNX3 gene methylation in epithelial ovarian cancer tissues and ovarian cancer cell lines. Omics A J. Integr. Biol. 2009, 13, 307–311. [Google Scholar] [CrossRef] [PubMed]
- Ojima, F.; Saito, Y.; Tsuchiya, Y.; Kayo, D.; Taniuchi, S.; Ogoshi, M.; Fukamachi, H.; Takeuchi, S.; Takahashi, S. Runx3 transcription factor regulates ovarian functions and ovulation in female mice. J. Reprod Dev. 2016, 62, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Barghout, S.H.; Zepeda, N.; Vincent, K.; Azad, A.K.; Xu, Z.; Yang, C.; Steed, H.; Postovit, L.M.; Fu, Y. RUNX3 contributes to carboplatin resistance in epithelial ovarian cancer cells. Gynecol. Oncol. 2015, 138, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Anglesio, M.S.; Wiegand, K.C.; Melnyk, N.; Chow, C.; Salamanca, C.; Prentice, L.M.; Senz, J.; Yang, W.; Spillman, M.A.; Cochrane, D.R.; et al. Type-specific cell line models for type-specific ovarian cancer research. PLoS ONE 2013, 8, e72162. [Google Scholar] [CrossRef]
- Beaufort, C.M.; Helmijr, J.C.; Piskorz, A.M.; Hoogstraat, M.; Ruigrok-Ritstier, K.; Besselink, N.; Murtaza, M.; van Ijcken, W.F.; Heine, A.A.; Smid, M.; et al. Ovarian cancer cell line panel (OCCP): Clinical importance of in vitro morphological subtypes. PLoS ONE 2014, 9, e103988. [Google Scholar] [CrossRef]
- Domcke, S.; Sinha, R.; Levine, D.A.; Sander, C.; Schultz, N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 2013, 4, 2126. [Google Scholar] [CrossRef]
- Shepherd, T.G.; Theriault, B.L.; Campbell, E.J.; Nachtigal, M.W. Primary culture of ovarian surface epithelial cells and ascites-derived ovarian cancer cells from patients. Nat. Protoc. 2006, 1, 2643–2649. [Google Scholar] [CrossRef]
- ANSI/ATCC. ASN-0002-2011. In Authentication of Human Cell Lines: Standardization of STR Profiling; ANSI eStandards Store: Washington, DC, USA, 2012. [Google Scholar]
- Dittmann, J.; Ziegfeld, A.; Jansen, L.; Gajda, M.; Kloten, V.; Dahl, E.; Runnebaum, I.B.; Durst, M.; Backsch, C. Gene expression analysis combined with functional genomics approach identifies ITIH5 as tumor suppressor gene in cervical carcinogenesis. Mol. Carcinog. 2017, 56, 1578–1589. [Google Scholar] [CrossRef]
- Kritsch, D.; Hoffmann, F.; Steinbach, D.; Jansen, L.; Mary Photini, S.; Gajda, M.; Mosig, A.S.; Sonnemann, J.; Peters, S.; Melnikova, M.; et al. Tribbles 2 mediates cisplatin sensitivity and DNA damage response in epithelial ovarian cancer. Int. J. Cancer 2017, 141, 1600–1614. [Google Scholar] [CrossRef]
- Melnikova, M.; Thomale, J. Visualization and Quantitative Measurement of Drug-Induced Platinum Adducts in the Nuclear DNA of Individual Cells by an Immuno-Cytological Assay. Methods Mol. Biol. 2018, 1655, 351–358. [Google Scholar]
- Cisyk, A.L.; Penner-Goeke, S.; Lichtensztejn, Z.; Nugent, Z.; Wightman, R.H.; Singh, H.; McManus, K.J. Characterizing the prevalence of chromosome instability in interval colorectal cancer. Neoplasia 2015, 17, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Penner-Goeke, S.; Lichtensztejn, Z.; Neufeld, M.; Ali, J.L.; Altman, A.D.; Nachtigal, M.W.; McManus, K.J. The temporal dynamics of chromosome instability in ovarian cancer cell lines and primary patient samples. PLoS Genet. 2017, 13, e1006707. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.L.; McManus, K.J. A novel multiplexed, image-based approach to detect phenotypes that underlie chromosome instability in human cells. PLoS ONE 2015, 10, e0123200. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.L.; Jeusset, L.M.; Lepage, C.C.; McManus, K.J. Evolving Therapeutic Strategies to Exploit Chromosome Instability in Cancer. Cancers 2017, 9, 151. [Google Scholar] [CrossRef] [PubMed]
- Lepage, C.C.; Morden, C.R.; Palmer, M.C.L.; Nachtigal, M.W.; McManus, K.J. Detecting Chromosome Instability in Cancer: Approaches to Resolve Cell-to-Cell Heterogeneity. Cancers 2019, 11, 226. [Google Scholar] [CrossRef] [PubMed]
- Spengler, K.; Grosse, S.; Kryeziu, N.; Knierim, A.; Heller, R. Studying the Role of AMPK in Angiogenesis. Methods Mol. Biol. 2018, 1732, 519–537. [Google Scholar] [PubMed]
- Ewels, P.; Magnusson, M.; Lundin, S.; Kaller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Kopylova, E.; Noe, L.; Touzet, H. SortMeRNA: Fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 2012, 28, 3211–3217. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, F.; Achuthan, P.; Akanni, W.; Allen, J.; Amode, M.R.; Armean, I.M.; Bennett, R.; Bhai, J.; Billis, K.; Boddu, S.; et al. Ensembl 2019. Nucleic Acids Res. 2019, 47, D745–D751. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Huntley, M.A.; Larson, J.L.; Chaivorapol, C.; Becker, G.; Lawrence, M.; Hackney, J.A.; Kaminker, J.S. ReportingTools: An automated result processing and presentation toolkit for high-throughput genomic analyses. Bioinformatics 2013, 29, 3220–3221. [Google Scholar] [CrossRef] [PubMed]
- Gerst, R.; Hölzer, M. PCAGO: An interactive web service to analyze RNA-Seq data with principal component analysis. bioRxiv 2018. [Google Scholar] [CrossRef]
- Varemo, L.; Nielsen, J.; Nookaew, I. Enriching the gene set analysis of genome-wide data by incorporating directionality of gene expression and combining statistical hypotheses and methods. Nucleic Acids Res. 2013, 41, 4378–4391. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef]
- Liao, Y.; Wang, J.; Jaehnig, E.J.; Shi, Z.; Zhang, B. WebGestalt 2019: Gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019, 47, W199–W205. [Google Scholar] [CrossRef]
- Supek, F.; Bosnjak, M.; Skunca, N.; Smuc, T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, R.S.; Abreu, R.B.; Velkova, A.; Marsillac, S.; Rodarte, R.S.; Suarez-Kurtz, G.; Iversen, E.S.; Monteiro, A.N.; Carvalho, M.A. Probing structure-function relationships in missense variants in the carboxy-terminal region of BRCA1. PLoS ONE 2014, 9, e97766. [Google Scholar] [CrossRef] [PubMed]
- Dumont, M.; Gamba, R.; Gestraud, P.; Klaasen, S.; Worrall, J.T.; De Vries, S.G.; Boudreau, V.; Salinas-Luypaert, C.; Maddox, P.S.; Lens, S.M.; et al. Human chromosome-specific aneuploidy is influenced by DNA-dependent centromeric features. EMBO J. 2020, 39, e102924. [Google Scholar] [CrossRef] [PubMed]
- Worrall, J.T.; Tamura, N.; Mazzagatti, A.; Shaikh, N.; van Lingen, T.; Bakker, B.; Spierings, D.C.J.; Vladimirou, E.; Foijer, F.; McClelland, S.E. Non-random Mis-segregation of Human Chromosomes. Cell Rep. 2018, 23, 3366–3380. [Google Scholar] [CrossRef]
- Chen, J.; Zhu, F.; Weaks, R.L.; Biswas, A.K.; Guo, R.; Li, Y.; Johnson, D.G. E2F1 promotes the recruitment of DNA repair factors to sites of DNA double-strand breaks. Cell Cycle 2011, 10, 1287–1294. [Google Scholar] [CrossRef]
- Karginova, O.; Siegel, M.B.; Van Swearingen, A.E.; Deal, A.M.; Adamo, B.; Sambade, M.J.; Bazyar, S.; Nikolaishvili-Feinberg, N.; Bash, R.; O’Neal, S.; et al. Efficacy of Carboplatin Alone and in Combination with ABT888 in Intracranial Murine Models of BRCA-Mutated and BRCA-Wild-Type Triple-Negative Breast Cancer. Mol. Cancer Ther. 2015, 14, 920–930. [Google Scholar] [CrossRef]
- Rego, M.A.; Harney, J.A.; Mauro, M.; Shen, M.; Howlett, N.G. Regulation of the activation of the Fanconi anemia pathway by the p21 cyclin-dependent kinase inhibitor. Oncogene 2012, 31, 366–375. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, X.; Xu, X.; Chen, L.; Li, W.; Yu, H.; Sun, Y.; Zeng, J.; Jia, J. RUNX3 inhibits survivin expression and induces cell apoptosis in gastric cancer. Eur. J. Cell Biol. 2014, 93, 118–126. [Google Scholar] [CrossRef]
- Marino, N.; Collins, J.W.; Shen, C.; Caplen, N.J.; Merchant, A.S.; Gokmen-Polar, Y.; Goswami, C.P.; Hoshino, T.; Qian, Y.; Sledge, G.W., Jr.; et al. Identification and validation of genes with expression patterns inverse to multiple metastasis suppressor genes in breast cancer cell lines. Clin. Exp. Metastasis 2014, 31, 771–786. [Google Scholar] [CrossRef]
- Sponziello, M.; Verrienti, A.; Rosignolo, F.; De Rose, R.F.; Pecce, V.; Maggisano, V.; Durante, C.; Bulotta, S.; Damante, G.; Giacomelli, L.; et al. PDE5 expression in human thyroid tumors and effects of PDE5 inhibitors on growth and migration of cancer cells. Endocrine 2015, 50, 434–441. [Google Scholar] [CrossRef]
- Cheon, D.J.; Tong, Y.; Sim, M.S.; Dering, J.; Berel, D.; Cui, X.; Lester, J.; Beach, J.A.; Tighiouart, M.; Walts, A.E.; et al. A collagen-remodeling gene signature regulated by TGF-beta signaling is associated with metastasis and poor survival in serous ovarian cancer. Clin. Cancer Res. 2014, 20, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Egawa, T.; Tillman, R.E.; Naoe, Y.; Taniuchi, I.; Littman, D.R. The role of the Runx transcription factors in thymocyte differentiation and in homeostasis of naive T cells. J. Exp. Med. 2007, 204, 1945–1957. [Google Scholar] [CrossRef] [PubMed]
- Lim, B.; Ju, H.; Kim, M.; Kang, C. Increased genetic susceptibility to intestinal-type gastric cancer is associated with increased activity of the RUNX3 distal promoter. Cancer 2011, 117, 5161–5171. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Category a | Affected Pathways | ngenes | % | p-Value | FDR | Genes b |
|---|---|---|---|---|---|---|
| I RUNX3 core (46 genes) | GO:0001764~neuron migration | 4 | 9.09 | 0.0018 | >0.05 | GATA2, TUBB2B, NAV1, DDIT4 |
| GO:0030574~collagen catabolic process | 3 | 6.82 | 0.0097 | >0.05 | COL6A2, COL15A1, COL5A1 | |
| GO:0030198~extracellular matrix organization | 4 | 9.09 | 0.0105 | >0.05 | ITGA5, SERPINE1, COL6A2, COL5A1 | |
| GO:0001525~angiogenesis | 4 | 9.09 | 0.0149 | >0.05 | NRP2, ITGA5, SERPINE1, COL15A1 | |
| GO:0035313~wound healing | 2 | 4.55 | 0.0207 | >0.05 | ITGA5, COL5A1 | |
| GO:0007155~cell adhesion | 5 | 11.36 | 0.0213 | >0.05 | NRP2, ITGA5, COL6A2, COL15A1, COL5A1 | |
| GO:0031589~cell–substrate adhesion | 2 | 4.55 | 0.0388 | >0.05 | CORO1A, ITGA5 | |
| I TV1-regulated (core+6 genes) | GO:0007165~signal transduction | 8 | 16.33 | 0.0238 | >0.05 | PSD, ARRB1, TLR1, PDE5A, SRGAP3, COL15A1, GDF15, DAPK1 |
| I TV2-regulated (core+7 genes) | GO:0030198~extracellular matrix organization | 5 | 9.8 | 0.0018 | >0.05 | ITGA5, SERPINE1, COL6A2, CDH1, COL5A1 |
| GO:0048565~digestive tract development | 3 | 5.88 | 0.0039 | >0.05 | GATA2, FAT4, DCHS1 | |
| GO:0007157~heterophilic cell–cell adhesion | 3 | 5.88 | 0.0079 | >0.05 | FAT4, ITGA5, DCHS1 | |
| GO:0072137~mesenchymal cell proliferation | 2 | 3.92 | 0.0080 | >0.05 | FAT4, DCHS1 | |
| GO:0007156~homophilic cell adhesion | 4 | 7.84 | 0.0087 | >0.05 | DCHS2, FAT4, CDH1, DCHS1 | |
| GO:0043931~ossification in bone maturation | 2 | 3.92 | 0.0186 | >0.05 | FAT4, DCHS1 | |
| II TV1/2 DEG (1070 genes) | GO:0043062~extracellular structure organization | 91 | 8.50 | 0 | 0 | CDH1, FN1, PDGFB, PECAM1, TNXB |
| GO:0050673~epithelial cell proliferation | 73 | 6.82 | 2.22 × 10−15 | 9.43 × 10−13 | CCL2, FGF1, GATA2, NGFR. WNT5A | |
| GO:0001525~angiogenesis | 84 | 7.85 | 3.99 × 10−14 | 1.05 × 10−11 | ANGPT1, EGF, GJA5, THBS2, VEGFC | |
| GO:0002009~morphogenesis of an epithelium | 83 | 7.76 | 4.95 × 10−14 | 1.05 × 10−11 | AREG, DLG3, ESRP2, VDR, WT1 | |
| GO:0001667~ameboidal-type cell migration | 71 | 6.64 | 8.37 × 10−14 | 1.42 × 10−11 | ANXA6, DPP4, MEF2C, TACSTD2, TWIST1 | |
| GO:0043588~skin development | 73 | 6.82 | 3.53 × 10−13 | 5.00 × 10−11 | BCL2, CDH3, DSC2, KDF1, KRT | |
| GO:0061564~axon development | 81 | 7.57 | 1.21 × 10−12 | 1.47 × 10−10 | APOE,CRABP2, CXCL12, GDNF, NCAM1 | |
| GO:0090130~tissue migration | 56 | 5.23 | 3.12 × 10−12 | 3.31 × 10−10 | ADAMTS9, CDH13, SEMA5A, SLIT2, ZEB2 | |
| GO:0001655~urogenital system development | 60 | 5.61 | 1.27 × 10−11 | 1.16 × 10−9 | ACE, AR, ESR1, FOXB1, IRX3 | |
| GO:0034330~cell junction organization | 55 | 5.14 | 1.37 × 10−11 | 1.16 × 10−9 | CADM1, GJA1, JUP, OCLN, PKP1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heinze, K.; Hölzer, M.; Ungelenk, M.; Gerth, M.; Thomale, J.; Heller, R.; Morden, C.R.; McManus, K.J.; Mosig, A.S.; Dürst, M.; et al. RUNX3 Transcript Variants Have Distinct Roles in Ovarian Carcinoma and Differently Influence Platinum Sensitivity and Angiogenesis. Cancers 2021, 13, 476. https://doi.org/10.3390/cancers13030476
Heinze K, Hölzer M, Ungelenk M, Gerth M, Thomale J, Heller R, Morden CR, McManus KJ, Mosig AS, Dürst M, et al. RUNX3 Transcript Variants Have Distinct Roles in Ovarian Carcinoma and Differently Influence Platinum Sensitivity and Angiogenesis. Cancers. 2021; 13(3):476. https://doi.org/10.3390/cancers13030476
Chicago/Turabian StyleHeinze, Karolin, Martin Hölzer, Martin Ungelenk, Melanie Gerth, Jürgen Thomale, Regine Heller, Claire R. Morden, Kirk J. McManus, Alexander S. Mosig, Matthias Dürst, and et al. 2021. "RUNX3 Transcript Variants Have Distinct Roles in Ovarian Carcinoma and Differently Influence Platinum Sensitivity and Angiogenesis" Cancers 13, no. 3: 476. https://doi.org/10.3390/cancers13030476
APA StyleHeinze, K., Hölzer, M., Ungelenk, M., Gerth, M., Thomale, J., Heller, R., Morden, C. R., McManus, K. J., Mosig, A. S., Dürst, M., Runnebaum, I. B., & Häfner, N. (2021). RUNX3 Transcript Variants Have Distinct Roles in Ovarian Carcinoma and Differently Influence Platinum Sensitivity and Angiogenesis. Cancers, 13(3), 476. https://doi.org/10.3390/cancers13030476