Simple Summary
Metabolic reprogramming is gaining attentions as a hallmark of cancers. However, lipid metabolism has been difficult to analyze due to technical problems. In recent years, lipidomics techniques such as mass spectrometry have advanced and allowed us to analyze detailed lipid profiles of cancers. Moreover, it has become clear that changes in lipid metabolism also play an important role in the interaction between the cancers and the surrounding microenvironment. This review summarizes the latest research progress of reprogrammed lipid metabolism and also lipidomics technologies applied in cancer research.
Abstract
Lipids in our body, which are mainly composed of fatty acids, triacylglycerides, sphingolipids, phospholipids, and cholesterol, play important roles at the cellular level. In addition to being energy sources and structural components of biological membranes, several types of lipids serve as signaling molecules or secondary messengers. Metabolic reprogramming has been recognized as a hallmark of cancer, but changes in lipid metabolism in cancer have received less attention compared to glucose or glutamine metabolism. However, recent innovations in mass spectrometry- and chromatography-based lipidomics technologies have increased our understanding of the role of lipids in cancer. Changes in lipid metabolism, so-called “lipid metabolic reprogramming”, can affect cellular functions including the cell cycle, proliferation, growth, and differentiation, leading to carcinogenesis. Moreover, interactions between cancer cells and adjacent immune cells through altered lipid metabolism are known to support tumor growth and progression. Characterization of cancer-specific lipid metabolism can be used to identify novel metabolic targets for cancer treatment, and indeed, several clinical trials are currently underway. Thus, we discuss the latest findings on the roles of lipid metabolism in cancer biology and introduce current advances in lipidomics technologies, focusing on their applications in cancer research.
1. Introduction
Lipids consist of numerous water-insoluble molecules and are mainly classified as fatty acids, triacylglycerides, phospholipids, or cholesterol. Lipids are widely distributed in cellular organelles and serve as building blocks of all the membranes. In addition to being components of biological membranes, lipids play an important role as energy sources, signaling molecules, and second messengers. Changes in metabolism have been considered a major characteristic of cancers. The most well-studied metabolic change is modification of glycolysis, the so-called Warburg effect [1]. Although research on the modification of cancer metabolism has increased since this discovery, it has mainly focused on the glycolytic system and glutamine metabolism. On the other hand, research on lipids, which are essential for living organisms, has been limited because they are water-insoluble; chemically unstable; and, unlike proteins, are not encoded by genes and must be analyzed directly using chemical methods. However, recent technological advances, such as mass spectrometry (MS), have fostered research into lipidomics. Lipidomics is the study of the structure and function of the complete set of lipids (the lipidome) produced in a given cell or organism, as well as their interaction with other lipids, proteins, and metabolites. Due to the wide range of polarities and the presence of structural analogs and isomers, lipid analysis requires advanced separation techniques. For the separation of lipids, not only the mass spectrometer alone but also the combination of various chromatography techniques have been used in different ways, depending on the target substance. Furthermore, new techniques known as mass spectrometry imaging (MSI) can be used for lipid localization to expand lipidomics further. The field of lipidomics, pioneered by these new technologies, is increasing our understanding of lipid metabolism reprogramming in cancers, and new therapeutic targets focusing on lipid metabolism have been identified. Therefore, in this review, we discuss the characteristic lipid metabolic changes in cancers and their pathological implications, as well as lipidomic techniques that have been applied.
2. Cell-Intrinsic Effects of Lipid Metabolic Reprogramming in Cancer Progression
2.1. Fatty Acids
2.1.1. Basics of Fatty Acid Metabolism
Fatty acids (FAs) are important as the basic backbone of many lipids. The de novo FA synthesis pathway converts citrate to palmitic acid through sequential enzymatic reactions catalyzed by ATP citrate lyase (ACLY), acetyl-CoA carboxylase (ACC), and fatty acid synthase (FASN). Palmitic acid can be elongated by elongases (ELOVLs). These FAs can also be saturated and converted to monounsaturated fatty acids (MUFAs) by stearoyl-CoA desaturase (SCD) [2,3,4,5]. In addition to de novo synthesis, cells acquire FAs through uptake from extracellular sources. Lipid uptake is conducted through membrane-associated transport proteins, including fatty acid transport protein-1 (FATP1), fatty acid translocase (CD36), and liver fatty acid-binding protein (L-FABP). The expression of these enzymes related to FA metabolism are mainly regulated by sterol regulatory element-binding protein 1 (SREBP-1), known as the master transcription factor of lipogenesis [6].
To be used in metabolic pathways, FAs must be activated by acyl-CoA synthetase (ACSLs), which converts free FAs to acyl-CoA. In the fatty acid oxidation (FAO) process, the rate-limiting step is the translocation of acyl-CoA across the mitochondrial membrane. Through this translocation, acyl-CoA is first converted to acylcarnitine via its conjugation to carnitine by carnitine palmitoyltransferase 1 (CPT1). Acylcarnitine is then translocated into the mitochondria via carnitine acylcarnitine translocase (CACT) and finally converted back to acyl-CoA by CPT2. Acyl-CoA then enters the FAO pathway and is followed by the tricarboxylic acid (TCA) cycle (Figure 1).
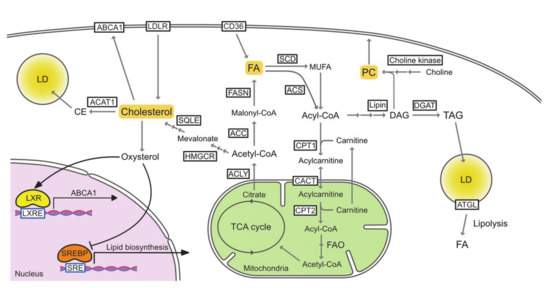
Figure 1.
The pathways of lipid metabolism. ABCA1, ATP-binding cassette transporter A1; ACAT1, acetyl-CoA acetyltransferase 1; ACC, acetyl-CoA carboxylase; Acetyl-CoA, acetyl-coenzyme A; ACLY, ATP citrate lyase; ACS, acyl-CoA synthetase; Acyl-CoA, acyl-coenzyme A; ATGL, adipose triglyceride lipase; CACT, carnitine acylcarnitine translocase; CE, cholesterol ester; CPT1, carnitine palmitoyltransferase 1; CPT2, carnitine palmitoyltransferase 2; DAG, diasylglycerol; DGAT, diglyceride acyltransferase; FA, fatty acid; FAO, fatty acid oxidation; FASN, fatty acid synthase; HMGCR, 3-hydroxy-3-methylglutaryl-CoA reductase; LD, lipid droplet; LDLR, low-density lipoprotein receptor; LXR, liver X receptor; LXRE, LXR response element; MUFA, monounsaturated fatty acid; PC, phosphatidylcholine; SCD, stearoly-CoA desaturase; SRE, sterol regulatory element; SREBP, sterol regulatory element-binding protein; SQLE, squalene epoxidase; TAG, triacylglycerol; TCA, tricarboxylic acid.
2.1.2. Reprogrammed Fatty Acid Metabolism in Cancer Cells
De novo fatty acid synthesis is activated in several cancers and has been extensively reviewed elsewhere [2,7,8,9]. Therefore, we briefly mention the change in de novo FA synthesis in cancer cells. Enzymes that catalyse de novo FA synthesis including ACLY, ACC, FASN, and SCD are upregulated in numerous cancers [9,10,11,12,13,14]. Several chemical inhibitors targeting these enzymes are currently in preclinical and clinical trials for cancer treatment. We summarize these inhibitors in Table 1.

Table 1.
Drugs targeting de novo FA synthesis pathway for cancer treatment.
In addition to de novo synthesis, interrupting FA uptake is known to be effective in cancer therapy. CD36 expression is upregulated in ovarian cancer, gastric cancer, glioblastoma, and oral squamous cell carcinoma [43]. The anti-tumor effect of SCD inhibitors can be reversed by exogenous oleic acid, but in CD36 knockdown breast cancer cells, the effect cannot be reversed [37]. Thus, combination therapy targeting lipogenesis and lipid uptake would be a promising approach. Two CD36-targeting drugs, ABT-510 and CVX-045, reached clinical trials but eventually both failed due to severe adverse effects and unsatisfactory efficacy [44]. Preclinical studies with other agents are currently underway [43].
Cancer cells frequently show changes in enzymes involved in FAO. FAO is an important bioenergetic pathway in many cancers that promotes proliferation, metastasis, stemness, and resistance to treatment [45,46]. Overexpression of CPT1 is associated with tumor progression in several types of cancers such as breast cancer, gastric cancer, and prostate cancer [47]. CPT1 inhibitors such as etomoxir and phexiline were expected to have antitumor effects but have not progressed to clinical studies due to their strong side effects [47]. Nevertheless, a more selective CPT1 inhibitor named ST1326 shows cytotoxic effects against acute myelogenous leukemia (AML) cells and is expected to be clinically applied [48]. On the other hand, suppression of FAO is sometimes beneficial for cancer cell growth. We previously reported that, in patients with nonalcoholic steatohepatitis (NASH), CPT2 downregulation-mediated suppression of FAO not only enables hepatocellular carcinoma (HCC) cells to escape lipotoxicity, but also promotes hepatocarcinogenesis through the accumulation of acylcarnitine as an oncometabolite [49]. Decreased expression of CPT2 in HCC have been confirmed by other recent studies [50,51]. In colon cancer, CPT2 expression is negatively correlated with tumor stage and low expression of CPT2 is associated with poor prognosis [52]. Glioblastoma cells store excess FAs into triglycerides and lipid droplets (LDs) by upregulating diacylglycerol-acyltransferase 1 (DGAT1), and inhibition of DGAT1 promotes glioblastoma cell death through excessive FAO-mediated reactive oxidative species (ROS) production [53]. These findings suggest that the role of FAO in carcinogenesis varies depending on cancer type and the surrounding microenvironment. Of note, we found that an altered serum acylcarnitine profile caused by downregulation of CPT2 is a useful marker for predicting HCC in patients with NASH [54]. Therefore, specific metabolites generated by the altered lipid metabolism may be biomarkers for cancer.
2.2. Cholesterol
2.2.1. Basics of Cholesterol Metabolism
Cholesterol is present in all cell membranes and plays an important role in the regulation of membrane function. The cholesterol content can control the fluidity and flexibility of the membrane. In addition, when present with sphingolipids, the two form clusters known as lipid rafts, regulating the two-dimensional distribution of membrane proteins. Typically, signal transduction-related proteins are believed to rely on these rafts. In mammalian cells, cholesterol is synthesized from acetyl-CoA through the mevalonate pathway. First, hydroxymethylglutaryl-CoA (HMG-CoA) is synthesized from three acetyl-CoA molecules. HMG-CoA is then reduced to mevalonate by HMG-CoA reductase (HMGCR). In a series of enzymatic reactions, mevalonate is converted to farnesyl pyrophosphate (FPP). The two FPP molecules are condensed to squalene and then oxidized by squalene epoxidase (SQLE) to 2,3-epoxy squalene, which is cyclized to lanosterol. Lanosterol is eventually converted to cholesterol [55].
Besides cholesterol biosynthesis, most cells acquire extracellular cholesterol from low-density lipoprotein (LDL) via the LDL receptor (LDLR) [56]. On the other hand, excess cholesterol is exported from cells by ATP-binding cassette (ABC) transporters, including ABCA1 and ABCGs, or converted to less toxic cholesteryl esters (CEs) by acyl CoA:cholesterol acyltransferases (ACATs). These CEs are stored in LDs or secreted into lipoproteins [57].
Cholesterol concentrations are tightly controlled by SREBP-2, liver X receptor (LXR), and nuclear factor erythroid 2-related factor-1 (NRF1). Accumulation of cholesterol and cholesterol-derived oxysterols inactivate the SREBP-2 pathway by inducing the retention of the SREBP cleavage-activating protein (SCAP)–SREBP-2 complex in the endoplasmic reticulum (ER) via the insulin-inducing gene (INSIG), which downregulates the biosynthesis and uptake of cholesterol [58]. On the other hand, desmosterol and oxysterol bind and activate LXRs, which enhances the expression of genes involved in cholesterol efflux [59]. High cholesterol levels inhibit nuclear translocation of NRF1 and restore the LXR pathway that is blocked by NRF1 [60]. In cholesterol deficiency, the three regulatory pathways work in a coordinated manner to increase cholesterol biosynthesis and uptake, while decreasing cholesterol efflux and esterification (Figure 1).
2.2.2. Reprogrammed Cholesterol Metabolism in Cancer Cells
Because of their rapid proliferation, cancer cells require high levels of cholesterol, and increased cholesterol biosynthesis is a hallmark of many cancers (Figure 2). SREBP2 and its target genes are markedly upregulated in prostate cancer, breast cancer, and HCC [61]. Cholesterol biosynthesis also plays an important role in the maintenance of cancer stem cells by activating sonic hedgehog and Notch pathway [62].
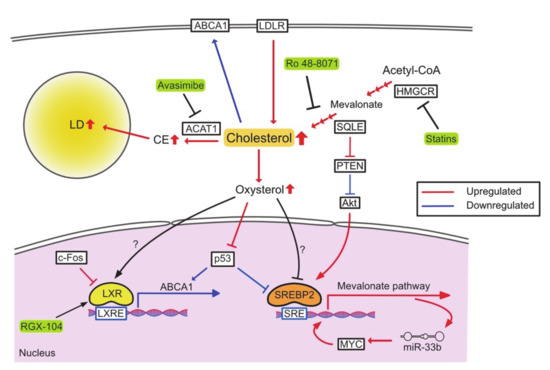
Figure 2.
Reprogrammed cholesterol metabolism and therapeutic targets. PTEN, phosphatase and tensin homolog deleted from chromosome 10.
Since de novo cholesterol synthesis is time- and energy-consuming, some cancers such as glioblastoma and pancreatic cancer utilize exogenous cholesterol [63,64]. Some anaplastic large cell lymphomas lack SQLE and aggressively upregulate LDLR to acquire cholesterol [65]. Furthermore, loss of SQLE leads to the upstream accumulation of squalene, which protects cells from ferroptosis by maintaining an appropriate composition of membranous polyunsaturated fatty acids (PUFAs).
Accumulation of CE is another characteristic of cancer. ACAT1 plays a tumor-promoting role in pancreatic cancer and lymphocytic leukemia [66,67]. Oxysterols are other cholesterol metabolites found abundantly in the tumor microenvironment (TME) [68]. In estrogen receptor-positive breast cancer patients, 27-hydroxycholesterol (27HC) is accumulated in both breast tissue and tumor [69]. 27HC promotes cell proliferation by enhancing the function of p53 E3 ligase MDM2 and inhibiting p53 activation in breast cancer cells [70]. On the other hand, oxidized sterols are known to be LXR ligands and inhibit cell proliferation [71]. 27HC, 24(R/S), and 25-epoxycholesterol inhibit growth and metastasis of gastric cancer by activating LXR signaling [72]. Thus, the roles of 27HC in cancers remain contradictory [73].
The acquisition of oncogenes and the loss of tumor suppressor genes are associated with changes in cholesterol metabolism. The oncogene MYC is required for upregulation of the mevalonate pathway in brain tumor cells, and the upregulation of this pathway forms a positive feedback that increases the expression of the microRNA miR-33b, which in turn increases expression of MYC [74]. In hepatocytes, transgenic expression of c-Fos, an oncogene, suppresses LXR signaling and increases the synthesis of cholesterol and cholesterol metabolites such as oxysterol and bile acids. This leads to increased inflammation and hepatocarcinogenesis [75]. On the other hand, p53 suppresses the mevalonate pathway by upregulating the expression of ABCA1 and limiting the activation of SREBP2 [76]. In addition to de novo cholesterol synthesis, tumor suppressor genes also restrict cholesterol uptake and esterification. In prostate cancer, loss of phosphatase and tensin homolog (PTEN) activates the PI3K-Akt pathway and increases cholesterol uptake and esterification, leading to CE accumulation [77]. SQLE expression is upregulated in NASH-derived HCC, which suppresses PTEN expression and activates Akt signaling, thereby increasing CE and inducing carcinogenesis [78].
Inhibition of cholesterol metabolism has been considered a feasible anti-tumor therapy [79,80]. Statins, well-known HMGCR inhibitors, reduce mortality in various cancer types, regardless of use before or after diagnosis [81,82,83,84], and have also been studied as anti-tumor drugs in clinical trials [85,86]. Another cholesterol synthetic enzyme, SQLE, is considered to be a target for anti-tumor therapy [87]. Several drugs for SQLE have been certified as antifungal agents and are being investigated for use as antitumor agents [55]. Ro 48-8071 (oxidosqualene cyclase inhibitor) significantly inhibits growth and metastasis of colorectal and pancreatic cancer [88]. Co-therapy of this inhibitor with 5-fluorouracil (5-FU) also shows a higher antitumor effect. Targeting cholesterol uptake with LXR agonists could cause LDLR degradation and increase the expression of ABCA1, promoting tumor cell death in numerous cancers [89,90,91]. Inhibition of cholesterol esterification is also an effective approach. CE inhibition by the ACAT1 inhibitor avasimibe suppresses tumor growth and restores imatinib sensitivity by downregulating MAPK signaling in imatinib-resistant myeloid leukemia [92]. In prostate cancer, avasimibe inhibits metastasis by blocking the Wnt-β-catenin pathway [93]. Avasimin (human albumin-capsulated avasimibe) specifically induces tumor apoptosis in a tumor xenograft model [94].
2.3. Triacylglycerol/Lipid Droplet
2.3.1. Basics of Triacylglycerol/Lipid Droplet Metabolism
Triacylglycerol (TAG) is composed of three fatty acids esterified to a glycerol molecule. The basic role of this molecule is storing fatty acids and promoting efficient transport. The largest source of TAG is the diet, and each cell takes up TAG as FAs. In adipocytes and hepatocytes, these FAs can be resynthesized into TAG. Acyl-CoA derived from FA is converted to TAG through a series of reactions catalyzed by glycerol-3-phosphate acyltransferase (GPAT), acylglycerol phosphate acyltransferase (AGPAT), phosphatidic acid phosphohydrolase (Lipin), and diacylglycerol acyltransferase (DGAT) [3]. TAGs are then sequestered within LDs.
LD is an organelle that has a neutral lipid core that is surrounded by a phospholipid monolayer. The internal lipid is mainly composed of TAG, but also contains CEs and acyl ceramides. The formation of LDs begins with de novo synthesis of TAGs and CEs between the lipid bilayers of the ER [95]. When sufficient lipids have accumulated in the ER intermembrane lens, they are released into the cytoplasm to form LDs. LDs are dynamically synthesized and degraded in response to the extracellular nutrient environment. LDs are mainly degraded by two mechanisms, namely, lipolysis and lipophagy [96,97]. In lipolysis, FAs can be supplied from TAGs in reactions with adipose triglyceride lipase (ATGL), hormone-sensitive lipase (HSL), and monoacylglycerol lipase (MAGL) [98,99] (Figure 1). Lipophagy is a recently discovered form of autophagy in which LDs are incorporated into autophagosomal membranes, fused with lysosomes, and hydrolyzed [100]. FAs supplied by the degradation of LDs fuel the mitochondrial oxidative metabolism during nutrient-deprived conditions in various cells [101]. On the other hand, lipolysis may lead to lipotoxicity. Excess lipolysis increases harmful free FAs in the cytoplasm and also increases FA oxidation in mitochondria, leading to the production of ROS [102]. Perilipin 5 inhibits ATGL-mediated lipolysis and protects tissues against these forms of oxidative stress [103].
2.3.2. Reprogrammed Triacylglycerol/Lipid Droplet Metabolism in Cancer Cells
During carcinogenesis, the demand for FA increases to support rapid proliferation. FA is supplied not only by increased de novo synthesis but also by increased uptake from extracellular sources and recycling of intracellular lipids through autophagy, which requires contribution of lipid droplets. The lipid droplet also plays an important role in defending against ROS and developing resistance to stress from the harsh surrounding environment, such as hypoxia and low nutrition [101,104,105]. Lipid droplets are known to accumulate in various types of cancer [106], and tumors with high concentrations of lipid droplets are associated with a poor prognosis in breast and pancreatic cancer [64,107]. Thus, changes in lipid droplet metabolism are also an important hallmark of cancer.
Hypoxia-associated lipid droplet production has been observed in brain, breast, renal, and prostate cancers [105,108,109,110,111,112,113]. HCC and cervical adenocarcinoma cells exposed to hypoxia accumulate lipid droplets by directly stimulating the expression of Lipin 1 in a HIF-1α-dependent manner [110]. Cancer cells exposed to hypoxia are known to upregulate FA uptake via FABP3/7 and accumulate lipid droplets, enabling ER and redox homeostasis during oxygen deprivation, supplying FA for mitochondrial energy production and promoting cell proliferation after reoxygenation [105].
Recent studies have shown that hypoxia can affect the expression of several lipid droplet-related proteins involved in lipid droplet degradation [114,115]. Hypoxia-inducible lipid droplet-associated protein (HILPDA)/hypoxia-inducible gene 2 (HIG2), localized on the lipid droplet surface, is an inhibitor of ATGLs, which are upregulated by HIF-1 during hypoxia [114,115,116]. HIG2 deficiency promoted lipid droplet degradation and β-oxidation, resulting in elevated ROS production and impaired xenograft tumor growth. Conversely, ATGL knockout (KO) reversed the effect of HIG2 KO, and ATGL inhibition by HIG2 inhibited lipid droplet degradation and isolated FA from mitochondrial oxidation and ROS production, allowing for cancer survival under hypoxic conditions [114]. Thus, lipid droplet degradation by ATGL may damage cancer cells under hypoxic conditions and increasing ATGL activity may be a novel therapeutic strategy. Indeed, ATGL overexpression represses proliferation of melanoma, colon carcinoma, HCC, and lung cancer cell lines [117,118], and also enhances the response of anti-cancer agents against HCC cell lines [118].
The role of lipophagy in cancer remains unclear. Although several reports have shown a tumor-promoting effect of lipophagy [119,120], most of them state that lipophagy inhibits tumorigenesis [121,122,123,124]. Microtubule-associated protein 1S (MAP1S)-mediated lipophagy promotes the elimination of lipid droplets, and high expression of MAP1S is associated with the suppression of tumor growth and metastasis and an improved prognosis in clear-cell renal cell carcinoma [121]. Another study showed that overexpression of ATG14 promotes lipid droplet degradation in HeLa cells and induces free FA accumulation, leading to ER stress and ROS-mediated apoptosis, while inhibition of lipophagy by 3-methyladenine (3-MA) and inhibition of lysosomal acid lipase (LAL) reverses these effects [122]. Recent studies have suggested that LAL plays a role as a tumor suppressor, and that impaired LAL function in mice is associated with spontaneous tumorigenesis. Re-expression of LAL in the liver and lungs improves inflammation and prevents metastasis to the same areas [123,124].
2.4. Phospholipid
2.4.1. Basics of Phospholipid Metabolism
Phospholipids (PLs) are components of the cell membrane and have diverse chemical structures and functions. PLs regulate various cellular functions such as homeostasis, cell adhesion and migration, signal transduction, vesicle transport, apoptosis, and post-translational modifications. Glycerophospholipids (GPLs) are classified into subclasses known as phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidylserine (PS), phosphatidylglycerol (PG), phosphatidylinositol (PI), and phosphatidic acid (PA), depending on the type of polar head group. The fatty acyl moieties of the membrane phospholipids show considerable diversity in chain length and saturation. These two parameters determine the biophysical properties of the cell membrane, such as fluidity, curvature, and subdomain structure. These factors affect membrane-related cellular processes, such as signal transduction, and molecular transport [125].
For de novo PL synthesis, FA is first taken up by phosphatidic acid (PA) as the main precursor of PL. The Kennedy pathway is the primary pathway for the synthesis of PC, which is the most abundant PL head group class [126]. The pathway contains three enzymatic reactions: choline phosphorylation by choline kinase, CDP-choline formation from phosphocholine and CTP catalyzed by CTP:phosphocholine cytidylyltransferase (CCT), and substitution of cytidine monophosphate by diasylglycerol (DAG) to produce PC catalyzed by CDP-choline:1,2-diacylglycerol cholinephosphotransferase (Figure 1). The next most common PL is PE, which can be synthesized de novo but can also be generated from PS by head group exchange. Besides de novo synthesis and head group exchange, the phospholipid composition is maintained through a deacylation and reacylation remodeling process called the Lands’ cycle [127]. Lysophosphatidylcholine acyltransferases (LPCATs) play an important role in lipid metabolism and homeostasis by regulating different PC species in multiple cells and tissue types. LPCAT1 is mainly expressed in alveolar type II cells and catalyzes the generation of dipalmitoyl-PC (DPPC) in lung surfactants [128]. LPCAT2 is highly expressed in inflammatory cells, such as macrophages and neutrophils, and is also present in the skin, colon, spleen, and brain [129]. In contrast, LPCAT3 is more widely expressed and abundant in the testes, kidneys, and metabolic tissues including liver, intestines, and fat. LPCAT4 is selectively expressed in the epididymis, brain, testes, and ovaries [130]. LPCAT1 and LPCAT2 are known to have lyso-platelet activating factor (PAF) acetyltransferase activity in PAF biosynthesis, in addition to LPCAT activity [129]. Most importantly, each LPCAT shows a different acyl-CoA preference. LPCAT1 prefers palmitoyl-CoA (16:0-acyl-CoA) as the acyl donor for synthesizing dipalmitoyl PC [128]. LPCAT2 shows the highest activity in the presence of acetyl-CoA or arachidonoyl CoA (20:4-acyl-CoA) [129]. In contrast, LPCAT3 and LPCAT4 prefer polyunsaturated fatty acyl-CoA (18:2-acyl-CoA or 20:4-acyl-CoA) and oleoyl-CoA (18:1-acyl-CoA) as substrates, respectively [130,131]. Thus, the different substrate preferences and tissue expression patterns of LPCAT contribute to tissue-selective remodeling of membrane PC species.
2.4.2. Reprogrammed Phospholipid Metabolism in Cancer Cells
Many of the enzymes involved in PL synthesis and remodeling are overexpressed in cancer. For example, Lipin-1, which regulates the rate-limiting step in PL synthesis and is a co-regulator of transcription factors such as peroxisome proliferator-activated receptors (PPARs) and SREBPs, is upregulated in a diverse subset of cancer types, including high-grade prostate, colon, lung, and breast cancer [131,132,133]. High levels of Lipin-1 expression are associated with a poor prognosis and inflammation, and downregulation of Lipin-1 induces ER stress and apoptosis and attenuates tumor growth in xenograft mouse models. Choline kinase alpha (ChoKalpha), the first committed enzyme of the Kennedy pathway for PC and PE synthesis, is overexpressed in a variety of tumor types and is activated by various oncogenic events. Activation and overexpression of ChoKalpha is associated with increased cellular requirement for PC. Knockdown or chemical inhibition of ChoKα causes cell death and attenuates tumor growth in vivo [134,135].
Another emerging class of enzymes that appear to be affected in many cancers is LPCAT. Upregulation of LPCAT1 has been observed in clear renal cell carcinoma, oral squamous cell carcinoma, hepatoma, esophageal cancers, gastric cancers, breast cancers, colorectal cancers, and prostate cancers [136]. LPCAT1 expression is correlated with prognosis and survival in clear cell renal cell carcinoma, breast cancer, and prostate cancer [137,138,139,140] and can be used as a diagnostic marker in prostate cancer and esophageal cancer [139,141]. Overexpression of LPCAT1 has been shown to increase cell proliferation, migration, and metastasis in clear cell renal cell carcinoma and HCC cell lines [137,142]. Consistent with LPCAT1 enzyme activity, the level of saturated phospholipids is increased in clear cell renal cell carcinoma, HCC, and gastric cancers [137,142,143]. However, it remains unclear as to how these changes in PC levels regulate the behavior of cancer cells. LPCAT1 may also contribute to tumor growth through lyso-PAF acetyltransferase activity to produce PAF, a lipid mediator that plays an important role in cell growth [144,145]. LPCAT2 has also been reported to be overexpressed in cervical and breast cancers [146], and has been identified as a susceptibility gene for aggressive prostate cancer in animal models and genome-wide association studies in human patients [147]. LPCAT2-mediated LD production is known to contribute to chemotherapy resistance in colorectal cancer. Thus, targeting LPCAT2-mediated intracellular LD formation may be a therapeutic approach to restore chemotherapy sensitivity in colorectal cancer [148]. Loss of LPCAT3 in the mouse gut reduces the composition of polyunsaturated phospholipids and promotes tumor development and growth in Apcmin mice [149]. LPCAT4 is associated with intestinal stem cell proliferation and tumorigenesis, and is also associated with high levels of PC (16:0/16:1) in colorectal cancer [135].
3. Altered Lipid Metabolism and Tumor Microenvironment
In addition to cancer cells, tumors contain a variety of immune-effector and immunosuppressive cells, termed tumor-infiltrating immune cells (TIIs). TIIs range from anti-tumor to tumor-promoting functions, and they depend on the type and stage of the tumor [150]. TIIs include T lymphocytes, B lymphocytes, tumor-associated macrophages (TAMs), dendritic cells (DCs), myeloid derived suppressor cells (MDSCs), neutrophils, and natural killer (NK) cells (Figure 3).
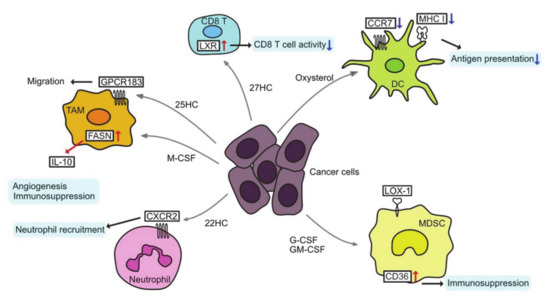
Figure 3.
Altered lipid metabolism and tumor microenvironment. CCR7, C-C chemokine receptor type 7; CXCR2, C-X-C chemokine receptor type 2; DC, dendritic cell; G-CSF, granulocyte colony-stimulating factor; GM-CSF, granulocyte macrophage colony-stimulating factor; GPCR 183, G-protein-coupled receptor 183; HC, hydroxycholesterol; IL-10, interleukin-10; LOX-1, lectin-like oxidized low-density lipoprotein (LDL) receptor-1; M−CSF, macrophage colony stimulating factor; MDSC, myeloid-derived suppressor cell; MHC I, major histocompatibility complex class I; TAM, tumor-associated macrophage.
3.1. Tumor-Associated Macrophages
TAMs can be reprogrammed as a result of changes in cholesterol metabolism. Tumor cells secrete hyaluronan oligomers that increase cholesterol efflux in the TAMs, leading the TAMs to the M2-like phenotype and accelerating tumor progression [151]. 25-Hydroxycholesterol (25HC) interacts with G-protein-coupled receptor 183 (GPCR183) to reconstitute the cytoskeletal protein vimentin, resulting in the migration of macrophages and monocytes [152]. Moreover, in the context of changes in FA metabolism, TAMs can be polarized to a pro-tumoral phenotype. Macrophage colony-stimulating factor (M-CSF) secreted from tumor cells upregulates FASN expression in TAMs, leading to PPARβ/δ activation and IL-10 expression, which is an anti-inflammatory cytokine [153].
3.2. T cells in TME
In CD8 T cells, SREBP2 signaling is essential for proliferation and effector function, whereas LXR signaling negatively regulates T cell activation [154,155]. Therefore, in oxysterol-rich TMEs, T cell tumor immunity may be inhibited by LXR activation. On the other hand, increased cholesterol synthesis and uptake by T cells may enhance the antitumor effect of T cells. Inhibition of ACAT1 in CD8 T cells alters cholesterol synthesis and leads to an accumulation of free cholesterol in the plasma membrane [156]. This cholesterol binds directly to T cell receptors and promotes nanoclustering, which triggers antigen-induced signals that increase cholesterol biosynthesis and uptake [157]. Furthermore, this cholesterol plays a role in the formation of mature immunological synapses for targeted killing of tumor cells. The ACAT1 inhibitor, avasimibe, enhances the therapeutic effect of chimeric antigen receptor (CAR)-T cells by increasing the ratio of cytotoxic CD8 T cells [158]. On the other hand, cholesterol accumulation in TME has been shown to induce ER stress and further increase T cell exhaustion [159]. Thus, the functions of endogenous and exogenous cholesterol may differ.
3.3. Tumor-Associated Dendritic Cells
Conditioned medium of multiple cancer cells activates LXR-alpha signaling in dendritic cells and reduces cell surface expression of CC chemokine receptor-7 (CCR7) [160]. As a result, the migration of dendritic cells from the tumor site to the lymph nodes is inhibited and the presentation of tumor antigens to T cells is suppressed. Inactivation of LXR-α ligand through the inhibition of cholesterol synthesis or expression of SULT2B1b (an enzyme that converts oxysterol to inactive sulfated oxysterol) in tumor-bearing mice restores dendritic cell function and antitumor response [160]. However, it remains unclear as to which oxysterol is responsible for this effect. Tumor-derived factors result in the accumulation of oxidized lipids such as CE, TAG, and FA, which reduce major histocompatibility complex (MHC) class I complexes on the surface of dendritic cells and decrease antigen presentation [161]. LD accumulation in DCs also inhibits their antigen-presenting ability in kidney, thyroid, ovarian, and head and neck cancer [162]. LPCAT-2-dependent lipid droplet accumulation in colorectal cancer causes calreticulin sequestration and prevents its exposure to the plasma membrane, thereby preventing DC maturation and subsequent CD8 T cell infiltration and immunogenic cell death under chemotherapy [148].
3.4. Immunosuppressive Cells
Neutrophils are considered an important immunosuppressive population in TME [163,164]. 22HC is abundant in the conditioned medium of various cancer cells and can mobilize CD11bhighGr1high neutrophils [165]. 22HC mobilizes neutrophils by binding to CXCR2, but not LXR. 24HC and 27HC can also mobilize neutrophils in other carcinomas. Pancreatic neuroendocrine tumors show that HIF1α-induced increases in 24S-HC levels induce neutrophils and angiogenesis [166]. 27HC promotes metastasis by attracting polymorphonuclear neutrophils and γδ T cells and reducing cytotoxic CD8 T cells in a high-cholesterol diet-fed breast cancer model [167].
MDSCs are very similar to neutrophils and are considered as immunosuppressive innate cell populations. They have unique characteristics, and LOX-1 (lectin-type oxidized LDL receptor-1) has recently been characterized as an important marker to distinguish them from neutrophils [168]. Overexpression of LOX-1 has been found in several cancers and is associated with a poor prognosis. LOX-1 is an LDL receptor, suggestive of altered cholesterol metabolism in MDSCs. LXR agonist RGX-104 depletes MDSCs and enhances T cell activity via upregulation of APOE [169]. LXR activation enhances the efficacy of immunotherapies such as immune checkpoint inhibitors and adoptive T cell transplantation [170,171]. In addition, granulocyte colony-stimulating factor (G-CSF) and granulocyte macrophage colony-stimulating factor (GM-CSF) secreted from cancer cells act via signal transducer and activator of transcription (STAT)3/5 in a paracrine manner on MDSCs, resulting in overexpression of CD36 and enhancement in uptake of FAs. Thus, STAT3 or STAT5 inhibition, or CD36 deletion, are known to downregulate lipid metabolism and prevent the immunosuppressive functions of MDSCs [172].
4. Lipidomic Research Techniques
Lipidomics is the large-scale profiling and quantification of lipid molecules, a study that comprehensively examines lipid pathways and interprets their physiological significance on the basis of analytical chemistry and statistical analysis [173,174]. Lipidomic research can be used to accumulate a vast amount of information that quantitatively describes spatial and temporal changes in the content and composition of lipid molecular species after perturbations such as diseases, drugs, and the environment. The goal of lipidomics is to explore the basic mechanisms of lipid metabolism and its changes under pathological conditions, which may reveal biomarkers that can be used to diagnose and treat diseases, target drug discovery, guide precision and personalized medicine, and intervene in dietary habits [175]. The commonly used operating procedures of lipidomics are graphically summarized in Figure 4 [175].
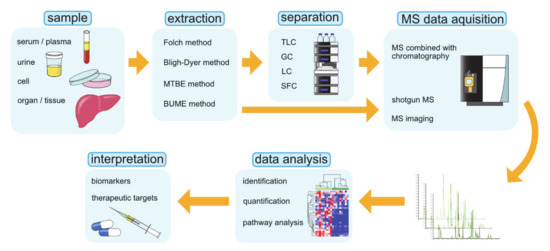
Figure 4.
The operating procedures in lipidomics research. BUME, butanol–methanol; GC, gas chromatography; LC, liquid chromatography; MS, mass spectrometry; MTBE, methyl-tert-butyl-ether; SFC, supercritical fluid chromatography; TLC, thin-layer chromatography.
The advances in techniques such as MS, NMR spectroscopy, and chromatography have contributed greatly to the development of lipidomics. Above all, MS is the fundamental technology that is widely used in lipidomics. MS-based lipidomics can be divided into two subfields, namely, non-targeting lipidomics and targeted lipidomics. Non-targeted lipidomics, or so-called global lipidomics, identifies and quantifies all detected lipids, whereas targeted lipidomics is focused on the analysis of the specific lipid class. While non-targeted lipidomics is a suitable method for capturing the rough profile of lipids in the sample, targeted lipidomics may be more applicable to solving specific biological problems. In lipidomics, three MS methods are mainly used: direct injection shotgun MS, MS combined with chromatographic separations, and mass spectrometric imaging (MSI) [175]. A summary of these techniques is given in Table 2.
Table 2.
Summary of the lipidomics techniques.
4.1. Shotgun MS
Shotgun MS is an analytical method in which lipid extracts are injected directly into the MS without prior chromatographic separation. It is a less time-consuming, more convenient, and more reproducible method compared to other methods. However, shotgun lipidomics is limited for detecting lipid molecules at relatively low concentrations because ionization is suppressed due to the complex matrix, and in many cases, they could not be detected. In addition, with shotgun lipidomics, lipid isomers are not distinguished by shotgun method. To address these problems, collaboration between chromatography equipment and MS is essential.
4.2. MS Coupled with Chromatography
Chromatographic methods are important for reducing matrix effects, separating lipid isomers, and condensing lipid molecules. These techniques are necessary for analysis of a wide variety of lipids from biological samples. Separation techniques include thin-layer chromatography (TLC), gas chromatography (GC), liquid chromatography (LC), and supercritical fluid chromatography (SFC).
4.2.1. TLC: Thin-Layer Chromatography
In TLC, a thin film of adsorbent such as silica gel on a glass or aluminum plate is used as the stationary phase, and liquid is used as the mobile phase. Sample materials can be separated by the variation in the transfer distance due to the difference in the strength of attachment to the adsorbent and the solubility to the solvent. TLC can separate lipid mixtures into individual lipid categories more quickly and inexpensively than LC or GC. However, the most problematic aspect of TLC analysis is separation efficiency and sensitivity. Low separation efficiency and sensitivity results in poor resolution and requires more sample. Therefore, it is not suitable for the separation of small amounts of biological samples. Furthermore, due to its structure, it is not possible to link TLC and MS detection directly to improve sensitivity. TLC and matrix assisted laser desorption/ionization (MALDI)–MS can, typically, be combined offline [186,187].
4.2.2. GC: Gas Chromatography
GC is a separation technique that uses a gas as the mobile phase and is used for the analysis of volatile components. He, N2, H2, etc. are used as the carrier gas, which is the mobile phase. The sample is heated and vaporized, and is introduced into the column by the carrier gas. The sample is separated by the difference in volatility and the interaction with the stationary phase, coated on the inner surface of the column. Therefore, GC is suitable for the separation of volatile compounds that are resistant to heat, and it has a high separation efficiency. For compounds that are difficult to volatilize, the derivatization process can be used for analysis. Derivatization is also used to improve the separation. This combined GC and MS method is currently the dominant method for profiling and quantification of FA and cholesterol [188,189,190]. Its relative low cost is another advantage. On the other hand, there is the disadvantage in that non-volatile substances and substances with low heat stability require prior derivatization processing.
4.2.3. LC: Liquid Chromatography
LC, with its high sensitivity and ease of connection with MS, is the most widely used chromatographic method. The separation modes of LC can be broadly divided into normal-phase and reversed-phase systems. In general, normal-phase separates lipids on the basis of the headgroup of the lipid compound. On the other hand, reversed-phase separates lipids according to the hydrophobicity, length, and number of double bonds of the FA chain. Thus, the separation mode should be chosen on the basis of where in the lipid structure there will be a focus. High-performance liquid chromatography (HPLC) is commonly used in lipidomics due to its high separation power and selectivity. In recent years, ultra high performance liquid chromatography (UHPLC) has been developed and rapidly spread. The use of high-pressure resistant columns and smaller size particles gives UHPLC higher separation efficiencies compared to conventional HPLC. UHPLC–MS enables high sensitivity, high resolution, and high speed of lipidomic analysis. The limitations of LC–MS are consumption of organic solvent, which is not environmentally friendly, requires pre-knowledge for targeted analysis, and involves complex data processing.
4.2.4. SFC: Supercritical Fluid Chromatography
SFC is a high-resolution, high-throughput separation method that uses supercritical fluid (a non-condensable fluid that exceeds the inherent gas–liquid critical point of a substance) as the mobile phase. It has the properties of both GC and HPLC; in addition, it is possible to select a wide range of separation modes by changing the state of the mobile phase. Although supercritical carbon dioxide, which is the most commonly used solvent, is low polarity, the polarity of the mobile phase can be changed by adding a polar organic solvent including methanol as a modifier. Therefore, a wide range of substances, from polar to nonpolar, can be separated. The disadvantage of this technology is that it is new, and thus there is little information about reproducibility, and it is not easily accessible.
4.3. MS Imaging
MSI is a relatively new MS-based imaging technique that uses direct ionization and MS detection to provide visualization and distribution of individual lipid molecules in tissue samples. MALDI, secondary ion mass spectrometry (SIMS), and desorption electrospray ionization (DESI) are the three ionization techniques used in MSI [191]. MALDI–MSI is the most commonly used technique, in which a matrix is applied to the sample surface to assist in the ionization. A wide range of molecules including lipids can be observed without fragmentation and with a spatial resolution of up to several micrometers. SIMS is a technique to obtain secondary ions of sample molecules by irradiating ionized noble gas or metal atoms. SIMS requires no pretreatment and has high spatial resolution on the submicron scale. Because it is a hard ionization method, fragmentation of lipid molecules is likely to occur, making it suitable for analyzing differences in fatty acid composition, headgroups, and other components of lipid molecules with high spatial resolution. DESI extracts, desorbs, and ionizes molecules by spraying charged microdroplets on the sample. It is a soft ionization technique that does not require matrix pretreatment, unlike MALDI, and can detect free fatty acids and lipid mediators, which are difficult to detect with MALDI. The disadvantage of DESI is its low spatial resolution (a few tens of micrometers). Eberlin et al. [183] showed that MUFA-PC is increased in HCC by using LC–MS and MALDI–MSI, and reported its diagnostic, prognostic, and therapeutic potential. Using DESI–MSI, Banerjee et al. [185] reported that cholesterol sulfate is increased in prostate cancer and its precancerous lesion, and can be used as a biomarker. Thus, MSI can be used to explore the visualization and distribution of individual lipid species, allowing us to investigate various biological processes and dynamic spatial distributions associated with lipid interactions.
5. Conclusions
At this time, cancer research has mainly focused on genetic mutations and their expression changes. Recent advances in next-generation sequencers have enabled comprehensive analysis of the genome and transcriptome, which has led to further advances in cancer research. On the other hand, although the importance of the relationship between cancer and lipid metabolism has been recognized, it has been difficult to analyze due to technical problems. However, advances in lipidomics techniques, as described in this review, have allowed us to analyze detailed lipid profiles. It has become clear that the reprogrammed lipid metabolism is not only involved in carcinogenesis through its relationship with oncogenes and tumor suppressor genes but also plays an important role in the interaction with the surrounding cancer environment. Therefore, techniques to analyze the localization of lipids such as MSI may play an important role in elucidating the pathogenesis of cancers. Of the more than 200,000 lipids in the body, only a small fraction can be analyzed at this time [5]. Further analysis of cancer lipid metabolism using more advanced technology is required to develop novel cancer treatments.
Author Contributions
Y.M. wrote the paper and prepared figures, H.N. revised manuscript and prepared figures, K.K. supervised the entire project. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Bristol Myers Squibb Research Grant; Takeda Science Foundation; MSD Life Science Foundation; The Naito Foundation; Life Science Foundation of Japan; The Cell Science Research Foundation; The Mitsubishi Foundation; The FUGAKU TRUST for Medical Research; KAKENHI grant numbers 18H02789 and 20K08376; and AMED under grant numbers JP19fk0210059, JP20fk0210059, JP18fk0210040, JP19fk0210040, and JP20fk0210040.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Currie, E.; Schulze, A.; Zechner, R.; Walther, T.C.; Farese, R.V. Cellular Fatty Acid Metabolism and Cancer. Cell Metab. 2013, 18, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, P. Fatty Acid Metabolism and Cancer Development. Sci. Bull. 2016, 61, 1473–1479. [Google Scholar] [CrossRef]
- Nakagawa, H.; Hayata, Y.; Kawamura, S.; Yamada, T.; Fujiwara, N.; Koike, K. Lipid Metabolic Reprogramming in Hepatocellular Carcinoma. Cancers 2018, 10, 447. [Google Scholar] [CrossRef] [PubMed]
- Butler, L.; Perone, Y.; Dehairs, J.; Lupien, L.E.; de Laat, V.; Talebi, A.; Loda, M.; Kinlaw, W.B.; Swinnen, J.V. Lipids and Cancer: Emerging Roles in Pathogenesis, Diagnosis and Therapeutic Intervention. Adv. Drug Deliv. Rev. 2020. [Google Scholar] [CrossRef] [PubMed]
- Shao, W.; Espenshade, P.J. Expanding Roles for SREBP in Metabolism. Cell Metab. 2012, 16, 414–419. [Google Scholar] [CrossRef]
- Menendez, J.A.; Lupu, R. Fatty Acid Synthase and the Lipogenic Phenotype in Cancer Pathogenesis. Nat. Rev. Cancer 2007, 7, 763–777. [Google Scholar] [CrossRef]
- Röhrig, F.; Schulze, A. The Multifaceted Roles of Fatty Acid Synthesis in Cancer. Nat. Rev. Cancer 2016, 16, 732–749. [Google Scholar] [CrossRef]
- Fhu, C.W.; Ali, A. Fatty Acid Synthase: An Emerging Target in Cancer. Molecules 2020, 25, 3935. [Google Scholar] [CrossRef]
- Icard, P.; Wu, Z.; Fournel, L.; Coquerel, A.; Lincet, H.; Alifano, M. ATP Citrate Lyase: A Central Metabolic Enzyme in Cancer. Cancer Lett. 2020, 471, 125–134. [Google Scholar] [CrossRef]
- Wang, C.; Ma, J.; Zhang, N.; Yang, Q.; Jin, Y.; Wang, Y. The Acetyl-CoA Carboxylase Enzyme: A Target for Cancer Therapy? Expert Rev. Anticancer 2015, 15, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Prins, R.M.; Dang, J.; Kuga, D.; Iwanami, A.; Soto, H.; Lin, K.Y.; Huang, T.T.; Akhavan, D.; Hock, M.B.; et al. EGFR Signaling Through an Akt-SREBP-1–Dependent, Rapamycin-Resistant Pathway Sensitizes Glioblastomas to Antilipogenic Therapy. Sci. Signal. 2009, 2, ra82. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.-W.; Lin, Y.-H.; Pai, M.-H.; Lo, A.-C.; Lee, Y.-C.; Fang, I.-C.; Lin, J.; Hsieh, R.-K.; Chang, Y.-F.; Chen, C.-L. Association between Phosphorylated AMP-Activated Protein Kinase and Acetyl-CoA Carboxylase Expression and Outcome in Patients with Squamous Cell Carcinoma of the Head and Neck. PLoS ONE 2014, 9, e96183. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, K.; Tsukamoto, H. Stearoyl-CoA Desaturase and Tumorigenesis. Chem-biol. Interact. 2019, 316, 108917. [Google Scholar] [CrossRef]
- Guais, A.; Baronzio, G.; Sanders, E.; Campion, F.; Mainini, C.; Fiorentini, G.; Montagnani, F.; Behzadi, M.; Schwartz, L.; Abolhassani, M. Adding a Combination of Hydroxycitrate and Lipoic Acid (METABLOCTM) to Chemotherapy Improves Effectiveness against Tumor Development: Experimental Results and Case Report. Invest. New Drug 2012, 30, 200–211. [Google Scholar] [CrossRef]
- Promkan, M.; Dakeng, S.; Chakrabarty, S.; Bögler, O.; Patmasiriwat, P. The Effectiveness of Cucurbitacin B in BRCA1 Defective Breast Cancer Cells. PLoS ONE 2013, 8, e55732. [Google Scholar] [CrossRef]
- Gao, Y.; Islam, M.S.; Tian, J.; Lui, V.W.Y.; Xiao, D. Inactivation of ATP Citrate Lyase by Cucurbitacin B: A Bioactive Compound from Cucumber, Inhibits Prostate Cancer Growth. Cancer Lett. 2014, 349, 15–25. [Google Scholar] [CrossRef]
- Guseva, N.V.; Rokhlin, O.W.; Glover, R.A.; Cohen, M.B. TOFA (5-Tetradecyl-Oxy-2-Furoic Acid) Reduces Fatty Acid Synthesis, Inhibits Expression of AR, Neuropilin-1 and Mcl-1 and Kills Prostate Cancer Cells Independent of P53 Status. Cancer Biol. Ther. 2011, 12, 80–85. [Google Scholar] [CrossRef]
- Li, S.; Qiu, L.; Wu, B.; Shen, H.; Zhu, J.; Zhou, L.; Gu, L.; Di, W. TOFA Suppresses Ovarian Cancer Cell Growth in Vitro and in Vivo. Mol. Med. Rep. 2013, 8, 373–378. [Google Scholar] [CrossRef]
- Beckers, A.; Organe, S.; Timmermans, L.; Scheys, K.; Peeters, A.; Brusselmans, K.; Verhoeven, G.; Swinnen, J.V. Chemical Inhibition of Acetyl-CoA Carboxylase Induces Growth Arrest and Cytotoxicity Selectively in Cancer Cells. Cancer Res. 2007, 67, 8180–8187. [Google Scholar] [CrossRef]
- Li, E.-Q.; Zhao, W.; Zhang, C.; Qin, L.-Z.; Liu, S.-J.; Feng, Z.-Q.; Wen, X.; Chen, C.-P. Synthesis and Anti-Cancer Activity of ND-646 and Its Derivatives as Acetyl-CoA Carboxylase 1 Inhibitors. Eur. J. Pharm. Sci. 2019, 137, 105010. [Google Scholar] [CrossRef] [PubMed]
- Svensson, R.U.; Parker, S.J.; Eichner, L.J.; Kolar, M.J.; Wallace, M.; Brun, S.N.; Lombardo, P.S.; Nostrand, J.L.V.; Hutchins, A.; Vera, L.; et al. Inhibition of Acetyl-CoA Carboxylase Suppresses Fatty Acid Synthesis and Tumor Growth of Non-Small-Cell Lung Cancer in Preclinical Models. Nat. Med. 2016, 22, 1108–1119. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.-W.; Addy, C.; Kusunoki, J.; Anderson, N.N.; Deja, S.; Fu, X.; Burgess, S.C.; Li, C.; Ruddy, M.; Chakravarthy, M.; et al. Acetyl CoA Carboxylase Inhibition Reduces Hepatic Steatosis but Elevates Plasma Triglycerides in Mice and Humans: A Bedside to Bench Investigation. Cell Metab. 2017, 26, 576. [Google Scholar] [CrossRef] [PubMed]
- Harriman, G.; Greenwood, J.; Bhat, S.; Huang, X.; Wang, R.; Paul, D.; Tong, L.; Saha, A.K.; Westlin, W.F.; Kapeller, R.; et al. Acetyl-CoA Carboxylase Inhibition by ND-630 Reduces Hepatic Steatosis, Improves Insulin Sensitivity, and Modulates Dyslipidemia in Rats. Proc. Natl. Acad. Sci. USA 2016, 113, E1796–E1805. [Google Scholar] [CrossRef]
- Falchook, G.; Patel, M.; Infante, J.; Arkenau, H.-T.; Dean, E.; Brenner, A.; Borazanci, E.; Lopez, J.; Moore, K.; Schmid, P.; et al. Abstract CT153: First in Human Study of the First-in-Class Fatty Acid Synthase (FASN) Inhibitor TVB-2640. Cancer Res. 2017, CT153. [Google Scholar] [CrossRef]
- Konkel, B.; Caflisch, L.D.; Duque, A.E.D.; Michalek, J.; Liu, Q.; Brenner, A.J. Prospective Phase II Trial in Patients with First Relapse of High-Grade Astrocytoma Using TVB-2640 in Combination with Bevacizumab versus Bevacizumab Alone. J. Clin. Oncol. 2019, 37, 2064. [Google Scholar] [CrossRef]
- Menendez, J.A.; Lupu, R. Fatty Acid Synthase (FASN) as a Therapeutic Target in Breast Cancer. Expert Opin. Ther. Targets 2017, 21, 1001–1016. [Google Scholar] [CrossRef]
- Schcolnik-Cabrera, A.; Chávez-Blanco, A.; Domínguez-Gómez, G.; Taja-Chayeb, L.; Morales-Barcenas, R.; Trejo-Becerril, C.; Perez-Cardenas, E.; Gonzalez-Fierro, A.; Dueñas-González, A. Orlistat as a FASN Inhibitor and Multitargeted Agent for Cancer Therapy. Expert Opin. Investig. Drug 2018, 27, 475–489. [Google Scholar] [CrossRef]
- Makowski, K.; Mir, J.F.; Mera, P.; Ariza, X.; Asins, G.; Hegardt, F.G.; Herrero, L.; García, J.; Serra, D. (−)-UB006: A New Fatty Acid Synthase Inhibitor and Cytotoxic Agent without Anorexic Side Effects. Eur. J. Med. Chem. 2017, 131, 207–221. [Google Scholar] [CrossRef]
- Zhou, W.; Simpson, P.J.; McFadden, J.M.; Townsend, C.A.; Medghalchi, S.M.; Vadlamudi, A.; Pinn, M.L.; Ronnett, G.V.; Kuhajda, F.P. Fatty Acid Synthase Inhibition Triggers Apoptosis during S Phase in Human Cancer Cells. Cancer Res. 2003, 63, 7330–7337. [Google Scholar]
- Hardwicke, M.A.; Rendina, A.R.; Williams, S.P.; Moore, M.L.; Wang, L.; Krueger, J.A.; Plant, R.N.; Totoritis, R.D.; Zhang, G.; Briand, J.; et al. A Human Fatty Acid Synthase Inhibitor Binds β-Ketoacyl Reductase in the Keto-Substrate Site. Nat. Chem. Biol. 2014, 10, 774–779. [Google Scholar] [CrossRef] [PubMed]
- Shaw, G.; Lewis, D.; Boren, J.; Ramos-Montoya, A.; Bielik, R.; Soloviev, D.; Kevin, B.; David, N. 509 Therapeutic Fatty Acid Synthase Inhibition in Prostate Cancer and the Use of 11c-Acetate to Monitor Therapeutic Effects. J. Urol. 2013, 189, e208–e209. [Google Scholar] [CrossRef]
- Alwarawrah, Y.; Hughes, P.; Loiselle, D.; Carlson, D.A.; Darr, D.B.; Jordan, J.L.; Xiong, J.; Hunter, L.M.; Dubois, L.G.; Thompson, J.W.; et al. Fasnall, a Selective FASN Inhibitor, Shows Potent Anti-Tumor Activity in the MMTV-Neu Model of HER2+ Breast Cancer. Cell Chem. Biol. 2016, 23, 678–688. [Google Scholar] [CrossRef] [PubMed]
- Tracz-Gaszewska, Z.; Dobrzyn, P. Stearoyl-CoA Desaturase 1 as a Therapeutic Target for the Treatment of Cancer. Cancers 2019, 11, 948. [Google Scholar] [CrossRef] [PubMed]
- von Roemeling, C.A.; Marlow, L.A.; Wei, J.J.; Cooper, S.J.; Caulfield, T.R.; Wu, K.; Tan, W.W.; Tun, H.W.; Copland, J.A. Stearoyl-CoA Desaturase 1 Is a Novel Molecular Therapeutic Target for Clear Cell Renal Cell Carcinoma. Clin. Cancer Res. 2013, 19, 2368–2380. [Google Scholar] [CrossRef]
- Pisanu, M.E.; Noto, A.; Vitis, C.D.; Morrone, S.; Scognamiglio, G.; Botti, G.; Venuta, F.; Diso, D.; Jakopin, Z.; Padula, F.; et al. Blockade of Stearoyl-CoA-Desaturase 1 Activity Reverts Resistance to Cisplatin in Lung Cancer Stem Cells. Cancer Lett. 2017, 406, 93–104. [Google Scholar] [CrossRef]
- Zhao, J.; Zhi, Z.; Wang, C.; Xing, H.; Song, G.; Yu, X.; Zhu, Y.; Wang, X.; Zhang, X.; Di, Y. Exogenous Lipids Promote the Growth of Breast Cancer Cells via CD36. Oncol. Rep. 2017, 38, 2105–2115. [Google Scholar] [CrossRef]
- Li, J.; Condello, S.; Thomes-Pepin, J.; Ma, X.; Xia, Y.; Hurley, T.D.; Matei, D.; Cheng, J.-X. Lipid Desaturation Is a Metabolic Marker and Therapeutic Target of Ovarian Cancer Stem Cells. Cell Stem. Cell 2017, 20, 303–314.e5. [Google Scholar] [CrossRef]
- Chen, L.; Ren, J.; Yang, L.; Li, Y.; Fu, J.; Li, Y.; Tian, Y.; Qiu, F.; Liu, Z.; Qiu, Y. Stearoyl-CoA Desaturase-1 Mediated Cell Apoptosis in Colorectal Cancer by Promoting Ceramide Synthesis. Sci. Rep. UK 2016, 6, 19665. [Google Scholar] [CrossRef]
- Fritz, V.; Benfodda, Z.; Rodier, G.; Henriquet, C.; Iborra, F.; Avancès, C.; Allory, Y.; de la Taille, A.; Culine, S.; Blancou, H.; et al. Abrogation of De Novo Lipogenesis by Stearoyl-CoA Desaturase 1 Inhibition Interferes with Oncogenic Signaling and Blocks Prostate Cancer Progression in Mice. Mol. Cancer Ther. 2010, 9, 1740–1754. [Google Scholar] [CrossRef]
- Chen, M.; Zhang, J.; Sampieri, K.; Clohessy, J.G.; Mendez, L.; Gonzalez-Billalabeitia, E.; Liu, X.-S.; Lee, Y.-R.; Fung, J.; Katon, J.M.; et al. An Aberrant SREBP-Dependent Lipogenic Program Promotes Metastatic Prostate Cancer. Nat. Genet. 2018, 50, 206–218. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.J.; Argus, J.P.; Zhu, Y.; Wilks, M.Q.; Marbois, B.N.; York, A.G.; Kidani, Y.; Pourzia, A.L.; Akhavan, D.; Lisiero, D.N.; et al. An Essential Requirement for the SCAP/SREBP Signaling Axis to Protect Cancer Cells from Lipotoxicity. Cancer Res. 2013, 73, 2850–2862. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, Y. CD36 Tango in Cancer: Signaling Pathways and Functions. Theranostics 2019, 9, 4893–4908. [Google Scholar] [CrossRef] [PubMed]
- Molckovsky, A.; Siu, L.L. First-in-Class, First-in-Human Phase I Results of Targeted Agents: Highlights of the 2008 American Society of Clinical Oncology Meeting. J. Hematol. Oncol. 2008, 1, 20. [Google Scholar] [CrossRef] [PubMed]
- Carracedo, A.; Cantley, L.C.; Pandolfi, P.P. Cancer Metabolism: Fatty Acid Oxidation in the Limelight. Nat. Rev. Cancer 2013, 13, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Temkin, S.M.; Hawkridge, A.M.; Guo, C.; Wang, W.; Wang, X.-Y.; Fang, X. Fatty Acid Oxidation: An Emerging Facet of Metabolic Transformation in Cancer. Cancer Lett. 2018, 435, 92–100. [Google Scholar] [CrossRef]
- Qu, Q.; Zeng, F.; Liu, X.; Wang, Q.J.; Deng, F. Fatty Acid Oxidation and Carnitine Palmitoyltransferase I: Emerging Therapeutic Targets in Cancer. Cell Death Dis. 2016, 7, e2226. [Google Scholar] [CrossRef]
- Ricciardi, M.R.; Mirabilii, S.; Allegretti, M.; Licchetta, R.; Calarco, A.; Torrisi, M.R.; Foà, R.; Nicolai, R.; Peluso, G.; Tafuri, A. Targeting the Leukemia Cell Metabolism by the CPT1a Inhibition: Functional Preclinical Effects in Leukemias. Blood 2015, 126, 1925–1929. [Google Scholar] [CrossRef]
- Fujiwara, N.; Nakagawa, H.; Enooku, K.; Kudo, Y.; Hayata, Y.; Nakatsuka, T.; Tanaka, Y.; Tateishi, R.; Hikiba, Y.; Misumi, K.; et al. CPT2 Downregulation Adapts HCC to Lipid-Rich Environment and Promotes Carcinogenesis via Acylcarnitine Accumulation in Obesity. Gut 2018, 67, 1493. [Google Scholar] [CrossRef]
- Lu, X.; Zhang, X.; Zhang, Y.; Zhang, K.; Zhan, C.; Shi, X.; Li, Y.; Zhao, J.; Bai, Y.; Wang, Y.; et al. Metabolic Profiling Analysis upon Acylcarnitines in Tissues of Hepatocellular Carcinoma Revealed the Inhibited Carnitine Shuttle System Caused by the Downregulated Carnitine Palmitoyltransferase 2. Mol. Carcinog. 2019, 58, 749–759. [Google Scholar] [CrossRef]
- Lin, M.; Lv, D.; Zheng, Y.; Wu, M.; Xu, C.; Zhang, Q.; Wu, L. Downregulation of CPT2 Promotes Tumorigenesis and Chemoresistance to Cisplatin in Hepatocellular Carcinoma. Oncotargets Ther. 2018, 11, 3101–3110. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.-L.; Zhang, Y.; Guo, Y.-C.; Yang, Z.-H.; Xu, Y.-C. A Prognostic Model Based on Six Metabolism-Related Genes in Colorectal Cancer. Biomed. Res. Int. 2020, 2020, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Geng, F.; Pan, M.; Wu, X.; Zhong, Y.; Wang, C.; Tian, Z.; Cheng, C.; Zhang, R.; Puduvalli, V.; et al. Targeting DGAT1 Ameliorates Glioblastoma by Increasing Fat Catabolism and Oxidative Stress. Cell Metab. 2020, 32, 229–242.e8. [Google Scholar] [CrossRef] [PubMed]
- Enooku, K.; Nakagawa, H.; Fujiwara, N.; Kondo, M.; Minami, T.; Hoshida, Y.; Shibahara, J.; Tateishi, R.; Koike, K. Altered Serum Acylcarnitine Profile Is Associated with the Status of Nonalcoholic Fatty Liver Disease (NAFLD) and NAFLD-Related Hepatocellular Carcinoma. Sci Rep. UK 2019, 9, 10663. [Google Scholar] [CrossRef]
- Huang, B.; Song, B.; Xu, C. Cholesterol Metabolism in Cancer: Mechanisms and Therapeutic Opportunities. Nat. Metab. 2020, 2, 1–10. [Google Scholar] [CrossRef]
- Goldstein, J.L.; Brown, M.S. The LDL Receptor. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 431–438. [Google Scholar] [CrossRef]
- Wang, Y.-J.; Bian, Y.; Luo, J.; Lu, M.; Xiong, Y.; Guo, S.-Y.; Yin, H.-Y.; Lin, X.; Li, Q.; Chang, C.C.Y.; et al. Cholesterol and Fatty Acids Regulate Cysteine Ubiquitylation of ACAT2 through Competitive Oxidation. Nat. Cell Biol. 2017, 19, 808–819. [Google Scholar] [CrossRef]
- Brown, M.S.; Radhakrishnan, A.; Goldstein, J.L. Retrospective on Cholesterol Homeostasis: The Central Role of Scap. Annu. Rev. Biochem. 2017, 87, 1–25. [Google Scholar] [CrossRef]
- Wang, B.; Tontonoz, P. Liver X Receptors in Lipid Signalling and Membrane Homeostasis. Nat. Rev. Endocrinol. 2018, 14, 452–463. [Google Scholar] [CrossRef]
- Widenmaier, S.B.; Snyder, N.A.; Nguyen, T.B.; Arduini, A.; Lee, G.Y.; Arruda, A.P.; Saksi, J.; Bartelt, A.; Hotamisligil, G.S. NRF1 Is an ER Membrane Sensor That Is Central to Cholesterol Homeostasis. Cell 2017, 171, 1094–1109.e15. [Google Scholar] [CrossRef]
- Xue, L.; Qi, H.; Zhang, H.; Ding, L.; Huang, Q.; Zhao, D.; Wu, B.J.; Li, X. Targeting SREBP-2-Regulated Mevalonate Metabolism for Cancer Therapy. Front. Oncol. 2020, 10, 1510. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.-Y. Therapeutic Targeting of Lipid Synthesis Metabolism for Selective Elimination of Cancer Stem Cells. Arch. Pharm. Res. 2019, 42, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Villa, G.R.; Hulce, J.J.; Zanca, C.; Bi, J.; Ikegami, S.; Cahill, G.L.; Gu, Y.; Lum, K.M.; Masui, K.; Yang, H.; et al. An LXR-Cholesterol Axis Creates a Metabolic Co-Dependency for Brain Cancers. Cancer Cell 2016, 30, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Guillaumond, F.; Bidaut, G.; Ouaissi, M.; Servais, S.; Gouirand, V.; Olivares, O.; Lac, S.; Borge, L.; Roques, J.; Gayet, O.; et al. Cholesterol Uptake Disruption, in Association with Chemotherapy, Is a Promising Combined Metabolic Therapy for Pancreatic Adenocarcinoma. Proc. Natl. Acad. Sci. USA 2015, 112, 2473–2478. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Bermudez, J.; Baudrier, L.; Bayraktar, E.C.; Shen, Y.; La, K.; Guarecuco, R.; Yucel, B.; Fiore, D.; Tavora, B.; Freinkman, E.; et al. Squalene Accumulation in Cholesterol Auxotrophic Lymphomas Prevents Oxidative Cell Death. Nature 2019, 567, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Gu, D.; Lee, S.S.-Y.; Song, B.; Bandyopadhyay, S.; Chen, S.; Konieczny, S.F.; Ratliff, T.L.; Liu, X.; Xie, J.; et al. Abrogating Cholesterol Esterification Suppresses Growth and Metastasis of Pancreatic Cancer. Oncogene 2016, 35, 6378–6388. [Google Scholar] [CrossRef] [PubMed]
- Mulas, M.F.; Abete, C.; Pulisci, D.; Pani, A.; Massidda, B.; Dessì, S.; Mandas, A. Cholesterol Esters as Growth Regulators of Lymphocytic Leukaemia Cells. Cell Prolif. 2011, 44, 360–371. [Google Scholar] [CrossRef]
- Kloudova, A.; Guengerich, F.P.; Soucek, P. The Role of Oxysterols in Human Cancer. Trends Endocrinol. Metab. 2017, 28, 485–496. [Google Scholar] [CrossRef]
- Wu, Q.; Ishikawa, T.; Sirianni, R.; Tang, H.; McDonald, J.G.; Yuhanna, I.S.; Thompson, B.; Girard, L.; Mineo, C.; Brekken, R.A.; et al. 27-Hydroxycholesterol Promotes Cell-Autonomous, ER-Positive Breast Cancer Growth. Cell Rep. 2013, 5, 637–645. [Google Scholar] [CrossRef]
- Raza, S.; Ohm, J.E.; Dhasarathy, A.; Schommer, J.; Roche, C.; Hammer, K.D.P.; Ghribi, O. The Cholesterol Metabolite 27-Hydroxycholesterol Regulates P53 Activity and Increases Cell Proliferation via MDM2 in Breast Cancer Cells. Mol. Cell Biochem. 2015, 410, 187–195. [Google Scholar] [CrossRef]
- Vedin, L.-L.; Lewandowski, S.A.; Parini, P.; Gustafsson, J.-Å.; Steffensen, K.R. The Oxysterol Receptor LXR Inhibits Proliferation of Human Breast Cancer Cells. Carcinogenesis 2009, 30, 575–579. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Hong, W.; Yang, M.; Xu, D.; Bai, Q.; Li, X.; Chen, Z. Upregulation of 24(R/S),25-Epoxycholesterol and 27-Hydroxycholesterol Suppresses the Proliferation and Migration of Gastric Cancer Cells. Biochem. Biophys. Res. Commun. 2018, 504, 892–898. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Zhou, S.; Tang, Q.; Xia, H.; Bi, F. Cholesterol Metabolism: New Functions and Therapeutic Approaches in Cancer. Biochim. Biophys. Acta BBA Rev. Cancer 2020, 1874, 188394. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Huang, Z.; Wu, Q.; Prager, B.C.; Mack, S.C.; Yang, K.; Kim, L.J.Y.; Gimple, R.C.; Shi, Y.; Lai, S.; et al. MYC-Regulated Mevalonate Metabolism Maintains Brain Tumor–Initiating Cells. Cancer Res. 2017, 77, 4947–4960. [Google Scholar] [CrossRef]
- Bakiri, L.; Hamacher, R.; Graña, O.; Guío-Carrión, A.; Campos-Olivas, R.; Martinez, L.; Dienes, H.P.; Thomsen, M.K.; Hasenfuss, S.C.; Wagner, E.F. Liver Carcinogenesis by FOS-Dependent Inflammation and Cholesterol DysregulationThe Functions of c-Fos in Hepatocarcinogenesis. J. Exp. Med. 2017, 214, 1387–1409. [Google Scholar] [CrossRef]
- Moon, S.-H.; Huang, C.-H.; Houlihan, S.L.; Regunath, K.; Freed-Pastor, W.A.; Morris, J.P.; Tschaharganeh, D.F.; Kastenhuber, E.R.; Barsotti, A.M.; Culp-Hill, R.; et al. P53 Represses the Mevalonate Pathway to Mediate Tumor Suppression. Cell 2018, 176, 564–580.e19. [Google Scholar] [CrossRef]
- Yue, S.; Li, J.; Lee, S.-Y.; Lee, H.J.; Shao, T.; Song, B.; Cheng, L.; Masterson, T.A.; Liu, X.; Ratliff, T.L.; et al. Cholesteryl Ester Accumulation Induced by PTEN Loss and PI3K/AKT Activation Underlies Human Prostate Cancer Aggressiveness. Cell Metab. 2014, 19, 393–406. [Google Scholar] [CrossRef]
- Liu, D.; Wong, C.C.; Fu, L.; Chen, H.; Zhao, L.; Li, C.; Zhou, Y.; Zhang, Y.; Xu, W.; Yang, Y.; et al. Squalene Epoxidase Drives NAFLD-Induced Hepatocellular Carcinoma and Is a Pharmaceutical Target. Sci. Transl. Med. 2018, 10, eaap9840. [Google Scholar] [CrossRef]
- Gruenbacher, G.; Thurnher, M. Mevalonate Metabolism in Immuno-Oncology. Front. Immunol. 2017, 8, 1714. [Google Scholar] [CrossRef]
- Thurnher, M.; Gruenbacher, G.; Nussbaumer, O. Regulation of Mevalonate Metabolism in Cancer and Immune Cells. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2013, 1831, 1009–1015. [Google Scholar] [CrossRef]
- Nielsen, S.F.; Nordestgaard, B.G.; Bojesen, S.E. Statin Use and Reduced Cancer-Related Mortality. New Engl. J. Med. 2012, 367, 1792–1802. [Google Scholar] [CrossRef] [PubMed]
- Cardwell, C.R.; Hicks, B.M.; Hughes, C.; Murray, L.J. Statin Use After Colorectal Cancer Diagnosis and Survival: A Population-Based Cohort Study. J. Clin. Oncol. 2014, 32, 3177–3183. [Google Scholar] [CrossRef] [PubMed]
- Larsen, S.B.; Dehlendorff, C.; Skriver, C.; Dalton, S.O.; Jespersen, C.G.; Borre, M.; Brasso, K.; Nørgaard, M.; Johansen, C.; Sørensen, H.T.; et al. Postdiagnosis Statin Use and Mortality in Danish Patients with Prostate Cancer. J. Clin. Oncol. 2017, 35, JCO.2016.71.898. [Google Scholar] [CrossRef] [PubMed]
- Sanfilippo, K.M.; Keller, J.; Gage, B.F.; Luo, S.; Wang, T.-F.; Moskowitz, G.; Gumbel, J.; Blue, B.; O’Brian, K.; Carson, K.R. Statins Are Associated with Reduced Mortality in Multiple Myeloma. J. Clin. Oncol. 2016, 34, 4008–4014. [Google Scholar] [CrossRef]
- Mullen, P.J.; Yu, R.; Longo, J.; Archer, M.C.; Penn, L.Z. The Interplay between Cell Signalling and the Mevalonate Pathway in Cancer. Nat. Rev. Cancer 2016, 16, 718–731. [Google Scholar] [CrossRef]
- Sondergaard, T.; Pedersen, P.; Andersen, T.; Søe, K.; Lund, T.; Østergaard, B.; Garnero, P.; Delaisse, J.; Plesner, T. A Phase II Clinical Trial Does Not Show That High Dose Simvastatin Has Beneficial Effect on Markers of Bone Turnover in Multiple Myeloma. Hematol. Oncol. 2009, 27, 17–22. [Google Scholar] [CrossRef]
- Cirmena, G.; Franceschelli, P.; Isnaldi, E.; Ferrando, L.; Mariano, M.D.; Ballestrero, A.; Zoppoli, G. Squalene Epoxidase as a Promising Metabolic Target in Cancer Treatment. Cancer Lett. 2018, 425, 13–20. [Google Scholar] [CrossRef]
- Maione, F.; Oliaro-Bosso, S.; Meda, C.; Nicolantonio, F.D.; Bussolino, F.; Balliano, G.; Viola, F.; Giraudo, E. The Cholesterol Biosynthesis Enzyme Oxidosqualene Cyclase Is a New Target to Impair Tumour Angiogenesis and Metastasis Dissemination. Sci. Rep. UK 2015, 5, 9054. [Google Scholar] [CrossRef]
- Boussac, H.D.; Alioui, A.; Viennois, E.; Dufour, J.; Trousson, A.; Vega, A.; Guy, L.; Volle, D.H.; Lobaccaro, J.-M.A.; Baron, S. Oxysterol Receptors and Their Therapeutic Applications in Cancer Conditions. Expert Opin. Ther. Targets 2013, 17, 1029–1038. [Google Scholar] [CrossRef]
- Lin, C.-Y.; Gustafsson, J.-Å. Targeting Liver X Receptors in Cancer Therapeutics. Nat. Rev. Cancer 2015, 15, 216–224. [Google Scholar] [CrossRef]
- Wu, G.; Wang, Q.; Xu, Y.; Li, J.; Zhang, H.; Qi, G.; Xia, Q. Targeting the Transcription Factor Receptor LXR to Treat Clear Cell Renal Cell Carcinoma: Agonist or Inverse Agonist? Cell Death Dis. 2019, 10, 416. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, S.; Li, J.; Traer, E.; Tyner, J.W.; Zhou, A.; Oh, S.T.; Cheng, J.-X. Cholesterol Esterification Inhibition and Imatinib Treatment Synergistically Inhibit Growth of BCR-ABL Mutation-Independent Resistant Chronic Myelogenous Leukemia. PLoS ONE 2017, 12, e0179558. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Li, J.; Vickman, R.E.; Li, J.; Liu, R.; Durkes, A.; Elzey, B.D.; Yue, S.; Liu, X.; Ratliff, T.L.; et al. Cholesterol Esterification Inhibition Suppresses Prostate Cancer Metastasis by Impairing the Wnt/β-Catenin Pathway. Mol. Cancer Res. 2018, 16, 974–985. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.S.-Y.; Li, J.; Tai, J.N.; Ratliff, T.L.; Park, K.; Cheng, J.-X. Avasimibe Encapsulated in Human Serum Albumin Blocks Cholesterol Esterification for Selective Cancer Treatment. ACS Nano 2015, 9, 2420–2432. [Google Scholar] [CrossRef]
- Wilfling, F.; Haas, J.T.; Walther, T.C.; Jr, R.V.F. Lipid Droplet Biogenesis. Curr. Opin. Cell Biol. 2014, 29, 39–45. [Google Scholar] [CrossRef]
- Lass, A.; Zimmermann, R.; Oberer, M.; Zechner, R. Lipolysis—A Highly Regulated Multi-Enzyme Complex Mediates the Catabolism of Cellular Fat Stores. Prog. Lipid Res. 2011, 50, 14–27. [Google Scholar] [CrossRef]
- Schulze, R.J.; Sathyanarayan, A.; Mashek, D.G. Breaking Fat: The Regulation and Mechanisms of Lipophagy. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2017, 1862, 1178–1187. [Google Scholar] [CrossRef]
- Zimmermann, R.; Strauss, J.G.; Haemmerle, G.; Schoiswohl, G.; Birner-Gruenberger, R.; Riederer, M.; Lass, A.; Neuberger, G.; Eisenhaber, F.; Hermetter, A.; et al. Fat Mobilization in Adipose Tissue Is Promoted by Adipose Triglyceride Lipase. Science 2004, 306, 1383–1386. [Google Scholar] [CrossRef]
- Grabner, G.F.; Zimmermann, R.; Schicho, R.; Taschler, U. Monoglyceride Lipase as a Drug Target: At the Crossroads of Arachidonic Acid Metabolism and Endocannabinoid Signaling. Pharmacol. Ther. 2017, 175, 35–46. [Google Scholar] [CrossRef]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy Regulates Lipid Metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef]
- Cabodevilla, A.G.; Sánchez-Caballero, L.; Nintou, E.; Boiadjieva, V.G.; Picatoste, F.; Gubern, A.; Claro, E. Cell Survival during Complete Nutrient Deprivation Depends on Lipid Droplet-Fueled β-Oxidation of Fatty Acids. J. Biol. Chem. 2013, 288, 27777–27788. [Google Scholar] [CrossRef] [PubMed]
- Osumi, T.; Kuramoto, K. Heart Lipid Droplets and Lipid Droplet-Binding Proteins: Biochemistry, Physiology, and Pathology. Exp. Cell Res. 2016, 340, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhao, Y.; Gao, X.; Li, L.; Yuan, Y.; Liu, F.; Zhang, L.; Wu, J.; Hu, P.; Zhang, X.; et al. Perilipin 5 Improves Hepatic Lipotoxicity by Inhibiting Lipolysis. Hepatology 2015, 61, 870–882. [Google Scholar] [CrossRef] [PubMed]
- Welte, M.A.; Gould, A.P. Lipid Droplet Functions beyond Energy Storage. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2017, 1862, 1260–1272. [Google Scholar] [CrossRef] [PubMed]
- Bensaad, K.; Favaro, E.; Lewis, C.A.; Peck, B.; Lord, S.; Collins, J.M.; Pinnick, K.E.; Wigfield, S.; Buffa, F.M.; Li, J.-L.; et al. Fatty Acid Uptake and Lipid Storage Induced by HIF-1α Contribute to Cell Growth and Survival after Hypoxia-Reoxygenation. Cell Rep. 2014, 9, 349–365. [Google Scholar] [CrossRef]
- Petan, T.; Jarc, E.; Jusović, M. Lipid Droplets in Cancer: Guardians of Fat in a Stressful World. Molecules 2018, 23, 1941. [Google Scholar] [CrossRef]
- de Gonzalo-Calvo, D.; López-Vilaró, L.; Nasarre, L.; Perez-Olabarria, M.; Vázquez, T.; Escuin, D.; Badimon, L.; Barnadas, A.; Lerma, E.; Llorente-Cortés, V. Intratumor Cholesteryl Ester Accumulation Is Associated with Human Breast Cancer Proliferation and Aggressive Potential: A Molecular and Clinicopathological Study. BMC Cancer 2015, 15, 460. [Google Scholar] [CrossRef]
- Koizume, S.; Miyagi, Y. Lipid Droplets: A Key Cellular Organelle Associated with Cancer Cell Survival under Normoxia and Hypoxia. Int. J. Mol. Sci. 2016, 17, 1430. [Google Scholar] [CrossRef]
- Qiu, B.; Ackerman, D.; Sanchez, D.J.; Li, B.; Ochocki, J.D.; Grazioli, A.; Bobrovnikova-Marjon, E.; Diehl, J.A.; Keith, B.; Simon, M.C. HIF2α-Dependent Lipid Storage Promotes Endoplasmic Reticulum Homeostasis in Clear-Cell Renal Cell Carcinoma. Cancer Discov. 2015, 5, 652–667. [Google Scholar] [CrossRef]
- Mylonis, I.; Sembongi, H.; Befani, C.; Liakos, P.; Siniossoglou, S.; Simos, G. Hypoxia Causes Triglyceride Accumulation by HIF-1-Mediated Stimulation of Lipin 1 Expression. J. Cell Sci. 2012, 125, 3485–3493. [Google Scholar] [CrossRef]
- Schlaepfer, I.R.; Nambiar, D.K.; Ramteke, A.; Kumar, R.; Dhar, D.; Agarwal, C.; Bergman, B.; Graner, M.; Maroni, P.; Singh, R.P.; et al. Hypoxia Induces Triglycerides Accumulation in Prostate Cancer Cells and Extracellular Vesicles Supporting Growth and Invasiveness Following Reoxygenation. Oncotarget 2015, 6, 22836–22856. [Google Scholar] [CrossRef] [PubMed]
- Laurenti, G.; Benedetti, E.; D’Angelo, B.; Cristiano, L.; Cinque, B.; Raysi, S.; Alecci, M.; Cerù, M.P.; Cifone, M.G.; Galzio, R.; et al. Hypoxia Induces Peroxisome Proliferator-activated Receptor α (PPARα) and Lipid Metabolism Peroxisomal Enzymes in Human Glioblastoma Cells. J. Cell Biochem. 2011, 112, 3891–3901. [Google Scholar] [CrossRef] [PubMed]
- Zoula, S.; Rijken, P.F.J.W.; Peters, J.P.W.; Farion, R.; der Sanden, B.P.J.V.; der Kogel, A.J.V.; Décorps, M.; Rémy, C. Pimonidazole Binding in C6 Rat Brain Glioma: Relation with Lipid Droplet Detection. Br. J. Cancer 2003, 88, 1439–1444. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Saarinen, A.M.; Hitosugi, T.; Wang, Z.; Wang, L.; Ho, T.H.; Liu, J. Inhibition of Intracellular Lipolysis Promotes Human Cancer Cell Adaptation to Hypoxia. Elife 2017, 6, e31132. [Google Scholar] [CrossRef] [PubMed]
- Gimm, T.; Wiese, M.; Teschemacher, B.; Deggerich, A.; Schödel, J.; Knaup, K.X.; Hackenbeck, T.; Hellerbrand, C.; Amann, K.; Wiesener, M.S.; et al. Hypoxia-inducible Protein 2 Is a Novel Lipid Droplet Protein and a Specific Target Gene of Hypoxia-inducible Factor-1. FASEB J. 2010, 24, 4443–4458. [Google Scholar] [CrossRef]
- Das, K.M.; Wechselberger, L.; Liziczai, M.; De la Rosa Rodriguez, M.; Grabner, G.F.; Heier, C.; Viertlmayr, R.; Radler, C.; Lichtenegger, J.; Zimmermann, R.; et al. Hypoxia-Inducible Lipid Droplet-Associated Protein Inhibits Adipose Triglyceride Lipase. J. Lipid Res. 2018, 59, 531–541. [Google Scholar] [CrossRef]
- Xie, H.; Heier, C.; Kien, B.; Vesely, P.W.; Tang, Z.; Sexl, V.; Schoiswohl, G.; Strießnig-Bina, I.; Hoefler, G.; Zechner, R.; et al. Adipose Triglyceride Lipase Activity Regulates Cancer Cell Proliferation via AMP-Kinase and MTOR Signaling. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2020, 1865, 158737. [Google Scholar] [CrossRef]
- Leo, L.D.; Vegliante, R.; Ciccarone, F.; Salvatori, I.; Scimeca, M.; Bonanno, E.; Sagnotta, A.; Grazi, G.L.; Aquilano, K.; Ciriolo, M.R. Forcing ATGL Expression in Hepatocarcinoma Cells Imposes Glycolytic Rewiring through PPAR-α/P300-Mediated Acetylation of P53. Oncogene 2019, 38, 1860–1875. [Google Scholar] [CrossRef]
- Kaini, R.R.; Sillerud, L.O.; Zhaorigetu, S.; Hu, C.A. Autophagy Regulates Lipolysis and Cell Survival through Lipid Droplet Degradation in Androgen-sensitive Prostate Cancer Cells. Prostate 2012, 72, 1412–1422. [Google Scholar] [CrossRef]
- Assumpção, J.A.F.; Magalhães, K.G.; Corrêa, J.R. The Role of Pparγ and Autophagy in Ros Production, Lipid Droplets Biogenesis and Its Involvement with Colorectal Cancer Cells Modulation. Cancer Cell Int. 2017, 17, 82. [Google Scholar] [CrossRef]
- Xu, G.; Jiang, Y.; Xiao, Y.; Liu, X.-D.; Yue, F.; Li, W.; Li, X.; He, Y.; Jiang, X.; Huang, H.; et al. Fast Clearance of Lipid Droplets through MAP1S-Activated Autophagy Suppresses Clear Cell Renal Cell Carcinomas and Promotes Patient Survival. Oncotarget 2015, 7, 6255–6265. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Schlaepfer, I.R.; Bergman, B.C.; Panda, P.K.; Praharaj, P.P.; Naik, P.P.; Agarwal, R.; Bhutia, S.K. ATG14 Facilitated Lipophagy in Cancer Cells Induce ER Stress Mediated Mitoptosis through a ROS Dependent Pathway. Free Radic. Biol. Med. 2017, 104, 199–213. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Zhao, T.; Ding, X.; Yan, C. Hepatocyte-Specific Expression of Human Lysosome Acid Lipase Corrects Liver Inflammation and Tumor Metastasis in Lal−/− Mice. Am. J. Pathol. 2015, 185, 2379–2389. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Ding, X.; Du, H.; Yan, C. Lung Epithelial Cell–Specific Expression of Human Lysosomal Acid Lipase Ameliorates Lung Inflammation and Tumor Metastasis in Lipa−/− Mice. Am. J. Pathol. 2016, 186, 2183–2192. [Google Scholar] [CrossRef]
- Harayama, T.; Riezman, H. Understanding the Diversity of Membrane Lipid Composition. Nat. Rev. Mol. Cell Biol. 2018, 19, 281–296. [Google Scholar] [CrossRef]
- Kennedy, E.P.; Weiss, S.B. The Function of Cytidine Coenzymes in the Biosynthesis of Phospholipides. J. Biol. Chem. 1956, 222, 193–214. [Google Scholar] [CrossRef]
- Lands, W.E. Metabolism of Glycerolipides; a Comparison of Lecithin and Triglyceride Synthesis. J. Biol. Chem. 1958, 231, 883–888. [Google Scholar] [CrossRef]
- Nakanishi, H.; Shindou, H.; Hishikawa, D.; Harayama, T.; Ogasawara, R.; Suwabe, A.; Taguchi, R.; Shimizu, T. Cloning and Characterization of Mouse Lung-Type Acyl-CoA:Lysophosphatidylcholine Acyltransferase 1 (LPCAT1) Expression in Alveolar Type II Cells and Possible Involvement in Surfactant Production. J. Biol. Chem. 2006, 281, 20140–20147. [Google Scholar] [CrossRef]
- Shindou, H.; Hishikawa, D.; Nakanishi, H.; Harayama, T.; Ishii, S.; Taguchi, R.; Shimizu, T. A Single Enzyme Catalyzes Both Platelet-Activating Factor Production and Membrane Biogenesis of Inflammatory Cells Cloning and Characterization of Acetyl-CoA:lyso-PAF Acetyltransferase. J. Biol. Chem. 2007, 282, 6532–6539. [Google Scholar] [CrossRef]
- Hishikawa, D.; Shindou, H.; Kobayashi, S.; Nakanishi, H.; Taguchi, R.; Shimizu, T. Discovery of a Lysophospholipid Acyltransferase Family Essential for Membrane Asymmetry and Diversity. Proc. Natl. Acad. Sci. USA 2008, 105, 2830–2835. [Google Scholar] [CrossRef]
- Rong, X.; Albert, C.J.; Hong, C.; Duerr, M.A.; Chamberlain, B.T.; Tarling, E.J.; Ito, A.; Gao, J.; Wang, B.; Edwards, P.A.; et al. LXRs Regulate ER Stress and Inflammation through Dynamic Modulation of Membrane Phospholipid Composition. Cell Metab. 2013, 18, 685–697. [Google Scholar] [CrossRef] [PubMed]
- Meana, C.; García-Rostán, G.; Peña, L.; Lordén, G.; Cubero, Á.; Orduña, A.; Győrffy, B.; Balsinde, J.; Balboa, M.A. The Phosphatidic Acid Phosphatase Lipin-1 Facilitates Inflammation-Driven Colon Carcinogenesis. JCI Insight 2018, 3, e97506. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Zhang, F.; Tay, L.W.R.; Boroda, S.; Nian, W.; Levental, K.R.; Levental, I.; Harris, T.E.; Chang, J.T.; Du, G. Lipin-1 Regulation of Phospholipid Synthesis Maintains Endoplasmic Reticulum Homeostasis and Is Critical for Triple-negative Breast Cancer Cell Survival. FASEB J. 2017, 31, 2893–2904. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Lopez, E.; Zimmerman, T.; del Pulgar, T.G.; Moyer, M.P.; Sanjuan, J.C.L.; Cebrian, A. Choline Kinase Inhibition Induces Exacerbated Endoplasmic Reticulum Stress and Triggers Apoptosis via CHOP in Cancer Cells. Cell Death Dis. 2013, 4, e933. [Google Scholar] [CrossRef] [PubMed]
- Arlauckas, S.P.; Popov, A.V.; Delikatny, E.J. Choline Kinase Alpha—Putting the ChoK-Hold on Tumor Metabolism. Prog. Lipid Res. 2016, 63, 28–40. [Google Scholar] [CrossRef]
- Wang, B.; Tontonoz, P. Phospholipid Remodeling in Physiology and Disease. Annu. Rev. Physiol. 2018, 81, 1–24. [Google Scholar] [CrossRef]
- Du, Y.; Wang, Q.; Zhang, X.; Wang, X.; Qin, C.; Sheng, Z.; Yin, H.; Jiang, C.; Li, J.; Xu, T. Lysophosphatidylcholine Acyltransferase 1 Upregulation and Concomitant Phospholipid Alterations in Clear Cell Renal Cell Carcinoma. J. Exp. Clin. Cancer Res. 2017, 36, 66. [Google Scholar] [CrossRef]
- Abdelzaher, E.; Mostafa, M.F. Lysophosphatidylcholine Acyltransferase 1 (LPCAT1) Upregulation in Breast Carcinoma Contributes to Tumor Progression and Predicts Early Tumor Recurrence. Tumor Biol. 2015, 36, 5473–5483. [Google Scholar] [CrossRef]
- Zhou, X.; Lawrence, T.J.; He, Z.; Pound, C.R.; Mao, J.; Bigler, S.A. The Expression Level of Lysophosphatidylcholine Acyltransferase 1 (LPCAT1) Correlates to the Progression of Prostate Cancer. Exp. Mol. Pathol. 2012, 92, 105–110. [Google Scholar] [CrossRef]
- Grupp, K.; Sanader, S.; Sirma, H.; Simon, R.; Koop, C.; Prien, K.; Hube-Magg, C.; Salomon, G.; Graefen, M.; Heinzer, H.; et al. High Lysophosphatidylcholine Acyltransferase 1 Expression Independently Predicts High Risk for Biochemical Recurrence in Prostate Cancers. Mol. Oncol. 2013, 7, 1001–1011. [Google Scholar] [CrossRef]
- Warnecke-Eberz, U.; Metzger, R.; Hölscher, A.H.; Drebber, U.; Bollschweiler, E. Diagnostic Marker Signature for Esophageal Cancer from Transcriptome Analysis. Tumor Biol. 2016, 37, 6349–6358. [Google Scholar] [CrossRef] [PubMed]
- Morita, Y.; Sakaguchi, T.; Ikegami, K.; Goto-Inoue, N.; Hayasaka, T.; Hang, V.T.; Tanaka, H.; Harada, T.; Shibasaki, Y.; Suzuki, A.; et al. Lysophosphatidylcholine Acyltransferase 1 Altered Phospholipid Composition and Regulated Hepatoma Progression. J. Hepatol. 2013, 59, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Uehara, T.; Kikuchi, H.; Miyazaki, S.; Iino, I.; Setoguchi, T.; Hiramatsu, Y.; Ohta, M.; Kamiya, K.; Morita, Y.; Tanaka, H.; et al. Overexpression of Lysophosphatidylcholine Acyltransferase 1 and Concomitant Lipid Alterations in Gastric Cancer. Ann. Surg. Oncol. 2016, 23, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Shida-Sakazume, T.; Endo-Sakamoto, Y.; Unozawa, M.; Fukumoto, C.; Shimada, K.; Kasamatsu, A.; Ogawara, K.; Yokoe, H.; Shiiba, M.; Tanzawa, H.; et al. Lysophosphatidylcholine Acyltransferase1 Overexpression Promotes Oral Squamous Cell Carcinoma Progression via Enhanced Biosynthesis of Platelet-Activating Factor. PLoS ONE 2015, 10, e0120143. [Google Scholar] [CrossRef]
- Xu, B.; Gao, L.; Wang, L.; Tang, G.; He, M.; Yu, Y.; Ni, X.; Sun, Y. Effects of Platelet-Activating Factor and Its Differential Regulation by Androgens and Steroid Hormones in Prostate Cancers. Br. J. Cancer 2013, 109, 1279–1286. [Google Scholar] [CrossRef][Green Version]
- Agarwal, A.K.; Garg, A. Enzymatic Activity of the Human 1-Acylglycerol-3-Phosphate-O-Acyltransferase Isoform 11: Upregulated in Breast and Cervical Cancers. J. Lipid Res. 2010, 51, 2143–2152. [Google Scholar] [CrossRef]
- Williams, K.A.; Lee, M.; Hu, Y.; Andreas, J.; Patel, S.J.; Zhang, S.; Chines, P.; Elkahloun, A.; Chandrasekharappa, S.; Gutkind, J.S.; et al. A Systems Genetics Approach Identifies CXCL14, ITGAX, and LPCAT2 as Novel Aggressive Prostate Cancer Susceptibility Genes. PLoS Genet 2014, 10, e1004809. [Google Scholar] [CrossRef]
- Cotte, A.K.; Aires, V.; Fredon, M.; Limagne, E.; Derangère, V.; Thibaudin, M.; Humblin, E.; Scagliarini, A.; de Barros, J.-P.P.; Hillon, P.; et al. Lysophosphatidylcholine Acyltransferase 2-Mediated Lipid Droplet Production Supports Colorectal Cancer Chemoresistance. Nat. Commun. 2018, 9, 322. [Google Scholar] [CrossRef]
- Wang, B.; Rong, X.; Palladino, E.N.D.; Wang, J.; Fogelman, A.M.; Martín, M.G.; Alrefai, W.A.; Ford, D.A.; Tontonoz, P. Phospholipid Remodeling and Cholesterol Availability Regulate Intestinal Stemness and Tumorigenesis. Cell Stem Cell 2018, 22. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-Related Inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Goossens, P.; Rodriguez-Vita, J.; Etzerodt, A.; Masse, M.; Rastoin, O.; Gouirand, V.; Ulas, T.; Papantonopoulou, O.; Eck, M.V.; Auphan-Anezin, N.; et al. Membrane Cholesterol Efflux Drives Tumor-Associated Macrophage Reprogramming and Tumor Progression. Cell Metab. 2019, 29, 1376–1389.e4. [Google Scholar] [CrossRef] [PubMed]
- Eibinger, G.; Fauler, G.; Bernhart, E.; Frank, S.; Hammer, A.; Wintersperger, A.; Eder, H.; Heinemann, A.; Mischel, P.S.; Malle, E.; et al. On the Role of 25-Hydroxycholesterol Synthesis by Glioblastoma Cell Lines. Implications for Chemotactic Monocyte Recruitment. Exp. Cell Res. 2013, 319, 1828–1838. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Lee, S.E.; Hur, J.; Hong, E.B.; Choi, J.-I.; Yang, J.-M.; Kim, J.-Y.; Kim, Y.-C.; Cho, H.-J.; Peters, J.M.; et al. M-CSF from Cancer Cells Induces Fatty Acid Synthase and PPARβ/δ Activation in Tumor Myeloid Cells, Leading to Tumor Progression. Cell Rep. 2015, 10, 1614–1625. [Google Scholar] [CrossRef] [PubMed]
- Kidani, Y.; Elsaesser, H.; Hock, M.B.; Vergnes, L.; Williams, K.J.; Argus, J.P.; Marbois, B.N.; Komisopoulou, E.; Wilson, E.B.; Osborne, T.F.; et al. Sterol Regulatory Element–Binding Proteins Are Essential for the Metabolic Programming of Effector T Cells and Adaptive Immunity. Nat. Immunol. 2013, 14, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Bensinger, S.J.; Bradley, M.N.; Joseph, S.B.; Zelcer, N.; Janssen, E.M.; Hausner, M.A.; Shih, R.; Parks, J.S.; Edwards, P.A.; Jamieson, B.D.; et al. LXR Signaling Couples Sterol Metabolism to Proliferation in the Acquired Immune Response. Cell 2008, 134, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Bai, Y.; Xiong, Y.; Zhang, J.; Chen, S.; Zheng, X.; Meng, X.; Li, L.; Wang, J.; Xu, C.; et al. Potentiating the Antitumour Response of CD8+ T Cells by Modulating Cholesterol Metabolism. Nature 2016, 531, 651–655. [Google Scholar] [CrossRef]
- Kidani, Y.; Bensinger, S.J. Modulating Cholesterol Homeostasis to Build a Better T Cell. Cell Metab. 2016, 23, 963–964. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Li, J.; Liu, Y.; Kang, L.; Chen, H.; Jin, Y.; Zhao, F.; Feng, J.; Fang, C.; Zhu, B.; et al. Cholesterol Esterification Enzyme Inhibition Enhances Antitumor Effects of Human Chimeric Antigen Receptors Modified T Cells. J. Immunother. 2018, 41, 45–52. [Google Scholar] [CrossRef]
- Ma, X.; Bi, E.; Lu, Y.; Su, P.; Huang, C.; Liu, L.; Wang, Q.; Yang, M.; Kalady, M.F.; Qian, J.; et al. Cholesterol Induces CD8+ T Cell Exhaustion in the Tumor Microenvironment. Cell Metab. 2019, 30, 143–156.e5. [Google Scholar] [CrossRef]
- Villablanca, E.J.; Raccosta, L.; Zhou, D.; Fontana, R.; Maggioni, D.; Negro, A.; Sanvito, F.; Ponzoni, M.; Valentinis, B.; Bregni, M.; et al. Tumor-Mediated Liver X Receptor-α Activation Inhibits CC Chemokine Receptor-7 Expression on Dendritic Cells and Dampens Antitumor Responses. Nat. Med. 2010, 16, 98–105. [Google Scholar] [CrossRef]
- Cao, W.; Ramakrishnan, R.; Tuyrin, V.A.; Veglia, F.; Condamine, T.; Amoscato, A.; Mohammadyani, D.; Johnson, J.J.; Zhang, L.M.; Klein-Seetharaman, J.; et al. Oxidized Lipids Block Antigen Cross-Presentation by Dendritic Cells in Cancer. J. Immunol. 2014, 192, 2920–2931. [Google Scholar] [CrossRef] [PubMed]
- Herber, D.L.; Cao, W.; Nefedova, Y.; Novitskiy, S.V.; Nagaraj, S.; Tyurin, V.A.; Corzo, A.; Cho, H.-I.; Celis, E.; Lennox, B.; et al. Lipid Accumulation and Dendritic Cell Dysfunction in Cancer. Nat. Med. 2010, 16, 880–886. [Google Scholar] [CrossRef] [PubMed]
- Dumitru, C.A.; Moses, K.; Trellakis, S.; Lang, S.; Brandau, S. Neutrophils and Granulocytic Myeloid-Derived Suppressor Cells: Immunophenotyping, Cell Biology and Clinical Relevance in Human Oncology. Cancer Immunol. Immunother. 2012, 61, 1155–1167. [Google Scholar] [CrossRef] [PubMed]
- Moses, K.; Brandau, S. Human Neutrophils: Their Role in Cancer and Relation to Myeloid-Derived Suppressor Cells. Semin. Immunol. 2016, 28, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Raccosta, L.; Fontana, R.; Maggioni, D.; Lanterna, C.; Villablanca, E.J.; Paniccia, A.; Musumeci, A.; Chiricozzi, E.; Trincavelli, M.L.; Daniele, S.; et al. The Oxysterol–CXCR2 Axis Plays a Key Role in the Recruitment of Tumor-Promoting NeutrophilsOxysterols and Migration of Protumor Neutrophils. J. Exp. Med. 2013, 210, 1711–1728. [Google Scholar] [CrossRef] [PubMed]
- Soncini, M.; Corna, G.; Moresco, M.; Coltella, N.; Restuccia, U.; Maggioni, D.; Raccosta, L.; Lin, C.-Y.; Invernizzi, F.; Crocchiolo, R.; et al. 24-Hydroxycholesterol Participates in Pancreatic Neuroendocrine Tumor Development. Proc. Natl. Acad. Sci. USA 2016, 113, E6219–E6227. [Google Scholar] [CrossRef] [PubMed]
- Baek, A.E.; Yu, Y.-R.A.; He, S.; Wardell, S.E.; Chang, C.-Y.; Kwon, S.; Pillai, R.V.; McDowell, H.B.; Thompson, J.W.; Dubois, L.G.; et al. The Cholesterol Metabolite 27 Hydroxycholesterol Facilitates Breast Cancer Metastasis through Its Actions on Immune Cells. Nat. Commun. 2017, 8, 864. [Google Scholar] [CrossRef]
- Condamine, T.; Dominguez, G.A.; Youn, J.-I.; Kossenkov, A.V.; Mony, S.; Alicea-Torres, K.; Tcyganov, E.; Hashimoto, A.; Nefedova, Y.; Lin, C.; et al. Lectin-Type Oxidized LDL Receptor-1 Distinguishes Population of Human Polymorphonuclear Myeloid-Derived Suppressor Cells in Cancer Patients. Sci. Immunol. 2016, 1, aaf8943. [Google Scholar] [CrossRef]
- Tavazoie, M.F.; Pollack, I.; Tanqueco, R.; Ostendorf, B.N.; Reis, B.S.; Gonsalves, F.C.; Kurth, I.; Andreu-Agullo, C.; Derbyshire, M.L.; Posada, J.; et al. LXR/ApoE Activation Restricts Innate Immune Suppression in Cancer. Cell 2018, 172, 825–840.e18. [Google Scholar] [CrossRef]
- Wenk, M.R. The Emerging Field of Lipidomics. Nat. Rev. Drug Discov. 2005, 4, 594–610. [Google Scholar] [CrossRef]
- Watson, A.D. Thematic Review Series: Systems Biology Approaches to Metabolic and Cardiovascular Disorders. Lipidomics: A Global Approach to Lipid Analysis in Biological Systems. J. Lipid Res. 2006, 47, 2101–2111. [Google Scholar] [CrossRef] [PubMed]
- Al-Khami, A.A.; Zheng, L.; Valle, L.D.; Hossain, F.; Wyczechowska, D.; Zabaleta, J.; Sanchez, M.D.; Dean, M.J.; Rodriguez, P.C.; Ochoa, A.C. Exogenous Lipid Uptake Induces Metabolic and Functional Reprogramming of Tumor-Associated Myeloid-Derived Suppressor Cells. Oncoimmunology 2017, 6, e1344804. [Google Scholar] [CrossRef]
- Han, X.; Yang, K.; Gross, R.W. Multi-dimensional Mass Spectrometry-based Shotgun Lipidomics and Novel Strategies for Lipidomic Analyses. Mass Spectrom. Rev. 2012, 31, 134–178. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wang, C.; Han, R.H.; Han, X. Novel Advances in Shotgun Lipidomics for Biology and Medicine. Prog. Lipid Res. 2016, 61, 83–108. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Zhang, J. Mass-spectrometry-based Lipidomics. J. Sep. Sci. 2018, 41, 351–372. [Google Scholar] [CrossRef] [PubMed]
- Marien, E.; Meister, M.; Muley, T.; del Pulgar, T.G.; Derua, R.; Spraggins, J.M.; Van de Plas, R.; Vanderhoydonc, F.; Machiels, J.; Binda, M.M.; et al. Phospholipid Profiling Identifies Acyl Chain Elongation as a Ubiquitous Trait and Potential Target for the Treatment of Lung Squamous Cell Carcinoma. Oncotarget 2016, 7, 12582–12597. [Google Scholar] [CrossRef]
- Dória, M.L.; Cotrim, C.Z.; Simões, C.; Macedo, B.; Domingues, P.; Domingues, M.R.; Helguero, L.A. Lipidomic Analysis of Phospholipids from Human Mammary Epithelial and Breast Cancer Cell Lines. J. Cell Physiol. 2013, 228, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Shen, M.; Li, Y.; Liu, C.; Zhou, K.; Hu, W.; Xu, B.; Xia, Y.; Tang, W. GC-MS-Based Metabolomic Analysis of Human Papillary Thyroid Carcinoma Tissue. Int. J. Mol. Med. 2015, 36, 1607–1614. [Google Scholar] [CrossRef]
- Kim, H.-Y.; Lee, K.-M.; Kim, S.-H.; Kwon, Y.-J.; Chun, Y.-J.; Choi, H.-K. Comparative Metabolic and Lipidomic Profiling of Human Breast Cancer Cells with Different Metastatic Potentials. Oncotarget 2016, 7, 67111–67128. [Google Scholar] [CrossRef]
- Li, J.; Ren, S.; Piao, H.; Wang, F.; Yin, P.; Xu, C.; Lu, X.; Ye, G.; Shao, Y.; Yan, M.; et al. Integration of Lipidomics and Transcriptomics Unravels Aberrant Lipid Metabolism and Defines Cholesteryl Oleate as Potential Biomarker of Prostate Cancer. Sci. Rep. UK 2016, 6, 20984. [Google Scholar] [CrossRef]
- Chen, X.; Chen, H.; Dai, M.; Ai, J.; Li, Y.; Mahon, B.; Dai, S.; Deng, Y. Plasma Lipidomics Profiling Identified Lipid Biomarkers in Distinguishing Early-Stage Breast Cancer from Benign Lesions. Oncotarget 2016, 7, 36622–36631. [Google Scholar] [CrossRef] [PubMed]
- Vallabhaneni, K.C.; Penfornis, P.; Dhule, S.; Guillonneau, F.; Adams, K.V.; Mo, Y.Y.; Xu, R.; Liu, Y.; Watabe, K.; Vemuri, M.C.; et al. Extracellular Vesicles from Bone Marrow Mesenchymal Stem/Stromal Cells Transport Tumor Regulatory MicroRNA, Proteins, and Metabolites. Oncotarget 2014, 6, 4953–4967. [Google Scholar] [CrossRef] [PubMed]
- Eberlin, L.S.; Dill, A.L.; Costa, A.B.; Ifa, D.R.; Cheng, L.; Masterson, T.; Koch, M.; Ratliff, T.L.; Cooks, R.G. Cholesterol Sulfate Imaging in Human Prostate Cancer Tissue by Desorption Electrospray Ionization Mass Spectrometry. Anal. Chem. 2010, 82, 3430–3434. [Google Scholar] [CrossRef] [PubMed]
- Angerer, T.B.; Magnusson, Y.; Landberg, G.; Fletcher, J.S. Lipid Heterogeneity Resulting from Fatty Acid Processing in the Human Breast Cancer Microenvironment Identified by GCIB-ToF-SIMS Imaging. Anal. Chem. 2016, 88, 11946–11954. [Google Scholar] [CrossRef]
- Banerjee, S.; Zare, R.N.; Tibshirani, R.J.; Kunder, C.A.; Nolley, R.; Fan, R.; Brooks, J.D.; Sonn, G.A. Diagnosis of Prostate Cancer by Desorption Electrospray Ionization Mass Spectrometric Imaging of Small Metabolites and Lipids. Proc. Natl. Acad. Sci. USA 2017, 114, 3334–3339. [Google Scholar] [CrossRef] [PubMed]
- Zanfini, A.; Dreassi, E.; Berardi, A.; Piomboni, P.; Costantino-Ceccarini, E.; Luddi, A. GC-EI-MS Analysis of Fatty Acid Composition in Brain and Serum of Twitcher Mouse. Lipids 2014, 49, 1115–1125. [Google Scholar] [CrossRef]
- Son, H.-H.; Moon, J.-Y.; Seo, H.S.; Kim, H.H.; Chung, B.C.; Choi, M.H. High-Temperature GC-MS-Based Serum Cholesterol Signatures May Reveal Sex Differences in Vasospastic Angina. J. Lipid Res. 2014, 55, 155–162. [Google Scholar] [CrossRef]
- Oh, S.F.; Vickery, T.W.; Serhan, C.N. Chiral Lipidomics of E-Series Resolvins: Aspirin and the Biosynthesis of Novel Mediators. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2011, 1811, 737–747. [Google Scholar] [CrossRef]
- Ling, Y.S.; Liang, H.; Lin, M.; Tang, C.; Wu, K.; Kuo, M.; Lin, C.Y. Two-dimensional LC-MS/MS to Enhance Ceramide and Phosphatidylcholine Species Profiling in Mouse Liver. Biomed. Chromatogr. 2014, 28, 1284–1293. [Google Scholar] [CrossRef]
- Wang, S.; Zhou, L.; Wang, Z.; Shi, X.; Xu, G. Simultaneous Metabolomics and Lipidomics Analysis Based on Novel Heart-Cutting Two-Dimensional Liquid Chromatography-Mass Spectrometry. Anal. Chim. Acta 2017, 966, 34–40. [Google Scholar] [CrossRef]
- Hall, Z.; Chiarugi, D.; Charidemou, E.; Leslie, J.; Scott, E.; Pelligrinet, L.; Allison, M.; Mocciaro, G.; Anstee, Q.M.; Evan, G.I.; et al. Lipid Remodelling in Hepatocyte Proliferation and Hepatocellular Carcinoma. Hepatology 2020. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).