

The Role and Therapeutic Targeting of JAK/STAT Signaling in Glioblastoma

Simple Summary
Abstract
1. Introduction
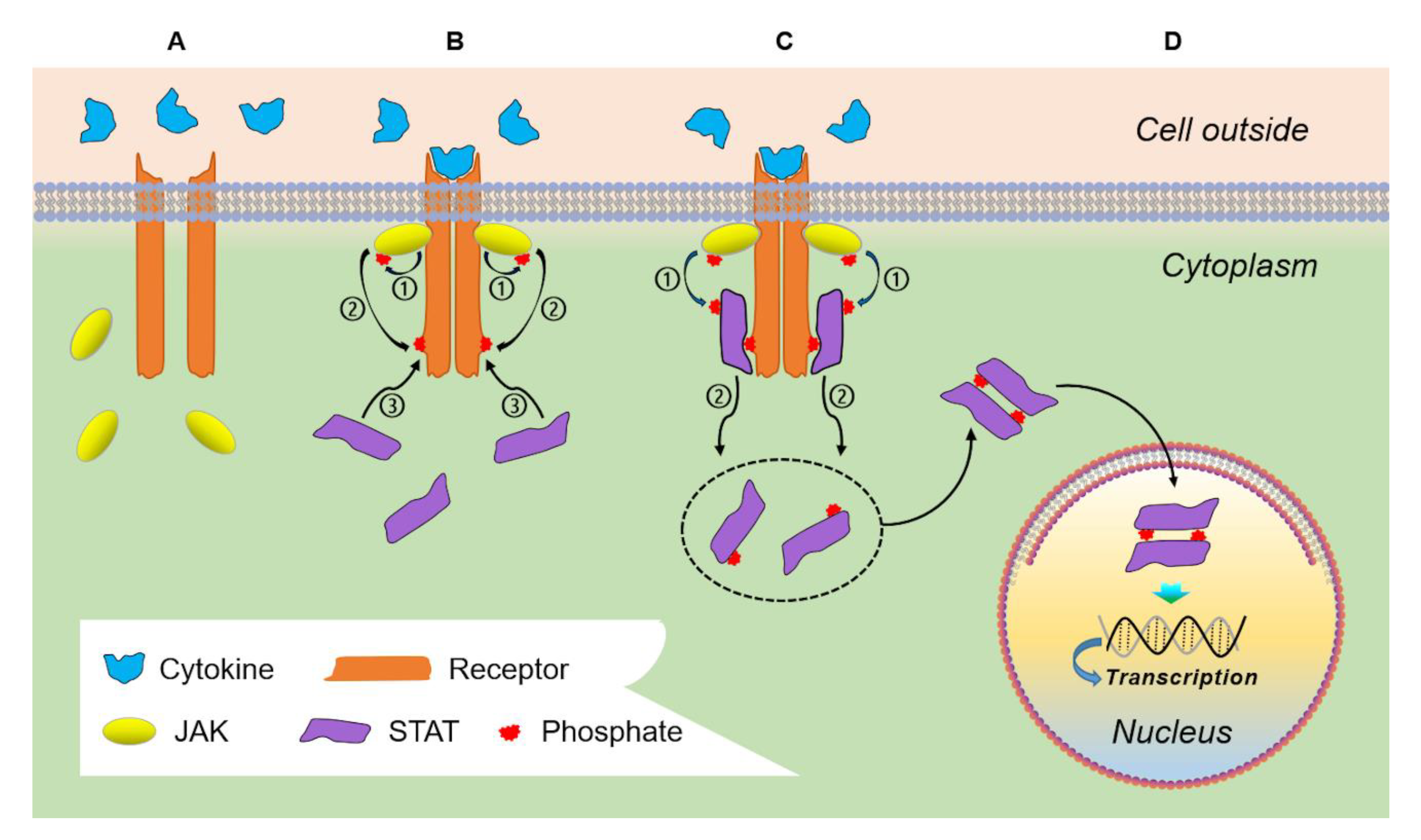
2. Physiologic JAK/STAT Signaling
3. Biological Principles in JAK/STAT Signaling in Glioblastoma Cells
3.1. STAT1 and STAT5 in GBM
3.2. STAT3 Dysregulation in GBM
3.3. Consequences of Dysregulated STAT3 Signaling in GBM
3.4. Consequences of Dysregulated STAT3 Signaling on the Immune Microenvironment
3.4.1. STAT3 Activation Generates an Immunosuppressive Cytokine Milieu
3.4.2. STAT3 Activation Impairs the Innate Immune System
3.4.3. STAT3 Activity Impairs the Adaptive Immune System
4. STAT3-Mediated Resistance to Therapeutic Modalities
Rationale for Combination Therapy
5. JAK/STAT Axis-Targeting Therapies
5.1. Nutraceutical Inhibition of STAT3
5.2. Pharmaceutical Inhibition of STAT3: JAK Inhibitors
5.3. Pharmaceutical Inhibition of STAT3: Peptide and Non-Peptide STAT3 Mimetics
5.4. Pharmaceutical Inhibition of STAT3: Oligonucleotide-Based Strategies
5.5. Pharmaceutical Inhibition of STAT3: Direct STAT3 Inhibitors
6. STAT3-Related Biomarkers
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Reich, N.C.; Liu, L. Tracking STAT nuclear traffic. Nat. Rev. Immunol. 2006, 6, 602–612. [Google Scholar] [CrossRef] [PubMed]
- Swiatek-Machado, K.; Kaminska, B. STAT signaling in glioma cells. Adv. Exp. Med. Biol. 2013, 986, 189–208. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, A.; Yasukawa, H.; Suzuki, A.; Kamizono, S.; Syoda, T.; Kinjyo, I.; Sasaki, M.; Johnston, J.A.; Yoshimura, A. Cytokine-inducible SH2 protein-3 (CIS3/SOCS3) inhibits Janus tyrosine kinase by binding through the N-terminal kinase inhibitory region as well as SH2 domain. Genes Cells 1999, 4, 339–351. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, A.; Ito, M.; Chikuma, S.; Akanuma, T.; Nakatsukasa, H. Negative Regulation of Cytokine Signaling in Immunity. Cold Spring Harb. Perspect. Biol. 2018, 10. [Google Scholar] [CrossRef]
- Shuai, K.; Liu, B. Regulation of gene-activation pathways by PIAS proteins in the immune system. Nat. Rev. Immunol. 2005, 5, 593–605. [Google Scholar] [CrossRef]
- O’Shea, J.J.; Gadina, M.; Schreiber, R.D. Cytokine signaling in 2002: New surprises in the Jak/Stat pathway. Cell 2002, 109, S121–S131. [Google Scholar] [CrossRef]
- Xu, D.; Qu, C.-K. Protein tyrosine phosphatases in the JAK/STAT pathway. Front. Biosci. 2008, 13, 4925–4932. [Google Scholar] [CrossRef]
- Roccograndi, L.; Binder, Z.A.; Zhang, L.; Aceto, N.; Zhang, Z.; Bentires-Alj, M.; Nakano, I.; Dahmane, N.; O’Rourke, D.M. SHP2 regulates proliferation and tumorigenicity of glioma stem cells. J. Neurooncol. 2017, 135, 487–496. [Google Scholar] [CrossRef]
- Hiroi, M.; Mori, K.; Sakaeda, Y.; Shimada, J.; Ohmori, Y. STAT1 represses hypoxia-inducible factor-1-mediated transcription. Biochem. Biophys. Res. Commun. 2009, 387, 806–810. [Google Scholar] [CrossRef]
- Chen, Z.; Mou, L.; Pan, Y. CXCL8 Promotes Glioma Progression By Activating The JAK/STAT1/HIF-1α/Snail Signaling Axis. OncoTargets Ther. 2019, 12, 8125–8138. [Google Scholar] [CrossRef]
- Thota, B.; Arimappamagan, A.; Kandavel, T.; Shastry, A.H.; Pandey, P.; Chandramouli, B.A.; Hegde, A.S.; Kondaiah, P.; Santosh, V. STAT-1 expression is regulated by IGFBP-3 in malignant glioma cells and is a strong predictor of poor survival in patients with glioblastoma. J. Neurosurg. 2014, 121, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Ju, H.; Li, X.; Li, H.; Wang, X.; Wang, H.; Li, Y.; Dou, C.; Zhao, G. Mediation of multiple pathways regulating cell proliferation, migration, and apoptosis in the human malignant glioma cell line U87MG via unphosphorylated STAT1: Laboratory investigation. J. Neurosurg. 2013, 118, 1239–1247. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Jin, G.; Zhang, J.; Mi, R.; Zhou, Y.; Fan, W.; Cheng, S.; Song, W.; Zhang, B.; Ma, M.; et al. Overexpression of STAT1 suppresses angiogenesis under hypoxia by regulating VEGF-A in human glioma cells. Biomed. Pharmacother. 2018, 104, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Duarte, C.W.; Willey, C.D.; Zhi, D.; Cui, X.; Harris, J.J.; Vaughan, L.K.; Mehta, T.; McCubrey, R.O.; Khodarev, N.N.; Weichselbaum, R.R.; et al. Expression signature of IFN/STAT1 signaling genes predicts poor survival outcome in glioblastoma multiforme in a subtype-specific manner. PLoS ONE 2012, 7, e29653. [Google Scholar] [CrossRef]
- Meng, D.; Chen, Y.; Yun, D.; Zhao, Y.; Wang, J.; Xu, T.; Li, X.; Wang, Y.; Yuan, L.; Sun, R.; et al. High expression of N-myc (and STAT) interactor predicts poor prognosis and promotes tumor growth in human glioblastoma. Oncotarget 2015, 6, 4901–4919. [Google Scholar] [CrossRef]
- Rajaraman, S.; Canjuga, D.; Ghosh, M.; Codrea, M.C.; Sieger, R.; Wedekink, F.; Tatagiba, M.; Koch, M.; Lauer, U.M.; Nahnsen, S.; et al. Measles Virus-Based Treatments Trigger a Pro-Inflammatory Cascade and a Distinctive Immunopeptidome in Glioblastoma. Mol. Ther. Oncolytics 2018, 12. [Google Scholar] [CrossRef]
- Haybaeck, J.; Obrist, P.; Schindler, C.U.; Spizzo, G.; Doppler, W. STAT-1 expression in human glioblastoma and peritumoral tissue. Anticancer Res. 2007, 27, 3829–3835. [Google Scholar]
- Cao, S.; Wang, C.; Zheng, Q.; Qiao, Y.; Xu, K.; Jiang, T.; Wu, A. STAT5 regulates glioma cell invasion by pathways dependent and independent of STAT5 DNA binding. Neurosci. Lett. 2011, 7, 228–233. [Google Scholar] [CrossRef]
- Liang, Q.C.; Xiong, H.; Zhao, Z.W.; Jia, D.; Li, W.X.; Qin, H.Z.; Deng, J.P.; Gao, L.; Zhang, H.; Gao, G.D. Inhibition of transcription factor STAT5b suppresses proliferation, induces G1 cell cycle arrest and reduces tumor cell invasion in human glioblastoma multiforme cells. Cancer Lett. 2009, 273, 164–171. [Google Scholar] [CrossRef]
- Roos, A.; Dhruv, H.D.; Peng, S.; Inge, L.J.; Tuncali, S.; Pineda, M.; Millard, N.; Mayo, Z.; Eschbacher, J.M.; Loftus, J.C.; et al. EGFRvIII-Stat5 Signaling Enhances Glioblastoma Cell Migration and Survival. Mol. Cancer Res. 2018, 16, 1185–1195. [Google Scholar] [CrossRef]
- Korzus, E.; Nagase, H.; Rydell, R.; Travis, J. The mitogen-activated protein kinase and JAK-STAT signaling pathways are required for an oncostatin M-responsive element-mediated activation of matrix metalloproteinase 1 gene expression. J. Biol. Chem. 1997, 10, 1188–1196. [Google Scholar] [CrossRef]
- Medina-Echeverz, J.; Haile, L.A.; Zhao, F.; Gamrekelashvili, J.; Ma, C.; Métais, J.Y.; Dunbar, C.E.; Kapoor, V.; Manns, M.P.; Korangy, F.; et al. IFN-γ regulates survival and function of tumor-induced CD11b+ Gr-1high myeloid derived suppressor cells by modulating the anti-apoptotic molecule Bcl2a1. Eur. J. Immunol. 2014, 44, 2457–2467. [Google Scholar] [CrossRef] [PubMed]
- Waight, J.D.; Netherby, C.; Hensen, M.L.; Miller, A.; Hu, Q.; Liu, S.; Bogner, P.N.; Farren, M.R.; Lee, K.P.; Liu, K.; et al. Myeloid-derived suppressor cell development is regulated by a STAT/IRF-8 axis. J. Clin. Investig. 2013, 123, 4464–4478. [Google Scholar] [CrossRef] [PubMed]
- Puram, S.V.; Yeung, C.M.; Jahani-Asl, A.; Lin, C.; de la Iglesia, N.; Konopka, G.; Jackson-Grusby, L.; Bonni, A. STAT3-iNOS Signaling Mediates EGFRvIII-Induced Glial Proliferation and Transformation. J. Neurosci. 2012, 6, 7806–7818. [Google Scholar] [CrossRef]
- Bromberg, J.F.; Wrzeszczynska, M.H.; Devgan, G. Stat3 as an oncogene. Cell 1999, 98, 295–303. [Google Scholar] [CrossRef]
- Lo, H.W.; Cao, X.; Zhu, H.; Ali-Osman, F. Constitutively activated STAT3 frequently coexpresses with epidermal growth factor receptor in high-grade gliomas and targeting STAT3 sensitizes them to Iressa and alkylators. Clin. Cancer Res. 2008, 14, 6042–6054. [Google Scholar] [CrossRef]
- Shao, H.; Cheng, H.Y.; Cook, R.G.; Tweardy, D.J. Identification and characterization of signal transducer and activator of transcription 3 recruitment sites within the epidermal growth factor receptor. Cancer Res. 2003, 15, 3923–3930. [Google Scholar]
- Kim, J.E.; Patel, M.; Ruzevick, J.; Jackson, C.M.; Lim, M. STAT3 activation in glioblastoma: Biochemical and therapeutic implications. Cancers 2014, 6, 376–395. [Google Scholar] [CrossRef]
- Sang, Y.; Li, Y.; Song, L.; Alvarez, A.A.; Zhang, W.; Lv, D.; Tang, J.; Liu, F.; Chang, Z.; Hatakeyama, S.; et al. TRIM59 Promotes Gliomagenesis by Inhibiting TC45 Dephosphorylation of STAT3. Cancer Res. 2018, 78, 1792–1804. [Google Scholar] [CrossRef]
- Oldrini, B. EGFR feedback-inhibition by Ran-binding protein 6 is disrupted in cancer. Nat. Commun. 2017, 8, 1–12. [Google Scholar] [CrossRef]
- Heimberger, A.B.; Hlatky, R.; Suki, D.; Yang, D.; Weinberg, J.; Gilbert, M.; Sawaya, R.; Aldape, K. Prognostic effect of epidermal growth factor receptor and EGFRvIII in glioblastoma multiforme patients. Clin. Cancer Res. 2005, 11, 1462–1466. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Feng, X.; Zhang, B.; Li, J.; Xu, X.; Liu, J.; Wang, X.; Wang, J.; Tong, X. Blocking the bFGF/STAT3 interaction through specific signaling pathways induces apoptosis in glioblastoma cells. J. Neurooncol. 2014, 120, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Li, Z.; Zhang, C.; Zhang, S.; Ji, Y.; Chen, F. Knockdown of PKCε expression inhibits growth, induces apoptosis and decreases invasiveness of human glioma cells partially through Stat3. J. Mol. Neurosci. 2015, 55, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Weissenberger, J.; Loeffler, S.; Kappeler, A.; Kopf, M.; Lukes, A.; Afanasieva, T.A.; Aguzzi, A.; Weis, J. IL-6 is required for glioma development in a mouse model. Oncogene 2004, 23, 3308–3316. [Google Scholar] [CrossRef]
- Liu, Q.; Li, G.; Li, R.; Shen, J.; He, Q.; Deng, L.; Zhang, C.; Zhang, J. IL-6 promotion of glioblastoma cell invasion and angiogenesis in U251 and T98G cell lines. J. Neurooncol. 2010. [Google Scholar] [CrossRef]
- Veeriah, S.; Brennan, C.; Meng, S.; Singh, B.; Fagin, J.A.; Solit, D.B.; Paty, P.B.; Rohle, D.; Vivanco, I.; Chmielecki, J.; et al. The tyrosine phosphatase PTPRD is a tumor suppressor that is frequently inactivated and mutated in glioblastoma and other human cancers. Proc. Natl. Acad. Sci. USA 2009, 9, 9435–9440. [Google Scholar] [CrossRef]
- Martini, M.; Pallini, R.; Luongo, G.; Cenci, T.; Lucantoni, C.; Larocca, L.M. Prognostic relevance of SOCS3 hypermethylation in patients with glioblastoma multiforme. Int. J. Cancer 2008, 15, 2955–2960. [Google Scholar] [CrossRef]
- Lindemann, C.; Hackmann, O.; Delic, S. SOCS3 promoter methylation is mutually exclusive to EGFR amplification in gliomas and promotes glioma cell invasion through STAT3 and FAK activation. Acta Neuropathol. 2011, 122, 241–251. [Google Scholar] [CrossRef]
- Zhou, H.; Miki, R.; Eeva, M.; Fike, F.M.; Seligson, D.; Yang, L.; Yoshimura, A.; Teitell, M.A.; Jamieson, C.A.; Cacalano, N.A. Reciprocal regulation of SOCS 1 and SOCS3 enhances resistance to ionizing radiation in glioblastoma multiforme. Clin. Cancer Res. 2007, 13, 2344–2353. [Google Scholar] [CrossRef]
- Shi, Y.; Guryanova, O.A.; Zhou, W. Ibrutinib inactivates BMX-STAT3 in glioma stem cells to impair malignant growth and radioresistance. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef]
- Brantley, E.C.; Nabors, L.B.; Gillespie, G.Y.; Choi, Y.H.; Palmer, C.A.; Harrison, K.; Roarty, K.; Benveniste, E.N. Loss of protein inhibitors of activated STAT-3 expression in glioblastoma multiforme tumors: Implications for STAT-3 activation and gene expression. Clin. Cancer Res. 2008, 14, 4694–4704. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.D.; Liao, J.; Liu, B.; Rao, X.; Jay, P.; Berta, P.; Shuai, K. Specific inhibition of Stat3 signal transduction by PIAS3. Science 1997, 278, 1803–1805. [Google Scholar] [CrossRef] [PubMed]
- Jiao, J.; Zhang, R.; Li, Z.; Yin, Y.; Fang, X.; Ding, X.; Cai, Y.; Yang, S.; Mu, H.; Zong, D.; et al. Nuclear Smad6 promotes gliomagenesis by negatively regulating PIAS3-mediated STAT3 inhibition. Nat. Commun. 2018, 9, 2504. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, J.V.; Mukherjee, N.; Chakravarti, A.; Robe, P.; Zhai, G.; Chakladar, A.; Loeffler, J.; Black, P.; Frank, D.A. A STAT3 Gene Expression Signature in Gliomas is Associated with a Poor Prognosis. Transl. Oncogenomics 2007, 2, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Birner, P.; Toumangelova-Uzeir, K.; Natchev, S.; Guentchev, M. STAT3 tyrosine phosphorylation influences survival in glioblastoma. J. Neurooncol. 2010, 100, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Tu, Y.; Zhong, Y.; Fu, J.; Cao, Y.; Fu, G.; Tian, X.; Wang, B. Activation of JAK/STAT signal pathway predicts poor prognosis of patients with gliomas. Med. Oncol. 2011, 28, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Abou-Ghazal, M.; Yang, D.S.; Qiao, W.; Reina-Ortiz, C.; Wei, J.; Kong, L.Y.; Fuller, G.N.; Hiraoka, N.; Priebe, W.; Sawaya, R.; et al. The incidence, correlation with tumor-infiltrating inflammation, and prognosis of phosphorylated STAT3 expression in human gliomas. Clin. Cancer Res. 2008, 15, 8228–8235. [Google Scholar] [CrossRef]
- Mizoguchi, M.; Betensky, R.A.; Batchelor, T.T.; Bernay, D.C.; Louis, D.N.; Nutt, C.L. Activation of STAT3, MAPK, and AKT in malignant astrocytic gliomas: Correlation with EGFR status, tumor grade, and survival. J. Neuropathol. Exp. Neurol. 2006, 65, 1181–1188. [Google Scholar] [CrossRef]
- Leventoux, N.; Augustus, M.; Azar, S.; Riquier, S.; Villemin, J.P.; Guelfi, S.; Falha, L.; Bauchet, L.; Gozé, C.; Ritchie, W.; et al. Transformation Foci in IDH1-mutated Gliomas Show STAT3 Phosphorylation and Downregulate the Metabolic Enzyme ETNPPL, a Negative Regulator of Glioma Growth. Sci. Rep. 2020, 26, 5504. [Google Scholar] [CrossRef]
- Park, A.K.; Kim, P.; Ballester, L.Y.; Esquenazi, Y.; Zhao, Z. Subtype-specific signaling pathways and genomic aberrations associated with prognosis of glioblastoma. Neuro-Oncology 2019, 1, 59–70. [Google Scholar] [CrossRef]
- De la Iglesia, N.; Konopka, G.; Lim, K.-L.; Nutt, C.L.; Bromberg, J.F.; Frank, D.A.; Mischel, P.S.; Louis, D.N.; Bonni, A. Deregulation of a STAT3-interleukin 8 signaling pathway promotes human glioblastoma cell proliferation and invasiveness. J. Neurosci. 2008, 28, 5870–5878. [Google Scholar] [CrossRef] [PubMed]
- De la Iglesia, N.; Konopka, G.; Puram, S.V.; Chan, J.A.; Bachoo, R.M.; You, M.J.; Levy, D.E.; Depinho, R.A.; & Bonni, A. Identification of a PTEN-regulated STAT3 brain tumor suppressor pathway. Genes Dev. 2008, 22, 449–462. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Li, J.; Shen, J.; Wang, Z.; Xu, H.; Wang, Q.; Chai, S.; Fu, P.; Huang, T.; Anas, O.; Zhao, H.; et al. ELTD1 facilitates glioma proliferation, migration and invasion by activating JAK/STAT3/HIF-1α signaling axis. Sci. Rep. 2019, 25, 13904. [Google Scholar] [CrossRef]
- Chen, X.; Xu, H.; Yuan, P. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell 2008, 133, 1106–1117. [Google Scholar] [CrossRef]
- Guryanova, O.A.; Wu, Q.; Cheng, L. Nonreceptor tyrosine kinase BMX maintains self-renewal and tumorigenic potential of glioblastoma stem cells by activating STAT3. Cancer Cell 2011, 19, 498–511. [Google Scholar] [CrossRef]
- Cheng, W.; Zhang, C.; Ren, X.; Jiang, Y.; Han, S.; Liu, Y.; Cai, J.; Li, M.; Wang, K.; Liu, Y.; et al. Bioinformatic analyses reveal a distinct Notch activation induced by STAT3 phosphorylation in the mesenchymal subtype of glioblastoma. J. Neurosurg. 2017, 126, 249–259. [Google Scholar] [CrossRef]
- Peñuelas, S.; Anido, J.; Prieto-Sánchez, R.M.; Folch, G.; Barba, I.; Cuartas, I.; García-Dorado, D.; Poca, M.A.; Sahuquillo, J.; Baselga, J.; et al. TGF-beta increases glioma-initiating cell self-renewal through the induction of LIF in human glioblastoma. Cancer Cell 2009, 7, 315–327. [Google Scholar] [CrossRef]
- Hossain, A.; Gumin, J.; Gao, F. Mesenchymal Stem Cells Isolated From Human Gliomas Increase Proliferation and Maintain Stemness of Glioma Stem Cells through the IL-6/gp130/STAT3 Pathway. Stem Cells 2015, 33, 2400–2415. [Google Scholar] [CrossRef]
- Wang, H.; Lathia, J.D.; Wu, Q.; Wang, J.; Li, Z.; Heddleston, J.M.; Eyler, C.E.; Elderbroom, J.; Gallagher, J.; Schuschu, J.; et al. Targeting interleukin 6 signaling suppresses glioma stem cell survival and tumor growth. Stem Cells 2009, 27, 2393–2404. [Google Scholar] [CrossRef] [PubMed]
- Che, S.; Sun, T.; Wang, J.; Jiao, Y.; Wang, C.; Meng, Q.; Qi, W.; Yan, Z. miR-30 overexpression promotes glioma stem cells by regulating Jak/STAT3 signaling pathway. Tumour. Biol. 2015, 36, 6805–6811. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Zhou, A.; Wu, Y.; Morris, S.A.; Lin, K.; Amin, S.; Verhaak, R.; Fuller, G.; Xie, K.; Heimberger, A.B.; et al. miR-182-5p Induced by STAT3 Activation Promotes Glioma Tumorigenesis. Cancer Res. 2016, 76, 4293–4304. [Google Scholar] [CrossRef] [PubMed]
- Zong, Z.; Pang, H.; Yu, R.; Jiao, Y. PCDH8 inhibits glioma cell proliferation by negatively regulating the AKT/GSK3β/β-catenin signaling pathway. Oncol. Lett. 2017, 14, 3357–3362. [Google Scholar] [CrossRef]
- Kim, E.; Kim, M.; Woo, D.H. Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell 2013, 23, 839–852. [Google Scholar] [CrossRef]
- Liu, H.; Sun, Y.; Qi, X. EZH2 Phosphorylation Promotes Self-Renewal of Glioma Stem-Like Cells Through NF-κB Methylation. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef]
- Li, G.H.; Wei, H.; Lv, S.Q.; Ji, H.; Wang, D.L. Knockdown of STAT3 expression by RNAi suppresses growth and induces apoptosis and differentiation in glioblastoma stem cells. Int. J. Oncol. 2010, 37, 103–110. [Google Scholar]
- Sherry, M.M.; Reeves, A.; Wu, J.K.; Cochran, B.H. STAT3 is required for proliferation and maintenance of multipotency in glioblastoma stem cells. Stem Cells 2009, 27, 2383–2392. [Google Scholar] [CrossRef]
- Villalva, C.; Martin-Lannerée, S.; Cortes, U.; Cortes, U.; Dkhissi, F.; Wager, M.; Le Corf, A.; Tourani, J.M.; Dusanter-Fourt, I.; Turhan, A.G.; et al. STAT3 is essential for the maintenance of neurosphere-initiating tumor cells in patients with glioblastomas: A potential for targeted therapy? Int. J. Cancer 2011, 15, 826–838. [Google Scholar] [CrossRef]
- Chen, F.; Xu, Y.; Luo, Y.; Zheng, D.; Song, Y.; Yu, K.; Li, H.; Zhang, L.; Zhong, W.; Ji, Y. Down-regulation of Stat3 decreases invasion activity and induces apoptosis of human glioma cells. J. Mol. Neurosci. 2010, 40, 353–359. [Google Scholar] [CrossRef]
- Gu, J.; Li, G.; Sun, T.; Su, Y.; Zhang, X.; Shen, J.; Tian, Z.; Zhang, J. Blockage of the STAT3 signaling pathway with a decoy oligonucleotide suppresses growth of human malignant glioma cells. J. Neurooncol. 2008, 89, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Weissenberger, J.; Priester, M.; Bernreuther, C.; Rakel, S.; Glatzel, M.; Seifert, V.; Kögel, D. Dietary curcumin attenuates glioma growth in a syngeneic mouse model by inhibition of the JAK1,2/STAT3 signaling pathway. Clin. Cancer Res. 2010, 16, 5781–5795. [Google Scholar] [CrossRef] [PubMed]
- Iwamaru, A.; Szymanski, S.; Iwado, E.; Aoki, H.; Yokoyama, T.; Fokt, I.; Hess, K.; Conrad, C.; Madden, T.; Sawaya, R.; et al. A novel inhibitor of the STAT3 pathway induces apoptosis in malignant glioma cells both in vitro and in vivo. Oncogene 2007, 26, 2435–2444. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, L.K.; Ren, Z.; Fuller, G.N.; Schaefer, T.S. Constitutive activation of Stat3alpha in brain tumors: Localization to tumor endothelial cells and activation by the endothelial tyrosine kinase receptor (VEGFR-2). Oncogene 2002, 21, 2058–2065. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.H.; Yu, M.O.; Park, K.J.; Chi, S.G.; Park, D.H.; Chung, Y.G. Activated STAT3 regulates hypoxia-induced angiogenesis and cell migration in human glioblastoma. Neurosurgery 2010, 67, 1386–1395. [Google Scholar] [CrossRef]
- Couto, M.; Coelho-Santos, V.; Santos, L.; Fontes-Ribeiro, C.; Silva, A.P.; Gomes CMF. The interplay between glioblastoma and microglia cells leads to endothelial cell monolayer dysfunction via the interleukin-6-induced JAK2/STAT3 pathway. J. Cell Physiol. 2019, 234, 19750–19760. [Google Scholar] [CrossRef]
- Li, R.; Li, G.; Deng, L.; Liu, Q.; Dai, J.; Shen, J.; Zhang, J. IL-6 augments the invasiveness of U87MG human glioblastoma multiforme cells via up-regulation of MMP-2 and fascin-1. Oncol. Rep. 2010, 23, 1553–1559. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Han, L.; Dong, Y.; Tian, J.; Huang, W.; Liu, Z.; Jia, X.; Jiang, T.; Zhang, J.; Li, X.; et al. JAK2/STAT3 targeted therapy suppresses tumor invasion via disruption of the EGFRvIII/JAK2/STAT3 axis and associated focal adhesion in EGFRvIII-expressing glioblastoma. Neuro-Oncology 2014, 16, 1229–1243. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.H.; Liu, Y.; Xiao, L.M.; Chen, L.K.; Zheng, S.Y.; Zeng, E.M.; Li, D.H.; Li, Y.P. Silencing microRNA-221/222 cluster suppresses glioblastoma angiogenesis by suppressor of cytokine signaling-3-dependent JAK/STAT pathway. J. Cell Physiol. 2019, 234, 22272–22284. [Google Scholar] [CrossRef] [PubMed]
- Hyun Hwang, J.; Smith, C.A.; Salhia, B.; Rutka, J.T. The Role of Fascin in the Migration and Invasiveness of Malignant Glioma Cells. Neoplasia 2008, 10, 149–159. [Google Scholar] [CrossRef]
- Ji, P.; Wang, L.; Liu, J.; Mao, P.; Li, R.; Jiang, H.; Lou, M.; Xu, M.; Yu, X. Knockdown of RPL34 inhibits the proliferation and migration of glioma cells through the inactivation of JAK/STAT3 signaling pathway. J. Cell Biochem. 2019, 120, 3259–3267. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Chen, F.Z.; Jia, Q.B.; Hu, D.F. Upregulation of microRNA-133a and downregulation of connective tissue growth factor suppress cell proliferation, migration, and invasion in human glioma through the JAK/STAT signaling pathway. IUBMB Life 2019, 71, 1857–1875. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Brandes, A.A.; Omuro, A.; Mulholland, P.; Lim, M.; Wick, A.; Baehring, J.; Ahluwalia, M.S.; Roth, P.; Bähr, O.; et al. Effect of Nivolumab vs Bevacizumab in Patients With Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1003. [Google Scholar] [CrossRef] [PubMed]
- Phase III CheckMate-548 Trial of Opdivo Fails Endpoint. Available online: https://www.thepharmaletter.com/article/phase-iii-checkmate-58-trial-of-opdivo-fails-endpoint (accessed on 23 December 2020).
- Johnson, L.A.; Scholler, J.; Ohkuri, T.; Kosaka, A.; Patel, P.R.; McGettigan, S.E.; Nace, A.K.; Dentchev, T.; Thekkat, P.; Loew, A.; et al. Rational development and characterization of humanized anti–EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci. Transl. Med. 2015, 7, 275ra22. [Google Scholar] [CrossRef]
- Ahmed, N.; Salsman, V.S.; Kew, Y.; Shaffer, D.; Powell, S.; Zhang, Y.J.; Grossman, R.G.; Heslop, H.E.; Gottschalk, S. HER2-specific T cells target primary Glioblastoma stem cells and induce regression of autologous experimental tumors. Clin. Cancer Res. 2010, 16, 474–485. [Google Scholar] [CrossRef] [PubMed]
- Bielamowicz, K.; Fousek, K.; Byrd, T.T.; Samaha, H.; Mukherjee, M.; Aware, N.; Wu, M.F.; Orange, J.S.; Sumazin, P.; Man, T.K.; et al. Trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma. Neuro-Oncology 2018, 20, 506–518. [Google Scholar] [CrossRef]
- Brown, C.E.; Starr, R.; Aguilar, B.; Shami, A.F.; Martinez, C.; D’Apuzzo, M.; Barish, M.E.; Forman, S.J.; Jensen, M.C. Stem-like tumor initiating cells isolated from IL13Rα2-expressing gliomas are targeted and killed by IL13-zetakine redirected T cells. Clin. Cancer Res. 2012, 18, 2199–2209. [Google Scholar] [CrossRef]
- Luoto, S.; Hermelo, I.; Vuorinen, E.M. Computational Characterization of Suppressive Immune Microenvironments in Glioblastoma. Cancer Res. 2018, 78, 5574–5585. [Google Scholar] [CrossRef]
- Wang, Q.; Hu, X.; Hu, B. Tumor Evolution of Glioma Intrinsic Gene Expression Subtype Associates with Immunological Changes in the Microenvironment. Cancer Biol. 2016. [Google Scholar] [CrossRef]
- Yu, H.; Kortylewski, M.; Pardoll, D. Crosstalk between cancer and immune cells: Role of STAT3 in the tumour microenvironment. Nat. Rev. Immunol. 2007, 7, 41–51. [Google Scholar] [CrossRef]
- Kortylewski, M.; Kujawski, M.; Wang, T. Inhibiting STAT3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat. Med. 2005, 11, 1314–1321. [Google Scholar] [CrossRef] [PubMed]
- Henrik Heiland, D.; Ravi, V.M.; Behringer, S.P.; Frenking, J.H.; Wurm, J.; Joseph, K.; Garrelfs, N.; Strähle, J.; Heynckes, S.; Grauvogel, J.; et al. Tumor-associated reactive astrocytes aid the evolution of immunosuppressive environment in glioblastoma. Nat. Commun. 2019, 10, 2541. [Google Scholar] [CrossRef] [PubMed]
- Chuang, H.-Y.; Su, Y.-K.; Liu, H.-W.; Chen, C.H.; Chiu, S.C.; Cho, D.Y.; Lin, S.Z.; Chen, Y.S.; Lin, C.M. Preclinical Evidence of STAT3 Inhibitor Pacritinib Overcoming Temozolomide Resistance via Downregulating miR-21-Enriched Exosomes from M2 Glioblastoma-Associated Macrophages. J. Clin. Med. 2019, 8, 959. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Ye, H.; Qi, Z.; Mo, L.; Yue, Q.; Baral, A.; Hoon, D.; Vera, J.C.; Heiss, J.D.; Chen, C.C.; et al. B7-H4(B7x)-Mediated Cross-talk between Glioma-Initiating Cells and Macrophages via the IL6/JAK/STAT3 Pathway Lead to Poor Prognosis in Glioma Patients. Clin. Cancer Res. 2016, 1, 2778–2790. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Eksioglu, E.A.; Zhou, J.; Zhang, L.; Djeu, J.; Fortenbery, N.; Epling-Burnette, P.; Van Bijnen, S.; Dolstra, H.; Cannon, J.; et al. Induction of myelodysplasia by myeloid-derived suppressor cells. J. Clin. Investig. 2013, 123, 4595–4611. [Google Scholar] [CrossRef]
- Jeon, S.-B.; Yoon, H.J.; Chang, C.Y.; Koh, H.S.; Jeon, S.-H.; Park, E.J. Galectin-3 exerts cytokine-like regulatory actions through the JAK-STAT pathway. J. Immunol. 2010, 185, 7037–7046. [Google Scholar] [CrossRef]
- Ko, H.J.; Kim, Y.J. Signal transducer and activator of transcription proteins: Regulators of myeloid-derived suppressor cell-mediated immunosuppression in cancer. Arch. Pharm Res. 2016, 39, 1597–1608. [Google Scholar] [CrossRef]
- Saraiva, M.; O’Garra, A. The regulation of IL-10 production by immune cells. Nat. Rev. Immunol. 2010, 10, 170–181. [Google Scholar] [CrossRef]
- Yu, H.; Liu, Y.; McFarland, B.C.; Deshane, J.S.; Hurst, D.R.; Ponnazhagan, S.; Benveniste, E.N.; Qin, H. SOCS3 Deficiency in Myeloid Cells Promotes Tumor Development: Involvement of STAT3 Activation and Myeloid-Derived Suppressor Cells. Cancer Immunol. Res. 2015, 727–740. [Google Scholar] [CrossRef]
- Wang, T.; Niu, G.; Kortylewski, M.; Burdelya, L.; Shain, K.; Zhang, S.; Bhattacharya, R.; Gabrilovich, D.; Heller, R.; Coppola, D.; et al. Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nat. Med. 2004, 10, 48–54. [Google Scholar] [CrossRef]
- Cheng, F.; Wang, H.-W.; Cuenca, A.; Huang, M.; Ghansah, T.; Brayer, J.; Kerr, W.G.; Takeda, K.; Akira, S.; Schoenberger, S.P.; et al. A critical role for Stat3 signaling in immune tolerance. Immunity 2003, 19, 425–436. [Google Scholar] [CrossRef]
- Wang, N.; Liang, H.; Zen, K. Molecular mechanisms that influence the macrophage m1-m2 polarization balance. Front. Immunol. 2014, 5, 614. [Google Scholar] [CrossRef] [PubMed]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A.; et al. Macrophage plasticity, polarization, and function in health and disease. J. Cell Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Wei, J.; Kong, L.Y.; Wang, Y.; Priebe, W.; Qiao, W.; Sawaya, R.; Heimberger, A.B. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro-Oncology 2010, 12, 1113–1125. [Google Scholar] [CrossRef] [PubMed]
- Huettner, C.; Czub, S.; Kerkau, S.; Roggendorf, W.; Tonn, J.C. Interleukin 10 is expressed in human gliomas in vivo and increases glioma cell proliferation and motility in vitro. Anticancer Res. 1997, 17, 3217–3224. [Google Scholar]
- O’Farrell, A.M.; Liu, Y.; Moore, K.W.; Mui, A.L. IL-10 inhibits macrophage activation and proliferation by distinct signaling mechanisms: Evidence for Stat3-dependent and -independent pathways. EMBO J. 1998, 16, 1006–1018. [Google Scholar] [CrossRef] [PubMed]
- Lang, R.; Patel, D.; Morris, J.J.; Rutschman, R.L.; Murray, P.J. Shaping gene expression in activated and resting primary macrophages by IL-10. J. Immunol. 2002, 169, 2253–2263. [Google Scholar] [CrossRef]
- Kortylewski, M.; Xin, H.; Kujawski, M.; Lee, H.; Liu, Y.; Harris, T.; Drake, C.; Pardoll, D.; Yu, H. Regulation of the IL-23 and IL-12 balance by Stat3 signaling in the tumor microenvironment. Cancer Cell 2009, 3, 114–123. [Google Scholar] [CrossRef]
- Hussain, S.F.; Yang, D.; Suki, D.; Aldape, K.; Grimm, E.; Heimberger, A.B. The role of human glioma-infiltrating microglia/macrophages in mediating antitumor immune responses. Neuro-Oncology 2006, 8, 261–279. [Google Scholar] [CrossRef]
- Molavi, O.; Ma, Z.; Hamdy, S.; Lavasanifar, A.; Samuel, J. Immunomodulatory and anticancer effects of intra-tumoral co-delivery of synthetic lipid A adjuvant and STAT3 inhibitor, JSI-124. Immunopharmacol. Immunotoxicol. 2009, 31, 214–221. [Google Scholar] [CrossRef]
- Poholek, C.H.; Raphael, I.; Wu, D.; Revu, S.; Rittenhouse, N.; Uche, U.U.; Majumder, S.; Kane, L.P.; Poholek, A.C.; McGeachy, M.J. Noncanonical STAT3 activity sustains pathogenic Th17 proliferation and cytokine response to antigen. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Klement, J.D.; Lu, C.; Ibrahim, M.L.; Liu, K. IFNAR1 Controls Autocrine Type I IFN Regulation of PD-L1 Expression in Myeloid-Derived Suppressor Cells. J. Immunol. 2018, 1, 264–277. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, P.C.; Quiceno, D.G.; Zabaleta, J.; Ortiz, B.; Zea, A.H.; Piazuelo, M.B.; Delgado, A.; Correa, P.; Brayer, J.; Sotomayor, E.M.; et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res. 2004, 15, 5839–5849. [Google Scholar] [CrossRef] [PubMed]
- Rajappa, P.; Cobb, W.S.; Vartanian, E.; Huang, Y.; Daly, L.; Hoffman, C.; Zhang, J.; Shen, B.; Yanowitch, R.; Garg, K.; et al. Malignant Astrocytic Tumor Progression Potentiated by JAK-mediated Recruitment of Myeloid Cells. Clin. Cancer Res. 2017, 15, 3109–3119. [Google Scholar] [CrossRef]
- Vasquez-Dunddel, D.; Pan, F.; Zeng, Q.; Gorbounov, M.; Albesiano, E.; Fu, J.; Blosser, R.L.; Tam, A.J.; Bruno, T.; Zhang, H.; et al. STAT3 regulates arginase-I in myeloid-derived suppressor cells from cancer patients. J. Clin. Investig. 2013, 123, 1580–1589. [Google Scholar] [CrossRef]
- Fujita, M.; Zhu, X.; Sasaki, K.; Ueda, R.; Low, K.L.; Pollack, I.F.; Okada, H. Inhibition of STAT3 promotes the efficacy of adoptive transfer therapy using type-1 CTLs by modulation of the immunological microenvironment in a murine intracranial glioma. J. Immunol. 2008, 15, 2089–2098. [Google Scholar] [CrossRef]
- Farren, M.R.; Carlson, L.M.; Netherby, C.S.; Lindner, I.; Li, P.K.; Gabrilovich, D.I.; Abrams, S.I.; Lee, K.P. Tumor-induced STAT3 signaling in myeloid cells impairs dendritic cell generation by decreasing PKCβII abundance. Sci. Signal. 2014, 18, ra16. [Google Scholar] [CrossRef]
- Chang, Q.; Bournazou, E.; Sansone, P.; Berishaj, M.; Gao, S.P.; Daly, L.; Wels, J.; Theilen, T.; Granitto, S.; Zhang, X.; et al. The IL-6/JAK/Stat3 feed-forward loop drives tumorigenesis and metastasis. Neoplasia 2013, 15, 848–862. [Google Scholar] [CrossRef]
- Zhang, L.; Alizadeh, D.; Van Handel, M.; Kortylewski, M.; Yu, H.; Badie, B. Stat3 inhibition activates tumor macrophages and abrogates glioma growth in mice. Glia 2009, 57, 1458–1467. [Google Scholar] [CrossRef]
- Hussain, S.F.; Kong, L.Y.; Jordan, J.; Conrad, C.; Madden, T.; Fokt, I.; Priebe, W.; Heimberger, A.B. A novel small molecule inhibitor of signal transducers and activators of transcription 3 reverses immune tolerance in malignant glioma patients. Cancer Res. 2007, 67, 9630–9636. [Google Scholar] [CrossRef] [PubMed]
- Yue, C.; Shen, S.; Deng, J.; Priceman, S.J.; Li, W.; Huang, A.; Yu, H. STAT3 in CD8+ T Cells Inhibits Their Tumor Accumulation by Downregulating CXCR3/CXCL10 Axis. Cancer Immunol. Res. 2015, 3, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Andaloussi, A.E.; Lesniak, M.S. An increase in CD4+CD25+FOXP3+ regulatory T cells in tumor-infiltrating lymphocytes of human glioblastoma multiforme. Neuro-Oncology 2006, 8, 234–243. [Google Scholar] [CrossRef] [PubMed]
- Zorn, E.; Nelson, E.A.; Mohseni, M.; Porcheray, F.; Kim, H.; Litsa, D.; Bellucci, R.; Raderschall, E.; Canning, C.; Soiffer, R.J.; et al. IL-2 regulates FOXP3 expression in human CD4+CD25+ regulatory T cells through a STAT-dependent mechanism and induces the expansion of these cells in vivo. Blood 2006, 108, 1571–1579. [Google Scholar] [CrossRef] [PubMed]
- Pallandre, J.-R.; Brillard, E.; Créhange, G.; Radlovic, A.; Remy-Martin, J.P.; Saas, P.; Rohrlich, P.S.; Pivot, X.; Ling, X.; Tiberghien, P.; et al. Role of STAT3 in CD4+CD25+FOXP3+ Regulatory Lymphocyte Generation: Implications in Graft-versus-Host Disease and Antitumor Immunity. J. Immunol. 2007, 179, 7593–7604. [Google Scholar] [CrossRef]
- Wei, J.; Wu, A.; Kong, L.Y.; Wang, Y.; Fuller, G.; Fokt, I.; Melillo, G.; Priebe, W.; Heimberger, A.B. Hypoxia potentiates glioma-mediated immunosuppression. PLoS ONE 2011, 20, e16195. [Google Scholar] [CrossRef]
- Roos, W.; Batista, L.; Naumann, S.; Wick, W.; Weller, M.; Menck, C.F.; Kaina, B. Apoptosis in malignant glioma cells triggered by the temozolomide-induced DNA lesion O6-methylguanine. Oncogene 2007, 26, 186–197. [Google Scholar] [CrossRef]
- Fu, D.; Calvo, J.; Samson, L. Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat. Rev. Cancer 2012, 12, 104–120. [Google Scholar] [CrossRef]
- Kohsaka, S.; Wang, L.; Yachi, K.; Mahabir, R.; Narita, T.; Itoh, T.; Tanino, M.; Kimura, T.; Nishihara, H.; Tanaka, S. STAT3 Inhibition Overcomes Temozolomide Resistance in Glioblastoma by Downregulating MGMT Expression. Mol. Cancer Ther. 2012, 1289–1299. [Google Scholar] [CrossRef]
- Kitange, G.J.; Carlson, B.L.; Schroeder, M.A. Induction of MGMT expression is associated with temozolomide resistance in glioblastoma xenografts. Neuro-Oncology 2009, 11, 281–291. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, L.; Bao, Z.; Li, S.; You, G.; Yan, W.; Shi, Z.; Liu, Y.; Yang, P.; Zhang, W.; et al. Inhibition of STAT3 reverses alkylator resistance through modulation of the AKT and β-catenin signaling pathways. Oncol. Rep. 2011, 26, 1173–1180. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.S.; Ko, K.K.; Joe, Y.A.; Kang, S.G.; Hong, Y.K. Inhibition of STAT3 reverses drug resistance acquired in temozolomide-resistant human glioma cells. Oncol. Lett. 2011, 2, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Chen, L.; Li, J.; Zhou, Q.; Huang, A.; Liu, W.W.; Wang, K.; Gao, L.; Qi, S.T.; Lu, Y.T. miR-519a enhances chemosensitivity and promotes autophagy in glioblastoma by targeting STAT3/Bcl2 signaling pathway. J. Hematol. Oncol. 2018, 11. [Google Scholar] [CrossRef] [PubMed]
- Heynckes, S.; Daka, K.; Franco, P.; Gaebelein, A.; Frenking, J.H.; Doria-Medina, R.; Mader, I.; Delev, D.; Schnell, O.; Heiland, D.H. Crosslink between Temozolomide and PD-L1 immune-checkpoint inhibition in glioblastoma multiforme. BMC Cancer 2019, 19, 117. [Google Scholar] [CrossRef] [PubMed]
- Sato, N.; Mizumoto, K.; Nakamura, M. Radiation-induced centrosome overduplication and multiple mitotic spindles in human tumor cells. Exp. Cell Res. 2000, 255, 321–326. [Google Scholar] [CrossRef]
- Rath, B.H.; Wahba, A.; Camphausen, K.; Tofilon, P.J. Coculture with astrocytes reduces the radiosensitivity of glioblastoma stem-like cells and identifies additional targets for radiosensitization. Cancer Med. 2015, 4, 1705–1716. [Google Scholar] [CrossRef]
- Yu, H.; Zhang, S.; Ibrahim, A.N.; Wang, J.; Deng, Z.; Wang, M. RCC2 promotes proliferation and radio-resistance in glioblastoma via activating transcription of DNMT1. Biochem. Biophys. Res. Commun. 2019, 27, 999–1006. [Google Scholar] [CrossRef]
- Mrowczynski, O.D.; Madhankumar, A.B.; Sundstrom, J.M. Exosomes impact survival to radiation exposure in cell line models of nervous system cancer. Oncotarget 2018, 9, 36083–36101. [Google Scholar] [CrossRef]
- Li, C.; Ran, H.; Song, S.; Liu, W.; Zou, W.; Jiang, B.; Zhao, H.; Shao, B. Overexpression of RPN2 suppresses radiosensitivity of glioma cells by activating STAT3 signal transduction. Mol. Med. 2020, 13, 43. [Google Scholar] [CrossRef]
- Ventero, M.P.; Fuentes-Baile, M.; Quereda, C.; Perez-Valeciano, E.; Alenda, C.; Garcia-Morales, P.; Saceda, M. Radiotherapy resistance acquisition in Glioblastoma. Role of SOCS1 and SOCS3. PLoS ONE 2019, 14, e0212581. [Google Scholar] [CrossRef]
- Maachani, U.B.; Shankavaram, U.; Kramp, T.; Tofilon, P.J.; Camphausen, K.; Tandle, A.T. FOXM1 and STAT3 interaction confers radioresistance in glioblastoma cells. Oncotarget 2016, 22, 77365–77377. [Google Scholar] [CrossRef] [PubMed]
- Zhong, C.; Tao, B.; Chen, Y. B7-H3 Regulates Glioma Growth and Cell Invasion Through a JAK2/STAT3/Slug-Dependent Signaling Pathway. Onco Targets Ther. 2020, 13, 2215–2224. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.-C.; Tsai, J.-T.; Chao, T.-Y.; Ma, H.-I.; Liu, W.-H. The STAT3/Slug Axis Enhances Radiation-Induced Tumor Invasion and Cancer Stem-like Properties in Radioresistant Glioblastoma. Cancers 2018, 10, 512. [Google Scholar] [CrossRef] [PubMed]
- Robe, P.A.; Martin, D.H.; Nguyen-Khac, M.T.; Artesi, M.; Deprez, M.; Albert, A.; Vanbelle, S.; Califice, S.; Bredel, M.; Bours, V. Early termination of ISRCTN45828668, a phase 1/2 prospective, randomized study of sulfasalazine for the treatment of progressing malignant gliomas in adults. BMC Cancer 2009, 9, 372. [Google Scholar] [CrossRef]
- Atkinson, G.P.; Nozell, S.E.; Benveniste, E.T. NF-kappaB and STAT3 signaling in glioma: Targets for future therapies. Expert Rev. Neurother. 2010, 10, 575–586. [Google Scholar] [CrossRef]
- Zhao, C.; Li, H.; Lin, H.-J.; Yang, S.; Lin, J.; Liang, G. Feedback Activation of STAT3 as a Cancer Drug-Resistance Mechanism. Trends Pharmacol. Sci. 2016, 37, 47–61. [Google Scholar] [CrossRef]
- Gao, L.; Li, F.; Dong, B.; Zhang, J.; Rao, Y.; Cong, Y.; Mao, B.; Chen, X. Inhibition of STAT3 and ErbB2 suppresses tumor growth, enhances radiosensitivity, and induces mitochondria-dependent apoptosis in glioma cells. Int. J. Radiat. Oncol. Biol. Phys 2010, 15, 1223–1231. [Google Scholar] [CrossRef]
- Jensen, K.V.; Hao, X.; Aman, A.; Luchman, H.A.; Weiss, S. EGFR blockade in GBM brain tumor stem cells synergizes with JAK2/STAT3 pathway inhibition to abrogate compensatory mechanisms in vitro and in vivo. Neuro-Oncol. Adv. 2020, 2, vdaa020. [Google Scholar] [CrossRef]
- De Groot, J.; Liang, J.; Kong, L.-Y.; Wei, J.; Piao, Y.; Fuller, G.; Qiao, W.; Heimberger, A.B. Modulating Antiangiogenic Resistance by Inhibiting the Signal Transducer and Activator of Transcription 3 Pathway in Glioblastoma. Oncotarget 2012, 3, 1036–1048. [Google Scholar] [CrossRef]
- Batchelor, T.T.; Reardon, D.A.; de Groot, J.F.; Wick, W.; Weller, M. Antiangiogenic therapy for glioblastoma: Current status and future prospects. Clin. Cancer Res. 2014, 20, 5612–5619. [Google Scholar] [CrossRef]
- Cruickshanks, N.; Zhang, Y.; Hine, S.; Gibert, M.; Yuan, F.; Oxford, M.; Grello, C.; Pahuski, M.; Dube, C.; Guessous, F.; et al. Discovery and Therapeutic Exploitation of Mechanisms of Resistance to MET Inhibitors in Glioblastoma. Clin. Cancer Res. 2019, 25, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.A.; Luo, X.; Lu, M.; An, Z.; Haas-Kogan, D.A.; Phillips, J.J.; Shokat, K.M.; Weiss, W.A.; Fan, Q.W. Cooperative Blockade of PKCα and JAK2 Drives Apoptosis in Glioblastoma. Cancer Res. 2020, 15, 709–718. [Google Scholar] [CrossRef] [PubMed]
- Ott, M.; Kassab, C.; Marisetty, A.; Hashimoto, Y.; Wei, J.; Zamler, D.; Leu, J.S.; Tomaszowski, K.H.; Sabbagh, A.; Fang, D.; et al. Radiation with STAT3 Blockade Triggers Dendritic Cell-T cell Interactions in the Glioma Microenvironment and Therapeutic Efficacy. Clin. Cancer Res. 2020, 26, 4983–4994. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, A.; Kortylewski, M.; Kujawski, M.; Zhang, C.; Reckamp, K.; Armstrong, B.; Wang, L.; Kowolik, C.; Deng, J.; Figlin, R.; et al. Targeting Stat3 in the myeloid compartment drastically improves the in vivo antitumor functions of adoptively transferred T cells. Cancer Res. 2010, 70, 7455–7464. [Google Scholar] [CrossRef]
- Kong, L.-Y.; Wei, J.; Fuller, G.N.; Schrand, B.; Gabrusiewicz, K.; Zhou, S.; Rao, G.; Calin, G.; Gilboa, E.; Heimberger, A.B. Tipping a favorable CNS intratumoral immune response using immune stimulation combined with inhibition of tumor-mediated immune suppression. Oncoimmunology 2016, 5, e1117739. [Google Scholar] [CrossRef]
- Stechishin, O.D.; Luchman, H.A.; Ruan, Y.; Blough, M.D.; Nguyen, S.A.; Kelly, J.J.; Cairncross, J.G.; Weiss, S. On-target JAK2/STAT3 inhibition slows disease progression in orthotopic xenografts of human glioblastoma brain tumor stem cells. Neuro-Oncology 2013, 15, 198–207. [Google Scholar] [CrossRef]
- Lamano, J.B.; Lamano, J.B.; Li, Y.D.; DiDomenico, J.D.; Choy, W.; Veliceasa, D.; Oyon, D.E.; Fakurnejad, S.; Ampie, L.; Kesavabhotla, K.; et al. Glioblastoma-Derived IL6 Induces Immunosuppressive Peripheral Myeloid Cell PD-L1 and Promotes Tumor Growth. Clin. Cancer Res. 2019, 25, 3643–3657. [Google Scholar] [CrossRef]
- Kudo, M.; Jono, H.; Shinriki, S.; Yano, S.; Nakamura, H.; Makino, K.; Hide, T.; Muta, D.; Ueda, M.; Ota, K.; et al. Antitumor effect of humanized anti–interleukin-6 receptor antibody (tocilizumab) on glioma cell proliferation: Laboratory investigation. J. Neurosurg. 2009, 111, 219–225. [Google Scholar] [CrossRef]
- Nellan, A.; McCully, C.M.L.; Cruz Garcia, R.; Jayaprakash, N.; Widemann, B.C.; Lee, D.W.; Warren, K.E. Improved CNS exposure to tocilizumab after cerebrospinal fluid compared to intravenous administration in rhesus macaques. Blood 2018, 132, 662–666. [Google Scholar] [CrossRef]
- Xiang, M.; Kim, H.; Ho, V.T.; Walker, S.R.; Bar-Natan, M.; Anahtar, M.; Liu, S.; Toniolo, P.A.; Kroll, Y.; Jones, N.; et al. Gene expression-based discovery of atovaquone as a STAT3 inhibitor and anticancer agent. Blood 2016, 128, 1845–1853. [Google Scholar] [CrossRef]
- Takabe, H.; Warnken, Z.N.; Zhang, Y.; Davis, D.A.; Smyth, H.; Kuhn, J.G.; Weitman, S.; Williams Iii, R.O. A Repurposed Drug for Brain Cancer: Enhanced Atovaquone Amorphous Solid Dispersion by Combining a Spontaneously Emulsifying Component with a Polymer Carrier. Pharmaceutics 2018, 10, 60. [Google Scholar] [CrossRef] [PubMed]
- Grimm, S.A.; Marymont, M.; Chandler, J.P.; Muro, K.; Newman, S.B.; Levy, R.M.; Jovanovic, B.; McCarthy, K.; Raizer, J.J. Phase I study of arsenic trioxide and temozolomide in combination with radiation therapy in patients with malignant gliomas. J. Neurooncol. 2012, 110, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Kumthekar, P.; Grimm, S.; Chandler, J.; Mehta, M.; Marymont, M.; Levy, R.; Muro, K.; Helenowski, I.; McCarthy, K.; Fountas, L.; et al. A phase II trial of arsenic trioxide and temozolomide in combination with radiation therapy for patients with malignant gliomas. J. Neurooncol. 2017, 133, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Zhang, S. Arsenic trioxide regulates the apoptosis of glioma cell and glioma stem cell via down-regulation of stem cell marker Sox2. Biochem. Biophys. Res. Commun. 2011, 410, 692–697. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Brown, C.; Buettner, R. Sorafenib induces growth arrest and apoptosis of human glioblastoma cells through the dephosphorylation of signal transducers and activators of transcription 3. Mol. Cancer Ther. 2010, 9, 953–962. [Google Scholar] [CrossRef] [PubMed]
- Carra, E.; Barbieri, F.; Marubbi, D.; Pattarozzi, A.; Favoni, R.E.; Florio, T.; Daga, A. Sorafenib selectively depletes human glioblastoma tumor-initiating cells from primary cultures. Cell Cycle 2013, 12, 491–500. [Google Scholar] [CrossRef]
- Hottinger, A.F.; Ben Aissa, A.; Espeli, V. Phase I study of sorafenib combined with radiation therapy and temozolomide as first-line treatment of high-grade glioma. Br. J. Cancer 2014, 110, 2655–2661. [Google Scholar] [CrossRef]
- Lee, E.Q.; Kuhn, J.; Lamborn, K.R.; Abrey, L.; DeAngelis, L.M.; Lieberman, F.; Robins, H.I.; Chang, S.M.; Yung, W.K.; Drappatz, J.; et al. Phase I/II study of sorafenib in combination with temsirolimus for recurrent glioblastoma or gliosarcoma: North American Brain Tumor Consortium study 05-02. Neuro-Oncology 2012, 14, 1511–1518. [Google Scholar] [CrossRef]
- Zustovich, F.; Landi, L.; Lombardi, G.; Porta, C.; Galli, L.; Fontana, A.; Amoroso, D.; Galli, C.; Andreuccetti, M.; Falcone, A.; et al. Sorafenib plus daily low-dose temozolomide for relapsed glioblastoma: A phase II study. Anticancer Res. 2013, 33, 3487–3494. [Google Scholar] [CrossRef]
- Gambini, J.; Inglés, M.; Olaso, G.; Lopez-Grueso, R.; Bonet-Costa, V.; Gimeno-Mallench, L.; Mas-Bargues, C.; Abdelaziz, K.M.; Gomez-Cabrera, M.C.; Vina, J.; et al. Properties of Resveratrol: In Vitro and In Vivo Studies about Metabolism, Bioavailability, and Biological Effects in Animal Models and Humans. Oxid Med. Cell Longev. 2015, 2015, 837042. [Google Scholar] [CrossRef]
- Xue, S.; Xiao-Hong, S.; Lin, S.; Jie, B.; Li-Li, W.; Jia-Yao, G.; Shun, S.; Pei-Nan, L.; Mo-Li, W.; Qian, W.; et al. Lumbar puncture-administered resveratrol inhibits STAT3 activation, enhancing autophagy and apoptosis in orthotopic rat glioblastomas. Oncotarget 2016, 7, 75790–75799. [Google Scholar] [CrossRef] [PubMed]
- Jhaveri, A.; Deshpande, P.; Pattni, B.; Torchilin, V. Transferrin-targeted, resveratrol-loaded liposomes for the treatment of glioblastoma. J. Control. Release 2018, 277, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Rocha, T.G.R.; de Lopes, S.C.A.; Cassali, G.D.; Ferreira, E.; Veloso, E.S.; Leite, E.A.; Tavitian, B. Evaluation of Antitumor Activity of Long-Circulating and pH-Sensitive Liposomes Containing Ursolic Acid in Animal Models of Breast Tumor and Gliosarcoma. Integr. Cancer Ther. 2016, 15, 512–524. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Yu, W.; Cai, G.; Zhu, J.; Zhang, C.; Li, S.; Guo, J.; Yin, G.; Chen, C.; Kong, L. A new synthetic derivative of cryptotanshinone KYZ3 as STAT3 inhibitor for triple-negative breast cancer therapy. Cell Death Dis. 2018, 9, 1–11. [Google Scholar] [CrossRef]
- Verdura, S.; Cuyàs, E.; Llorach-Parés, L.; Pérez-Sánchez, A.; Micol, V.; Nonell-Canals, A.; Joven, J.; Valiente, M.; Sánchez-Martínez, M.; Bosch-Barrera, J.; et al. Silibinin is a direct inhibitor of STAT3. Food Chem. Toxicol. 2018, 116 Pt B, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Elhag, R.; Mazzio, E.A.; Soliman, K.F.A. The Effect of Silibinin in Enhancing Toxicity of Temozolomide and Etoposide in p53 and PTEN-mutated Resistant Glioma Cell Lines. Anticancer Res. 2015, 35, 1263–1269. [Google Scholar]
- Wang, C.; He, C.; Lu, S.; Wang, X.; Wang, L.; Liang, S.; Wang, X.; Piao, M.; Cui, J.; Chi, G.; et al. Autophagy activated by silibinin contributes to glioma cell death via induction of oxidative stress-mediated BNIP3-dependent nuclear translocation of AIF. Cell Death Dis. 2020, 11, 1–16. [Google Scholar] [CrossRef]
- Bosch-Barrera, J.; Sais, E.; Cañete, N.; Marruecos, J.; Cuyàs, E.; Izquierdo, A.; Porta, R.; Haro, M.; Brunet, J.; Pedraza, S.; et al. Response of brain metastasis from lung cancer patients to an oral nutraceutical product containing silibinin. Oncotarget 2016, 7, 32006–32014. [Google Scholar] [CrossRef]
- Flaig, T.W.; Glodé, M.; Gustafson, D.; van Bokhoven, A.; Tao, Y.; Wilson, S.; Su, L.J.; Li, Y.; Harrison, G.; Agarwal, R.; et al. A study of high-dose oral silybin-phytosome followed by prostatectomy in patients with localized prostate cancer. Prostate 2010, 70, 848–855. [Google Scholar] [CrossRef]
- Lu, L.; Li, C.; Li, D.; Wang, Y.; Zhou, C.; Shao, W.; Peng, J.; You, Y.; Zhang, X.; Shen, X. Cryptotanshinone inhibits human glioma cell proliferation by suppressing STAT3 signaling. Mol. Cell Biochem. 2013, 381, 273–282. [Google Scholar] [CrossRef]
- Cai, Y.; Zhang, W.; Chen, Z.; Shi, Z.; He, C.; Chen, M. Recent insights into the biological activities and drug delivery systems of tanshinones. Int. J. Nanomed. 2016, 11, 121–130. [Google Scholar] [CrossRef]
- Wang, X.; Yu, Z.; Wang, C.; Cheng, W.; Tian, X.; Huo, X.; Wang, Y.; Sun, C.; Feng, L.; Xing, J.; et al. Alantolactone, a natural sesquiterpene lactone, has potent antitumor activity against glioblastoma by targeting IKKβ kinase activity and interrupting NF-κB/COX-2-mediated signaling cascades. J. Exp. Clin. Cancer Res. 2017, 36. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Yi, F.; Rasul, A.; Li, T.; Wang, N.; Gao, H.; Gao, R.; Ma, T. Alantolactone induces apoptosis in glioblastoma cells via GSH depletion, ROS generation, and mitochondrial dysfunction. IUBMB Life 2012, 64, 783–794. [Google Scholar] [CrossRef] [PubMed]
- Matias, D.; Balça-Silva, J.; Dubois, L.G.; Pontes, B.; Ferrer, V.P.; Rosário, L.; do Carmo, A.; Echevarria-Lima, J.; Sarmento-Ribeiro, A.B.; Lopes, M.C.; et al. Dual treatment with shikonin and temozolomide reduces glioblastoma tumor growth, migration and glial-to-mesenchymal transition. Cell Oncol. (Dordr.) 2017, 40, 247–261. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Kretschmer, N.; Bauer, R.; Efferth, T. Shikonin and its derivatives inhibit the epidermal growth factor receptor signaling and synergistically kill glioblastoma cells in combination with erlotinib. Int. J. Cancer 2015, 137, 1446–1456. [Google Scholar] [CrossRef] [PubMed]
- Miao, Z.; Yu, F.; Ren, Y.; Yang, J. d,l-Sulforaphane Induces ROS-Dependent Apoptosis in Human Gliomablastoma Cells by Inactivating STAT3 Signaling Pathway. Int. J. Mol. Sci. 2017, 18, 72. [Google Scholar] [CrossRef] [PubMed]
- Karmakar, S.; Weinberg, M.S.; Banik, N.L.; Patel, S.J.; Ray, S.K. Activation of multiple molecular mechanisms for apoptosis in human malignant glioblastoma T98G and U87MG cells treated with sulforaphane. Neuroscience 2006, 141, 1265–1280. [Google Scholar] [CrossRef]
- Bijangi-Vishehsaraei, K.; Saadatzadeh, M.R.; Wang, H.; Nguyen, A.; Kamocka, M.M.; Cai, W.; Cohen-Gadol, A.A.; Halum, S.L.; Sarkaria, J.N.; Pollok, K.E.; et al. Sulforaphane suppresses the growth of glioblastoma cells, glioblastoma stem cell–like spheroids, and tumor xenografts through multiple cell signaling pathways. J. Neurosurg. 2017, 127, 1219–1230. [Google Scholar] [CrossRef]
- Kim, B.; Lee, K.Y.; Park, B. Crocin Suppresses Constitutively Active STAT3 through Induction of Protein Tyrosine Phosphatase SHP-1. J. Cell Biochem. 2017, 118, 3290–3298. [Google Scholar] [CrossRef]
- Colapietro, A.; Mancini, A.; Vitale, F.; Martellucci, S.; Angelucci, A.; Llorens, S.; Mattei, V.; Gravina, G.L.; Alonso, G.L.; Festuccia, C. Crocetin Extracted from Saffron Shows Antitumor Effects in Models of Human Glioblastoma. Int. J. Mol. Sci. 2020, 21, 423. [Google Scholar] [CrossRef]
- Lautenschläger, M.; Sendker, J.; Hüwel, S.; Galla, H.J.; Brandt, S.; Düfer, M.; Riehemann, K.; Hensel, A. Intestinal formation of trans-crocetin from saffron extract (Crocus sativus L.) and in vitro permeation through intestinal and blood brain barrier. Phytomedicine 2015, 22, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Liu, J.; Zhao, X.; Yan, Z.; Jiang, B.; Wang, L.; Cao, S.; Shi, D.; & Lin, X. Cardamonin induces apoptosis by suppressing STAT3 signaling pathway in glioblastoma stem cells. Tumour Biol. 2015, 36, 9667–9676. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Sharma, A.; Shukla, M.; Lal, J. Gender-related pharmacokinetics and bioavailability of a novel anticancer chalcone, cardamonin, in rats determined by liquid chromatography tandem mass spectrometry. J. Chromatogr. B 2015, 986–987, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Yang, Y.; Zhang, H.; Che, Y.; Wang, W.; Lv, H.; Li, J.; Wang, Y.; Hou, S. Serenoa Repens Induces Growth Arrest, Apoptosis and Inactivation of STAT3 Signaling in Human Glioma Cells. Technol. Cancer Res. Treat. 2015, 14, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.; Ma, R.; Huang, S.; Liao, Y.; Ding, Y.; Li, Z.; Guo, Q.; Tan, R.; Zhang, L.; Zhao, L. Oroxylin A increases the sensitivity of temozolomide on glioma cells by hypoxia-inducible factor 1α/hedgehog pathway under hypoxia. J. Cell Physiol. 2019, 234, 17392–17404. [Google Scholar] [CrossRef]
- Zou, M.; Hu, C.; You, Q.; Zhang, A.; Wang, X.; Guo, Q. Oroxylin A induces autophagy in human malignant glioma cells via the mTOR-STAT3-Notch signaling pathway. Mol. Carcinog. 2015, 54, 1363–1375. [Google Scholar] [CrossRef] [PubMed]
- Ren, G.; Chen, H.; Zhang, M.; Yang, N.; Yang, H.; Xu, C.; Li, J.; Ning, C.; Song, Z.; Zhou, S.; et al. Pharmacokinetics, tissue distribution and excretion study of Oroxylin, A.; Oroxylin A 7-O-glucuronide and Oroxylin A sodium sulfonate in rats after administration of Oroxylin, A. Fitoterapia 2020, 142, 104480. [Google Scholar] [CrossRef]
- Michaud-Levesque, J.; Bousquet-Gagnon, N.; Béliveau, R. Quercetin abrogates IL-6/STAT3 signaling and inhibits glioblastoma cell line growth and migration. Exp. Cell Res. 2012, 1, 925–935. [Google Scholar] [CrossRef]
- Fujiwara, Y.; Komohara, Y.; Kudo, R.; Tsurushima, K.; Ohnishi, K.; Ikeda, T.; Takeya, M. Oleanolic acid inhibits macrophage differentiation into the M2 phenotype and glioblastoma cell proliferation by suppressing the activation of STAT3. Oncol. Rep. 2011, 26, 1533–1537. [Google Scholar] [CrossRef]
- Guo, G.; Yao, W.; Zhang, Q.; Bo, Y. Oleanolic acid suppresses migration and invasion of malignant glioma cells by inactivating MAPK/ERK signaling pathway. PLoS ONE 2013, 8, e72079. [Google Scholar] [CrossRef]
- Jiang, Q.; Yang, X.; Du, P.; Zhang, H.; Zhang, T. Dual strategies to improve oral bioavailability of oleanolic acid: Enhancing water-solubility, permeability and inhibiting cytochrome P450 isozymes. Eur. J. Pharm. Biopharm. 2016, 99, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Perry, M.-C.; Demeule, M.; Régina, A.; Moumdjian, R.; Béliveau, R. Curcumin inhibits tumor growth and angiogenesis in glioblastoma xenografts. Mol. Nutr. Food Res. 2010, 54, 1192–1201. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.H.; Shen, C.Y.; Chien, Y.C.; Chang, W.S.; Tsai, C.W.; Lin, Y.H.; Hwang, J.J. Validation of Enhancing Effects of Curcumin on Radiotherapy with F98/FGT Glioblastoma-Bearing Rat Model. Int. J. Mol. Sci. 2020, 19, 4385. [Google Scholar] [CrossRef] [PubMed]
- Dei Cas, M.; Ghidoni, R. Dietary Curcumin: Correlation between Bioavailability and Health Potential. Nutrients 2019, 11, 2147. [Google Scholar] [CrossRef]
- Cho, H.J.; Park, J.H.; Nam, J.H.; Chang, Y.C.; Park, B.; Hoe, H.S. Ascochlorin Suppresses MMP-2-Mediated Migration and Invasion by Targeting FAK and JAK-STAT Signaling Cascades. J. Cell Biochem. 2018, 119, 300–313. [Google Scholar] [CrossRef]
- Magae, J.; Tsuruga, M.; Maruyama, A.; Furukawa, C.; Kojima, S.; Shimizu, H.; Ando, K. Relationship between peroxisome proliferator-activated receptor-γ activation and the ameliorative effects of ascochlorin derivatives on type II diabetes. J. Antibiot. 2009, 62, 365–369. [Google Scholar] [CrossRef][Green Version]
- Ge, W.; Chen, X.; Han, F.; Liu, Z.; Wang, T.; Wang, M.; Chen, Y.; Ding, Y.; Zhang, Q. Synthesis of Cucurbitacin B Derivatives as Potential Anti-Hepatocellular Carcinoma Agents. Molecules 2018, 23, 3345. [Google Scholar] [CrossRef]
- Premkumar, D.R.; Jane, E.P.; Pollack, I.F. Cucurbitacin-I inhibits Aurora kinase A.; Aurora kinase B and survivin, induces defects in cell cycle progression and promotes ABT-737-induced cell death in a caspase-independent manner in malignant human glioma cells. Cancer Biol. Ther. 2015, 16, 233–243. [Google Scholar] [CrossRef]
- Almeida, L.; Vaz-da-Silva, M.; Falcão, A.; Soares, E.; Costa, R.; Loureiro, A.I.; Fernandes-Lopes, C.; Rocha, J.F.; Nunes, T.; Wright, L.; et al. Pharmacokinetic and safety profile of trans-resveratrol in a rising multiple-dose study in healthy volunteers. Mol. Nutr. Food Res. 2009, 53 (Suppl. 1), S7–S15. [Google Scholar] [CrossRef]
- Baskin, R.; Park, S.O.; Keserű, G.M.; Bisht, K.S.; Wamsley, H.L.; Sayeski, P.P. The Jak2 small molecule inhibitor, G6, reduces the tumorigenic potential of T98G glioblastoma cells in vitro and in vivo. PLoS ONE 2014, 27, e105568. [Google Scholar] [CrossRef]
- Mukthavaram, R.; Ouyang, X.; Saklecha, R. Effect of the JAK2/STAT3 inhibitor SAR317461 on human glioblastoma tumorspheres. J. Transl. Med. 2015, 13. [Google Scholar] [CrossRef] [PubMed]
- Nefedova, Y.; Nagaraj, S.; Rosenbauer, A.; Muro-Cacho, C.; Sebti, S.M.; Gabrilovich, D.I. Regulation of dendritic cell differentiation and antitumor immune response in cancer by pharmacologic-selective inhibition of the janus-activated kinase 2/signal transducers and activators of transcription 3 pathway. Cancer Res. 2005, 15, 9525–9535. [Google Scholar] [CrossRef] [PubMed]
- McFarland, B.C.; Gray, G.K.; Nozell, S.E.; Hong, S.W.; Benveniste, E.N. Activation of the NF-κB pathway by the STAT3 inhibitor JSI-124 in human glioblastoma cells. Mol. Cancer Res. 2013. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Li, G.; Zhang, X.; Gu, J.; Zhang, C.; Tian, Z.; Zhang, J. JSI-124 Inhibits Glioblastoma Multiforme Cell Proliferation through G(2)/M Cell Cycle Arrest and Apoptosis Augment. Cancer Biol. Ther. 2008, 1243–1249. [Google Scholar] [CrossRef]
- Plimack, E.R.; Lorusso, P.M.; McCoon, P.; Tang, W.; Krebs, A.D.; Curt, G.; Eckhardt, S.G. AZD1480: A phase I study of a novel JAK2 inhibitor in solid tumors. Oncologist 2013, 18, 819–820. [Google Scholar] [CrossRef]
- McFarland, B.C.; Ma, J.Y.; Langford, C.P.; Gillespie, G.Y.; Yu, H.; Zheng, Y.; Nozell, S.E.; Huszar, D.; Benveniste, E.N. Therapeutic Potential of AZD1480 for the Treatment of Human Glioblastoma. Mol. Cancer Ther. 2011, 2384–2393. [Google Scholar] [CrossRef]
- Jensen, K.V.; Cseh, O.; Aman, A.; Weiss, S.; Luchman, H.A. The JAK2/STAT3 inhibitor pacritinib effectively inhibits patient-derived GBM brain tumor initiating cells in vitro and when used in combination with temozolomide increases survival in an orthotopic xenograft model. PLoS ONE 2017, 18, e0189670. [Google Scholar] [CrossRef]
- Delen, E.; Doganlar, O.; Doganlar, Z.B.; Delen, O. Inhibition of the Invasion of Human Glioblastoma U87 Cell Line by Ruxolitinib: A Molecular Player of miR-17 and miR-20a Regulating JAK/STAT Pathway. Turk. Neurosurg. 2020, 30, 182–189. [Google Scholar] [CrossRef]
- Haile, W.B.; Gavegnano, C.; Tao, S.; Jiang, Y.; Schinazi, R.F.; Tyor, W.R. The Janus kinase inhibitor ruxolitinib reduces HIV replication in human macrophages and ameliorates HIV encephalitis in a murine model. Neurobiol. Dis. 2016, 92 Pt B, 137–143. [Google Scholar] [CrossRef]
- Kurokawa, C.; Iankov, I.D.; Anderson, S.K.; Aderca, I.; Leontovich, A.A.; Maurer, M.J.; Oberg, A.L.; Schroeder, M.A.; Giannini, C.; Greiner, S.M.; et al. Constitutive Interferon Pathway Activation in Tumors as an Efficacy Determinant Following Oncolytic Virotherapy. J. Natl. Cancer Inst. 2018, 110, 1123–1132. [Google Scholar] [CrossRef]
- Turkson, J.; Ryan, D.; Kim, J.S.; Zhang, Y.; Chen, Z.; Haura, E.; Laudano, A.; Sebti, S.; Hamilton, A.D.; Jove, R. Phosphotyrosyl peptides block Stat3-mediated DNA binding activity, gene regulation, and cell transformation. J. Biol. Chem. 2001, 276, 45443–45455. [Google Scholar] [CrossRef] [PubMed]
- Fuh, B.; Sobo, M.; Cen, L. LLL-3 inhibits STAT3 activity, suppresses glioblastoma cell growth and prolongs survival in a mouse glioblastoma model. Br. J. Cancer 2009, 100, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Ball, S.; Li, C.; Li, P.K.; Lin, J. The small molecule, LLL12, inhibits STAT3 phosphorylation and induces apoptosis in medulloblastoma and glioblastoma cells. PLoS ONE 2011, 19, e18820. [Google Scholar] [CrossRef] [PubMed]
- Ashizawa, T.; Akiyama, Y.; Miyata, H.; Iizuka, A.; Komiyama, M.; Kume, A.; Omiya, M.; Sugino, T.; Asai, A.; Hayashi, N.; et al. Effect of the STAT3 inhibitor STX-0119 on the proliferation of a temozolomide-resistant glioblastoma cell line. Int. J. Oncol. 2014, 45, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, Y.; Nonomura, C.; Ashizawa, T.; Iizuka, A.; Kondou, R.; Miyata, H.; Sugino, T.; Mitsuya, K.; Hayashi, N.; Nakasu, Y.; et al. The anti-tumor activity of the STAT3 inhibitor STX-0119 occurs via promotion of tumor-infiltrating lymphocyte accumulation in temozolomide-resistant glioblastoma cell line. Immunol. Lett. 2017, 190, 20–25. [Google Scholar] [CrossRef]
- Kortylewski, M.; Swiderski, P.; Herrmann, A.; Wang, L.; Kowolik, C.; Kujawski, M.; Lee, H.; Scuto, A.; Liu, Y.; Yang, C.; et al. In vivo delivery of siRNA to immune cells by conjugation to a TLR9 agonist enhances antitumor immune responses. Nat. Biotechnol. 2009, 27, 925–932. [Google Scholar] [CrossRef]
- Wei, J.; Wang, F.; Kong, L.Y.; Xu, S.; Doucette, T.; Ferguson, S.D.; Yang, Y.; McEnery, K.; Jethwa, K.; Gjyshi, O.; et al. miR-124 inhibits STAT3 signaling to enhance T cell-mediated immune clearance of glioma. Cancer Res. 2013, 1, 3913–3926. [Google Scholar] [CrossRef]
- Yaghi, N.K.; Wei, J.; Hashimoto, Y.; Kong, L.Y.; Gabrusiewicz, K.; Nduom, E.K.; Ling, X.; Huang, N.; Zhou, S.; Kerrigan, B.C.; et al. Immune modulatory nanoparticle therapeutics for intracerebral glioma. Neuro-Oncology 2017, 19, 372–382. [Google Scholar] [CrossRef]
- Linder, B.; Weirauch, U.; Ewe, A.; Uhmann, A.; Seifert, V.; Mittelbronn, M.; Harter, P.N.; Aigner, A.; Kögel, D. Therapeutic Targeting of Stat3 Using Lipopolyplex Nanoparticle-Formulated siRNA in a Syngeneic Orthotopic Mouse Glioma Model. Cancers 2019, 11, 333. [Google Scholar] [CrossRef]
- Hendruschk, S.; Wiedemuth, R.; Aigner, A.; Töpfer, K.; Cartellieri, M.; Martin, D.; Kirsch, M.; Ikonomidou, C.; Schackert, G.; Temme, A. RNA interference targeting survivin exerts antitumoral effects in vitro and in established glioma xenografts in vivo. Neuro-Oncology 2011, 13, 1074–1089. [Google Scholar] [CrossRef]
- Masliantsev, K.; Pinel, B.; Balbous, A.; Guichet, P.O.; Tachon, G.; Milin, S.; Godet, J.; Duchesne, M.; Berger, A.; Petropoulos, C.; et al. Impact of STAT3 phosphorylation in glioblastoma stem cells radiosensitization and patient outcome. Oncotarget 2017, 9, 3968–3979. [Google Scholar] [CrossRef] [PubMed]
- Leidgens, V.; Proske, J.; Rauer, L.; Moeckel, S.; Renner, K.; Bogdahn, U.; Riemenschneider, M.J.; Proescholdt, M.; Vollmann-Zwerenz, A.; Hau, P.; et al. Stattic and metformin inhibit brain tumor initiating cells by reducing STAT3-phosphorylation. Oncotarget 2017, 8, 8250–8263. [Google Scholar] [CrossRef] [PubMed]
- Debnath, B.; Xu, S.; Neamati, N. Small molecule inhibitors of signal transducer and activator of transcription 3 (Stat3) protein. J. Med. Chem. 2012, 55, 6645–6668. [Google Scholar] [CrossRef] [PubMed]
- Sanseverino, I.; Purificato, C.; Gauzzi, M.C.; Gessani, S. Revisiting the Specificity of Small Molecule Inhibitors: The Example of Stattic in Dendritic Cells. Chem. Biol. 2012, 19, 1213–1214. [Google Scholar] [CrossRef] [PubMed]
- Senft, C.; Priester, M.; Polacin, M.; Schröder, K.; Seifert, V.; Kögel, D.; Weissenberger, J. Inhibition of the JAK-2/STAT3 signaling pathway impedes the migratory and invasive potential of human glioblastoma cells. J. Neurooncol. 2011, 101, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Haftchenary, S.; Luchman, H.A.; Jouk, A.O.; Veloso, A.J.; Page, B.D.; Cheng, X.R.; Dawson, S.S.; Grinshtein, N.; Shahani, V.M.; Kerman, K.; et al. Potent Targeting of the STAT3 Protein in Brain Cancer Stem Cells: A Promising Route for Treating Glioblastoma. ACS Med. Chem. Lett. 2013, 4, 1102–1107. [Google Scholar] [CrossRef]
- Yue, P.; Lopez-Tapia, F.; Paladino, D.; Li, Y.; Chen, C.H.; Namanja, A.T.; Hilliard, T.; Chen, Y.; Tius, M.A.; Turkson, J. Hydroxamic acid and benzoic acid-based Stat3 inhibitors suppress human glioma and breast cancer phenotypes in vitro and in vivo. Cancer Res. 2016, 76, 652–663. [Google Scholar] [CrossRef]
- Cui, P.; Wei, F.; Hou, J.; Su, Y.; Wang, J.; Wang, S. STAT3 inhibition induced temozolomide-resistant glioblastoma apoptosis via triggering mitochondrial STAT3 translocation and respiratory chain dysfunction. Cell. Signal. 2020, 71, 109598. [Google Scholar] [CrossRef]
- Sai, K.; Wang, S.; Balasubramaniyan, V.; Conrad, C.; Lang, F.F.; Aldape, K.; Szymanski, S.; Fokt, I.; Dasgupta, A.; Madden, T.; et al. Induction of cell-cycle arrest and apoptosis in glioblastoma stem-like cells by WP1193, a novel small molecule inhibitor of the JAK2/STAT3 pathway. J. Neurooncol. 2012, 107, 487–501. [Google Scholar] [CrossRef]
- Ferrajoli, A.; Faderl, S.; Van, Q.; Koch, P.; Harris, D.; Liu, Z.; Hazan-Halevy, I.; Wang, Y.; Kantarjian, H.M.; Priebe, W.; et al. WP1066 disrupts Janus kinase-2 and induces caspase-dependent apoptosis in acute myelogenous leukemia cells. Cancer Res. 2007, 1, 11291–11299. [Google Scholar] [CrossRef]
- Han, D.; Yu, T.; Dong, N.; Wang, B.; Sun, F.; Jiang, D. Napabucasin, a novel STAT3 inhibitor suppresses proliferation, invasion and stemness of glioblastoma cells. J. Exp. Clin. Cancer Res. 2019, 5, 289. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Rogoff, H.A.; Keates, S.; Gao, Y.; Murikipudi, S.; Mikule, K.; Leggett, D.; Li, W.; Pardee, A.B.; Li, C.J. Suppression of cancer relapse and metastasis by inhibiting cancer stemness. Proc. Natl. Acad. Sci. USA 2015, 112, 1839–1844. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.; Zhong, Z.; Darnell, J.E., Jr. Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell 1995. [Google Scholar] [CrossRef]
- Tan, M.S.Y.; Sandanaraj, E.; Chong, Y.K. A STAT3-based gene signature stratifies glioma patients for targeted therapy. Nat. Commun. 2019, 10, 3601. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Natural Compound | Mechanism | Preclinical Evidence of Efficacy in Glioma | Limitations | References |
|---|---|---|---|---|
| Silibinin | STAT3 inhibition, autophagy, chemosensitization | In vitro | Poor oral bioavailability, low potency | [176,177,178,179,180] |
| Cryptotanshinone | STAT3 inhibition | In vitro | Poor bioavailability | [181,182] |
| Alantolactone | STAT3, NF-κB inhibition | In vitro | Poor bioavailability, rapid metabolism, low potency | [183,184] |
| Shikonin | STAT3, EGFR inhibition | In vitro | Poor bioavailability, low potency | [185,186] |
| Sulforaphane | JAK2, STAT3, NF-κB inhibition | In vitro | Poor bioavailability, moderate potency | [187,188,189] |
| Crocetin | STAT3 inhibition (SHP-1 induction) | In vitro | Poor bioavailability, limited BBB penetrance, low potency | [190,191,192] |
| Cardamonin | STAT3 inhibition | In vitro | Poor bioavailability | [193,194] |
| Serenoa repens (Saw palmetto) | STAT3 inhibition | In vitro | Poor bioavailability | [195] |
| Oroxylin A | mTOR, STAT3 inhibition | In vitro | Poor bioavailability, rapid metabolism, low potency | [196,197,198] |
| Quercetin | IL-6, STAT3 inhibition | In vitro | Poor bioavailability, rapid metabolism, low potency | [199] |
| Oleanolic acid | STAT3 inhibition, IL-10 inhibition | In vitro | Poor bioavailability, rapid metabolism, low potency | [200,201,202] |
| Cucurmin | JAK1, JAK2, STAT3 inhibition | In vitro | Poor bioavailability, low potency | [203,204,205] |
| Ascochlorin | FAK, STAT3 inhibition | In vitro | Poor bioavailability, low potency | [206,207] |
| Cucurbitacin | JAK, STAT3, PI3K, MAPK inhibition | In vitro | Poor bioavailability, specificity, high toxicity | [208,209] |
| Resveratrol | STAT3 inhibition | In vitro In vivo | Poor bioavailability, low potency | [171,172,173,210] |
| Agent | Mechanism | Evidence of Efficacy in Glioma | Notes | References |
|---|---|---|---|---|
| G6 | JAK2 | In vitro | In vivo studies lacking, therapeutic requirement for JAK2 overexpression | [211] |
| SAR317461 | JAK2 | In vitro | In vivo studies lacking, compensatory autophagy | [212] |
| AZD1480 | JAK1/JAK2 | In vitro In vivo | Unacceptable dose-limiting toxicities | [150,216,217] |
| JSI-124 | JAK2 | In vitro In vivo | Anti-proliferative, immune modulatory | [213,214,215] |
| Pacritinib | JAK2 | In vitro In vivo | BBB penetrant, chemosensitizing | [94,218] |
| * Ruxolitinib | JAK1/JAK2 | In vitro In vivo | BBB penetrant, anti-proliferative, radiosensitizing, immune modulatory | [219,220] |
| PY * LKTK | STAT3 | In vitro | In vivo studies lacking, low potency | [222] |
| LLL12 | STAT3 | In vitro In vivo | Potent, low solubility/poor bioavailability, unclear BBB penetrance | [223,224] |
| STX-0119 | STAT3 | In vitro In vivo | Minimal growth inhibition of GBM in mouse model | [225,226] |
| AG490 | STAT3, JAK2 | In vitro | Low potency, in vivo efficacy lacking | [73,236] |
| Stattic | STAT3, STAT1, STAT2 | In vitro | Susceptible to intracellular modification, in vivo efficacy lacking, low specificity | [232,233,234,235] |
| WP1193 | STAT3, JAK2 | In vitro | In vivo efficacy data lacking | [240] |
| SH-4-54 | STAT3 STAT5 | In vitro In vivo | BBB penetrant, potent, specific, in vivo studies in subcutaneously implanted GBMs | [237,238,239] |
| * Napabucasin (BBI608) | STAT3 | In vitro In vivo | Bioavailable, BBB penetrant | [242,243] |
| * WP1066 | STAT3, JAK2 | In vitro In vivo | Bioavailable, BBB penetrant, immune modulatory | [73,122,241] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ou, A.; Ott, M.; Fang, D.; Heimberger, A.B. The Role and Therapeutic Targeting of JAK/STAT Signaling in Glioblastoma. Cancers 2021, 13, 437. https://doi.org/10.3390/cancers13030437
Ou A, Ott M, Fang D, Heimberger AB. The Role and Therapeutic Targeting of JAK/STAT Signaling in Glioblastoma. Cancers. 2021; 13(3):437. https://doi.org/10.3390/cancers13030437
Chicago/Turabian StyleOu, Alexander, Martina Ott, Dexing Fang, and Amy B. Heimberger. 2021. "The Role and Therapeutic Targeting of JAK/STAT Signaling in Glioblastoma" Cancers 13, no. 3: 437. https://doi.org/10.3390/cancers13030437
APA StyleOu, A., Ott, M., Fang, D., & Heimberger, A. B. (2021). The Role and Therapeutic Targeting of JAK/STAT Signaling in Glioblastoma. Cancers, 13(3), 437. https://doi.org/10.3390/cancers13030437